

Abstract
A complete loss of merosin, which is encoded by LAMA2, causes congenital muscular dystrophy with leukoencephalopathy. Partial merosin deficiency can be caused not only by primarily LAMA2 mutations, but also secondarily by dystroglycanopathy. Although it can be molecularly diagnosed based on a genetic analysis, this method is labor-intensive because of its huge genome size. A 26-year-old male patient presented with mild muscular weakness, joint contractures, and epilepsy. Double immunofluorescence staining of a muscle biopsy specimen showed mislocalization of merosin, and a genetic analysis revealed a homozygous c.818G>A (p.Arg273Lys) mutation in LAMA2. Double immunofluorescence staining and whole exome sequencing were useful for the diagnosis of partial merosin deficiency.
Keywords: merosin, LAMA2, partial merosin deficiency, leukoencephalopathy, lissencephaly
Introduction
Merosin, also known as laminin subunit α2, is encoded by LAMA2. In skeletal muscle, merosin together with the subunits β1 and γ1 constitute laminin-211, which plays an important role in anchoring the cytoskeleton to the basal membrane (1). Laminin-211 is also expressed in the blood vessels of the brain, Schwann cells, the epidermis, and the kidney (2,3), and it is considered that - rather than merely acting as a mechanical support of the sarcoplasmic membrane - it also functions in the internal signaling pathways (4).
A complete loss of merosin due to LAMA2 mutations causes merosin-deficient congenital muscular dystrophy (MDC1A) (5), which is one of the most common forms of congenital muscular dystrophy in Western countries, but which is very rare in Japan (6). A partial loss of merosin can be seen in both patients with LAMA2 mutations and in those with other muscular dystrophies (7,8). Distinctive leukoencephalopathy could be a clue to the diagnosis of partial merosin deficiency; however, finding causative mutations in LAMA2 remains a challenge due to the gene's huge size and numerous exons (9).
We herein report the case of a Japanese patient with partial merosin deficiency caused by a homozygous mutation of c.818G>A, p.(Arg273Lys), in LAMA2.
Case Report
The patient's parents were consanguineous; his father was his mother's uncle (Fig. 1). His mother had two spontaneous abortions prior to the patient's birth. Both spontaneous abortions occurred at 8 to 12 weeks, and neither the mother nor doctor noticed any abnormality before the abortion. The patient was born uneventfully at 39 weeks, but his mother felt that his body was “stiff” at birth. He could walk at 1 year of age, and his motor milestones were normal until 1 year of age, at which time he tended to fall and started to walk on his toes.
Figure 1.
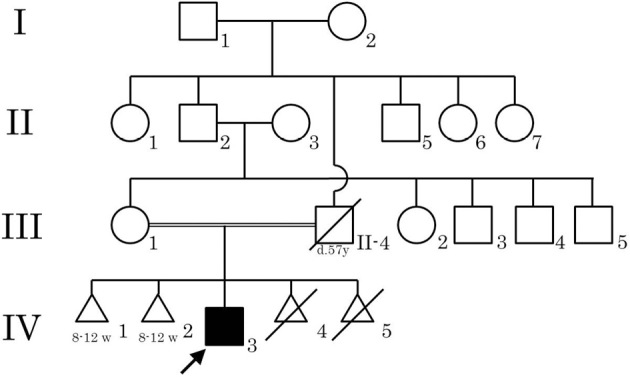
The family tree of the patient. None of the other family members had muscle disease or epilepsy. The patient’s father died of leukemia at 57 years of age. The patient’s mother had two spontaneous abortions (IV-1 and IV-2) and two induced abortions (IV-4 and IV-5).
His mother brought him to a hospital, and neurological examination revealed that he had underdeveloped musculature, muscle weakness, and multiple joint contractures in his ankles, knees, and fingers. His serum creatine kinase (CK) level was elevated to approximately 600 U/L, and needle electromyography (nEMG) showed myopathic changes. No abnormalities were detected in a nerve conduction study (NCS), electrocardiography (ECG), or electroencephalography (EEG). He underwent muscle biopsy of the left biceps brachii muscle, and a histopathological examination revealed a myopathic change with scattered necrotic and regenerating fibers. He was diagnosed with progressive muscular dystrophy; however, a detailed diagnosis was not made.
The patient's school performance was average to below average until he graduated from an ordinary high school. He was a very slow runner, but was able to play baseball in junior high school. He remained ambulant with rehabilitation, walking braces, and three Achilles tenotomies to correct severe club feet. At 18 years of age, he developed epilepsy, and treatment with antiepileptic drugs was started. The seizures could not be controlled, despite the administration of multiple antiepileptic drugs.
The patient visited our hospital when he was 26 years of age. A neurological examination revealed diffuse muscle atrophy and mild muscle weakness [grade 4 on the medical research council (MRC)] scale. His masseter muscles and sternocleidomastoid muscles showed mild weakness, but his extraocular muscles, facial muscles, and tongue were not affected, and he did not have dysarthria or dysphagia. He also had multiple joint contractures and deformities in the ankles, spine, elbows, and fingers. Despite muscle weakness and joint deformities, he was able to walk unassisted, and his gait seemed normal except for instability due to ankle contracture. He had no limb ataxia in a finger-to-nose test or in a heel-to-shin test. His tendon reflexes were attenuated, but he did not complain of sensory loss. The patient's plantar reflexes were bilaterally flexor, and no signs of pyramidal dysfunction were observed. He scored 29/30 on a Mini-Mental State Examination, The Wechsler Adult Intelligence Scale-Revised (WAIS-R) scale revealed that he had a low intelligence quotient (IQ 58), but his memory function, as determined using the WMS-R scale, was normal.
Laboratory tests revealed normal serum CK levels. Although his arterial carbon dioxide partial pressure was slightly elevated (46 mmHg), his oxygen partial pressure and arterial blood pH were both normal. A pulmonary function test showed a decreased vital capacity (2.51 L, 60%) with a normal forced expiratory volume (FEV) of 1.0% (96.4, 112.8%), suggesting moderate restrictive lung disease. The patient's cardiac function was normal. Brain magnetic resonance imaging (MRI) revealed leukoencephalopathy sparing the corpus callosum, brainstem, and internal capsule, and lissencephaly in the temporal and occipital lobes (Fig. 2). Epileptic discharges were detected in the temporal and occipital lobes by EEG; the sites were coincident with the sites of lissencephaly. NCS showed normal conduction velocities, but decreased sensory nerve action potentials (2-4 μV).
Figure 2.
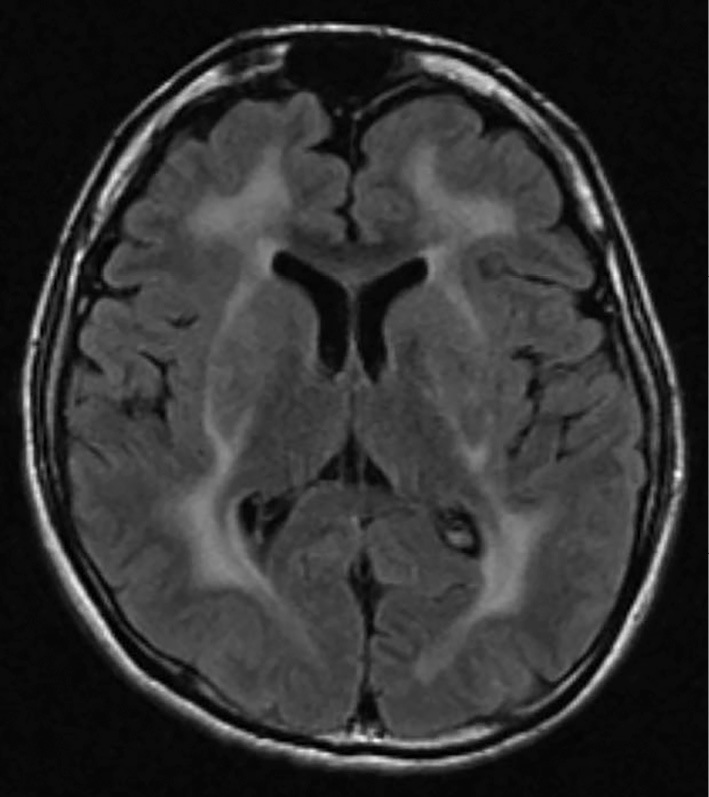
Brain MRI at 26 years of age. Fluid-attenuated inversion recovery (FLAIR) images clearly revealed leukoencephalopathy, sparing the corpus callosum and internal capsule. The brain sulci in the temporal and occipital lobes were scarce and shallow, showing lissencephaly.
Frozen biopsied muscle tissue samples were obtained from his previous medical institution, and immunohistochemistry was performed by a conventional method using 3,3-diaminobenzidine (DAB). Normal immunostaining patterns were observed on immunostaining with antibodies to dystrophin, dysferlin, dystroglycan, sarcoglycans (α-, β-, γ-, and δ-), and emerin. Three different anti-merosin antibodies (NCL-merosin, Novocastra, 1:100; MAB1922, Millipore, 1:100; 4H8-2, Sigma-Aldrich, 1:100) were tested, and the pattern of merosin staining showed a very subtle patchy change. Double immunofluorescence staining was performed using anti-merosin (NCL-Merosin, Novocastra, 1:100) and anti-dystrophin (ab15227, Abcam, 1:50) antibodies. The immunostained samples were examined under a confocal microscope (LSM510, Zeiss), which proved the partial merosin deficiency on the sarcomeric membrane (Fig. 3). Western blotting was also carried out by a conventional method. Briefly, after homogenization and denaturation in lysis buffer with 2-mercaptoethanol, protein samples (50 μg each) extracted from a control subject and the present case were separated through a 5-20% polyacrylamide gel sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with skim milk, followed by incubation with an anti-merosin antibody that recognizes an 80 kDa fragment at the C terminus (MAB1922, Millipore, 1:100) or an anti-β actin antibody (MAB1501, Millipore, 1:1,000) at 4°C overnight. After incubation with a secondary antibody (anti-mouse IgG antibody conjugated with horseradish peroxidase), the membrane was developed using ECL reagents (GE Healthcare, Little Chalfont, UK), and observed under a CCD imager (LAS3000, Fujifilm, Tokyo, Japan). The amount of merosin fragment normalized to that of β-actin was comparable in the control subject and the patient (Fig. 4).
Figure 3.

Immunohistochemistry of muscle biopsy specimens. Frozen muscle samples of the control subject (A, C-E) and the patient (B, F-H) were sliced (thickness: 10 μm) and subjected to immunostaining with the conventional DAB method using anti-merosin antibodies (A, B). Double immunofluorescence staining was performed using anti-merosin antibodies (C, F) and anti-dystrophin antibodies (D, G), and composite images of double immunofluorescence staining were obtained (E, H). No staining pattern abnormalities were detected on conventional DAB staining (B), but partial merosin deficiency was apparent on double immunofluorescence staining (H). Bars indicate 20 μm.
Figure 4.
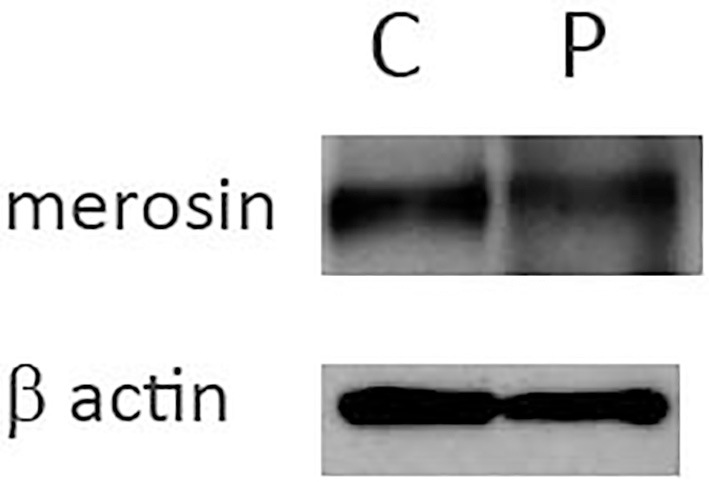
Western blotting. Fifty micrograms each of the biopsied muscle tissue samples of the control subject (indicated as C) and the patient (indicated as P) was subjected to Western blotting. The amount of merosin normalized to that of β-actin was comparable between the patient and the control subject.
A genetic analysis of the patient and his mother was performed after acquiring written informed consent. After obtaining the single nucleotide polymorphisms (SNP) genotypes of the proband and his mother using Genome-Wide Human SNP array 6.0 (Affymetrix, Santa Clara, USA), a parametric linkage analysis was performed using SNP-HiTLink (10) and Allegro version 2 (11). A region on chromosome 6 flanked by rs567564 and rs9386195, which includes the LAMA2 locus, showed a positive LOD score with runs of homozygous SNPs (Supplementary material 1a). A copy number analysis using Genome-Wide Human SNP array 6.0 revealed no copy number alteration in the locus (Supplementary material 1b), confirming the homozygosity of the locus and excluding the possibility of apparent homozygosity due to a large deletion of one allele. A whole exome analysis was performed using a SureSelect V6+UTRs kit (Agilent) and a HiSeq2500 sequencer (Illumina). Variant calling was performed as previously described (12). Whole exome sequencing revealed a homozygous mutation of hg19 chr6:129465224G>A [c.818G>A, p.(Arg273Lys)] in LAMA2, and no other pathogenic mutations were found in POMT1, POMT2, FKTN, FKRP, LARGE1, POMGNT1, or ISPD. Sanger sequencing of the polymerase chain reaction (PCR) products was performed by a conventional method using a primer set: forward 5'-CGCTCGCTATATTCGCCTGA-3'; reverse 5'-TGTTTCAAAGCCCCTGCACT-3'. A direct nucleotide sequence analysis confirmed that the patient carried the mutation in a homozygous state, and that his mother was heterozygous for the mutation (Fig. 5A-C). The mutation was not found in the in-house database of 803 controls or in the ExAC database (http://exac.broadinstitute.org/). The mutation was located in laminin-N-terminal domain VI, which plays an important role in laminin polymerization (13) and integrin α1β1 binding (14). An in silico analysis predicted that the mutation would be deleterious [combined annotation dependent depletion (CADD) score (15): 31.00], and the arginine at the mutation site is well preserved among vertebral species (Fig. 5D).
Figure 5.

An electropherogram acquired by Sanger sequencing. Sanger sequencing was performed for the control subject (A), the patient’s mother (B), and the patient (C). His mother was heterozygous (B), and the patient was homozygous (C) for the LAMA2 mutation of c.818G>A.
Discussion
Merosin-deficient congenital muscular dystrophy (MDC1A) is characterized by profound muscle weakness soon after birth and distinctive central nervous system manifestations. In classical MDC1A, a diagnosis is relatively easy to make, based on the presence of leukoencephalopathy and the complete loss of merosin on immunohistochemical staining. However, residual merosin staining, which is termed partial merosin deficiency is observed in some patients with LAMA2 mutations. Such patients often manifest milder muscle phenotypes with a later onset in comparison to patients with classical MDC1A (16), and show variable residual merosin staining, which ranges from faint staining to subtly attenuated staining. In previous studies, immunohistochemistry using two anti-merosin antibodies to recognize different epitopes located in the N and C termini was reported to be useful for detecting partial merosin deficiency (17). However, the partial loss of merosin staining is not only seen in MDC1A, but also in other muscular dystrophies, including dystroglycanopathy (7,18). A genetic analysis is necessary to establish a diagnosis of partial merosin deficiency; however, this is laborious due to the huge size of LAMA2, which contains 65 exons, and the size of merosin complimentary DNA exceeds 9,000 base pairs.
In this patient, a homozygous mutation of c.818G>A (p.Arg273Lys) was detected by whole exome sequencing. The same mutation was previously reported in the Leiden Open Variation Database (http://www.dmd.nl). Patient #0019153 with the MDC1A phenotype had the same mutation (p.R273K) and a nonsense mutation (p.R1326*) in a compound heterozygous state. This strongly supported the pathogenicity of the c.818G>A (p.R273K) mutation. Furthermore, it is hypothesized that the c.818G>A mutation itself causes partial merosin deficiency in a homozygous state, whereas it causes the MDC1A phenotype when it is present in a heterozygous state combined with a nonsense mutation.
In our patient, we tried to prove merosin deficiency by conventional DAB immunohistochemistry using three anti-merosin antibodies; however, the staining abnormality was too subtle to conclude that the specimen showed partial merosin deficiency. Furthermore, Western blotting, failed to reveal any change in the amount of merosin. However, double immunofluorescence staining using anti-merosin and anti-dystrophin antibodies clearly showed the mislocalization of merosin. The results of immunohistochemical staining and Western blotting suggest that the mutation impairs the normal protein function without altering the amount of protein. The mutation was located in laminin-N-terminal domain VI, which has multiple functions including laminin polymerization (13). It is hypothesized that the mislocalization of merosin seen in the present case could be due to disturbed laminin polymerization.
In comparison to classical MDC1A, the clinical features of this patient were atypical in terms of the muscle phenotype. In contrast, the patient had full-blown central nervous system manifestations. Patients with classical MDC1A show weakness soon after birth, and almost never gain the ability to walk. However, several reports have revealed that merosin deficiency can cause more divergent muscle phenotypes, especially in cases of partial deficiency. As was also seen in our patient, patients with partial merosin deficiency often develop muscle weakness later in life, resembling limb-girdle muscular dystrophy (18-20), and multiple joint contractures can be more prominent than muscle weakness (21). Although some patients show normal or only subtle changes on MRI, leukoencephalopathy seems to show less variation than muscle weakness (19). In most cases, leukoencephalopathy itself remains subclinical. Gyral malformation, epilepsy, and mental retardation are sometimes associated with merosin deficiency. It is not clear whether our patient had mental retardation due to merosin deficiency or regression as a sequel to uncontrolled epilepsy. Interestingly, there is no relationship between muscle weakness and the severity of central nervous system involvement (19,22), which was also seen in this patient.
MDC1A is a common type of congenital muscular dystrophy in Western countries, but is very rare in Japan (6,23,24). Thus far, all reported cases have involved congenital muscular dystrophies. Our case report suggests that there might be more undiagnosed Japanese patients with merosin deficiency, especially among those with late-onset muscular dystrophy. Although leukoencephalopathy is common and diagnostic in cases of merosin deficiency, it can be subclinical, and MRI should be performed, even if the patient shows no symptoms or signs of central nervous involvement.
We reported the case of a Japanese patient with partial merosin deficiency due to a homozygous LAMA2 mutation of c.818G>A. We clinically and pathologically characterized the effect of the p.R273K mutation. Double immunofluorescence staining and whole exome sequencing were helpful in establishing the diagnosis of partial merosin deficiency.
The authors state that they have no Conflict of Interest (COI).
Supplementary Material
Parametric linkage study of chromosome 6 and copy number analysis of LAMA2 gene.
References
- 1.Wewer UM, Engvall E. Merosin/laminin-2 and muscular dystrophy. Neuromuscul Disord 6: 409-418, 1996. [DOI] [PubMed] [Google Scholar]
- 2.Leivo I, Engvall E. Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci U S A 85: 1544-1548, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villanova M, Malandrini A, Toti P, et al. . Localization of merosin in the normal human brain: implications for congenital muscular dystrophy with merosin deficiency. J Submicrosc Cytol Pathol 28: 1-4, 1996. [PubMed] [Google Scholar]
- 4.Holmberg J, Durbeej M. Laminin-211 in skeletal muscle function. Cell Adh Migr 7: 111-121, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allamand V, Guicheney P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of laminin). Eur J Hum Genet 10: 91-94, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi YK, Koga R, Tsukahara T, et al. . Deficiency of laminin alpha 2-chain mRNA in muscle in a patient with merosin-negative congenital muscular dystrophy. Muscle Nerve 18: 1027-1030, 1995. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi YK, Engvall E, Arikawa-Hirasawa E, et al. . Abnormal localization of laminin subunits in muscular dystrophies. J Neurol Sci 119: 53-64, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Di Blasi C, Piga D, Brioschi P, et al. . LAMA2 gene analysis in congenital muscular dystrophy: new mutations, prenatal diagnosis, and founder effect. Arch Neurol 62: 1582-1586, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Tezak Z, Prandini P, Boscaro M, et al. . Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha 2 (LAMA2) deficiency. Hum Mutat 21: 103-111, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda Y, Nakahara Y, Date H, et al. . SNP HiTLink: a high-throughput linkage analysis system employing dense SNP data. BMC Bioinformatics 10: 121, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A. Allegro version 2. Nat Genet 37: 1015-1016, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Ichikawa Y, Ishiura H, Mitsui J, et al. . Exome analysis reveals a Japanese family with spinocerebellar ataxia, autosomal recessive 1. J Neurol Sci 331: 158-160, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Yurchenco PD, Cheng YS. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J Biol Chem 268: 17286-17299, 1993. [PubMed] [Google Scholar]
- 14.Colognato-Pyke H, O'Rear JJ, Yamada Y, Carbonetto S, Cheng YS, Yurchenco PD. Mapping of network-forming, heparin-binding, and alpha 1 beta 1 integrin-recognition sites within the alpha-chain short arm of laminin-1. J Biol Chem 270: 9398-9406, 1995. [DOI] [PubMed] [Google Scholar]
- 15.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310-315, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geranmayeh F, Clement E, Feng LH, et al. . Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 20: 241-250, 2010. [DOI] [PubMed] [Google Scholar]
- 17.Sewry CA, Naom I, D'Alessandro M, et al. . Variable clinical phenotype in merosin-deficient congenital muscular dystrophy associated with differential immunolabelling of two fragments of the laminin alpha 2 chain. Neuromuscul Disord 7: 169-175, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Chan SH, Foley AR, Phadke R, et al. . Limb girdle muscular dystrophy due to LAMA2 mutations: diagnostic difficulties due to associated peripheral neuropathy. Neuromuscul Disord 24: 677-683, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Jones KJ, Morgan G, Johnston H, et al. . The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet 38: 649-657, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gavassini BF, Carboni N, Nielsen JE, et al. . Clinical and molecular characterization of limb-girdle muscular dystrophy due to LAMA2 mutations. Muscle Nerve 44: 703-709, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Nelson I, Stojkovic T, Allamand V, et al. . Laminin alpha2 deficiency-related muscular dystrophy mimicking emery-dreifuss and collagen VI related diseases. J Neuromuscul Dis 2: 229-240, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marques J, Duarte ST, Costa S, et al. . Atypical phenotype in two patients with LAMA2 mutations. Neuromuscul Disord 24: 419-424, 2014. [DOI] [PubMed] [Google Scholar]
- 23.Tachi N, Kamimura S, Ohya K, Chiba S, Sasaki K. Congenital muscular dystrophy with partial deficiency of merosin. J Neurol Sci 151: 25-27, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Fujii Y, Sugiura C, Fukuda C, Maegaki Y, Ohno K. Sequential neuroradiological and neurophysiological studies in a Japanese girl with merosin-deficient congenital muscular dystrophy. Brain Dev 33: 140-144, 2011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Parametric linkage study of chromosome 6 and copy number analysis of LAMA2 gene.