

Abstract
Synthetic transformations that functionalize unactivated, aliphatic C–H bonds in an intermolecular fashion offer unique strategies for the synthesis and late-stage derivatizations of complex molecules. Herein, we report a general approach to the intermolecular functionalization of aliphatic C–H bonds using an acridinium photoredox catalyst and phosphate salt under blue LED irradiation. This strategy encompasses a range of valuable C–H transformations, including the direct conversions of a C–H bond to C–N, C–F, C–Br, C–Cl, C–S, and C–C bonds, in all cases using the alkane substrate as the limiting reagent. Detailed mechanistic studies are consistent with the intermediacy of a putative oxygen-centered radical as the hydrogen atom abstracting species in these processes.
Graphical abstract
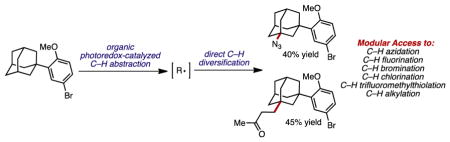
The strategic functionalization of the ubiquitous, unactivated aliphatic carbon–hydrogen (C–H) bond unlocks unique strategies for the synthesis of natural products, pharmaceuticals, agrochemicals, and materials.1 For example, in the context of drug discovery, late-stage C–H functionalization allows for expedient access to structural analogs of targets, which can result in bolstered physicochemical properties and structure-activity relationships (SARs) without the need for de novo synthesis.2 Both the abundance and the low reactivity of these C–H bonds also present major challenges in the development of site-selective, intermolecular functionalization reactions.
Recent efforts have led to a variety of useful intermolecular C–H transformations of unactivated alkanes, notably including many that use the hydrocarbon substrate as limiting reagent. Several methods for C–H oxidation,3 halogenation,4 and azidation5 have been reported, but new reaction systems are typically required to promote each transformation (Figure 1). Additionally, a photoredox manifold has been used by MacMillan to achieve C–H alkylation6 and arylation7 of C–H bonds at positions activated via hyperconjugation, such as those adjacent to π systems or heteroatoms.8,9 To date, there are few reports detailing the use of photoredox catalysis to functionalize unactivated, aliphatic C–H bonds.10
Figure 1.
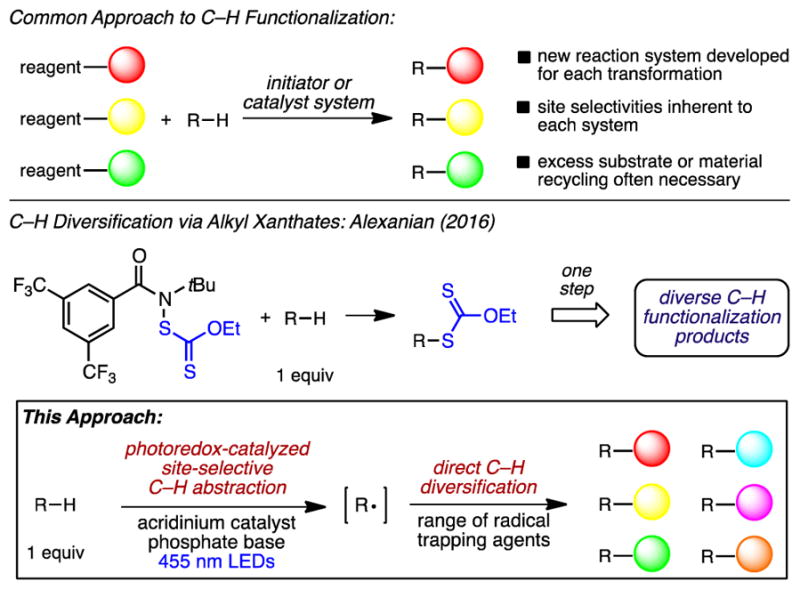
Approaches to site-selective, intermolecular C–H functionalization.
Previous efforts in the Alexanian group led to the development of N-haloamide and N-xanthylamide reagents enabling site-selective C–H halogenation11 and xanthylation,12 respectively, of unactivated aliphatic C–H bonds with substrate as limiting reagent. The alkyl xanthate products arising from the latter reaction can be transformed into a variety of derivatives, enabling net C–S, C–C, C–N, C–D, and C–O bond constructions from unactivated C–H sites (Figure 1). While this approach constitutes a two-step diversification of unactivated C–H bonds, we sought a strategy that would facilitate the direct, one-step conversion of aliphatic C–H bonds into a variety of functional groups. Such a modular system for radical-mediated aliphatic C–H functionalization would decouple the C–H abstraction step from the radical trapping step, allowing different products to be accessed simply by substitution of the added radical trapping agent.
We hypothesized that highly oxidizing acridinium photoredox catalyst 1 (E1/2(cat*/cat•) = +2.08 V vs SCE)13 could be used to oxidatively generate heteroatom-centered radicals capable of abstracting unactivated aliphatic C–H bonds. Coupled with the use of a sulfonyl group transfer reagent, we conjectured that the C–H functionalization could be rendered catalytic. To this end, we screened a variety of anionic, inorganic bases using acridinium photooxidant 1, sulfonyl azide 3, and cyclooctane as a substrate.14 We found that the use of either K3PO4 in 1,2-dichloroethane (DCE) or a DCE/pH 8 phosphate buffer system led to productive C–H azidation. Following further optimization, we discovered that the use of K3PO4 in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) was the most efficient system for this transformation.
With these conditions in hand, we explored the substrate scope of the C–H azidation, with the alkane substrate as limiting reagent in all cases (Figure 2). Cyclic hydrocarbons provided azides 4–6 in moderate to good yields (57–70%). Trans-decalin afforded secondary azides 7 in 57% combined yield, with regioselectivity for the secondary C–H sites. Adamantane underwent azidation exclusively at the more sterically accessible tertiary C–H site to give azide 8 in 75% yield.
Figure 2.

Products of C–H azidations using 0.1 mmol substrate. Yields refer to isolated yields. aNMR yield with hexamethyldisiloxane (HMDS) as an internal standard.
Azidation of benzylic C–H bonds was also successful, as the functionalization of propylbenzene afforded azide 9 in moderate yield. Tert-butylcyclohexane and cis-4-methylcyclohexyl pivalate reacted exclusively at the tertiary C–H sites in 51% and 45% yield, respectively, despite the presence of additional secondary C–H bonds. The azidation of isobutylbenzene delivered tertiary azide 12 in moderate yield; this product serves as a precursor to the psychostimulant pharmaceutical phentermine.
We next sought to examine the site selectivity of the C–H azidation using acyclic substrates with multiple reactive sites. Methyl 6-methylheptanoate produced tertiary azide 13 in 71% yield as a single regioisomer. However, methyl hexanoate, a substrate containing several electronically deactivated secondary C–H bonds but no tertiary sites, did not undergo functionalization with this system, consistent with a strong sensitivity to the BDE of the C–H bonds in the substrate. In order to examine the electronic site selectivity of the C–H azidation further, we surveyed several derivatives of dihydrocitronellol, each containing two tertiary C–H sites. In all cases, azidation was favored at the site distal to the other functionality present, with no secondary C–H functionalization observed. Acetate and benzoate esters delivered products 14 and 15 in 73% and 91% yield respectively, with good levels of regioselectivity. Derivatives containing protected primary amines and halide substituents provided similar site selectivities, affording 16 and 17 in good yields. Dihydrocitronellol itself was also a competent substrate, producing 18 in 63% yield with no detectable oxidation of the free alcohol, highlighting the mild conditions associated with the system. Azidation of a substrate bearing a phenoxy group, which could be competitively oxidized by the acridinium catalyst, proceeded to give 19, albeit in lower yield and selectivity. We also studied the C–H azidation of a substrate containing a nitrogen heterocycle, which can be problematic for C–H functionalizations involving high-valent metal-oxo systems.15 The azidation of a pyridyl ketone substrate delivered 20 in moderate yield, further highlighting the functional group compatibility of our catalytic system.
Having demonstrated an efficient C–H azidation, we investigated the use of other reagents to develop a modular C–H functionalization. Using cyclooctane as substrate, we sought to access a diverse array of C–H functionalizations (Figure 3). In these studies, we used DCE as solvent with pH 8 phosphate buffer rather than K3PO4 to ensure complete solubility of all reagents. Several strategies for aliphatic C–H fluorination have been developed, often involving ultraviolet irradiation or transition metal catalysts.4b–f Using our catalytic photoredox approach, substituting N-fluorobenzenesulfonimide (NFSI) for the sulfonyl azide delivered fluorocyclooctane in moderate yield. We could also extend our strategy to C–H bromination or chlorination by using diethyl bromomalonate or N-chlorosuccinimide, respectively. Trifluoromethylthiol group installation has proven to be useful in medicinal chemistry owing to the beneficial effects this moiety has on molecular polarity and lipophilicity.16 The use of a radical trifluoromethylthiolating reagent developed by Shen17 enabled C–H trifluoromethylthiolation of cyclooctane in low yield, possibly due to product instability under the reaction conditions. We note that in each system, both the phosphate base and photocatalyst were necessary for productive reactivity.14 In all cases, these transformations proceed simply by changing the radical trap reagent; although these reactions are not fully optimized, they provide proof-of-concept for the modularity of our approach.
Figure 3.
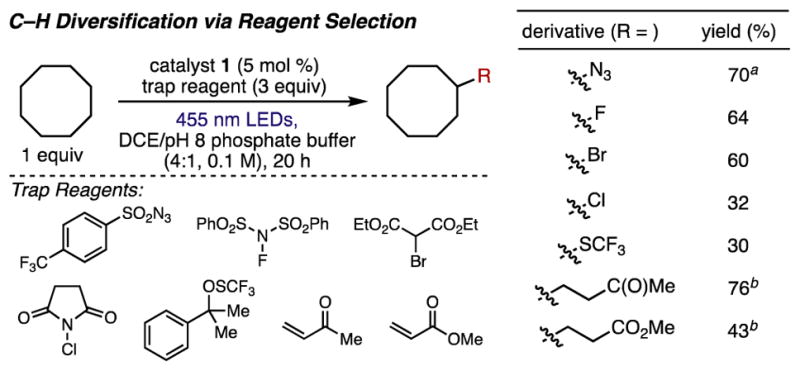
Products of modular C–H functionalization using 0.1 mmol substrate. Yields refer to NMR yields with HMDS or fluorobenzene as an internal standard. aSee Figure 2 for azidation conditions. bReactions use catalyst 2 and an alternate solvent system, see Supporting Information for details.
We next sought to develop a C–H alkylation using electron-poor alkenes as coupling partners. The use of a more reducing acridinium catalyst 2 and a solvent mixture of DCE and 2,2,2-trifluoroethanol (TFE) enabled the use of both methyl vinyl ketone and methyl acrylate as radical traps, delivering ketone and ester products in 76% and 43% yield, respectively. Recent studies have reported intramolecular C–H alkylations using activated alkenes and iridium photoredox catalysis proceeding by 1,5-hydrogen atom abstraction.18 To the best of our knowledge, this is the first report of a visible light-mediated, intermolecular unactivated C–H alkylation using the substrate as limiting reagent.19
Our studies continued with applications to several complex substrates with multiple C–H sites (Figure 4). The functionalization of (−)-menthol benzoate afforded azide 21 in 54% yield favoring functionalization at the most electron-rich tertiary position. Owing to the presence of adamantyl cores in several pharmaceuticals, we studied the reactivity of these substrates using several C–H transformations. The azidation of N-phthalimide protected memantine, an NMDA receptor antagonist used to treat Alzheimer’s disease, occurred at the tertiary position to deliver 22 in 55% yield. Similarly, the fluorination of this substrate was successful, delivering 23 in 86% yield with complete site selectivity for the tertiary position. A precursor to differin, a topical retinoid, was transformed to azide 24 in moderate yield despite the presence of an electron-rich aromatic ring, underscoring the mild nature of our system. This substrate also coupled effectively with methyl vinyl ketone, delivering adduct 25 in 45% yield as a single regioisomer.20 Ibuprofen methyl ester–containing both tertiary and benzylic sites–slightly favored benzylic azidation, providing a mixture of regioisomers (26) in 57% yield. Other reported strategies for the C–H azidation of this substrate provide only the benzylic azide,5a highlighting the complementary nature of our protocol that allows access to new derivatives of important pharmaceuticals. Steroid 5α-cholestan-3-one, containing 46 aliphatic C–H bonds, underwent functionalization at the C17 and C25 positions providing 27 in 37% yield, with similar site selectivity to that observed by Curci in the context of C–H hydroxylation.21
Figure 4.
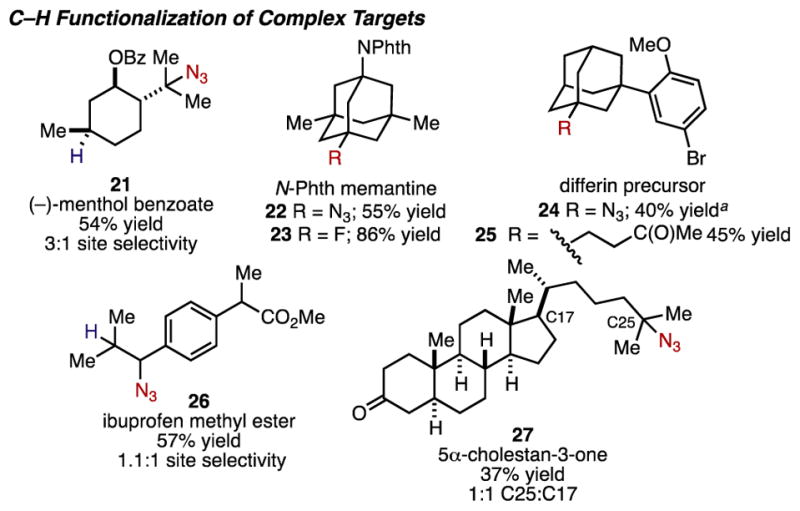
Complex products of modular C–H functionalization. For reaction details see the Supporting Information. Yields refer to isolated yields. aNMR yield with HMDS as an internal standard.
In order to shed light on the mechanism, we sought to identify the active hydrogen atom abstracting species. We initially focused on the C–H azidation for these studies. A Stern-Volmer analysis determined that sulfonyl azide 3 did not quench the acridinium fluorescence.14 Owing to the acidity of HFIP and the use of stoichiometric base, we considered that oxidation of the resultant alkoxide could produce an oxygen-centered radical. We also found that sodium 1,1,1,3,3,3-hexafluoroisopropoxide in HFIP did not quench the catalyst excited state.14 We next considered the role of the base. Although either K3PO4 or pH 8 phosphate buffer was competent in these reactions, we were unable to obtain redox potentials of either substance in MeCN owing to insolubility or immiscibility. While attempting to study Stern-Volmer fluorescence quenching with these bases, our efforts were similarly hampered regardless of the solvent used. To circumvent this issue, we synthesized a more soluble dibasic phosphate 28 from the corresponding phosphoric acid 29.22 We found that the dibasic phosphate quenched the excited state of the acridinium and the protonated acid did not (Figure 5), highlighting the necessity of the anionic base. Addition of the dibasic phosphate also leads to a shift in the resonances of the 1H NMR of acridinium catalyst 1 in CDCl3, suggesting the possibility of an acridinium-phosphate complex in solution.14
Figure 5.
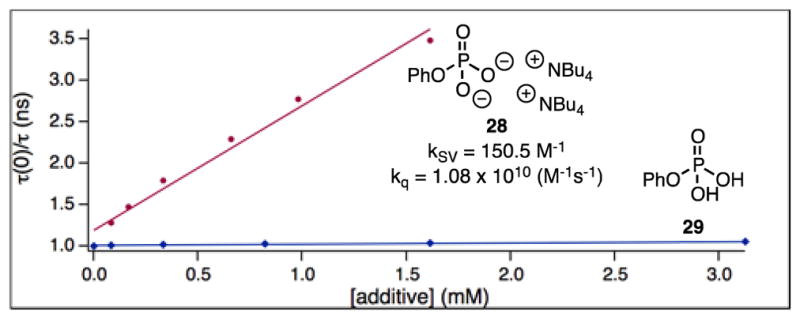
Stern-Volmer fluorescence quenching of 1 in DCE.
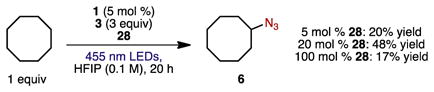
We also examined the reactivity of dibasic phosphate 28 under the azidation conditions with cyclooctane as substrate (eq 1). The use of substoichiometric 28 (5 or 20 mol %) produced up to 48% yield of azide 6, consistent with catalytic activity of the base. However, the use of stoichiometric 28 led to a diminished yield of 17%. Using methyl 6-methylheptanoate as substrate with 20 mol % 28 produced azide 13 in 20% yield. Importantly, only functionalization at the tertiary C–H bond occurred, suggesting that 28 and K3PO4 serve the same role in the transformation.
 |
(1) |
 |
(2) |
We provide a mechanistic hypothesis in Figure 6 consistent with this data. The acridinium photoredox catalyst is excited by the 455 nm LEDs and can undergo single-electron transfer from the phosphate salt, generating an oxygen-centered radical. Abstraction of the most electron-rich aliphatic C–H bond of the substrate generates a carbon-centered radical, which, upon trapping by the sulfonyl azide, affords the desired product and a sulfonyl radical. Based on previous work in the Nicewicz lab,23 we hypothesize that the sulfonyl radical is capable of oxidizing Mes-Acr•, regenerating the acridinium catalyst.24
Figure 6.
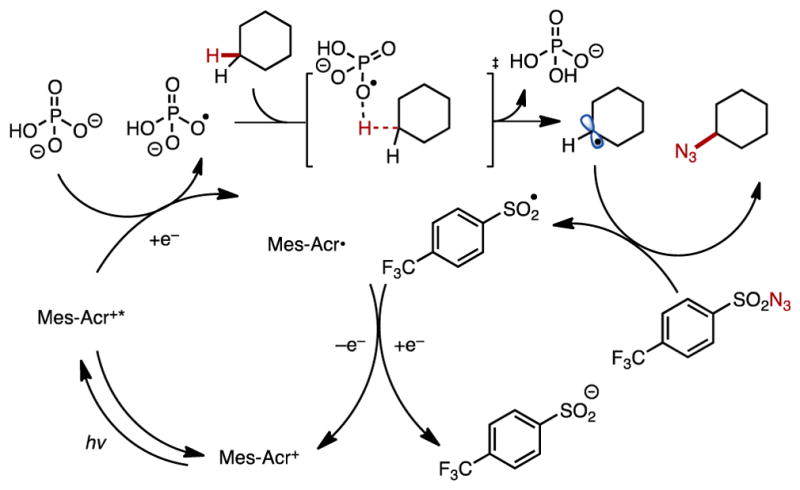
Proposed mechanism of the C–H azidation.
The versatility of the current approach in accessing a diverse array of C–H functionalizations highlights the unique capabilities of organic photoredox catalysis to generate reactive intermediates in a controlled manner. The present catalytic system enables the decoupling of the C–H abstracting species from the C–X forming step. This allows for the development of a modular system to access a range of functionalized products directly, thus obviating the need to develop new methodology for each specific C–H transformation. We anticipate that the synthetic utility and mild conditions of this strategy will lead to applications in complex molecule synthesis and late-stage functionalization across a wide range of contexts.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS) Award No. R01 GM 120163 (E.J.A.) and an Eli Lilly Grantee Award (D.A.N.). This research made use of an Edinburgh FLS920 emission spectrometer funded by the UNC EFRC: Center for Solar Fuels, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DESC0001011. We thank the UNC Department of Chemistry Mass Spectrometry Core Laboratory for its assistance with MS analysis.
Footnotes
Supporting Information. Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; (b) Newhouse T, Baran PS. Angew Chem Int Ed. 2011;50:3362. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) White MC. Science. 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 2.Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW. Chem Soc Rev. 2016;45:546. doi: 10.1039/c5cs00628g. [DOI] [PubMed] [Google Scholar]
- 3.(a) Brodsky BH, Du Bois J. J Am Chem Soc. 2005;127:15391. doi: 10.1021/ja055549i. [DOI] [PubMed] [Google Scholar]; (b) Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (c) Chen MS, White MC. Science. 2010;327:566. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]; (d) Gormisky PE, White MC. J Am Chem Soc. 2013;135:14052. doi: 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]; (e) Moteki SA, Usui A, Zhang T, Alvarado CRS, Maruoka K. Angew Chem Int Ed. 2013;52:8657. doi: 10.1002/anie.201304359. [DOI] [PubMed] [Google Scholar]; (f) Canta M, Font D, Gómez L, Ribas X, Costas M. Adv Synth Catal. 2014;356:818. [Google Scholar]; (g) Kawamata Y, Yan M, Liu Z, Bao DH, Chen J, Starr JT, Baran PS. J Am Chem Soc. 2017;139:7448. doi: 10.1021/jacs.7b03539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Liu W, Groves JT. J Am Chem Soc. 2010;132:12847. doi: 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]; (b) Liu W, Huang X, Cheng M-J, Nielsen RJ, Goddard WA, III, Groves JT. Science. 2012;337:1322. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]; (c) Bloom S, Pitts CR, Miller DC, Haselton N, Holl MG, Urheim E, Lectka T. Angew Chem Int Ed. 2012;51:10580. doi: 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]; (d) Halperin SD, Fan H, Chang S, Martin RE, Britton R. Angew Chem Int Ed. 2014;53:4690. doi: 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]; (e) Bloom S, Knippel JL, Lectka T. Chem Sci. 2014;5:1175. [Google Scholar]; (f) Kee CW, Chin KF, Wong MW, Tan CH. Chem Comm. 2014;50:8211. doi: 10.1039/c4cc01848f. [DOI] [PubMed] [Google Scholar]
- 5.(a) Huang X, Bergsten TM, Groves JT. J Am Chem Soc. 2015;137:5300. doi: 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]; (b) Sharma A, Hartwig JF. Nature. 2015;517:600. doi: 10.1038/nature14127. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Karimov RR, Sharma A, Hartwig JF. ACS Cent Sci. 2016;2:715. doi: 10.1021/acscentsci.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Huang X, Groves JT. ACS Catal. 2016;6:751. [Google Scholar]
- 6.(a) Hager D, MacMillan DWC. J Am Chem Soc. 2014;136:16986. doi: 10.1021/ja5102695. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jeffrey JL, Terrett JA, MacMillan DWC. Science. 2015;349:1532. doi: 10.1126/science.aac8555. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Le C, Liang Y, Evans RW, Li X, MacMillan DWC. Nature. 2017;547:79. doi: 10.1038/nature22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Qvortrup K, Rankic DA, MacMillan DWC. J Am Chem Soc. 2014;136:626. doi: 10.1021/ja411596q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cuthbertson JD, MacMillan DWC. Nature. 2015;519:74. doi: 10.1038/nature14255. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jin J, MacMillan DWC. Angew Chem Int Ed. 2015;54:1565. doi: 10.1002/anie.201410432. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Science. 2016;352:1304. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mendes J, Zhou CW, Curran HJ. J Phys Chem A. 2014;118:1300. doi: 10.1021/jp412496g. [DOI] [PubMed] [Google Scholar]
- 9.For examples achieving the same bond formation via mechanisms that do not involve direct C–H abstraction, see: McNally A, Prier CK, MacMillan DWC. Science. 2011;334:1114. doi: 10.1126/science.1213920.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525.Noble A, MacMillan DWC. J Am Chem Soc. 2014;136:11602. doi: 10.1021/ja506094d.Prier CK, MacMillan DWC. Chem Sci. 2014;5:4173. doi: 10.1039/C4SC02155J.Zhou R, Liu H, Tao H, Yu X, Wu J. Chem Sci. 2017;8:4654. doi: 10.1039/c7sc00953d.
- 10.Mukherjee S, Maji B, Tlahuext-Aca A, Glorius F. J Am Chem Soc. 2016;138:16200. doi: 10.1021/jacs.6b09970. [DOI] [PubMed] [Google Scholar]
- 11.(a) Schmidt VA, Quinn RK, Brusoe AT, Alexanian EJ. J Am Chem Soc. 2014;136:14389. doi: 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]; (b) Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD, Alexanian EJ. J Am Chem Soc. 2016;138:696. doi: 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Czaplyski WL, Na CG, Alexanian EJ. J Am Chem Soc. 2016;138:13854. doi: 10.1021/jacs.6b09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi-Pangu A, Lévesque F, Roth HG, Oliver SF, Campeau LC, Nicewicz D, DiRocco DA. J Org Chem. 2016;81:7244. doi: 10.1021/acs.joc.6b01240. [DOI] [PubMed] [Google Scholar]
- 14.See Supporting Information for details.
- 15.Howell JM, Feng K, Clark JR, Trzepkowski LJ, White MC. J Am Chem Soc. 2015;137:14590. doi: 10.1021/jacs.5b10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landelle G, Panossian A, Leroux FR. Curr Top Med Chem. 2014;14:941. doi: 10.2174/1568026614666140202210016. [DOI] [PubMed] [Google Scholar]
- 17.Shao X, Xu C, Lu L, Shen Q. Acc Chem Res. 2015;48:1227. doi: 10.1021/acs.accounts.5b00047. [DOI] [PubMed] [Google Scholar]
- 18.(a) Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR. Nature. 2016;539:268. doi: 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chu JCK, Rovis T. Nature. 2016;539:272. doi: 10.1038/nature19810. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen DF, Chu JCK, Rovis T. J Am Chem Soc. 2017;139:14897. doi: 10.1021/jacs.7b09306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For an example of C–H alkylation mediated by UV irradiation, see: Kamijo S, Takao G, Kamijo K, Tsuno T, Ishiguro K, Murafuji T. Org Lett. 2016;18:4912. doi: 10.1021/acs.orglett.6b02391.
- 20.Alkylation of another substrate containing tertiary C–H bonds (cis-4-methylcyclohexyl pivalate) using methyl vinyl ketone proceeded in low yield (17%). Efforts to improve the scope of the alkylation are underway.
- 21.Bovicelli P, Lupattelli P, Mincione E, Prencipe T, Curci R. J Org Chem. 1992;57:5052. [Google Scholar]
- 22.Under the optimized conditions with K3PO4/HFIP or pH 8 phosphate buffer, dibasic phosphate is the most abundant phosphate in solution, with less tribasic and negligible monobasic to be expected.
- 23.Perkowski A, Nicewicz DA. J Am Chem Soc. 2013;135:10334. doi: 10.1021/ja4057294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.We cannot completely discount the possibility of free radical chain processes in these transformations. Further mechanistic studies to elucidate reaction pathways are underway.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.