

Abstract
Background
Glioblastoma (GBM) is the most common and most aggressive primary malignant brain tumor. Standard-of-care treatment involves maximal surgical resection of the tumor followed by radiation and chemotherapy (temozolomide [TMZ]). The 5-year survival rate of patients with GBM is <10%, a colossal failure that has been partially attributed to intrinsic and/or acquired resistance to TMZ through O6-methylguanine DNA methyltransferase (MGMT) promoter methylation status in the tumor.
Methods
A drug screening aimed at evaluating the potential recycling and repurposing of known drugs was conducted in TMZ-resistant GBM cell lines and primary cultures of newly diagnosed GBM with different MGMT promoter methylation status, phenotypic/genotypic background and subtype, and validated with sphere formation, cell migration assays, and quantitative invasive orthotopic in vivo models.
Results
We identified hydroxyurea (HU) to synergize with TMZ in GBM cells in culture and in vivo, irrespective of MGMT promoter methylation status, subtype, and/or stemness. HU acts specifically on the S-phase of the cell cycle by inhibiting the M2 unit of enzyme ribonucleotide reductase. Knockdown of this enzyme using RNA interference and other known chemical inhibitors exerted a similar effect to HU in combination with TMZ both in culture and in vivo.
Conclusions
We demonstrate preclinical efficacy of repurposing hydroxyurea in combination with TMZ for adjuvant GBM therapy. This combination benefit is of direct clinical interest given the extensive use of TMZ and the associated problems with TMZ-related resistance and treatment failure.
Keywords: cancer stem cells, drug combination, glioblastoma, hydroxyurea, temozolomide resistance
Importance of the study
Through repurposing drug screening, we demonstrated that the FDA-approved drug hydroxyurea synergizes with TMZ in GBM cells and patient-derived stemlike cells from newly diagnosed and recurrent tumors and in different orthotopic models, irrespective of MGMT promoter methylation status, molecular subtype, and stemness. The combination of HU and TMZ as an adjuvant therapy for GBM could provide a major benefit to a broad population of patients, leading to an increase in their overall survival. These results warrant further evaluation of this combination for treating glioblastoma patients in a clinical trial.
Over the last two decades, the major improvement in the treatment for glioblastoma (GBM) has been the addition of the DNA alkylating agent temozolomide (TMZ) to the standard of care (surgery and radiation), yielding an increase in 2-year overall survival from 10.9% to 27.2%.1,2 Despite this success, over 90% of patients receiving both TMZ and radiation after surgery die within 5 years after diagnosis, a colossal failure that partly can be attributed to intrinsic or acquired drug resistance.2 One of the major predictors of GBM response to TMZ is the intrinsic O6-methylguanine DNA methyltransferase (MGMT) promoter DNA methylation status.2 TMZ induces methylation of guanine at the O6 position, a change that causes a futile cycle of attempted DNA repair, resulting in cell apoptosis. MGMT removes the DNA adduct caused by this alkylating agent, resulting in resistance to TMZ therapy. Nearly all GBMs sooner or later recur and, in cases where the tumor is not already resistant to TMZ, will acquire resistance. A large-scale cancer genome sequencing analysis of malignant gliomas identified a hypermutation phenotype and somatic truncating/inactivating mutations in the MutS homolog 6 (MSH6) mismatch repair gene in recurrent (post-TMZ treated) GBM, particularly those growing more rapidly during TMZ therapy.3,4 Loss of MSH6 was found to occur in a subset of recurrent GBMs and was associated with tumor progression and TMZ resistance.3,4 These studies lead us to believe that novel therapies that could overcome intrinsic and/or acquired TMZ resistance in GBM could have a therapeutic benefit.
The repurposing of drugs builds upon a significant previous research and development effort detailing that the drug pharmacology, formulation, target, and potential toxicity has several advantages, since parameters such as bioavailability, toxicity, manufacturing, and pharmacology have already all been examined. Here, we aimed at recycling known drugs that could enhance or synergize the response to TMZ in GBM. Through screening of known anticancer agents on TMZ-resistant GBM cells and patient-derived stemlike neurospheres, we identified hydroxyurea (HU), an FDA-approved drug, to sensitize newly diagnosed and recurrent GBM to TMZ, both in culture and in invasive orthotopic GBM in vivo models, irrespective of MGMT promoter methylation status, genotypic alterations, molecular subtype, and stemness. Our results highlight the power of drug screening technologies in repurposing/recycling old drugs for new therapeutic applications.
Materials and Methods
Cell Culture, Reagents, and Viability Assay
Human GBM cell lines Hs683, U87, and LNZ308 and their resistant sublines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The generation of TMZ-resistant sublines was previously described.5 Patient-derived GBM cells from newly diagnosed and recurrent patient tumor specimens were cultured from discarded tissues at the Massachusetts General Hospital with approval of the institutional review board and were previously characterized.6–8 These cells were grown as neural spheres in neurobasal medium as we previously described.8 Knockdown cells of ribonucleotide reductase subunit M2 (RRM2) were created by transfecting cells with plasmids containing short hairpin (sh)RNA against human RRM2 gene. Temozolomide and HU (Sigma-Aldrich) were both dissolved in dimethyl sulfoxide (DMSO) to prepare a stock concentration of 100 mM, which was further diluted in 5% dextrose (pH = 4). For in vivo applications, HU was dissolved in phosphate buffered saline (PBS). Fludarabine and gemcitabine (Sigma-Aldrich) were used in the same way for comparison analysis. An FDA-approved anticancer drug library was arbitrarily assembled from compounds (Selleck Chemicals) and dissolved in DMSO. The Gluc cDNA and the green fluorescent protein expression cassette separated by an internal ribosomal entry site element were cloned into a lentivirus vector under the control of the strong constitutive cytomegalovirus promoter.9 Similar vector has been generated to express Fluc and mCherry fluorescent protein.10–12 Gluc activity was determined using a FlexStation3 microplate reader (Molecular Device). Cell proliferation was also confirmed by scratching a straight line in GBM cells grown as monolayer and measuring the gap closure (see Supplementary material for further details).
Sphere Formation Assay
Neurospheres were dissociated into single cells, and 1000 cells/well in 500 µL stem cell medium were plated in 48-well ultra-low adherent culture plates. After one day, cells were treated with DMSO, TMZ, HU, or combination of TMZ and HU. Four days posttreatment, phase-contrast images were obtained to visualize the morphology of sphere. Spheres were then dissociated into single cells with PBS-EDTA and 1000 cells/well were seeded to image and count the secondary sphere formation 5 days later. Stem cell frequency was compared with sphere limiting dilution analysis and calculated using the extreme limiting dilution analysis algorithm13 (see Supplementary material for further details).
In Vivo GBM Models
All animal studies were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care following guidelines set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. GBM cells were first transduced with a lentivirus vector to stably express firefly luciferase (Fluc).12 Athymic mice were anesthetized (100 mg/kg ketamine and 5 mg/kg xylazine) and stereotactically implanted with 50000 (unless otherwise noted) GBM cells using the following coordinates from the bregma in millimeters: anterior-posterior +0.5 mm, medio-lateral +2.0 mm, dorso-ventral −2.5 mm, as we previously described.11,12 One week (or as noted) post-implantation, mice were randomized into different treatment groups. Tumor growth was monitored over time by in vivo Fluc bioluminescence imaging using the Xenogen IVIS 200 Imaging System (PerkinElmer) as we previously described11,12 (see Supplementary material for further details).
Statistical Analysis
GraphPad Prism v6.01 software was used for statistical analysis of all data. A P-value less than 0.05 was considered to be statistically significant. For analysis between multiple groups, a 2-tailed Student’s t-test (unpaired), ANOVA, and Tukey’s post-hoc test were performed as indicated. The half-maximal inhibitory concentration (IC50) was calculated using nonlinear regression analysis. The interaction indices of IC50 OBS/IC50 IND were then calculated, and synergy was concluded when these indices were significantly lower than 1. Survival was analyzed using Kaplan–Meier curves and log-rank (Mantel–Cox) tests.
Results
Recycling Drug Screen to Identify Adjuvant GBM Therapeutics
We first assembled a panel of 7 patient-derived GBM stemlike cell cultures from newly diagnosed tumors grown as neurospheres with different MGMT status and representing the most commonly found genetic alterations, molecular subclasses, and stemness (Table 1). A subset of 2 cultures created from recurrent GBM tissues were tested in parallel for comparison. Six TMZ-resistant cell lines—generated by long-term exposure of U87, LNZ308, and Hs683 GBM cells to a steady concentration of TMZ (MGMT methylation and DNA mismatch repair do not play a role in this resistance)5—as well as their parental cell lines were also added, resulting in a total of 18 GBM cell cultures. Clinical, histopathological, molecular characteristics and GBM subtypes of the patient-derived GBM stemlike neurospheres are summarized in Table 1. We engineered these GBM cultures to stably express the naturally secreted Gaussia luciferase (Gluc) as a bioluminescent reporter for cell viability. The level of Gluc secretion to the conditioned medium is linearly related with respect to cell number and proliferation;9,10,12 thus, cell viability and drug kinetics can be monitored over time by assaying aliquots of conditioned medium for Gluc activity. We screened a library of 21 drugs selected by neuro-oncologists as either promising targeted agents against cancer or traditional chemotherapeutic agents most common in use. The concentrations used in preliminary tests were based on previously published literature (Fig. 1A). Readout was cell viability 72 hours posttreatment using the Gluc high-throughput screening assay, which we have previously described,14 in the presence and absence of 100 µM TMZ. The cell viability screen results are shown in Fig. 1A as a heatmap with gradations of red to white, where red means no cell survived and white depicts no cell death (cell viability is the same as control wells treated with vehicle). This initial screen revealed that (i) MGG4, MGG6, MGG8 neurospheres and all 3 parental GBM cell lines, U87, LNZ308, and Hs683, were sensitive to TMZ treatment; (ii) 3 of the 21 drugs (crizotinib, imatinib, and methotrexate) demonstrated strong inhibition in cell viability in almost all cell cultures; (iii) certain drugs, such as cyclophosphamide, daunorubicin, irinotecan, and isotretinoin, exhibited inhibitory effects against 3 or more GBM cultures, mostly TMZ-sensitive GBM cell lines and patient-derived neurospheres with methylated MGMT promoter; (iv) among the TMZ-resistant cultures, chlorambacil and topotecan (2/21 compounds) demonstrated moderate synergistic effect in 3 or more cultures; (v) however, a single compound, hydroxyurea, sensitized 6 of the 7 patient-derived neurospheres (except MGG29), both recurrent GBM primary cultures, and all 6 TMZ-resistant cell sublines to TMZ, while having minimal effect at the tested dose in the absence of TMZ (Fig. 1A). We therefore selected HU for further evaluation.
Table 1.
Summary of patient-derived GBM
| GBM Cells | In Vivo Phenotype | Genetic Alteration | CD44 | PTEN | p-Akt (Ser473) | p-Akt (Thr308) |
MGMT | Main Subtype | TMZ IC50 (µM) |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Invasiveness | Pathological Feature | Methylation | Protein | ||||||||
| MGG4 | N | Endothel. proliferation | MYC# | - | + | + | + | M | − | Not clear | 32 |
| MGG6 | I | Conventional | EGFR#, MDM4#, PI3KC2B#, CDKN2A&B- - | NT | + | + | + | M | − | Classical | 36 |
| MGG8 | I | PNET-like component | MYCN#, MDM2#, PDGFRA#, CDKN2A&B- - |
+/- | + | − | + | M | − | PN | 6 |
| MGG18 | N | Giant cell | NT | ++ | − | ++ | ++ | M | − | MES | 315 |
| MGG23 | I | Gemistocytic | CDKN2A&B- - | ++ | − | ++ | + | U | + | MES | 355 |
| MGG24 | I | Conventional | EGFR# | ++ | + | + | + | U/M | + | Classical | 166 |
| MGG29 | N | Oligo-like component | NT | −/+ | + | ++ | + | U | − | Not clear | >3000 |
Abbreviations: N, nodular; I, invasive; #, copy number amplifications; - -, homozygous deletion; PNET, primitive neuroectodermal tumor; PTEN, phosphatase and tensin homolog; +, positive; ++, strongly positive; −, negative; +/-, heterogeneous; M, methylated MGMT promoter; U, unmethylated MGMT promoter; U/M, a major unmethylated band with a weak methylated band; PN, proneural; MES, mesenchymal; NT, not tested.
Fig. 1.

HU, identified through repurposing drug screening, restores TMZ sensitivity in resistant GBM cells in culture. (A) Heatmap from the “repurposing/recycling” chemical screen demonstrating the sensitivity of 18 GBM cell cultures to TMZ in the presence of one of the 21 most commonly prescribed chemotherapy drugs; parental cell lines U87, LNZ308, and Hs683 and 2 corresponding TMZ-resistant sublines (R1 and R2), patient-derived cells from newly diagnosed (MGG4, MGG6, MGG8, MGG13, MGG23, MGG24, MGG29) and recurrent GBM tumors (rGBMa and rGBMb). The relative decrease in cell viability at 72 h posttreatment is shown as gradations of red to white; red, no cell alive; white, same proportion of live cells as in vehicle control (0.1% DMSO). (B) All 9 patient-derived cultures were treated with different concentrations of TMZ (100 nM to 1 mM) in the presence or absence of 30 μM HU. Cell viability was measured 3 days later using the Gluc assay and normalized to the vehicle control (0.1% DMSO), which was set at 100%. Results are shown as the mean ± SD of 8 wells. Nonlinear regression of the data was performed to calculate the IC50 and the synergic indices.
Hydroxyurea Sensitizes Resistant Glioblastoma Cells to TMZ in Culture
In a more profound dose-response assay on newly diagnosed and recurrent patient-derived GBM neurospheres, we confirmed that MGG4, MGG6, and MGG8 were sensitive to TMZ treatment, with IC50 values of 38.5 µM, 23.1 µM, and 7.9 µM, respectively. MGG18, MGG23, and MGG24 had mediate sensitivity, with IC50 values of 338.1 µM, 351.0 µM, and 85.7 µM, respectively. MGG29, rGBMa, and rGBMb were resistant to TMZ treatment, with IC50 values of 3087 µM, 1527 µM, and 750 µM, respectively (Supplementary Figure S1). The presence of 30 µM HU effectively restored the sensitivity to TMZ, where IC50 values were lowered to 8.4 µM, 7.0 µM, 3.8 µM, 44.0 µM, 12.7 µM, 10.6 µM, 491.4 µM, 61.9 µM, and 39.7 µM in MGG4, MGG6, MGG8 MGG18, MGG23, MGG24 MGG29, rGBMa, and rGBMb, respectively. Even though MGG29 is very resistant to TMZ, in the presence of 30 µM HU, the IC50 was lowered to <500 µM from >3000 µM (Fig. 1B). All of the 6 TMZ-resistant sublines showed significant TMZ resistance (IC50 values in 300–1000 µM range). However, in the presence of HU, the IC50 values were restored to double-digit micromolar levels, indicating strong synergistic effects (Supplementary Figures S2, S3).
To evaluate whether TMZ and/or HU could regulate the motility of these cells, we performed a scratch assay on a monolayer of MGG8, MGG23, and MGG29 cells. Twenty-four hours post-scratch, all GBM cells treated with TMZ or HU alone had the ability to move and fill large parts of the scratch. However, cells treated with HU in combination with TMZ showed a significant decrease in cell motility (Supplementary Figure S4). These results were confirmed with U87, U87R1, and U87R2 cell cultures (Supplementary Figure S5).
The ability of HU and/or TMZ to induce apoptosis was analyzed by flow cytometry using annexin V/propidium iodide (PI) staining. In MGG8 cells, the addition of HU increased the percentage of late apoptotic cells (PI+/annexin V+) from 39.4% ± 2.5% (TMZ) to 72.0% ± 5.4% (HU+TMZ, P < 0.01; Supplementary Figure S6). Similar results were obtained in the parental U87 cell line. In the TMZ-resistant U87R1 and U87R2 sublines, 30 µM TMZ caused a minor ~10% increase in apoptotic cells compared with control; however, when treated with HU and TMZ, apoptosis increased up to ~25% (P < 0.01, HU+TMZ vs TMZ alone; Supplementary Figure S7).
Hydroxyurea Sensitizes Resistant Glioblastoma Cells to TMZ In Vivo
We then validated the anti-GBM effect of HU and TMZ in an in vivo orthotopic model using U87 cells expressing the bioluminescent reporter Fluc.12 One week post-implantation of 50000 U87 GBM cells in the striatum of mice brains, mice were randomized into 4 different groups (n = 6–10/group) receiving (i) DMSO vehicle control; (ii) intraperitoneal (i.p) injection of 5 mg/kg body weight of TMZ; (iii) 50 mg/kg HU i.p; and (iv) combination of similar doses of TMZ+HU 4 days/week for 2 weeks. HU alone had no significant effect on U87 tumor growth and mice survival (median survival = 23 days for control group, 22.5 days for HU group). TMZ alone had a moderate effect on parental U87 tumors and mice survival (median survival = 37.5 days, P = 0.0391 vs control). On the other hand, combination of HU+TMZ reduced U87 tumor growth significantly (P < 0.001 vs TMZ; Figure 2A–C), leading to a significant increase in mice survival (median survival = 56.5 days, P = 0.0165 vs TMZ). This experiment was repeated with U87R1 cells using similar dose of HU and i.p injection of 30 mg/kg body weight of TMZ, 4 days/week for 2 weeks. HU or TMZ alone had no significant effect on U87R1 tumor growth (P > 0.3 vs control, for both drugs) but had moderate effect on mice survival (median survival = 29.5 days [P = 0.0224] and 32.5 days [P = 0.01093] for HU and TMZ groups, respectively, vs 19 days for control group). Combined treatment with HU and TMZ reduced U87R1 tumor growth significantly (P < 0.0001 vs TMZ; Fig. 2D–F and Supplementary Figure S8A), leading to a significant increase in mice survival (median survival = 51.5 days, P < 0.0001 vs TMZ).
Fig. 2.

HU sensitizes GBM cells to TMZ in vivo. (A–F) Fifty thousand U87 (A–C) or U87R1 (D–F) cells expressing Fluc were implanted in the striatum of athymic mice brains (day 1). One week later, mice were randomized into 4 groups (n = 6–10/group) and treated with either DMSO vehicle, HU (30 mg/kg), TMZ (5 mg/kg for U87 cells and 50 mg/kg for U87R1 cells), or similar doses of TMZ+HU. Tumor growth was monitored over time by Fluc imaging and survival was recorded generating Kaplan–Meier plots. Images of a representative mouse from each group are shown (A and D), tumor-associated bioluminescence signal was quantified (B and E; **P < 0.001, TMZ+HU vs TMZ only), and survival was analyzed; # P < 0.05 vs control, * P < 0.05 (C), ** P < 0.01 (F), TMZ+HU vs TMZ only. (G–I) Two hundred thousand U87R1 cells expressing Fluc were implanted in the brain of nude mice (day 1). Two weeks later, mice were divided into 2 groups (n = 10/group) and treated with either TMZ (50 mg/kg) or TMZ (50 mg/kg) + HU (30 mg/kg) for one week. Tumor volume (P > 0.05) and mice survival (**P = 0.0031) were analyzed. (J–L) U87R1 cells expressing Fluc were implanted in the brain of nude mice (day 1). One week later, mice were divided into 6 groups (n = 10–20/group) and treated with either DMSO vehicle, radiation (R; 3 Gy), HU, TMZ, R+TMZ or R+TMZ+HU. Tumor growth was monitored over time by Fluc imaging (**P < 0.01, R+TMZ+HU vs R+TMZ) and survival was recorded to generate Kaplan–Meier plots (#P < 0.05, ##P < 0.01 vs control, **P < 0.01 R+TMZ+HU vs R+TMZ by log-rank test).
In another experiment, we challenged this therapeutic strategy by implanting a higher number (200000) of TMZ-resistant U87R1 cells (day 1) and allowing 2 weeks for tumor growth before initiating the treatment of these large tumors (30 mg/kg body weight of TMZ, or 30 mg/kg TMZ + 50 mg/kg HU) for 1 week (day 15–day 22). Based on Fluc imaging, tumors had reached saturation signal and mice were expected to die within a few days. All mice treated with TMZ alone died within 2–6 days after the treatment was stopped, while the HU+TMZ-treated mice survived for another 6–14 days, showing the efficacy of this combined therapy (median survival of 32 days for HU+TMZ vs 26 days for control, P = 0.0002; Fig. 2G–I). We then tested the effect of HU on U87R1 cells in combination with standard of care (radiation and TMZ) using the same intracranial model (50000 U87R1 cells implanted and therapy initiated 1 week later). The triple therapy (6 Gy radiation + 30 mg/kg TMZ + 50 mg/kg HU, 4 d/wk for 2 wk) yielded an enhanced and significant inhibition of tumor growth (P < 0.001 vs radiation, TMZ, or HU only) and increased survival rate, with 50% of mice remaining alive 53.5 days post-implantation (median survival for control = 20 days; radiation only = 28 days [P = 0.004 vs control]; HU only = 27.5 days [P = 0.004 vs control]; TMZ only = 32.5 days [P = 0.0007 vs control]; radiation+TMZ = 37.5 days [P = 0.0004 vs control]; radiation+HU+TMZ, 53.5 days [P < 0.0001 vs radiation, TMZ, HU, or radiation+TMZ]; n = 8/group; Fig. 2J–L).
Hydroxyurea Sensitizes Patient-Derived GBM Neurospheres to TMZ Irrespective of MGMT Promoter Methylation Status, Genetic Background, Molecular Subclass, and Stemness
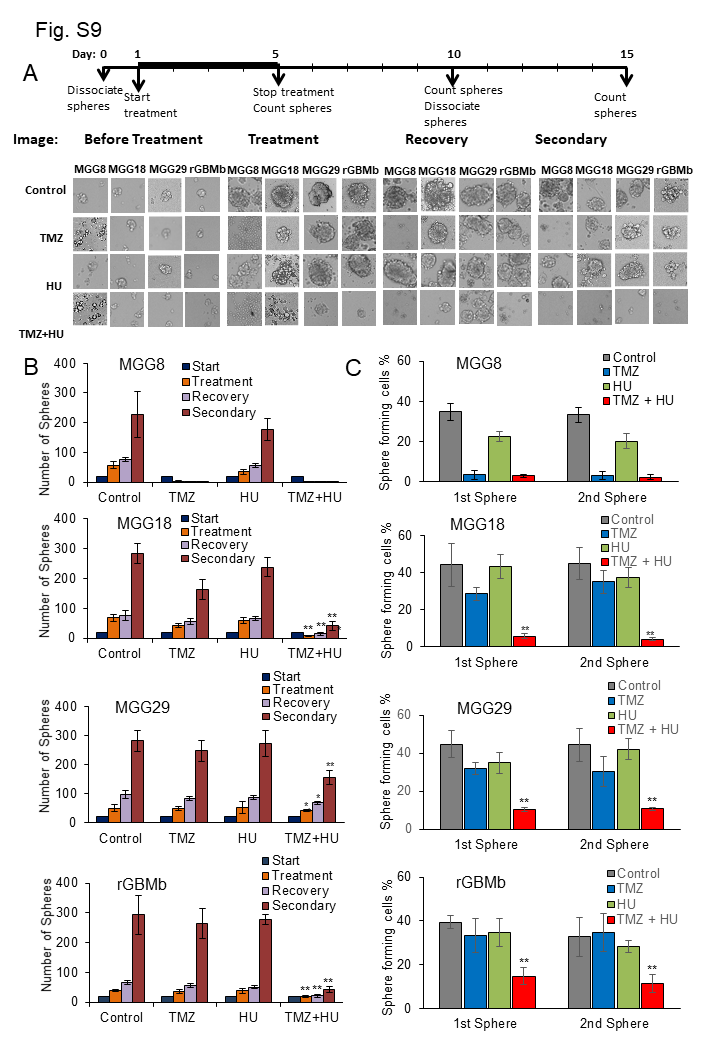
We analyzed the effect of HU on TMZ response in GBM neurospheres from newly diagnosed patients with different MGMT status, genotypic/phenotypic background, and molecular subtype. Neurosphere formation, growth/recovery, and secondary sphere formation (by dissociating spheres posttreatment and replating single cells in a new well) were monitored in culture for the different mono (HU or TMZ) or combined (HU+TMZ) therapies (Figure 3). Combined HU+TMZ yielded an enhanced therapeutic effect on all GBM stem cells obtained from newly diagnosed tumors with methylated (MGG6-invasive tumor with epidermal growth factor receptor [EGFR] amplification), unmethylated (MGG23-invasive with cyclin-dependent kinase inhibitor [CDKN] 2A and B deletion and cluster of differentiation [CD]44 positive, a feature of the mesenchymal phenotype), or mixed (MGG24-invasive with EGFR amplification, a feature of the classical phenotype) MGMT promoter, as well as from recurrent (rGBMa) tumors8 (Fig. 3A, B). Combined HU+TMZ yielded a significant inhibition in sphere numbers in all 4 cultures (P < 0.05). The secondary sphere formation responded to the combined treatment more profoundly in all cultures (P < 0.01; Fig. 3A, B). These results were confirmed using sphere limiting dilution assay to compare the in vitro self-renewal ability. The HU+TMZ combination caused a remarkable decrease in the proportion of cells that could form new spheres (P < 0.01; Fig. 3C). These results were consistent in another set of 4 cultures (Supplementary Figure S9).
Fig. 3.

Effect of HU+TMZ on GBM neurospheres with different MGMT status, genetic background, and molecular subtype. (A–B) Neurospheres from newly diagnosed GBM tumors with methylated (MGG6), unmethylated (MGG23), or mixed (MGG24) MGMT promoter and from recurrent (rGBMa) tumors were treated with either DMSO, HU, TMZ, or HU+TMZ for 4 days. Spheres were counted and left without treatment for another 5 days to allow recovery. Recovered spheres were dissociated and 1000 cells were plated in new 48-well plates to measure secondary sphere formation 5 days later. The experiment was repeated 3 times, and a representative image from 4 replicates in each treatment group is displayed (A). Scale bar, 200 μm. Total sphere numbers in the well recorded at each event; *P < 0.05, **P < 0.01 vs TMZ alone (A). (C) Sphere limiting dilution analysis comparing stem cell frequency. Different amounts (1 to 1000 cells) of GBM stem cells were plated as single cells. The number of cells that could form a sphere were quantified at day 5 (first sphere) and day 15 (second sphere) as described in the Materials and Methods section; *P < 0.05 TMZ+HU vs TMZ.
In Vivo Effect of HU in Combination with TMZ on Patient-Derived GBM Stemlike Xenograft Models
We next evaluated the combined effect of HU and TMZ in an in vivo intracranial GBM model that infiltrates the brain of mice similar to human tumors using 2 different patient-derived cells.7 Fifty thousand cells of primary MGG8 neurospheres expressing Fluc were implanted in the left mid-striatum of nude mice. Four weeks after implantation, when brain tumors started to grow rapidly (as observed by Fluc imaging), mice were randomized into 2 groups, which received DMSO vehicle (n = 6) or i.p injection of 30 mg/kg TMZ (n = 34), 4 days/week over 2 weeks. All mice in the control (DMSO-treated) group died by week 7 after i mplantation of MGG8 cells (median survival = 41 days). On the other hand, and as expected, MGG8 tumors carrying methylated MGMT promoter responded very well to TMZ (Fig. 4A–C). Subsequently, mice were left off TMZ to facilitate the outgrowth of TMZ-resistant neoplasms, recapitulating the patient scenario. At this point, the TMZ group was divided into 4 subgroups (n = 8–10/group), each receiving DMSO, HU, TMZ, or HU+TMZ 4 days/week over 3 weeks. The second round of TMZ treatment and monotherapy with HU had a moderate effect on tumor volume and mice survival (median survival = 51, 65, and 70.5 days for DMSO, HU, and TMZ groups, respectively, P < 0.01 HU vs DMSO, P < 0.001 TMZ vs DMSO). On the other hand, the combined HU+TMZ therapy had a highly significant effect on both tumor growth (P < 0.00001, TMZ+HU vs TMZ; Fig. 4C) and survival (P < 0.0001, TMZ+HU vs TMZ), with 50% of mice surviving over 145 days and remaining tumor free, while all mice treated with a second round of TMZ alone died by day 140. We also repeated this combined therapy using similar groups of mice on TMZ-resistant MGG23 tumors carrying unmethylated MGMT promoter with homozygous deletion for CDKN2A/B and strongly positive for CD44, a feature of the mesenchymal subtype (Table 1).8 We observed that these tumors also responded very well to the combined HU+TMZ therapy (4 d/wk for 2 wk), leading to a significant tumor regression (P < 0.00001, TMZ+HU vs TMZ) and significant increase in mice survival (median survival for control, TMZ, HU, and TMZ+HU groups [n = 8–10] were 32.5, 45, 50.5, and 79.5 days, respectively; P < 0.0001, TMZ+HU vs TMZ; Fig. 4D–F, Supplementary Figure S8B). Altogether, these data support our hypothesis that combinatorial treatment with HU+TMZ could be used to treat newly diagnosed and recurrent GBMs, irrespective of their MGMT status, genetic alterations, molecular subtype, and stemness.
Fig. 4.

HU sensitizes patient-derived GBM tumors to TMZ in vivo irrespective of MGMT status. (A–C) 5×104 MGG8 cells with methylated MGMT promoter expressing Fluc were stereotactically injected into the left mid-striatum of nude mice brain as small neurospheres (day 1). Four weeks later, mice were divided into 2 groups and treated with either TMZ (n = 34) or vehicle (n = 6) for 2 weeks, and then left off-treatment for 3 weeks. The TMZ-treated group was then divided into 4 subgroups, which received vehicle, HU, TMZ, or HU+TMZ (n = 8–10/group). Representative Fluc images of a single mouse from each group at different time points are shown (A). Tumor-associated Fluc signal was quantified (B); **P < 0.01, TMZ+HU vs TMZ only. Animal survival was recorded in a Kaplan–Meier plot (C); +P < 0.05 control vs no-treatment, #P < 0.05 vs control, **P < 0.01 TMZ+HU vs TMZ only by log-rank test. (D–F) MGG23 cells with unmethylated MGMT promoter were implanted in the brain of nude mice and treated with DMSO, HU, TMZ, or TMZ+HU. (D) Representative Fluc images of a single mouse from each group are shown over time. (E) Quantitation of tumor-associated Fluc signal; **P < 0.01 TMZ+HU vs TMZ. (F) Kaplan–Meier plots analyzing survival, #P < 0.05 vs control, **P < 0.01 TMZ+HU vs TMZ only.
Next, we assessed toxicity of this combined therapy by treating mice with either DMSO, TMZ, HU, or HU+TMZ 4 days/week for 2 weeks and analyzing their blood for white blood cells, hematocrit, red blood cells, mean corpuscular volume, red cell distribution width, hemoglobin, mean corpuscular hemoglobin, platelets, and mean platelet volume. We did not observe significant difference between the treatment groups at the evaluated dose/time point (Supplementary Table S1).
Inhibition of Ribonucleotide Reductase M2 Sensitizes GBM Cells and Patient-Derived Stem Cells to TMZ
We sought to understand the mechanism by which HU synergizes with TMZ. Since HU is known to target RRM2, an enzyme involved in nucleotide metabolism,15 a knockdown experiment was performed in different GBM cells and patient-derived GBM stemlike cells, using lentivirus vectors carrying an expression cassette for shRNA against RRM2 (shRRM2) or scrambled control (shScram). Quantitative real time PCR showed that shRRM2 could downregulate RRM2 mRNA levels by ~50% in these cells (Supplementary Figure S10). Cell viability assays showed that knockdown of RRM2 could sensitize GBM cells to TMZ (P < 0.01 shRRM2 vs shScram; Fig. 5A, B). Further, we evaluated the effect of knocking down RRM2 on TMZ response in brain tumors by implanting U87 cells expressing either shRRM2 or shScram. Treatment with TMZ 4 days/week for 2 weeks had a significantly greater suppressive effect on the growth of tumors expressing shRRM2 compared with tumors expressing shScram (P < 0.001, shRRM2+TMZ vs shScram+DMSO, shScram+TMZ, or shRRM2+DMSO) and a significant increase in mice survival (median survival for shScram+DMSO, shScram+TMZ, shRRM2+DMSO, and shRRM2+TMZ = 22.5, 33, 28.5, and >60 days, respectively, P < 0.01, shRRM2+TMZ vs shScram+DMSO, shScram+TMZ, or shRRM2+DMSO; Fig. 5C–E). We also evaluated other known ribonucleotide reductase (RNR) inhibitors to determine if a similar effect as HU is obtained on GBM cells. TMZ-resistant GBM cells cultured from recurrent tumors expressing Gluc were treated with either fludarabine or gemcitabine—classical RNR inhibitors—and cell viability was assessed using the Gluc assay. TMZ alone had a moderate to no effect on these cells. Both RNR inhibitors sensitized recurrent GBM cells to TMZ, suggesting that RNR is one of the potential key players in the synergistic effect of HU toward TMZ (Fig. 5F).
Fig. 5.

Knockdown/inhibition of RNR sensitizes GBM cells to TMZ. (A–B) U87 and TMZ-resistant subline U87R1 (A), TMZ-sensitive MGG6 with methylated MGMT promoter and TMZ-resistant MGG23 with unmethylated MGMT promoter (B) were infected with a lentivirus vector expressing scrambled shRNA (shScram) or shRNA against RRM2 (shRRM2) and treated with different doses (0–100 µM) of TMZ. Cell viability was assessed 4 days later using the Gluc assay. Data presented as Gluc relative light unit (mean ± SD, n = 8; **P < 0.01 shScram vs shRRM2). (C–E) The left forebrains of mice were implanted with 2×104 U87 cells expressing Fluc and either shScram or shRRM2. Each group of mice was divided into 2 subgroups, which received i.p. injection (3 times/wk over 3 wk) of either DMSO or TMZ (30 mg/kg body weight). (C) Fluc imaging was performed once/week to monitor tumor growth. Representative images at 0, 10, and 14 days post-TMZ treatment are shown. (D) Tumor-associated signal is quantified; **P < 0.01 vs shScram+DMSO, ##P < 0.01, shRRM2+TMZ vs shRRM2+DMSO, ++P < 0.01, shRRM2+TMZ vs shScram+TMZ by ANOVA and Tukey’s post-hoc test. (E) Kaplan–Meier plots analyzing mice survival in different groups; #P < 0.05, shScram+TMZ or shRRM2+DMSO vs shScram+DMSO; **P < 0.01 shRRM2+TMZ vs shRRM2+DMSO. (F) rGBMa cells cultured from recurrent GBM tumors were treated with different doses of either gemcitabine or fludarabine in the presence or absence of TMZ (100 μM). Three days later, cell viability was assessed using the Gluc assay; **P < 0.01 gemcitabine+TMZ or fludarabine+TMZ vs gemcitabine or fludarabine only.
Discussion
The development of new drug therapies is a long, arduous, and costly process filled with hurdles and high failure rate. Despite increased investments from both academia and industry, minimal success in improving patient outcome has been accomplished, mostly due to unexpected clinical side effects.16,17 In recent years, we have observed an increased interest in repositioning known drugs that are approved for other conditions for the treatment of different diseases and disorders.16,18–20 Through “repurposing/recycling” screening of known drugs, we identified the FDA-approved drug hydroxyurea to sensitize newly diagnosed and recurrent GBM to TMZ, independent of MGMT promoter methylation status, genetic alterations, molecular subtype, and stemness.
Hydroxyurea is a simple organic compound that acts specifically on the S-phase of the cell cycle by inhibiting the enzyme ribonucleotide reductase, thereby hindering the reductive conversion of ribonucleotides to deoxyribonucleotides, limiting de novo DNA synthesis.21 In addition, HU has been shown to inhibit the repair of DNA damage induced by chemicals or irradiation, which offers potential synergy between HU and irradiation or DNA-damaging agents.22 HU is FDA approved and has been used to treat myeloproliferative diseases23 and sickle cell anemia,24 as well as some neoplasms such as melanoma, ovarian carcinoma, squamous cell carcinoma, head and neck carcinoma, and brain tumors.25 Although HU has been previously evaluated in malignant gliomas in combination with radiation or cytotoxic chemotherapy and has shown limited efficacy,26,27 it has never been evaluated with an agent with validated efficacy in GBM such as TMZ. To our knowledge, this is the first demonstration of HU as a potential adjuvant therapy for GBM patients in combination with TMZ. An additional advantage of this strategy is that both drugs have already cleared several key steps in the development process, giving a strong starting point to accelerate the pace into the clinic.
Major obstacles for the treatment of brain tumors, including GBM, are the blood–brain barrier (BBB) and the blood–tumor barrier (BTB), which hamper delivery of chemotherapeutics to the brain and/or brain tumor. One advantage for using HU for GBM is its ability to efficiently bypass these biological barriers.28,29 A study in rats showed that HU can increase the BTB permeability of certain chemotherapeutics by means of nitric oxide production.30 However, a preclinical analysis revealed that HU had no effect on permeability of imatinib mesylate (Gleevec)31 across the BBB. It would be interesting to evaluate whether or not HU increases the penetration of TMZ to the brain and/or to GBM tumors, potentially leading to a decrease in the dose needed to achieve therapeutic effect, and helping to reduce TMZ-associated toxicities and progression-related phenotypes.
One of the major predictors of GBM response to TMZ is the intrinsic MGMT promoter methylation status.2,32 MGMT activity can remove the DNA adducts caused by TMZ, thereby conferring TMZ resistance. Thus, patients with a methylated MGMT promoter are more likely to benefit from TMZ treatment. In this study, we observed tumor regrowth around 2 weeks after the 2-week treatment window was discontinued. It would be of great interest to evaluate whether longer treatment period might give better therapeutic outcome as well as mechanism of resistance to TMZ/HU combined therapy in future studies.
Conclusions
Through a recycling/repurposing drug screen, we demonstrate for the first time the potential use of hydroxyurea as a promising adjuvant GBM therapy in combination with TMZ. Since HU, an inhibitor of RNR, is FDA approved and has already been used for the treatment of cancer, including gliomas, it should be readily translatable to the clinic. The combination of HU and TMZ as an adjuvant therapy for GBM could provide a major benefit to a broad population of GBM patients, leading to an increase in their overall survival. A phase I clinical trial testing HU and dose-intense TMZ in patients with recurrent glioblastoma should be initiated to identify the maximum tolerated dose for this combination and to explore the quantitative (frequency, duration) and qualitative (organ-specific) nature of toxicities.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by grants from the National Institutes of Health, the National Institute of Neurological Disorders and Stroke (R01NS064983, 1R01NS095647, and P30NS04776), the National Cancer Institute (R01CA166077 [B.A.T.]), and Stichting Stophersentumoren.nl (T.W. and P.W.).
Acknowledgments
The authors acknowledge the MGH Neuroscience Image Analysis Core (for confocal microscopy), and the MGH Vector Core (for producing the viral vector) supported by NIH/NINDS P30NS04776 as well as 1S10RR025504 Shared Instrumentation grant for the IVIS imaging system.
Conflict of interest statement. A patent application for the use of hydroxyurea and other ribonucleotide reductase inhibitors in combination with TMZ for cancer therapy has been filed by Massachusetts General Hospital (MGH).
References
- 1. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Hegi ME, Mason WP, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. [DOI] [PubMed] [Google Scholar]
- 3. Cahill DP, Codd PJ, Batchelor TT, Curry WT, Louis DN. MSH6 inactivation and emergent temozolomide resistance in human glioblastomas. Clin Neurosurg. 2008;55:165–171. [PubMed] [Google Scholar]
- 4. Yip S, Miao J, Cahill DP, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15(14):4622–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hiddingh L, Tannous BA, Teng J, et al. EFEMP1 induces γ-secretase/Notch-mediated temozolomide resistance in glioblastoma. Oncotarget. 2014;5(2):363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Teng J, Carla da Hora C, Kantar RS, et al. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017;19(6):820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wakimoto H, Kesari S, Farrell CJ, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69(8):3472–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wakimoto H, Mohapatra G, Kanai R, et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012;14(2):132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11(3):435–443. [DOI] [PubMed] [Google Scholar]
- 10. Tannous BA. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat Protoc. 2009;4(4):582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teng J, Hejazi S, Badr CE, Tannous BA. Systemic anticancer neural stem cells in combination with a cardiac glycoside for glioblastoma therapy. Stem Cells. 2014;32(8):2021–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wurdinger T, Badr C, Pike L, et al. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat Methods. 2008;5(2):171–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1-2):70–78. [DOI] [PubMed] [Google Scholar]
- 14. Badr CE, Wurdinger T, Tannous BA. Functional drug screening assay reveals potential glioma therapeutics. Assay Drug Dev Technol. 2011;9(3):281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chapman TR, Kinsella TJ. Ribonucleotide reductase inhibitors: a new look at an old target for radiosensitization. Front Oncol. 2011;1:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strittmatter SM. Overcoming drug development bottlenecks with repurposing: old drugs learn new tricks. Nat Med. 2014;20(6):590–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arrowsmith J, Miller P. Trial watch: phase II and phase III attrition rates 2011–2012. Nat Rev Drug Discov. 2013;12(8):569. [DOI] [PubMed] [Google Scholar]
- 18. Shchors K, Massaras A, Hanahan D. Dual targeting of the autophagic regulatory circuitry in gliomas with repurposed drugs elicits cell-lethal autophagy and therapeutic benefit. Cancer Cell. 2015;28(4):456–471. [DOI] [PubMed] [Google Scholar]
- 19. Huang X, He Y, Dubuc AM, et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat Neurosci. 2015;18(9):1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yuen T, Stachnik A, Iqbal J, et al. Bisphosphonates inactivate human EGFRs to exert antitumor actions. Proc Natl Acad Sci U S A. 2014;111(50):17989–17994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koç A, Wheeler LJ, Mathews CK, Merrill GF. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem. 2004;279(1):223–230. [DOI] [PubMed] [Google Scholar]
- 22. Yarbro JW. Mechanism of action of hydroxyurea. Semin Oncol. 1992;19(3 Suppl 9):1–10. [PubMed] [Google Scholar]
- 23. Streiff MB, Smith B, Spivak JL. The diagnosis and management of polycythemia vera in the era since the Polycythemia Vera Study Group: a survey of American Society of Hematology members’ practice patterns. Blood. 2002;99(4):1144–1149. [DOI] [PubMed] [Google Scholar]
- 24. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332(20):1317–1322. [DOI] [PubMed] [Google Scholar]
- 25. Madaan K, Kaushik D, Verma T. Hydroxyurea: a key player in cancer chemotherapy. Expert Rev Anticancer Ther. 2012;12(1):19–29. [DOI] [PubMed] [Google Scholar]
- 26. Kyritsis AP, Yung WK, Jaeckle KA, et al. Combination of 6-thioguanine, procarbazine, lomustine, and hydroxyurea for patients with recurrent malignant gliomas. Neurosurgery. 1996;39(5):921–926. [DOI] [PubMed] [Google Scholar]
- 27. Reardon DA, Dresemann G, Taillibert S, et al. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br J Cancer. 2009;101(12):1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dogruel M, Gibbs JE, Thomas SA. Hydroxyurea transport across the blood-brain and blood-cerebrospinal fluid barriers of the guinea-pig. J Neurochem. 2003;87(1):76–84. [DOI] [PubMed] [Google Scholar]
- 29. Syvänen S, Barletta J, Blomquist G, Långström B, Bergström M. PET-evaluated transport of [11C]hydroxyurea across the rat blood-brain barrier–lack of influence of cyclosporin and probenecid. Drug Metab Lett. 2007;1(3):189–194. [DOI] [PubMed] [Google Scholar]
- 30. Yin D, Wang X, Konda BM, et al. Increase in brain tumor permeability in glioma-bearing rats with nitric oxide donors. Clin Cancer Res. 2008;14(12):4002–4009. [DOI] [PubMed] [Google Scholar]
- 31. Bihorel S, Camenisch G, Gross G, Lemaire M, Scherrmann JM. Influence of hydroxyurea on imatinib mesylate (gleevec) transport at the mouse blood-brain barrier. Drug Metab Dispos. 2006;34(12):1945–1949. [DOI] [PubMed] [Google Scholar]
- 32. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.