

Abstract
Canine parvovirus (CPV) is a fast-evolving single-stranded DNA virus that causes one of the most significant infectious diseases of dogs. Although the virus dispersed over long distances in the past, current populations are considered to be spatially confined and with only a few instances of migration between specific localities. It is unclear whether these dynamics occur in South America where global studies have not been performed. The aim of this study is to analyze the patterns of genetic variability in South American CPV populations and explore their evolutionary relationships with global strains. Genomic sequences of sixty-three strains from South America and Europe were generated and analyzed using a phylodynamic approach. All the obtained strains belong to the CPV-2a lineage and associate with global strains in four monophyletic groups or clades. European and South American strains from all the countries here analyzed are representative of a widely distributed clade (Eur-I) that emerged in Southern Europe during 1990–98 to later spread to South America in the early 2000s. The emergence and spread of the Eur-I clade were correlated with a significant rise in the CPV effective population size in Europe and South America. The Asia-I clade includes strains from Asia and Uruguay. This clade originated in Asia during the late 1980s and evolved locally before spreading to South America during 2009–10. The third clade (Eur-II) comprises strains from Italy, Brazil, and Ecuador. This clade appears in South America as a consequence of an early introduction from Italy to Ecuador in the middle 1980s and has experienced extensive local genetic differentiation. Some strains from Argentina, Uruguay, and Brazil constitute an exclusive South American clade (SA-I) that emerged in Argentina in the 1990s. These results indicate that the current epidemiological scenario is a consequence of inter- and intracontinental migrations of strains with different geographic and temporal origins that set the conditions for competition and local differentiation of CPV populations. The coexistence and interaction of highly divergent strains are the main responsible for the drastic epidemiological changes observed in South America in the last two decades. This highlights the threat of invasion from external sources and the importance of whole-genome resolution to robustly infer the origin and spread of new CPV variants. From a taxonomic standpoint, the findings herein show that the classification system that uses a single amino acid to identify variants (2a, 2b, and 2c) within the CPV-2a lineage does not reflect phylogenetic relationships and is not suitable to analyze CPV evolution. In this regard, the identification of clades or sublineages within circulating CPV strains is the first step towards a genetic and evolutionary classification of the virus.
Keywords: canine parvovirus, genomic evolution, South America, phylodynamics
1. Introduction
The parvoviruses (Parvoviridae family) are small (25 nm), non-enveloped viruses with icosahedral symmetry that infect a wide range of animal species. The subfamily Densovirinae infects invertebrates, while the subfamily Parvovirinae has diverse vertebrate hosts, including humans, pigs, rodents, cattle, and several domestic and wild carnivore species (Allison et al. 2013). Canine parvovirus (CPV) belongs to the species Carnivore protoparvovirus 1 (genus Protoparvovirus), a highly genetically related cluster of viruses that also comprises raccoon parvovirus, mink enteritis virus (MEV), and feline panleukopenia virus (FPV) (Cotmore et al. 2013).
CPV is a well-recognized cause of acute and often fatal hemorrhagic gastroenteritis in puppies and needs to be controlled by early vaccination (Decaro and Buonavoglia 2012). The virus has a linear single-stranded DNA genome (5.2 kb) with two open reading frames (ORFs) (Reed et al. 1988). The left ORF encodes nonstructural proteins 1 and 2 (NS1 and NS2), which are involved in DNA replication, capsid assembly, and viral cellular transport (Wang et al. 1998). The N-terminal region of NS1 and NS2 are identical in sequence, whereas the C-terminal region of NS2 is generated from differential splicing and is translated in a different reading frame. The right ORF encodes the viral capsid proteins 1 and 2 (VP1 and VP2), which are the main antigens that induce protective antibodies (Tsao et al. 1991; Agbandje et al.1995; Nelson et al. 2007). VP1 and VP2 are splice variants and are identical in sequence, except for a 143-amino-acid N-terminal region that is unique to VP1 and essential for viral cell infection (Tattersall and Ward 1976). In infectious virions, VP2 may be proteolytically cleaved near the amino terminus to form VP3 (Weichert et al. 1998). At both ends of the genome, there are non-translated regions with hairpin structures that prime viral DNA replication (Tattersall and Ward 1976).
CPV emerged in the late 1970s as a consequence of an interspecies jump from other carnivores to dogs, a process that involved a small number of amino acid changes in the VPs (Parrish 1999). The newly emerged CPV, referred as CPV type-2 (CPV-2), rapidly spread worldwide causing a pandemic in the new canine host (Decaro and Buonavoglia 2012). In 1979 appeared CPV-2a, a new genetic lineage that differs at five residues in VP2 and regained the ability to infect cats and other carnivores (Parrish et al. 1988; Truyen et al. 1996; Decaro et al. 2010, 2011). This CPV-2a lineage replaced the original CPV-2 strains within two years and became the ancestor of all the variants that currently circulate worldwide. This early period of CPV emergence was characterized by global spreading and strain replacement (Hoelzer et al. 2008).
The CPV-2a lineage underwent further evolution, showing an intrinsically high substitution rate that resembled fast-evolving RNA viruses (Shackelton et al. 2005). Changes in the residue 426 of VP2 have received considerable interest because they alter the antigenic properties of the capsid and have a geographic and temporal pattern. According to the amino acid in the 426 position, variants are denoted 2a (Asn), 2b (Asp), and 2c (Glu). The CPV-2a lineage was comprised of only 2a variants until the 2b variant was detected in the United States in 1984 (Parrish et al. 1991). The newly emerged 2b variant reached high prevalence in some countries and is currently circulating worldwide, but with different frequencies (Decaro and Buonavoglia 2012; Miranda and Thompson 2016). The 2c variant was identified around 2000 in Italy and is now detected mainly in Europe and America (Buonavoglia et al. 2001; Miranda and Thompson 2016).
Although the virus dispersed over long distances in the past, several current CPV populations have acquired particular nucleotide substitutions, which is evidence of local differentiation. This fact leads to the hypothesis that current populations are spatially confined and show only a few instances of migration between specific localities (Doki et al. 2006; Hoelzer et al. 2008; Clegg et al. 2011). However, it is unclear whether these dynamics persist in other scenarios due to the difficulty of comparing strains using only the 2a/2b/2c classification and partial VP2 sequences. To infer the emergence and spread of the genetic variants, it is critical to extend the analysis to other genomic regions and focus on virus populations from different places and periods.
In South America (Argentina, Brazil, Colombia, Ecuador, and Uruguay), CPV has shown singular characteristics that supports a significant temporal and spatial genetic variability (Pereira et al. 2007; Streck et al. 2009; Gallo Calderón et al. 2012, 2015; Pérez et al. 2012; Aldaz et al. 2013; Duque-García et al. 2017). Events of local diversification were detected in Brazil during 1980–2000 by analyzing positively selected residues in VP1 (Pereira et al. 2007). In the early 2000s, the emergence and spread of 2c strains seemingly changed the epidemiological scenario of CPV in South America (Pérez et al. 2007). More recently, a divergent 2a strain emerged in the Uruguayan 2c homogenous population and reached a frequency of 85% within 2 years (Maya et al. 2013).
Questions remain about the origin and spread of South American variants as most strains have been only partially sequenced; complete genomes available in South America are exclusively from Uruguay (Pérez et al. 2014).
In this study, the complete coding genome of global CPV populations was analyzed to infer the origin and patterns of genetic variability in South America.
2. Materials and methods
2.1 Strains
Viral sequences came from sixty-three fecal samples collected from puppies (age 1–12 months) of diverse breeds that were treated in different veterinarian clinics from South America and Europe (Supplementary Table S1). Dogs were clinically diagnosed with a parvovirus infection and tested positive for CPV by PCR amplification of partial VP2 sequences (Pérez et al. 2012). The South American strains (n = 46) belonged to Argentina (n = 9), Brazil (n = 12), Ecuador (n = 18), Paraguay (n = 1), and Uruguay (n = 6). European strains (n = 17) came from Albania (n = 1), France (n = 1), and Italy (n = 15). Some partial VP sequences have been previously reported (Calderón et al. 2011; Pérez et al. 2012; Puentes et al. 2012; Aldaz et al. 2013) (Supplementary Table S1).
2.2 Genome dataset
To generate a comprehensive genome dataset, most full-length sequences available in the GenBank database were retrieved (Supplementary Table S2). The only genomes available from South America corresponded to forty-one Uruguayan strains that were previously reported by our research group (Pérez et al. 2014).
2.3 DNA extraction, PCR, and sequencing
Viral DNA was extracted from dog feces using the QIAamp DNA Mini kit (Qiagen, Valencia, CA, USA). Full-length genome amplification was performed with a Long PCR Enzyme Mix (Thermo Scientific, Waltham, MA, USA), and PCR products were directly sequenced using internal primers according to Pérez et al. (2014).
2.4 Sequence assembly and alignment
Sequence assembly and consensus sequences were obtained using the Lasergene Genomics Suite (DNASTAR). DNA sequences were aligned using the MAFFT algorithm (Katoh et al. 2002).
2.5 Recombination analysis
Identification of potential recombinant, parental sequences and localization of possible recombinant breakpoints were performed using the recombination detection program software, which implements seven distinct algorithms for characterization of recombinant sequences (Martin et al. 2015). Only potential recombination events detected by two or more algorithms, coupled with phylogenetic evidence of recombination, were considered significant using the highest acceptable P-value cutoff of 0.05.
2.6 Phylogenetic inferences
For DNA alignments, the best-fit model of nucleotide substitution (TPMuf+I+G) was selected under the Akaike and the Bayesian information criteria in jModelTest (Posada 2008). Maximum-likelihood trees, with 1000-replicate bootstrap for the support of internal nodes, were inferred using PhyML (Guindon and Gascuel 2003). Phylogenetic trees were visualized and edited with FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
2.7 Evolutionary rates and population dynamics
The evolutionary rates (measured as nucleotide substitutions per site per year, s/s/y), the ages of the most recent common ancestor (tMRCA) and internal nodes, and the geographic movement patterns of the CPV population were jointly estimated using the Bayesian Markov Chain Monte Carlo approach implemented in BEAST v1.7.5 package (Drummond and Rambaut 2007; Drummond et al. 2012). In order to minimize sampling bias, some countries were pooled together: China, India, and Japan were pooled as Asia, Albania, and Italy as Southern Europe and France and Germany as Western Europe. A single sample from Paraguay was pooled with Argentina. The Bayesian skyline plot method was used as the coalescent tree prior, under a GTR+I+G4 substitution rate model and a relaxed uncorrelated lognormal molecular clock model (Drummond et al. 2006). Migratory events were inferred from a Continuous Time Markov Chain model for the reconstruction of discrete ancestral characters. For comparison of reversible and non-reversible matrices of geographic movement, path, and stepping-stone samplings were used to determine Bayes factors (BFs). The Bayesian stochastic search variable selection approach was used to identify significant migratory events over non-zero rates. Markov Chain Monte Carlo was ran for 100 million generations and convergence of all parameters was checked by observing Effective Sample Size > 200 with TRACER v1.5 (Drummond and Rambaut 2007). The maximum clade credibility tree was obtained from the posterior distribution of trees using TreeAnnotator v1.8 (part of the BEAST package) after discarding 10% of steps as burn-in. For each node, posterior probability (PP) and posterior state probability (PSP) was collected. Phylogenetic uncertainty for each parameter was informed as 95% highest probability density (HPD) values. Maps, migration routes, trees and population effective number through time were plotted using in-house R scripts.
2.8 Informative amino acid residues and selective pressures
Identification of variable residues in the protein alignments was performed using DIVEIN web server (Deng et al. 2010). To evaluate selective pressures on individuals sites of the codon alignments, the synonymous (dS) and non-synonymous (dN) substitution rates per site were estimated using different methods available in DATAMONKEY web server: Single-Likelihood Ancestor Counting (SLAC), Fixed Effects Likelihood (FEL), Fast Unconstrained Bayesian AppRoximation (FUBAR), Internal Fixed Effects Likelihood (IFEL) and Mixed Effects Model of Evolution (MEME) (Pond and Frost 2005).
3. Results
3.1 Strain sequencing
Sequences of sixty-three CPV genomes (4629 nt) were obtained from South American and European strains (Supplementary Table S1). The sixty-three newly sequenced genomes were added to the genomes retrieved from the GenBank (n = 110) (Supplementary Table S2), to compile a dataset of 173 full-length sequences of Carnivore protoparvovirus 1. Sequences came from strains isolated mainly from dogs but also from raccoons (n = 9), felines (n = 11), and minks (n = 7).
3.2 Recombination analysis
As recombination may lead to confounding the signal of vertical evolution when reconstructing phylogenetic trees, the first approach was to analyze the strains to detect recombination events. Thirteen recombinant strains were detected and characterized (Supplementary Tables S2 and S3).
3.3 Phylogenetic analysis using full-length genomes
An initial phylogenetic analysis was performed with the non-recombinant dataset that included 160 genomes of Carnivore protoparvovirus 1. This analysis shows two distinct and highly supported genetic groups, which are referred as the FPV- and the CPV-like viruses (Hoelzer and Parrish 2010) (Fig. 1). The FPV-like group contains the viruses from felines and minks going back to the first isolates that were collected in the early 1960s. The CPV-like group splits into two branches: the original pandemic CPV-2 strains that were collected in the USA during 1978–79 and the CPV-2a lineage that includes the variants collected from 1980 onward (Fig. 1).
Figure 1.

Condensed phylogenetic tree obtained using the maximum-likelihood method with full-length genomes of 160 Carnivore protoparvovirus 1. The FPV-like and CPV-like clades are displayed. Support for each node (expressed as percentage), done with 1000-replicate bootstrap.
3.4 Phylodynamic analysis and identification of amino acid signatures
The dataset for the phylodynamic analysis (n = 144) included only the viruses from the CPV-like group (CPV-2 and CPV-2a lineage). Most of the strains of the CPV-2a lineage come from domestic dogs but there are also some raccoon strains that appear grouped in the phylogeny. The tMRCA of the 2a lineage was estimated to be 36 years, calculated from the most recent analyzed strain (2014) with a 95% HPD ranging from 34 to 38 years. South American CPVs associate with global strains in four monophyletic groups or clades (Europe I, Asia I, Europe II, and South America I) with high PP (Fig. 2A, Supplementary Figs S1 and S2).
Figure 2.
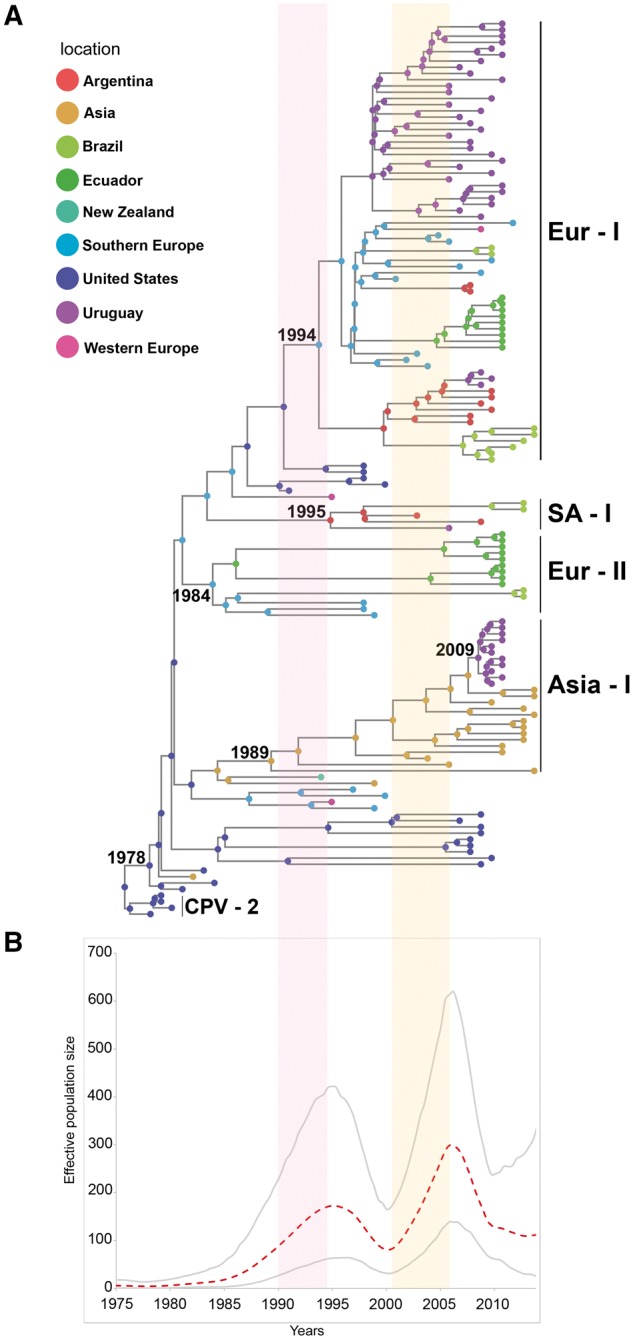
CPV phylogeny and population dynamics. (A) Phylogram of the sampled CPV population using full-length genomes. The tips and internal nodes of the tree are colored according to the actual or ancestral geographic region inferred under a symmetric discrete phylogeographic model. Evolutionary groups (clades) are indicated as follows: Eur-I, European/South American (all countries) strains; Asia-I, Asiatic/South American (Uruguayan) strains; SA-I, South American strains (exclusive South American group from Argentina, Brazil, and Uruguay); Eur-II, European/South American (Ecuadorian and Brazilian) strains. (B) Bayesian skyline plot showing the size fluctuations in CPV population along time. The dashed line indicates the effective population size median and the gray lines delimit the 95% HPD interval. Red vertical shade areas associate the rapid-grow phase observed in the Bayesian skyline plot with CPV-2c emergence in Europe and spreading in South America.
The Europe I clade (Eur-I) includes European (Italy, France, and Albania) and South American (Argentina, Brazil, Ecuador, Paraguay, and Uruguay) strains (Fig. 2A). This clade has Southern Europe as its most probable ancestral geographic location (PSP = 0.57) (Supplementary Fig. S3); and has strains from the United States and Western Europe (Germany) as sister branches (Fig. 2A). The tMRCA of this clade was estimated to be 20 years with a 95% HPD interval of 16–24 years. The strains from this clade are relatively homogenous (p-distance: 2.7 × 10−3 and 1.6 × 10−3 for nucleotides and amino acids, respectively). This clade has a characteristic combination of residues that includes the 426E, which identifies strains originally denoted as 2c, but there is not a single amino acid in the NS and VP sequences that is unique (i.e. exclusively found) for the Eur-I clade (Table 1). The strains of the Eur-I clade group with no geographic pattern, with the exception of Ecuadorian strains that cluster in a single branch and associate with Southern European strains (Fig. 2A).
Table 1.
Relevant amino acid changes of the protein sequence of NS1, NS2, VP1/VP2 for each of the evolutionary groups.
| Protein | NS1 | NS2 | VP1/VP2 | ||||
|---|---|---|---|---|---|---|---|
| Position | 544a | 572a | 152a | 267 | 297a | 324a | 426a |
| Eur-I clade | Y | E | V | F | A | Y | E |
| Asia-I clade | Y | K | V | Y | A | I | N |
| SA-I clade | Y | E | M | F | N | Y | N/D |
| Eur-II clade | F | E | V | F | A | Y | N/D/E |
Evolutionary groups (clades) are indicated as follows: Eur-I, European/South American (all countries) strains; Asia-I, Asiatic/South American (Uruguayan) strains, SA-I: South American strains (exclusive South American group from Argentina, Brazil, and Uruguay); Eur-II, European/South American (Ecuadorian and Brazilian) strains.
aPositively selected amino acids.
The Asia I clade (Asia-I) includes strains from China and India dated from 2004 to 2014 and Uruguayan strains collected during 2010–2011. The Asia-I clade has the Asiatic continent as the likeliest geographical origin (PSP = 0.98) (Supplementary Fig. S3) and a tMRCA of 25 years (95% HPD: 20–29 years) (Fig. 2A). The most related Uruguayan and Asiatic strains differ in only two single-nucleotide synonymous substitutions; the overall genetic variability of the clade is 3.1 × 10−3 (nucleotides) and 2.4 × 10−3 (amino acids). There are characteristic residues in NS1 (572K) and VP2 (267Y, 324I, 426N) (Table 1).
The South America I clade (SA-I) includes strains collected during 2003–13 from Argentina and Brazil, and a single strain collected in Uruguay during 2006 (Fig. 2A). This exclusively South American clade has Argentina as the likeliest ancestral location (PSP = 0.77) (Supplementary Fig. S3) and a tMRCA of 19 years (95% HPD: 15–24 years). The nucleotide and amino acid variability is 3.8 × 10−3 and 2.4 × 10−3, respectively. All strains share a 152M residue in NS2 and a 297N residue in VP2 (Table 1); the two Brazilian strains have unique changes in NS1 (585H), NS2 (95R), and VP2 (324L) (Supplementary Table S4).
The Europe II clade (Eur-II) comprises strains from Europe (Italy) and South American strains from Ecuador and Northeast Brazil. The Eur-II clade has Southern Europe as the geographic origin (PSP = 0.98) (Supplementary Fig. S3) and a tMRCA of 30 years with a 95% HPD interval of 25–31 years (Fig. 2A). This clade has high variability (5.6 × 10−3 and 4.4 × 10−3 for nucleotide and amino acids, respectively). The NS1 residue 544F is shared by all the Italian and South American strains (Table 1). Brazilian strains have characteristic residues in NS1 (369K, 583K, and 616N) and NS2 (92K, 125K); they also show a one nucleotide (G) deletion at position 2343 of the intronic region of VP. Ecuadorian strains have a 668E substitution in NS1, and a T440S change in VP2; some Ecuadorian strains have 139I and 426E residues in VP2 (Supplementary Table S4).
3.5 Effective population size through time
The global full-length sequences were also analyzed using the Bayesian skyline plot method to determine the population size fluctuations that occurred during the 1978–2014 period of the CPV epidemic. The Bayesian skyline plot reveals that CPV has undergone two relatively recent events of population size expansion. The first event occurred in the 1990s and the second one in the 2000s (Fig. 2B).
3.6 Significant dispersal pathways involving South American strains
The analysis herein dated the emergence of the CPV-2a lineage in the USA during 1978 (95% HPD: 1976–80). From then, the virus began expanding across continents, including Asia (BF = 62) and Southern Europe (BF = 56) (Fig. 3A and B). Southern Europe was a secondary hub from where the virus disseminated to Western Europe and South America (Fig. 3A). There are significant inter- and intracontinental migration events that involved South American strains (Fig. 3C). Highly significant intercontinental movements include the migration from Southern Europe to Ecuador (BF = 139) and Argentina (BF = 12), and the migration of the Asia-I clade from Asia to Uruguay (BF = 91). The most significant regional movements (i.e. across countries borders) were from Argentina to Uruguay (BF = 91) and Brazil (BF = 49) (Fig. 3B and C).
Figure 3.

Main routes of CPV migration. (A) CPV movements worldwide. Line thickness is according to BF values. (B) Migration matrix from the symmetric phylogeographic model. BFs values > 5 are displayed. (C) Zoom into CPV dynamics in South America. Significant migration routes are highlighted according to BF values, dated and annotated with the clade involved in the migration event. South American migrations are supported by BF > 5.
4. Discussion
The original pandemic CPV (type 2) was described from dog samples collected simultaneously in 1978 from the USA, Europe and Australia (Appel et al. 1979). By 1980, the disease had spread worldwide because of the transportation of dogs or contaminated fomites (Hoelzer and Parrish 2010). CPV-2 was replaced 2 years later by the CPV-2a lineage (Parrish et al. 1988). The CPV-2a lineage has been incredibly successful and originated all the strains that are actually circulating in the field (Hoelzer and Parrish 2010). Data here obtained indicate that the CPV-2a lineage emerged in the USA (PSP = 1) (Supplementary Fig. S3) during the late 1970 s and spread to Eurasia; this constitutes the first report of the geographic origin and the dispersal routes that this lineage underwent during its intercontinental spreading (Fig. 3A). Europe served as a second dispersion hub from where certain CPV strains arrived in South America (Figs 2A and 3C).
Early records of CPV in South America dated back to 1980 in Brazil and Chile (Abalos et al. 1982; Pereira et al. 2007). Brazilian CPV strains belonged to the 2a lineage and were extremely similar to strains collected in other continents during the same period, representing a clear example of the global dispersion occurred during the early period of the CPV-2a lineage emergence (Pereira et al. 2007; Hoelzer et al. 2008). The present pattern of genetic variability in the continent is remarkably different; the heterogeneity in the viral population and the phylogenetic relationship with foreign strains suggests that long migrations and local differentiation have been driving forces for CPV evolution in South America in the recent past. Viral migrations seem to be acquiring a more predominant role also in other continents, as evidenced by the increase in the number of reports of strains that share high homology with variants from distant geographic regions (Zhao et al. 2015; Zhou et al. 2017).
4.1 Eur-I clade: origin and spread in South America
The findings here reported indicate that some European and South American strains from all the countries here analyzed are representative of a relevant clade (Eur-I) within the 2a lineage. The Eur-I clade was circulating in Europe during 1990–98 to later spread to South American territories. Southern Europe appears as the ancestral location of this clade. The first representative strains of this clade were reported in Italy in 2001 (Buonavoglia et al. 2001) and were denoted as ‘2c strains’ based on a single amino acid position (426E). Notable, the Eur-I clade is not related with Italian strains collected during the late 1990s (1997–2000) (Fig. 2A), suggesting that Eur-I clade would have emerged in another European country and later introduced in Italy around 2001. Strains with the 426E residue were circulating in Germany in 1996 and in Poland in 1995 but genome sequences are not available to confirm their precise origin (Decaro et al. 2007; Majer-Dziedzic et al. 2011).
Soon after emerging, strains of the Eur-I clade successfully spread in several European dog populations in few years, replacing previous variants (Martella et al. 2005; Decaro et al. 2007, 2009; Decaro and Buonavoglia 2012). Interestingly, there was a significant rise in the CPV effective population size just before the emergence of this clade in Europe (Fig. 2B). It is possible that the ancestor of the Eur-I clade had a better fitness than previously circulating variants and infect dogs more effectively, leading to virus immune escape, vaccine failure and the subsequent population increase (Decaro et al. 2008, Decaro and Buonavoglia 2012). To our knowledge, there has been no previous phylodynamic evidence of a rapid expansion growth in the global CPV population associated with the emergence of a CPV strain with particular amino acid combinations (Fig. 2B, Table 1). Similar expansion events have been observed in other viruses with worldwide distribution, such as HIV and influenza, associated with the emergence of genetic variants (Ekong et al. 2017; Li et al. 2017).
The Eur-I strains invaded and spread in all South American countries analyzed so far (Argentina, Brazil, Ecuador, Paraguay, and Uruguay) (Pérez et al. 2007, 2012; Streck et al. 2009; Calderón et al. 2011; Pinto et al. 2012; Aldaz et al. 2013). Some regions of Uruguay, Argentina and Brazil have seen Eur-I strains becoming the only variant circulating in some period during the last decade. This spreading and replacement behavior was even more prominent than in Europe, where this clade has been circulating in some countries for almost 20 years but remains scarce or absent in others (Decaro and Buonavoglia 2012). The introduction in South America of the Eur-I clade was soon after its emergence in Europe and occurred independently in Argentina and Ecuador (Fig. 3C). From Argentina, the strains migrated to Uruguay and Brazil, revealing their successful spread and invasion capability. The Argentinean origin is coincident with epidemiological data, as strains from this clade were reported in Argentina during 2003 (Calderón et al. 2009), before the first description in Uruguay (2006) and Brazil (2008) (Pérez et al. 2007; Streck et al. 2009). The second migration wave to Ecuador of the Eur-I clade is evidenced by an Ecuadorian subclade that is closely related with some strains of the ancestral South European CPV population.
The invasion and replacement dynamics of the Eur-I clade in South America are unique and had been only observed for the CPV-2a lineage that primary replaced CPV-2 and, in a lesser extent, for the 2b antigenic type that reached high frequencies in the USA during the 1980s (Parrish et al. 1991). Notable, the South American spread of the Eur-I clade correlates with a second CPV population expansion during the early 2000s (Fig. 2B). The occurrence of the population expansion inferred from genomic data is supported by an apparent increase in the number of cases in South America (Gallo Calderón et al. 2012).
The Glu426 residue, which was used to describe the variant 2c, is a retained ancestral character for the Eur-I clade but also occurs in unrelated strains of the Eur-II clade from Ecuador (Table 1) and therefore cannot be used as a diagnostic character for the group. The group identity is based on non-coding changes and particular residue combinations (molecular signature) in the NS and VP viral proteins (Table 1).
4.2 The introduction of the Asia-I clade in South America
Some Uruguayan strains are representative of a large Asiatic clade (Asia-I) within the CPV-2a lineage (Fig. 2A). This clade originated in Asia during the late 1980s and evolved in this continent by the acquisition of particular nucleotide and amino acid changes. The amino acid signature of the Asia-I clade is very characteristic and includes unique residues in the NS and VP (Table 1).
After emerging and evolving in Asia, the Asia-I clade was introduced to South America around 2009 (Fig. 3C). This scenario is completely concordant with previous studies; Asiatic strains from this clade were already reported in 2004 (Ying et al. 2009) but in Uruguay, they were first detected in 2010 (Pérez et al. 2012). Interestingly, the Uruguayan strains reached a frequency of 85% in 2 years, replacing the previous variant of the Eur-I clade that was prevalent in the territory (Pérez et al. 2012). Although the spreading was fast, this strain has not been described in the neighboring country of Argentina (Gallo Calderón et al. 2015). This could be indicating that migration and replacement events are restricted to certain strains with particular characteristics and that not all strains have the same fitness in different regions. CPV dynamics seem to follow the changing connection between the countries as the introduction of the Asiatic clade in South America was associated with an increase in the cultural and economic connections between China and Uruguay.
The Uruguayan strains of the Asia-I clade are genetically homogenous, most likely because its presence in South America is the consequence of a single and recent introduction event. During the co-circulation of the Eur-I and Asia-I clades in South America, one recombinant strain was detected in Uruguay (Pérez et al. 2014). This strain did not increase its frequency in the population to acquire a more significant role in CPV evolution. Similar sporadic events of recombination have been identified within CPV strains or between CPV and FPV or MEV (Mochizuki et al. 2008; Ohshima and Mochizuki 2009; Wang et al. 2012) (Supplementary Table S3). Remarkably, nine of the recombinant strains here detected are closely related and should be considered as circulating recombinant forms (Supplementary Table S3). These strains were collected in China from 2009 to 2014 and originated from a single recombinant event between an Asiatic strain of the Asia-I clade and another undetermined strain of the CPV-2a lineage (Wang et al. 2016). This circulating recombinant form is a clear evidence that recombination could strongly influence CPV evolution by creating new lineages.
4.3 Early introduction of European strains to South America
Some strains from Ecuador, Italy, and Brazil form a third clade (Eur-II) within the CPV-2a lineage. This clade appears in South America as a consequence of an early introduction from Europe to Ecuador in the middle 1980s (Fig. 3B and C). The strains of the Eur-II clade have significant levels of variability (5.6 × 10−3 for nucleotide and 4.4 × 10−3 for amino acids) and signs of local differentiation (Table 1). The existence of relevant amino acid changes indicates that some viruses were undergoing diversification that may be related to selection pressures, a fact that is supported by the presence of positively selected residues in the molecular signature (Table 1) (Truyen et al. 1995; Decaro et al. 2009; Yoon et al. 2009). One remarkable change is the occurrence of the 426E residue in some Ecuadorian strains of the Eur-II clade. This residue emerged de novo in Ecuador and does not have the same ancestral origin that the 426E change of the Eur-I clade (Fig. 2A, Table 1). Notably, Brazilian strains also show a conserved deletion in the VP1 intron that occurs in most isolates after 1990 but was not present in strains from other regions of the world (Pereira et al. 2007).
4.4 An exclusive South American genetic clade
Some South American strains from Argentina, Brazil and Uruguay constitute a clade (SA-I) that has evolved exclusively in South America and harbors a particular amino acid signature (Table 1). The strains of the SA-I clade can be considered as excellent representatives of the evolution of CPV strains in the continent. The strains of the SA-I clade acquired changes by local differentiation and may have been circulating before the Eur-I and Asia-I introductions to South America. A similar situation occur in other countries that show spatial clustering and a limited international spread in different continents (Doki et al. 2006; Hoelzer et al. 2008; Clegg et al. 2011).
4.5 CPV classification and phylodynamics
The classification system that uses a single amino acid to identify variants (2a, 2b, and 2c) within the CPV-2a lineage does not reflect phylogenetic relationships of the strains and it is not suitable to analyze CPV evolution. In this regard, the identification of clades or sublineages within circulating CPV strains is the first step towards a genetic and evolutionary classification of the virus. The new classification system should take into account the phylogenetic clustering using the coding genome, the presence of unique amino acid signatures, and the ancestral geographic location of the genetic clade. The clade nomenclature can be based on the continent or territory of origin to refer to them in a consistent and standardized way. The well-characterized complete genome sequences of CPV here analyzed will serve as the reference or representative sequences for future studies that deal with newly identified CPV strains.
5. Conclusion
CPV evolution in South America is the consequence of the coexistence of different strains from multiple origins that sets the condition for competition and local genetic differentiation. Evolutionary dynamics involve locally differentiated populations that spread within or between continents and began to interact with CPV strains of the receptive population. One potential advantage of this type of evolution is the coexistence of highly divergent strains in the same territory that can compete and even recombine to allow the virus to evolve into more adapted forms. This could eventually produce impactful evolutionary events that can drastically change the epidemiological scenario in a short period. From a taxonomic standpoint, the results here obtained support that the amino acid changes that have been traditionally used for CPV typing (i.e. 426 for 2a/2b/2c strains) occurred independently in the populations and are therefore not suitable markers to study CPV evolution. Viral epidemiology requires a new classification system that takes into account phylodynamic data and the inclusion of recombinant circulating forms.
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest: None declared.
Supplementary Material
Acknowledgements
This work was supported in part by ‘Comisión Sectorial de Investigación Científica’ (CSIC), ‘Programa de Desarrollo de las Ciencias Básicas’ (PEDECIBA), and ‘Agencia Nacional de Investigación e Innovación’ (ANII) from Uruguay. We thank the clinical practitioners from the participating Uruguayan veterinary clinics for generously providing samples for analysis.
References
- Abalos P. et al. (1982) ‘Aislamiento De Parvovirus Canino En Perros Con Gastroenteritis’, Archivos de Medicina Veterinaria, 14: 47–9.] [Google Scholar]
- Agbandje M. et al. (1995) ‘The Structure of Parvoviruses’, Seminars in Virology, 6: 299–309. [Google Scholar]
- Aldaz J. et al. (2013) ‘High Local Genetic Diversity of Canine Parvovirus from Ecuador’, Veterinary Microbiology, 166: 214–9. [DOI] [PubMed] [Google Scholar]
- Allison A. B. et al. (2013) ‘Frequent Cross-Species Transmission of Parvoviruses among Diverse Carnivore Hosts’, Journal of Virology, 87: 2342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel M. et al. (1979) ‘Isolation and Immunisation Studies of a Canine Parco-like Virus from Dogs with Haemorrhagic Enteritis’, Veterinary Record, 105: 156–9. [DOI] [PubMed] [Google Scholar]
- Buonavoglia C. et al. (2001) ‘Evidence for Evolution of Canine Parvovirus Type 2 in Italy’, The Journal of General Virology, 82: 3021–5. [DOI] [PubMed] [Google Scholar]
- Calderón M. G. et al. (2009) ‘Molecular Characterization of Canine Parvovirus Strains in Argentina: Detection of the Pathogenic Variant CPV2c in Vaccinated Dogs’, Journal of Virological Methods, 159: 141–5. [DOI] [PubMed] [Google Scholar]
- Calderón M. G. et al. (2011) ‘Evolution of Canine Parvovirus in Argentina between Years 2003 and 2010: CPV2c Has Become the Predominant Variant Affecting the Domestic Dog Population’, Virus Research, 157: 106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg S. R. et al. (2011) ‘Molecular Epidemiology and Phylogeny Reveal Complex Spatial Dynamics in Areas Where Canine Parvovirus Is Endemic’, Journal of Virology, 85: 7892–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotmore S. F. et al. (2013) ‘Rationalization and Extension of the Taxonomy of the Family Parvoviridae’, ICTV Official Taxonomy: Updates since the 8th Report, code 2013.001a-aaaV. International Committee on Taxonomy of Viruses (ICTV). Available from: http://ictvonline.org/proposals/2013.001a-aaaV.A.v4.Parvoviridae.pdf. [Google Scholar]
- Decaro N. et al. (2007) ‘Molecular Epidemiology of Canine Parvovirus, Europe’, Emerging Infectious Diseases, 13: 1222–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N. et al. (2008) ‘Evidence for Immunisation Failure in Vaccinated Adult Dogs Infected with Canine Parvovirus Type 2c’, New Microbiologica, 31: 125–30. [PubMed] [Google Scholar]
- Decaro N. et al. (2009) ‘Genetic Analysis of Canine Parvovirus Type 2c’, Virology, 385: 5–10. [DOI] [PubMed] [Google Scholar]
- Decaro N. et al. (2010) ‘Characterisation of Canine Parvovirus Strains Isolated from Cats with Feline Panleukopenia’, Research in Veterinary Science, 89: 275–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N. et al. (2011) ‘Canine Parvovirus Type 2c Infection in a Kitten Associated with Intracranial Abscess and Convulsions’, Journal of Feline Medicine and Surgery, 13: 231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., Buonavoglia C. (2012) ‘Canine Parvovirus–a Review of Epidemiological and Diagnostic Aspects, with Emphasis on Type 2c’, Veterinary Microbiology, 155: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W. et al. (2010) ‘DIVEIN: A Web Server to Analyze Phylogenies, Sequence Divergence, Diversity, and Informative Sites’, BioTechniques, 48: 405–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doki M. et al. (2006) ‘Sequence Analysis of VP2 Gene of Canine Parvovirus Isolated from Domestic Dogs in Japan in 1999 and 2000’, Comparative Immunology, Microbiology and Infectious Diseases, 29: 199–206. [DOI] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2006) ‘Relaxed Phylogenetics and Dating with Confidence’, PLoS Biology, 4: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A. (2007) ‘BEAST: Bayesian Evolutionary Analysis by Sampling Trees’, BMC Evolutionary Biology, 7: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2012) ‘Bayesian Phylogenetics with BEAUti and the BEAST 1.7’, Molecular Biology and Evolution, 29: 1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque-García Y. et al. (2017) ‘Prevalence and Molecular Epidemiology of Canine Parvovirus 2 in Diarrheic Dogs in Colombia, South America: A Possible New CPV-2a Is Emerging?’, Veterinary Microbiology, 201: 56–61. [DOI] [PubMed] [Google Scholar]
- Ekong P. S. et al. (2017) ‘Spatiotemporal Evolutionary Epidemiology of H5N1 Highly Pathogenic Avian Influenza in West Africa and Nigeria, 2006–2015’, Transboundary and Emerging Diseases, 1–13. [DOI] [PubMed] [Google Scholar]
- Gallo Calderón M. et al. (2012) ‘Study of Canine Parvovirus Evolution: Comparative Analysis of Full-Length VP2 Gene Sequences from Argentina and International Field Strains’, Virus Genes, 44: 32–9. [DOI] [PubMed] [Google Scholar]
- Gallo Calderón M. et al. (2015) ‘Resurgence of Canine Parvovirus 2a Strain in the Domestic Dog Population from Argentina’, Journal of Virological Methods, 222: 145–9. [DOI] [PubMed] [Google Scholar]
- Guindon S., Gascuel O. (2003) ‘A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood’, Systematic Biology, 52: 696–704. [DOI] [PubMed] [Google Scholar]
- Hoelzer K. et al. (2008) ‘Phylogenetic Analysis Reveals the Emergence, Evolution and Dispersal of Carnivore Parvoviruses’, The Journal of General Virology, 89: 2280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelzer K., Parrish C. R. (2010) ‘The Emergence of Parvoviruses of Carnivores’, Veterinary Research, 41: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K. (2002) ‘MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform’, Nucleic Acids Research, 30: 3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. (2017) ‘Tracing the Epidemic History of HIV-1 CRF01_AE Clusters Using near-Complete Genome Sequences’, Scientific Reports, 7: 4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majer-Dziedzic B. et al. (2011) ‘Phylogenetic Analysis of Canine Parvovirus CPV-2 Strains and Its Variants Isolated in Poland’, Polish Journal of Veterinary Sciences, 14: 379–84. [DOI] [PubMed] [Google Scholar]
- Martella V. et al. (2005) ‘Surveillance Activity for Canine Parvovirus in Italy’, Journal of Veterinary Medicine. B, Infectious Diseases and Veterinary Public Health, 52: 312–5. [DOI] [PubMed] [Google Scholar]
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya L. et al. (2013) ‘Phylodynamics Analysis of Canine Parvovirus in Uruguay: Evidence of Two Successive Invasions by Different Variants’, Archives of Virology, 158: 1133–41. [DOI] [PubMed] [Google Scholar]
- Miranda C., Thompson G. (2016) ‘Canine Parvovirus: The Worldwide Occurrence of Antigenic Variants’, Journal of General Virology, 97: 2043–57. [DOI] [PubMed] [Google Scholar]
- Mochizuki M. et al. (2008) ‘Recombination between Vaccine and Field Strains of Canine Parvovirus Is Revealed by Isolation of Virus in Canine and Feline Cell Cultures’, The Journal of Veterinary Medical Science, 70: 1305–14. [DOI] [PubMed] [Google Scholar]
- Nelson C. D. S. et al. (2007) ‘Different Mechanisms of Antibody-Mediated Neutralization of Parvoviruses Revealed Using the Fab Fragments of Monoclonal Antibodies’, Virology, 361: 283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T., Mochizuki M. (2009) ‘Evidence for Recombination Between Feline Panleukopenia Virus and Canine Parvovirus Type 2’, Journal of Veterinary Medical Science, 71: 403–8. [DOI] [PubMed] [Google Scholar]
- Parrish C. R. et al. (1988) ‘The Global Spread and Replacement of Canine Parvovirus Strains’, Journal of General Virology, 69: 1111–6. [DOI] [PubMed] [Google Scholar]
- Parrish C. R. et al. (1991) ‘Rapid Antigenic-Type Replacement and DNA Sequence Evolution of Canine Parvovirus’, Journal of Virology, 65: 6544–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish C. R. (1999) ‘Host Range Relationships and the Evolution of Canine Parvovirus’, Veterinary Microbiology, 69: 29–40. [DOI] [PubMed] [Google Scholar]
- Pereira C. A. D. et al. (2007) ‘Selective Regimen Shift and Demographic Growth Increase Associated with the Emergence of High-Fitness Variants of Canine Parvovirus’, Infection, Genetics and Evolution: journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 7: 399–409. [DOI] [PubMed] [Google Scholar]
- Pérez R. et al. (2007) ‘First Detection of Canine Parvovirus Type 2c in South America’, Veterinary Microbiology, 124: 147–52. [DOI] [PubMed] [Google Scholar]
- Pérez R. et al. (2012) ‘Recent Spreading of a Divergent Canine Parvovirus Type 2a (CPV-2a) Strain in a CPV-2c Homogenous Population’, Veterinary Microbiology, 155: 214–9. [DOI] [PubMed] [Google Scholar]
- Pérez R. et al. (2014) ‘Phylogenetic and Genome-Wide Deep-Sequencing Analyses of Canine Parvovirus Reveal Co-Infection with Field Variants and Emergence of a Recent Recombinant Strain’, PLoS One, 9: e111779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L. D. et al. (2012) ‘Typing of Canine Parvovirus Strains Circulating in Brazil between 2008 and 2010’, Virus Research, 165: 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond S. L. K., Frost S. D. W. (2005) ‘Datamonkey: Rapid Detection of Selective Pressure on Individual Sites of Codon Alignments’, Bioinformatics, 21: 2531–3. [DOI] [PubMed] [Google Scholar]
- Posada D. (2008) ‘jModelTest: Phylogenetic Model Averaging’, Molecular Biology and Evolution, 25: 1253–6. [DOI] [PubMed] [Google Scholar]
- Puentes R. et al. (2012) ‘Isolation and Characterization of Canine Parvovirus Type 2C (CPV-2C) from Symptomatic Puppies’, Brazilian Journal of Microbiology, 43: 1005–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed A. P. et al. (1988) ‘Nucleotide Sequence and Genome Organization of Canine Parvovirus’, Journal of Virology, 62: 266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton L. A. et al. (2005) ‘High Rate of Viral Evolution Associated with the Emergence of Carnivore Parvovirus’, Proceedings of the National Academy of Sciences of the United States of America, 102: 379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streck A. F. et al. (2009) ‘First Detection of Canine Parvovirus Type 2c in Brazil’, Brazilian Journal of Microbiology, 40: 465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattersall P., Ward D. C. (1976) ‘Rolling Hairpin Model for Replication of Parvovirus and Linear Chromosomal DNA’, Nature, 263: 106–9. [DOI] [PubMed] [Google Scholar]
- Truyen U. et al. (1995) ‘Evolution of the Feline-Subgroup Parvoviruses and the Control of Canine Host Range in Vivo’, Journal of Virology, 69: 4702–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truyen U. et al. (1996) ‘Evolution of Canine Parvovirus Involved Loss and Gain of Feline Host Range’, Virology, 215: 186–9. [DOI] [PubMed] [Google Scholar]
- Tsao J. et al. (1991) ‘The Three-Dimensional Structure of Canine Parvovirus and Its Functional Implications’, American Association for the Advancement of Science Stable, 251: 1456–64. [DOI] [PubMed] [Google Scholar]
- Wang D. et al. (1998) ‘Nonstructural Protein-2 and the Replication of Canine Parvovirus’, Virology, 240: 273–81. [DOI] [PubMed] [Google Scholar]
- Wang H. et al. (2016) ‘Isolation and Sequence Analysis of the Complete NS1 and VP2 Genes of Canine Parvovirus from Domestic Dogs in 2003 and 2004 in China’, Archives of Virology, 161: 385–93. [DOI] [PubMed] [Google Scholar]
- Wang J. et al. (2012) ‘Evidence for Natural Recombination between Mink Enteritis Virus and Canine Parvovirus’, Virology Journal, 9: 252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichert W. et al. (1998) ‘Assaying for Structural Variation in the Parvovirus Capsid and Its Role in Infection’, Virology, 250: 106–17. [DOI] [PubMed] [Google Scholar]
- Ying H. et al. (2009) ‘Expression or Subcellular Targeting of Virus Capsid Proteins with Cloning Genome of a Canine Parvovirus from China’, Research in Veterinary Science, 87: 239–41. [DOI] [PubMed] [Google Scholar]
- Yoon S. H. et al. (2009) ‘Molecular Insights into the Phylogeny of Canine Parvovirus 2 (CPV-2) with Emphasis on Korean Isolates: A Bayesian Approach’, Archives of Virology, 154: 1353–60. [DOI] [PubMed] [Google Scholar]
- Zhao H. et al. (2015) ‘Typing of Canine Parvovirus Strains Circulating in North-East China’, Transboundary and Emerging Diseases, 64: 495–503. [DOI] [PubMed] [Google Scholar]
- Zhou P. et al. (2017) ‘The Genetic Evolution of Canine Parvovirus – a New Perspective’, PLoS One, 12: e0175035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.