

Abstract
Over a decade of genome-wide association studies have made great strides toward the detection of genes and genetic mechanisms underlying complex traits. However, the majority of associated loci reside in non-coding regions that are functionally uncharacterized in general. Now, the availability of large-scale tissue and cell type-specific transcriptome and epigenome data enables us to elucidate how non-coding genetic variants can affect gene expressions and are associated with phenotypic changes. Here we provide an overview of this emerging field in human genomics, summarizing available data resources and state-of-the-art analytic methods to facilitate in-silico prioritization of non-coding regulatory mutations. We also highlight the limitations of current approaches and discuss the direction of much needed future research.
Keywords: functional annotation, regulatory mutation, gene regulation, non-coding variants, in-silico annotation
INTRODUCTION
Recent successes in genome-wide association studies (GWAS) have established that risk for many common and complex diseases in humans is influenced by non-coding genetic mutations that regulate gene expression(Edwards 2013). Specifically, single nucleotide polymorphisms (SNPs), which represent common substitutions of a single nucleotide at a specific base pair position, are at the forefront of such discoveries as most recent GWAS findings represent non-coding SNPs (Figure 1). Moreover, rapid advances in high-throughput sequencing technology now enable us to study extremely rare or even private genome-wide genetic mutations. Despite small sample sizes, a growing list of whole genome sequencing studies have uncovered rare regulatory mutations that increase disease liability(Smedley 2016; Takata 2016).
Figure 1. Functional category of SNPs registered in the GWAS Catalog.
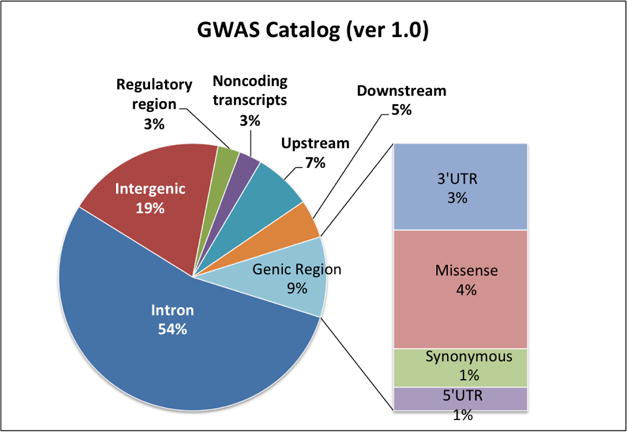
The 3′ untranslated regions (3′ UTRs) of mRNAs contain binding sites for microRNAs and have a role in mRNA stability and translation. 5′ UTRs contain regulatory elements for both transcription and translation stages, such as the 5′‐cap structure, translation initiation motifs and internal ribosome entry sites.
However, the issue of how to best annotate and prioritize functionally relevant non-coding mutations still remains unresolved(Zhang 2015). While much of the human genome consists of introns, pseudo-genes, non-coding RNAs, repetitive segments, and loci with potentially regulatory elements, the precise roles of many of these sequences remain unknown(Taher 2015). In-silico data mining is often considered an inevitable prerequisite to laborious and expensive functional validation. With the enormous scale and the complexity of genetic regulatory mechanisms, most databases and prediction methods are evolving rapidly to characterize and represent the functional impact of potentially deleterious mutations. Yet, functional interpretation of non-coding variants poses a daunting task for many biomedical researchers in the field as it requires integrative analyses of genomic, transcriptomic, and epigenomic data(Degner 2012; Trynka 2013).
In this review, we discuss the principles of and recent progress in annotating non-coding single nucleotide variants (SNVs) with an array of genomics and bioinformatics resources. We start with a brief overview of how regulatory variations confer their effects on gene expression. Specifically, we review the molecular mechanisms of gene regulation and the potential impact of genetic variants in the contexts of transcription, alternative splicing, non-coding RNAs, and epigenetic mechanisms. Next, we introduce two major types of data resources that play fundamental roles in the functional annotation of non-coding variants, the field of comparative genomics, and the use of high-throughput sequencing/assay data. Finally, we describe collective databases that integrate diverse annotation sources. These resources either map variants to functional regulatory elements or provide quantifiable scores representing the functional impact of non-coding mutations on gene regulation. We conclude with a discussion of much needed future research directions.
I. Basic mechanisms in regulation of gene expression
Gene regulation, which involves a broad range of genetic mechanisms that work to increase or decrease the production of specific proteins or RNAs, is a central feature of living organisms. The major steps of gene regulation include transcription, post-transcriptional RNA processing, translation, and post-translational modification. Various genetic regulatory elements work additively, synergistically, or competitively in the above processes depending on specific cell types, developmental periods, and environmental stimuli. Here, we briefly review the basic concepts and mechanisms of gene regulation in the context of non-coding variation studies.
The first step in gene regulation, transcription is coordinated through the precise and intricate interaction of multiple proteins and regulatory genetic elements including promoters, enhancers, and silencers (Figure 2). Promoters are located immediately upstream of transcription start sites; here, the pre-initiation complex (PIC), which includes RNA polymerase II and general transcription factors, binds to and initiates the transcription of a target gene. On average, the core promoter in eukaryotes encompasses about 100 base pairs, while some genes have extended promoter regions (also known as proximal promoters) of up to 1000 base pairs where cell-specific transcription factors can bind.
Figure 2. Schematic model illustrating major regulatory elements.
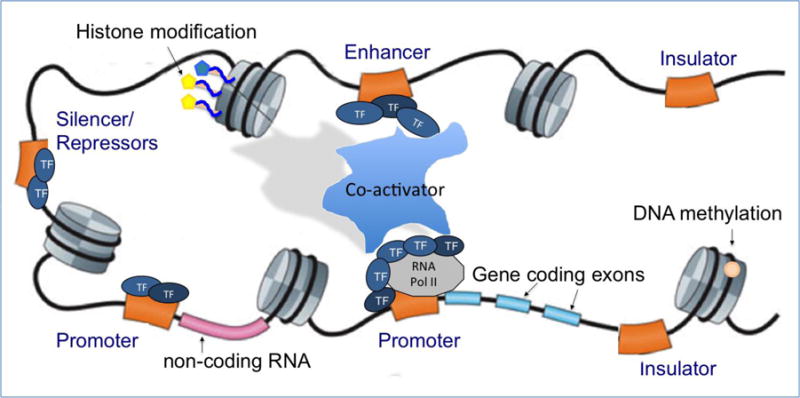
1) transcription factors interacting with RNA polymerase II and the promoter sequence to initiate the transcription of gene coding exons, 2) Co-activators binding to transcription factors to recruit enhancers, 3) histone modifications and DNA methylation, 4) silencers, repressors, and insulators, and 5) non-coding RNA.
There also exist distal DNA regulatory elements, including enhancers, silencers, and insulators, which govern the dynamic control of spatiotemporal gene expression. Enhancers, silencers, and insulators are DNA regulatory sequences (also known as cis-regulatory modules), where a number of transcription factors can bind and regulate expression rates of target genes. Enhancers represent about 50-1500 base pairs of DNA sequences to which transcription factors bind. Unlike promoters, the location of enhancers varies dramatically and may apart a few mega bases from the target genes. A complex of transcription factor and co-activators, mediated by enhancers, induce a conformational change of the chromatin structure, allowing the rapid production of specific genes depending on tissue/cell-type and development-specific contexts. This lies in contrast to co-repressors, which serve to reduce gene expression by attaching to silencers. Insulators are another type of long-range regulatory DNA elements that are typically 300-2000 base pairs. By mediating loop formation and nucleosome modifications, insulators establish boundaries of gene expression and thus prevent unneeded interactions of both enhancers and silencers with promoters(West 2003).
Mutations that occur in any of these key regulatory DNA elements could have broad, over-arching effects on gene regulation. In particular, SNPs that influence gene expression are referred to as expression quantitative trait loci (eQTLs). Nearly 80% of genetic variants that influence gene regulation occur through cis-acting eQTLs, which reside within 1M base pairs from their target genes (Albert 2015; Battle 2014; Wittkopp and Kalay 2011). In particular, transcription factors show differential allelic occupancy, preferentially binding to a specific allele over another allele of an SNP, near the transcription start sites. Cis-eQTLs are often found near genomic sites of such differential allelic occupancy and feature significant enrichment among disease-associated variants(Albert 2015; Battle 2014). For example, non-coding SNP rs522444 is associated with amyotrophic lateral sclerosis (ALS)(Landers et al. 2009). Functional analysis revealed that by altering the SP1 transcription-factor binding sites, the SNP changes the expression of multiple genes in various tissues, including brain and nerve cells. Similarly, non-coding SNP rs7163757, which is associated with type 2 diabetes(Yamauchi et al. 2010) and insulin response(Grarup et al. 2011), is a strong eQTL marker in the adipose tissue. The SNP changes the affinity of NFAT transcription factor binding sites, which are key regulators of glucose and insulin homeostasis(Yang et al. 2006).
A growing number of studies have also reported enriched clustering of disease-susceptibility SNPs in distal regulatory elements (Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014). An interesting example is SNP rs9349379 on the 6p24 region. The locus has been associated with five vascular diseases, including coronary artery disease, migraine headache, cervical artery dissection, fibromuscular dysplasia, and hypertension. In a recent study by Gupta et al.(Gupta et al. 2017), genetic fine-mapping prioritized SNP rs9349379, a common non-coding SNP in the third intron of the PHACTR1 gene as a putative causal variant. Epigenomic profiling revealed the presence of enhancer (H3K27ac) signature at rs9349379 exclusively in aortic artery samples, suggesting that a gene regulatory function for this SNP exists in vascular tissue. CRISPR/Cas9 genome editing in endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) showed that loss of the 88 bp flanking region containing rs9349379 resulted in higher expression of the endothelin-1 (EDN1), a gene located 600 kb upstream of PHACTR1 and its protein product ET1, a potent vasoconstrictor peptide. This combined evidence illustrates an example of distal regulatory SNP (in the enhancer) that could increase genetic risk for five related diseases.
Once the transcription process is initiated, RNA polymerase II moves along the DNA strand and creates a complementary RNA copy. These RNA products undergo extensive modification in eukaryotes including capping, addition of Poly(A) tails, splicing, and transport to the cytoplasm(Nainar 2016). Alternative splicing has drawn particular interest because genetic mutations in splice sites can impinge on the splicing process, and, as a result, exons or introns can be unnecessarily added or removed. Several groups have recently coined the term of splicing quantitative trait loci (sQTLs), which refer to SNPs of which the alleles are associated with the inclusion or loss of certain exons/introns. Most sQTLs are found within or proximal to primary splice sites, but some studies also found their enrichment near 5′ UTRs or transcription start sites, suggesting potential interactions with transcriptional regulation(Battle A 2014). Consistently, sQTLs were reported to alter gene expression and lead to changes in the corresponding phenotypes(Cieply 2015; Monlong et al. 2014). Altered expression of mis-spliced mRNAs has been identified as a causal mechanism in several diseases, including acute myeloid leukemia, spinal muscular atrophy, and Parkinson’s disease(Cieply 2015).
Beyond mRNAs, transcription also produces an abundant amount of non-coding RNAs (Figure 2). Non-coding RNAs (ncRNAs) are functional RNA molecules that are transcribed from DNA but not translated into proteins. ncRNAs regulate gene expression at the transcriptional and post-transcriptional level. Many of these non-coding RNAs are less than 200 nucleotides long, and are thus collectively called small RNAs. MicroRNAs (miRNAs), small interfering RNAs (siRNAs), or small nucleolar RNAs (snoRNAs) belong in this category. Small RNA products perform various regulatory roles including alternative splicing, mRNA turnover, and translation of mRNAs(Parts. L et al. 2012). Moreover, some non-coding RNAs show correlated expression with mRNA synthesis from nearby genes, suggesting that they may directly alter transcription of the target genes by activating promoters, facilitating the adoption of an open chromatin structure, or even binding to other enhancers(Kim 2010; Wang. K 2011). There also exist large non-coding RNAs (lncRNAs) that are longer than 200 nucleotides and often found in genomic sequences that do not encode a protein. Recent GWAS studies have reported that variations in lncRNA expression can influence changes in the levels of protein-coding gene expression(Cao 2014; Popadin. K 2013). lncRNAs can regulate gene transcription through epigenetic regulation (Mercer. T 2009) or by acting as enhancers in a highly tissue specific manner(Popadin. K 2013).
Lastly, epigenetic changes represent heritable differences in gene activity and expression that occur without alterations to the DNA sequence. Two major mechanisms driving epigenetics are DNA methylation and histone modification, both of which govern spatiotemporal gene regulation(Handy Diane 2011). DNA methylation has been extensively studied in recent years. Methylation is predominantly found in CpG sites in the genome, where a cytosine nucleotide is followed by a guanine nucleotide(Bilian Jin 2011). Typically, DNA methyltransferases (DNMTs) convert cytosine to 5-methylcytosine, which has been linked to the silencing of transposable elements and repeat sequences. In humans, 70-80% of all CpG dinucleotides are methylated in heterochromatic regions across tissues, while unmethylated CpGs are mainly confined to CpG islands, typically 200-2000 base pairs, in promoter-proximal regions. These unmethylated CpG sites are also found at certain distal enhancers and contribute to tissue specific gene expression(Taudt. A 2016). Similar to eQTLs, SNPs that alter methylation of genomic sites are called mQTLs(Grundberg E 2013). Interestingly, mQTLs are known to reside more often in enhancers and insulators(Banovich 2014; Maria Gutierrez-Arcelus 2015). How the complex relationship between DNA methylation and gene expression can be illuminated and how such genomic and epigenetic variation together determine cellular phenotypes are important questions of active research.
Histone modifications are critical for the regulation of chromatin structure and function, two key components of transcription and recombination(Burgess 2012). Genomic DNA is tightly packed into chromatin, which consists of protein/DNA complexes called nucleosomes that are coiled with histones. Due to this compact structure, the accessibility of genomic regions dynamically changes based on the state of the nucleosomes, which can be altered significantly through histone modification(Taudt. A 2016). The two most common types of histone modification are acetylation and methylation at histone tails. Histone acetylation is carried out through the modification of the enzyme histone acetyltransferase, which is heavily involved in the regulation of key cellular processes (Eric L. Greer 2012). As a result of acetylation, chromatin loses its condensed structure, allowing for easier transcription. Another key regulatory epigenomic mechanism is histone methylation. Histone methylation occurs when histone methyltransferase (HMT) enzymes add methyl groups to the lysine or arginine residues of histone proteins (Eric L. Greer 2012). Signatures of histone modification have been extensively used to predict DNA elements with potential regulatory function. Regulatory variants affecting histone modifications can thus affect chromatin structure or lead to improper chromosome conformation(Grubert F 2015; Kilpinen H 2013; McVicker G 2013a; McVicker G 2013b; Waszak SM 2015).
II. Comparative genomics data to infer functionally significant regulatory elements
Early efforts to identify functional regulatory elements began with comparative genomics studies(Siepel. A et al. 2005). In principle, functional elements are expected to evolve under negative selection(Liu 2016), and thus comparison of DNA sequences among diverse species could provide direct insights into the biology and evolution of genomic elements(Burgess 2011). Multiple comparisons of human genomes with mouse and dog genomes reveal that at least 5% of the human genome is conserved(Chinwalla et al. 2002; Consortium 2002; Gibbs RA 2004; Lindblad-Toh K 2005), suggesting that there exists a common, base level of likely functional regulatory regions. We review here some key resources that provide evolutionary conserved scores for genetic loci on a genome-wide scale (Table 1).
Table 1.
Comparative genomics data to infer functionally significant regulatory elements
| Program | Mechanisms | Target Regulatory Elements | URL |
|---|---|---|---|
| fitCons | Compares the selective pressure consequences to indicate potential for genomic function | Any Non-Coding Genetic Variant | http://compgen.cshl.edu/fitCons/ |
| GERP++ | Compares p-values in order to predict genetic conservation in terms of rejected substitution (RS), which is the difference between neutral rates of substitution and the observed rate as estimated by maximum likelihood . | TF Binding Sites, Promoters, Enhancers, Silencers | http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html |
| Phastcons/PhyoP | Computes conservation or acceleration p-values based on alignments with respect to a neutral model of evolution | TF Binding Sites, Promoters, Enhancers, Silencers | http://compgen.cshl.edu/phast/index.php |
| SiPHY | Models biased nucleotide substitutions and characterizes substitution patterns from sequence alignments | TF Binding Sites, Promoters, Enhancers, Silencers | http://portals.broadinstitute.org/genome_bio/siphy/ |
| dbPSHP | Combines data from over 15,000 loci from publications and 15 statistical terms to identify genetic regions with potential positive selection | Any Single Nucleotide Variant | http://jjwanglab.org/dbpshp |
The PhastCons resource(Siepel. A et al. 2005) is a component of the widely used PHAST (Phylogenetic Analysis with Space/Time models) package, providing a base-by-base evolutionary conservation score that denotes the probability that a base belongs to a conserved element. PhastCons uses a phylogenetic Hidden Markov Model (HMM), a statistical model in which the system being modeled is assumed to be a Markov process with unobserved states. Despite the base-by-base resolution, the PhastCons score, which ranges from 0 to 1, is calculated as an average of surrounding DNA elements in a multiple alignment; this makes PhastCons better suited for detecting a region of functional DNA elements. In contrast, PhyloP, another component of the PHAST package, measures evolutionary conservation at individual alignment sites to determine if these sites are expected to be conserved or fast-evolving. PhyloP scores are thus useful to evaluate signatures of selection at specific nucleotides or classes of nucleotides.
The GERP++ (Genomic Evolutionary Rate Profiling)(Davydov. E 2010) is another widely used program that quantifies position-specific constraints in terms of rejected substitution (RS). RS is the difference between the expected neutral rates of substitution and the observed rate as estimated by maximum likelihood. GERP++ uses the position-specific RS score to generate candidates of conserved elements and uses the element’s length and score to compute a significance p-value. Users can download pre-computed base-wise scores and predicted elements for the human genome (hg19) on the UCSC genome browser. Compared to PhastCons, GERP++ is known to predict longer and more conserved elements as being potentially functional(Lizio. M 2014).
The dbPSHP (database of recent Positive Selection across Human Populations(Burgess 2012) is another comparative genomics database that aims to help researchers identify, validate and visualize positively selected loci in human evolution. Distinct from other resources, the database has more than 15,000 loci compiled under positive selection from 132 publications. Based on genomic information from the literature, a number of methods were used for measuring positive selection with genotype data from the HapMap3(Sebastian M. Waszak 2015) and the 1000 Genomes(Grubert F 2015) projects. Positive selection skews allelic frequency when compared to a neutral model of evolution, and this can be detected by various methods including the fixation index(Holsinger 2009), increased identity-by-descent (IBD)(Browning 2008), the cross population composite likelihood ratio test (XP-CLR)(Chen 2010), and the cross population extended haplotype homozygosity test (XP-EHH)(Zhong et al. 2011). Other auxiliary information available at dbPSHP includes variant allele/genotype frequency, heterozygosity, and pairwise population differentiation. Like other computational genomics resources, users can download pre-computed datasets from the web server(Burgess 2012).
The fitCons (fitness Consequences of functional annotation)(Gulko B 2015) is distinguished from the aforementioned computational genomics programs as it integrates functional assay data with selective pressures to identify genomic regions with potential function. The fitCons resource first identifies potential regulatory loci based on cell-type-specific functional data and then compares these sites with a known pattern of neutral evolution. The resulting fitCons score for the class and the nucleotide-specific score (0-1) indicate the fraction of genomic positions likely under selection and reveals the probability that it could affect fitness. In the comparison study(Gulko B 2015), fitCons significantly outperformed other methods in predictions for putative cis-regulatory elements.
III. Public resources with high-throughput functional data
Due to rapid advances in high-throughput biotechnology and international collaborative efforts, there has been a dramatic increase in public functional genomics data(Jennifer Harrow et al. 2012). These experimental data, in combination with the comparative genomics resources described earlier, have been essential in the development of numerous secondary databases and algorithms that have facilitated a broader understanding of gene regulation.
The ENCODE (Encyclopedia of DNA Elements) project (https://www.encodeproject.org/)(ENCODE Project Consortium 2012) was initiated in 2003 with the ambitious goal of cataloging all functional elements in the human genome. The pilot phase, which focused on the mapping of about 1% of the human genome, revealed that a substantial portion of the non-protein coding human genome is indeed transcribed. Such transcription activity can be gauged by chromatin accessibility and/or histone modification data whereas the transcribed DNA sequences are evolutionarily constrained in only half of the cases. To date, the ENCODE project has characterized that as much as 80% of the human genome features the signatures of biochemical function1, potentially involved in DNA-protein interactions, DNA methylation, and chromatin accessibility and structure in addition to coding and non-coding transcription. The ENCODE project has also expanded to major model organisms including mice, flies, and worms. Included on the ENCODE website is a “materials and methods” section that summarizes the software tools used to obtain the data and different file formats accepted by the tools, including FASTQ, BAM, bigWIG, and bigBED. There is also the option to upload the data as a bigWIG file and visualize the genomic regions in the UCSC genome browser. The multitude of tools available on the ENCODE portal make it a valuable resource for investigating the genome. One drawback that researchers must keep in mind though is that batch effects may exist, as multiple laboratories have contributed to the ENCODE project, and thus need to be accounted for when analyzing various ENCODE datasets together.
Table 2 summarizes high-throughput experimental data from a variety of functional assays, including ones generated from the ENCODE project(Diehl 2016). Popular tools for capturing open chromosome regions include DNase HyperSensitivity (DHS) assays, ATAC-seq, and FAIRE-seq. DNase footprinting combines deeply sequenced DNase-seq data with transcription factor (TF) motif information and thus can help narrow down the sites for TF occupancy at a high-resolution. ChIP-seq can identify genomic regions where specific transcription factors bind to. These loci are typically implicated in DNA methylation and histone modifications. RNA-seq, RNA-Pet, and CAGE (cap analysis of gene expression) data can measure the amount of transcriptional output. Techniques like 3C, 4C, Capture-C(Hughes 2014; Rao 2014)(Hughes 2014; Rao 2014)(Hughes 2014; Rao 2014)(Hughes 2014; Rao 2014), Hi-C, or ChIA-Pet can assess the 3D interactions of local chromatin looping or long-range chromosomal contact(Heidari 2014).
Table 2.
High-throughput functional genomics and epigenomics data
| Category | Technology | Description of Experimental Approaches | Database/Resource | |
|---|---|---|---|---|
| ENCODE | RoadMap | |||
| Open Chromatin Structure | FAIRE-seq | uses formaldehyde cross linking to segregate DNAs in open chromatin; sensitive to enhancers; | Yes | |
| ATAC-seq | uses mutated hyperactive transposases to identify Dnase I hypersensitive sites | Yes | ||
| MNase-seq | sequences micrococcal nuclease sensitive sites to identify accessible DNA regions | Yes | ||
| DNase-seq | identifies chromatin regions sensitive to cleavage by Dnase 1 enzyme marking trascriptionally active regions | Yes | Yes | |
| DNase-DGF | analyzes dense mapping of DNase1 cleave sites from Dnase-seq and helps identify which proteins bind to the DNA regions | Yes | Yes | |
| Histone Modifications | ChIP-seq | determines whether given chromatin associated transcription factors interact with the DNA | Yes | Yes |
| Transcription Factors & Other DNA Binding Proteins | ChIP-seq | determines whether given chromatin associated transcription factors interact with the DNA | Yes | Yes |
| DNase-DGF | analyzes dense mapping of DNase1 cleave sites from Dnase-seq and help identify which proteins bind to the DNA regions | Yes | Yes | |
| Transcription Levels | RNA-seq | Assesses the levels of mRNA transcripts and RNAs | Yes | Yes |
| smRNA-seq | idenitifes expression levels of small non-coding RNAs | Yes | Yes | |
| CAGE | utilzes the initial 20 nucleotides from 5′ end mRNAs to profile TSS’s as well as promoter usage analysis | Yes | ||
| RNA Protein Interactions | CLIP-seq | uses crosslinking immuno-precipitation to identify RNA-protein interactions and locate RNA modifications. | Yes | |
| iCLIP-seq | extends CLIP techniques to the resolution of individual nucleotides | Yes | ||
| eCLIP-seq | enhancs iCLIP by discarding PCR duplicate reads while maintaing individual nucleotide resolution | Yes | ||
| RIP-chip | uses immunoprecipiation of an RNA binding proteinfor the identification of RNA subsets associated with proteins | Yes | ||
| RIP-seq | utilizes high-throughput sequencing instead of a micro-array to analyze RNA-protein binding | Yes | ||
| 3D Chromatin Interactions | ChIA-PET | uses HI-C and ChIP-seq to detect all interactions medicated by a protein of interest. | Yes | |
| 5C | detects interactions between all restriction fragments within a given region, the region of the which is typically less than a megabase | Yes | ||
| Hi-C | uses high-throughput sequencing to find all possible pairwise interactions between fragments genomewide | Yes | ||
| DNA Methylation | MeDIP-seq | isolates and sequences methylated DNA fragments using an antibody against 5-methycytosine to identify DNA methylation | Yes | Yes |
| Bisulfite-seq | assesses high-resolution methylation data from a small number of reference epi-genomes. | Yes | Yes | |
| RR Bisulfite-seq | uses restriction enzymes in addition to bisulfate to identify regions of the genome high in CpG content | Yes | Yes | |
The FANTOM5 project (http://fantom.gsc.riken.jp/5/)(Kasowski. M 2010) is an international collaborative effort that attempts to determine the regulatory mechanisms for specific cell types in human and mice. FANTOM5 complements ENCODE by using CAGE technology to quantify transcriptional activities of genetic elements such as promoters and enhancers. CAGE produces a snapshot of the 5′ end of mRNAs from biological samples, outputs a short set of nucleotide sequences (tags), and assesses the expression levels of the RNA sequences. In contrast to ENCODE, which uses immortalized cell lines, FANTOM5 uses primary human cells and tissues to analyze the process of initializing and maintaining cell identity. Utilizing CAGE analysis data (in addition to complementary experimental data), FANTOM5 can detect and characterize transcriptional start sites (TSS) for both coding and non-coding transcripts alike. The latest release of the FANTOM5 project includes information about active promoters and enhancers to characterize gene expression in more than 1,000 mouse and human cell lines and tissues(de Hoon 2015; Lizio. M 2014). The next release in the process, FANTOM6, will be based on high throughput screenings of lncRNAs and utilizing CAGE with molecular phenotyping in order to distinguish lncRNA function from the genomic background.
While the ENCODE and FANTOM5 projects aim to catalog functional regulatory elements on the genome, the GTEx (Genotype Tissue Expression) project (http://www.gtexportal.org/home/)(The GTEX Consortium 2013) was initialized with the goal of identifying tissue-specific regulatory eQTLs and elucidating their regulatory mechanisms. By partnering with organ procurement organizations, the GTEx project obtained and consolidated many healthy tissue samples using highly standardized procedures; RNA-seq was conducted, along with SNP genotyping, and eQTL mapping was performed using the GENCODE (v12) annotation(Jennifer Harrow et al. 2012). As of July 2017, GTEx contains 8,555 total tissue samples, 7,051 of which have undergone eQTL analysis. Search options on the GTEx web portal include tools to investigate genetic association using single tissue eQTLs whereas a separate eQTL browser tool allows for comparison of different tissue tracks. There are also query options to look up gene expression as well as to search variants involved in alternative splicing and protein truncation.
After the successful completion of the pilot phase of the ENCODE project and various international initiatives, the NIH Roadmap Epigenomics project (http://www.roadmapepigenomics.org/0)(Bradley E Bernstein 2010) was launched to investigate epigenetic modifications of the human genome. As summarized in Table 1, epigenomic data can be obtained using various next-generation sequencing and assay technologies. DNA methylation was measured using several sequencing methods, including bisulfite sequencing, MethylC-seq, BS-seq, MeDIP-seq, and MRE-seq, while histone modifications were assessed using ChIP-seq with various antibodies. Six major histone modification sites were assessed: H3K4me1, H3K4me3, H3K9me3, H3K9ac, H3K27me3, and H3K36me3. It should be noted that H3K4me3 are often associated with active promoters, whereas H3K27ac are aligned with active promoters and enhancers(Khurana E 2016). Chromatin accessibility was assessed using DNase-seq, and small RNA transcripts were measured using RNA-seq and microarrays. The Human Epigenome Atlas website (http://www.genboree.org/epigenomeatlas/) provides the epigenomic maps of major tissues and cell lines as well as their batch downloads.
Along with the NIH Roadmap Epigenomics project, various epigenomics programs were launched in recent years worldwide. The largest of these projects include Blueprint (http://www.blueprint-epigenome.eu/), the Canadian Epigenetics, Environment, and Health Research Consortium (CEEHRC) (http://www.epigenomes.ca/), and Deep (Das Deutsche Epigenome Programm)(http://www.deutsches-epigenom-programm.de/)(Bae 2013). The International Human Epigenome Consortium (IHEC) aims to unify these various international efforts to better understand how the epigenome is relevant to health and disease. The primary goals of the IHEC include generating the epigenomic reference map of at least 1,000 subjects as well as creating international standards and data sharing policy. The IHEC data portal (http://epigenomesportal.ca/ihec/index.html) currently provides more than 7,000 high-throughput epigenomes and transcriptomes.
Finally, the BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) provides a useful data portal service for researchers interested in transcriptome and epigenomic data analysis. It compiles information from large scale functional genomics research projects, which include genome sequencing and assembly, transcriptome sequencing and expression, epigenomic data, QTL studies, and various functional genomics experimental data(Barret 2012). Primary submission projects are those directly related to the submitted data while umbrella projects may be part of a collaborative effort but have specific methodologies that are distinct from the larger group effort. In addition to browsing and searching the web portal, investigators have access to additional tools and the option to download the entire database of Entrez programming utilities and thereby access the BioProject records.
IV. Public databases integrating various annotation sources for non-coding variants of potential regulatory effects
With the wealth of high-throughput experimental data and comparative genomics resources, in-silico prediction of functional non-coding variants has evolved rapidly. Basic annotation starts with tagging whether non-coding variants are located within specific functional regulatory elements, such as promoters, enhancers, TF binding sites (TFBS), non-coding RNAs (ncRNA), or CpG dinucleotides. We here review some of the public databases that provide such primary annotation for non-coding variants (Table 3; Supplementary Table 1).
Table 3.
Public databases integrating various annotation sources for non-coding variants of potential regulatory effects
| Database | Input | Target Regulatory Elements | Batch Data | Pubmed Reference |
|---|---|---|---|---|
| TRAP | FASTA sequence < 5kb | TFBS | No | 22051799 |
| HaploReg | dbSNP rsIDs, CC, GWAS from NHGRI | Proximal-Distal Regulation, Epigenomics | Yes | 22064851 |
| GWASrap | dbSNP rsIDs, CC, PLINK | Proximal-Distal Regulation, TFBS, miRNA Regulation | Yes | 22801476 |
| RegulomeDB | dbSNP rsIDs, CC, BED, VCF, GFF3 | Proximal-Distal Regulation, TFBS, eQTL, Epigenomics | Yes | 22955989 |
| GWAS3D | dbSNP rsIDs, CC, VCF, PLINK | Proximal-Distal Regulation, Epigenomics | No | 23723249 |
| ChroMoS | dbSNP IDs, GWAS SNPs, VCF | Proximal-Distal Regulation | yes | 23782616 |
| rSNPBase (Now rVarBase) | dbSNP rsIDs, CC | Proximal-Distal Regulation, miRNA Regulation, RNA Binding, eQTL | Yes | 24285297 |
| Enlight | dbSNP rsIDs, CC, BED, HitSpec, HiC Plot | TFBS, eQTL, Epigenomics | No | 25262152 |
| Spidex | dbSNP IDs | Alternative Splicing Variants | yes | 25525159 |
| GREGOR | dbSNP rsIDs, CC, BED | Epigenomics | Yes | 25886982 |
| motifbreakR | dbSNP rsIDs, CC, BED, VCF | TFBS | Yes | 26272984 |
RegulomeDB(Boyle. A et al. 2012) serves as one of the most popular databases that identify the regulatory potential of a variant by incorporating information from high-throughput, experimental datasets such as ENCODE, computational predictions, and manual annotations. Specifically, the database uses a combination of four input data sources to categorize regulatory SNPs into different functional categories; large ChiP-seq datasets, eQTL analyses, manually annotated regions, and chromatin state datasets. It is critical to note that RegulomeDB currently only curates common SNPs with a minor allelic frequency greater than 1%.
In addition to RegulomeDB(Boyle. A et al. 2012), several other gene regulation databases are worthy of attention. The HaploReg resource(Lucas Ward and Kellis 2012) provides functional annotations of non-coding genomic variants including conservation scores, promoter/enhancer histone marks, open chromatin positions, and eQTL markers. Unique features of HaploReg include its haplotype block-based annotation in which the query SNP and nearby SNPs and indels in high linkage disequilibrium (LD r2>0.8) are displayed together. HaploReg also provides transcription factor binding motifs that the input variant could affect. The rSNPBase(Liyuan Guo 2013) is another widely used database that provides a diverse array of functional data for regulatory SNPs. Given a dbSNP identifier, it provides information about whether the variant is located within proximal or distal regulatory sites, its putative engagement with miRNAs or RNA binding proteins, and relevant eQTL data. The TRAP resource(Morgane Thomas-Chollier 2011) is a useful web service by which users can investigate the changes in transcription factor binding affinity given different allelic information. Similarly, the motifbreakR (Simon G. Coetzee 2015) assesses the effects of genetic variants on target TFBSs, whereas the ChroMoS database(Maxim Barenboim 2013) predicts the functional effect of SNPs on chromatin modification.
The Spidex resource(Michael K. K. Leung 2014) focuses on the potential impact of genetic variants on human splicing and provides computational predictions for ~328 million SNPs. The software assesses how the sequence change may affect RNA splicing in humans, including rare or even de novo mutations. Another database for splicing variants is dbNSFP(Liu 2016), which provides functional annotations for non-synonymous and splice-site SNVs at a genome-wide level. The coverage of dbNSFP is notable that the database provides annotations for all possible SNVs that are derived theoretically, thus not restricted to those that have been observed in existing sequencing data.
The latest development in gene regulation resources enables users to directly input the results of GWAS or sequencing data analysis; the output provides annotations and regional plots with functional regulatory information for the input data. For example, the Enlight web service(Yunfei Guo 2014) plots functional genomics data including TFBSs, epigenetic modifications, DNase sensitivity sites, eQTL data, and HiC-seq interaction data on standard regional plots, which facilitates the fine mapping of potentially causal regulatory variants. The GWAS3D service(Mulin Jun Li 2013) also integrates GWAS results but improves over other methods by providing a probability score that measures the likelihood of genetic variants to affect examined phenotypes. This is done using a combination of various functional genomics assay data, sequence motifs, evolutionary conservation scores, and epigenetic information. Another useful tool developed by the same group is SNVrap(Mulin Jun Li 2012), which provides annotation and presentation data for GWAS. The GREGOR resource (Genomic Regulatory Elements and GWAS Overlap algorithm)(Schmidt EM 2015) is a stand-alone software tool that provides enrichment analysis of trait-associated GWAS SNPs using a list of experimentally annotated regulatory loci. Other tools with similar purposes are FORGE (Functional element Overlap analysis of the Results of Genome wide association study Experiments(Dunham et al. 2014) and FunciSNP(Coetzee S 2012).
V. Data resources providing quantitative scores for the deleterious impact of non-coding variants on gene regulation
In the previous section, we introduced a multitude of primary databases that use a combination of functional data and annotation sources to identify non-coding variants residing in potential regulatory elements. Given the genomic locus or the sequence of target mutations, users can crosscheck if a mutation lies in regions of the genome annotated for regulatory elements. Here we focus on a new generation of databases that take this further, providing a unified score that quantifies the deleterious effects of mutations on gene regulation and the potential downstream phenotypic impact (Table 4; Supplementary Table 2).
Table 4.
Data resources providing quantitative scores for the deleterious impact of non-coding variants on gene regulation
| Resource | Training Methods | INDEL | Stand-alone Tools | Web Service | Batch Data Download | Pubmed Reference |
|---|---|---|---|---|---|---|
| Basset | Deep Convolutional Neural Network | No | Python | No | No | 27197224 |
| CADD | Linear Kernel Support Vector Machine | Yes | Python | Yes | Yes | 24487276 |
| CScape | Sequential Learning | No | Python | Yes | Yes | 28912487 |
| DANN | Deep Neural Network | No | Python | No | Yes | 25338716 |
| DanQ | Deep Learning | No | Python | No | No | 27084946 |
| DeepSEA | Deep Convolutional Neural Network | Yes | – | Yes | Yes | 26301843 |
| deltaSVM | Gapped k-mer Support Vector Machine | No | R & C++ | No | No | 26075791 |
| EIGEN | Unsupervised Spectral Learning | No | R | No | Yes | 26727659 |
| FATHMM-indel | Gaussian Kernel Support Vector Machine | Yes | – | Yes | Yes | 28985712 |
| FATHMM-MKL | Multiple Kernel Learning | No | Python, Perl | Yes | Yes | 25583119 |
| FATHMM-XF | Extended Features | No | Python | Yes | Yes | 28968714 |
| FunSeq2 | Weighted Scoring | No | Perl | Yes | Yes | 25273974 |
| GAVIN | Gene-specific Calibration | Yes | Java, MOLGENIS | Yes | No | 28093075 |
| GenoCanyon | Unsupervised Learning | No | R | Yes | Yes | 26015273 |
| GWAVA | Random Forest Classifier | Yes | Python | Yes | Yes | 24487584 |
| LINSIGHT | Probabilistic Modeling/GLM | No | C++ | Yes | Yes | 28288115 |
| PredictSNP2 | Consensus Scoring | No | N/A | Yes | Yes (disease-specific) | 27224906 |
| PRVCS | Composite Likelihood | No | Java, Perl | No | Yes | 27273672 |
| RSVP | Ensemble of Decision Trees | No | C++ | No | Upon Request | 27406314 |
| SNVrap | Integration of 18 Resources | No | N/A | Yes | No | 25308971 |
Many of the scoring systems utilize supervised machine learning methods by contrasting features of simulation or previously identified pathogenic SNVs from the ClinVar or HGMD with carefully selected neutral SNVs. The CADD (Combined Annotation Dependent Depletion)(Kircher M 2014) was one of the earliest genome-wide scoring algorithms designed to integrate various annotation data to assess the deleterious effects of both coding and non-coding variants. It uses a linear kernel support vector machine (SVM), which is trained on two groups of variants: one with evolutionarily derived, likely benign variants and the other with simulated de novo variants labeled as deleterious. Here a major assumption is that under natural selection, observed fixed alleles are depleted of deleterious variants compared to simulated variants. The outputted Phred-scale C score represents the potential deleterious effect of an SNV or an indel. It is also relatively easy to expand new annotations in CADD. With the rapid growth of functional data resources such as the ENCODE project(ENCODE Project Consortium 2012), CADD’s expandability is critical in remaining up-to-date.
Somewhat similar to the CADD is the DANN (Deleterious Annotation using Neural Networks)(Quang D 2015), which was trained with the same features as the CADD but uses a Deep Neural Network (DNN) algorithm. In contrast to classic neural networks that build on the layers of features manually selected by human experts, DNNs automatically identify complex structures in high-dimensional datasets using a general-purpose learning procedure(LeCun et al. 2015). This mechanism thus allows learning of informative and generic features that can generalize beyond training datasets, which represents a key advantage of DNNs compared to other machine learning techniques.
The GWAVA resource (Genome Wide Annotation of Variants)(Ritchie GR 2014) is another functional annotation database that specifically focuses on the assessment of non-coding variants. The GWAVA scoring system accepts chromosomal coordinates as inputs and uses an extended random forest classifier trained with more than 180 variant attributes (e.g., GC content, CpG islands, TFBS, and histone modifications). GWAVA scores are defined as a matrix of scores in which the chromosome locations are listed in the first column and the scores for each specific variant attribute are listed in the following columns. Unlike the CADD and the DANN, the GWAVA provides not only a numerical score of functional impact, but also a binary value of 0 or 1 to designate absolute classification of the functional attribute.
Similar to the GWAVA is the SNVrap(Li M.J. 2015), which takes either a dbSNP ID or a chromosomal coordinate as an input and outputs over 40 variant annotation types. Also provided is an annotation tree, which contains and ranks all scores for each annotation type. The data types include information about the genomic locus (e.g., gene locations, validated/predicted enhancer regions, etc.), disease annotations (e.g., GWAS references, phenotypical relations, published papers, etc.), and functional predictions (e.g., miRNA target site affinities, TFBS affinity predictions, splicing sites predictions, putative protein domains, etc.). The final functional score is assigned to each SNP as a normalization of the various scores obtained from various annotation databases.
After the successful application of these genome-wide scoring systems for regulatory SNVs, various extensions and new additions were proposed in the bioinformatics field. A series of SVM model predictors include deltaSVM(Ghandi M 2014a; Ghandi M 2014b; Lee D and Gorkin DU 2015) and FATHMM-MKL(Shihab 2015), which quantifies the functional effect of variants as changes in the SVM scores of different alleles. FATHMM-MKL has been expanded by two recent developments called CScape(Rogers et al. 2017a) and FATHMM-XF(Rogers et al. 2017b). CScape uses greedy sequential learning with leave-one-chromosome-out cross-validation (LOCO-CV) to identify the optimal features for predicting pathogenic single-point mutations in the cancer genome, whereas FATHMM-XF applies the model with an extended feature set to further enhance prediction accuracy. Scoring systems extending the deep neural network learning include DeepSEA(Zhou J 2015), Basset(Kelley 2016), and DanQ(Quang 2016). The resource DeepSEA(Zhou J 2015) applies deep convolutional neural networks (CNNs) to predict the effects of non-coding variants on TF binding, DNA accessibility, and histone marks using DNA sequence information, whereas Basset focuses on the application of CNNs to the DNase-seq functional assay data in 164 cell types obtained from the ENCODE and the Roadmap Epigenomics Consortium. Another development, DanQ(Quang 2016), proposes a hybrid neural network framework that combines CNNs with bi-directional long short-term memory recurrence to predict the regulatory functional impact of DNA sequences. Finally, RSVP(Peterson 2016) employs an ensemble of decision trees with a diverse list of features. The distinguishing idea of RSVP compared to all aforementioned methods is to include features not only for those directly annotated to the interrogated SNV, but also for those annotated to the genes within whose transcription regulatory region the SNV lies. Incorporating such gene-based features is shown to be key in predicting the effect of variants on the regulation of genes in relation to disease.
Despite steady improvement of these supervised machine-learning methods over the years, the performance of the classifiers still depends largely on a properly labeled training dataset. Unsupervised learning approaches, on the other hand, could be advantageous when such labeled data are limited. Lu et al.(Cieply 2015) developed an unsupervised learning framework called GenoCanyon to predict the functional potential of each position in the human genome. GenoCanyon assumes that there exists a latent class variable indicating the functional status of a given genomic location, and given the functional status, all the 22 different annotations are conditionally independent. Although not being designed as a variant classifier, GenoCanyon provides insights on prediction of known functional regions, and can be generalized to predict tissue-specific functional regions(Stenson PD 2008). Ionita-Laza et al.(Ionita-Laza I 2016) also noted the inaccuracy and the bias of supervised approaches and proposed an unsupervised scoring method called EIGEN to quantify the functional effects of variants. By assuming block-wise conditional independence among functional scores of similar categories (e.g., evolutionary conservation, regulatory information, and allele frequencies), the EIGEN method first quantifies the prediction accuracy of each annotation without labeled training data. The scoring system then accounts for the prediction accuracy for each annotation and outputs a weighted sum of scores. Another tool, LINSIGHT(Huang et al. 2017) is a fast and scalable integrative method for predicting deleterious noncoding variants. LINSIGHT combines a generalized linear model for genomic features with a probabilistic INSIGHT-fitCons model of molecular evolution to predict the fitness consequences of noncoding mutations in the human genome. This type of unsupervised approach fits well for non-coding variants, the vast majority of which have functional impacts that are not clearly understood(Quang 2016). Yet, the improvement in power of this unsupervised scoring system is often marginal for regulatory variants over individual annotation data, which leaves ample room for further development.
While the aforementioned scoring systems incorporate a combination of various high-throughput genomic and epigenomic data with evolutionary conservation or sequence-specific features, some groups have proposed an integrative approach to combine the third-party generated functional scores themselves. This is because individual scores alone have not shown clear sensitivity for the majority of regulatory variants underlying complex traits(Ionita-Laza I 2016). One development of such integrative systems is PredictSNP2(Bendl 2014), which combines six scoring methods: CADD, DANN, FATHMM-MKL, GWAVA, FunSeq2(Lizio. M 2014), and FitCons. The PredictSNP2, however, does not consider dependencies between the integrated methods due to shared data resources. PRVCS is another method that takes an integrative approach for the prediction of functional regulatory variants(Li et al. 2016). This tool integrates eight scoring algorithms: CADD, GWAVA, FunSeq(Khurana E 2013), FunSeq2, GWAS3D, SuRFR(Ryan et al. 2014), DANN and FATHMM-MKL, and computes the composite likelihood of a given variant playing a regulatory role. This composite model takes advantage of possible complementarities among different tools, and is shown to have improved prediction performance compared to individual tools.
Lastly, we highlight recent developments in the scoring systems that focus on tissue/cell-type/disease-specific predictions. These tools directly address that the functional impact of regulatory variants varies dynamically within and across a wide range of tissues and cell types. For instance, GenoSkyline(Stenson PD 2008) extends the GenoCanyon framework by fitting a two-component mixture model of multiple binary epigenetic marks, and calculates the posterior probability of a variant being in the functional class for each tissue and cell types separately. FUN-LDA(Backenroth et al. 2017) is another recently developed method that predicts tissue and cell-type-specific effects of non-coding variants. By using the latent Dirichlet allocation model, this method performs unsupervised modeling of tissue/cell-type specific genetic effects at a genome-wide level. The calculated scores are publicly available for all ENCODE/RoadMap Epigenomics tissues. Another tissue/cell-type prediction method, cepip(Li et al. 2017) builds on the PRVCS framework by integrating cell type-specific chromatin state data along with general functional data. The method computes the probability of a causal regulatory variant by a composite likelihood model in a context-dependent way, demonstrating improved prediction accuracy compared to other cell-type specific methods.
With the emergence of the methods focusing on tissue/cell type specific prediction, some groups have extended the approach to calibrate the pathogenicity of genetic variants for a specific phenotypic outcome. DIVAN(Chen et al. 2016) is a recent feature selection-based ensemble leaning framework that identifies disease-specific non-coding risk variants by leveraging a comprehensive collection of genome-wide epigenomic profiles across multiple cell types and factors, along with other static genomic features. DIVAN incorporates a total of 1806 epigenomic features as the initial feature set, and uses a p-value threshold defined by cross-validation to select informative features. DIVAN identified the depletion of H3K9me3 as the most prominent mark around risk variants, demonstrating the enhanced predictive performance for detecting disease-specific, non-coding risk variants, compared to CADD, GWAVA, GenoCanyon, and EIGEN.
CONCLUSION
Most known risk factors for complex disorders consist of non-coding variants, whose role in gene regulation is only beginning to be understood. In this paper, we reviewed the emerging list of public data resources and state-of-the-art bioinformatics methods to infer how non-coding genetic variation may affect gene regulatory mechanisms relevant to health and disease. In particular, we focused on annotation resources that provide the potential regulatory impact of non-coding variants. The resources offering a unified, quantitative scoring metric are of great practical value to whole genome sequencing studies, which require systematic and computational prioritization of genetic variants.
This emerging field, however, also includes inherent complexities and major challenges for further improvement. First, although many annotation resources provide scoring metrics to quantify the deleterious functional effect of regulatory variants, few of them provide development-specific effects in gene regulation. It is well established that the functional impact of regulatory variants varies dynamically not only within and across a wide range of tissues and cell types but also at different developmental stages. For instance, for early-onset neurodevelopmental disorders, it is essential to consider specific developmental times that are most relevant to the disease pathology. A current norm in functional annotation is, however, to combine all-available high-throughput datasets. To calibrate pathogenicity or predict adverse phenotypic effect, it will be crucial to winnow most informative data in gene regulation relevant to a specific phenotypic outcome.
Secondly, we found few systematic approaches for aggregating the functional impact of a mutation in the context of cellular processes. Mutations and genes do not act in isolation. Indeed, studies have consistently suggested that common and complex disorders result from collaborative effects of multiple genetic alterations that disrupt common bio-molecular function or cellular processes(Gonzalez-Perez A 2013; Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium 2015). Furthermore, regulatory mutations that disrupt highly connected genes in protein–protein interaction networks confer a more deleterious functional impact than those occurring in non-central genes in the network(Khurana E 2013). A promising trend is taking a systems biology approach, which involves the integration of the knowledge of biological pathways and network analyses in the functional annotation and scoring of regulatory mutations.
Similarly, there has been relatively little progress made in the study of insertions/deletions and structural variations in the non-coding genome. This is partly because of technical challenges in variant calling for these complex variant types; repetitive DNA sequences fill in approximately half of the human genome, which specifically complicates the reliable detection of structural variations. Furthermore, there has been a lack of consensus about how to functionally annotate complex variant types. Some existing attempts on predicting functional effects of indels include FATHMM-indel(Ferlaino et al. 2017), CADD, and GAVIN(van der Velde et al. 2017). We anticipate further developments of in-silico functional prediction for complex variant types as sequencing and variant calling technologies advance.
Another active area of research is linking distal regulatory SNPs to their target genes. Unlike promoters, distal regulatory elements can reside as far as several millions of base pairs away(Lettice LA and G 2003) or even on different chromosomes from their target genes. Distal elements also show considerable tissue-specificity(Petretto 2006), rapid evolutionary turnover(Schmidt 2010), and little consensus in regulatory motifs; all of these characteristics make the identification and mapping of distal regulatory elements to their target genes extremely challenging. Nevertheless, more than half of the non-coding variants registered in the GWAS catalog are located at least 20kb from the nearest TSS, and thus are potentially involved in distal regulatory mechanisms(Taher 2015). As we increase general understanding of the complex 3D interactions between distal regulatory elements and their target genes, it will be important to develop advanced analytic approaches that integrate each of these rapidly evolving datasets.
Lastly, we emphasize the importance of unbiased evaluation frameworks established at the community level. Very often, performance results reported for new prediction methods are not comparable due to differences in training/test datasets, parameter settings, and selective presentation of evaluation measures(Vihinen 2012). Benchmark datasets for genetic variations, such as VariBench(Nair 2013), have been proposed, but the use of the resource has been limited in the field. Some initiatives like CAGI (Critical Assessment of Genome Interpretation) (Hoskins 2017) represent a promising direction. The CAGI challenges (https://genomeinterpretation.org) provide valuable opportunities to objectively assess the performance of in-silico methods for predicting the phenotypic impacts of genetic variation. Furthermore, the experiments highlight much-needed innovations and progress in the field, and chart the directions for future efforts. Similarly, we emphasize the importance of standardized vocabularies for functional annotation. Use of common notations, such as variation ontology (http://www.variationontology.org) or sequence ontology (http://www.sequenceontology.org), will facilitate direct comparison of different database resources.”
The coming years offer an exciting opportunity to improve our understanding of the genetic regulatory mechanisms that underlie complex human traits. Along with improvements in computational prioritization approaches, functional and biological validation of non-coding variants will be essential. Genome editing using the CRISPR/Cas9(Shalem 2015) technology, along with patient-derived iPSCs(Paull 2015), will lead the forefront. Our enhanced understanding of non-coding human genome will ultimately help us move towards the era of personalized genomic medicine(Bombard 2014; Burska 2014).
Supplementary Material
Acknowledgments
This work was supported by NIMH grants R00MH101367 (PHL) and U01MH111660 (MJD).
Footnotes
ORC id: 0000-0003-1770-3100
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
It though remains an unresolved question how much of the human genome is “actually” functional. Typically, the mutations of “functional” elements are expected to result in a biological consequence. Since the ENCODE study, comparative genomics studies have suggested that at most 8-25% of the human genome may carry a biological function (Graur 2017; Rands et al. 2014). Refer to (Chi 2016) for further discussion of this issue.
References
- Albert F, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Reviews Genet. 2015;16:197–212. doi: 10.1038/nrg3891. [DOI] [PubMed] [Google Scholar]
- Backenroth D, et al. FUN-LDA: A latent Dirichlet allocation model for predicting tissue-specific functional effects of noncoding variation. bioRxiv. 2017:069229. doi: 10.1016/j.ajhg.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae J. Perspectives of International Human Epigenome Consortium. Genomics & Informatics. 2013;11:7–14. doi: 10.5808/GI.2013.11.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banovich N, Lan X, McVicker G, van de Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK, Gilad Y. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 2014;10:e1004663. doi: 10.1371/journal.pgen.1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barret T, et al. BioProject and BioSample databases at NCBI:Facilitating capture and organization of metadata. Nucleic Acids Research. 2012;40:d57–d63. doi: 10.1093/nar/gkr1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Mostafavi S, Zhu X, Potash JB, Weissman MM, McCormick C, Haudenschild CD, Beckman KB, Shi J, Mei R, Urban AE, Montgomery SB, Levinson DF, Koller D. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle AMS, Zhu X, Potash JB, Weissman MM, McCormick C, Haudenschild CD, Beckman KB, Shi J, Mei R, Urban AE. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome research. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Damborsky J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PloS Computational biology. 2014;10 doi: 10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilian Jin YL, Robertson Keith D. DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes & Cancer. 2011;2:607–617. doi: 10.1177/1947601910393957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombard Y, Rozmovits L, Trudeau M, Leighl NB, Deal K, Marshall DA. Access to personalized medicine: factors influencing the use and value of gene expression profiling in breast cancer treatment. Curr Oncol. 2014;21:e426–433. doi: 10.3747/co.21.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AELH, Hariharan Manoj, Cheng Yong, Schaub Marc A, Maya Kasowski KJK, Park Julie, Hitz Benjamin C, Weng Shuai, Michael Cherry J, S M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Research. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein Bradley EJAS, Costello Joseph F, Ren Bing, Milosavljevic Aleksandar, Meissner Alexander, Kellis Manolis, Marra Marco A, Beaudet Arthur L, Ecker Joseph R, Farnham Peggy J, Hirst Martin, Lander Eric S, Mikkelsen Tarjei S, Thomson James A. The NIH Roadmap Epigenomics Mapping Consortium. Nature Biotechnology. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR. Estimation of pairwise identity by descent from dense genetic marker data in a population sample of haplotypes. Genetics. 2008;178:2123–2132. doi: 10.1534/genetics.107.084624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DJ. Comparative genomics: Mammalian alignments reveal human functional elements. Nature Review Genetics. 2011;12:806–807. doi: 10.1038/nrg3112. [DOI] [PubMed] [Google Scholar]
- Burgess DJ. Cancer genomics: Histone modification at the gene level. Nature Review Genetics. 2012;12:153–156. doi: 10.1038/nrc3233. [DOI] [PubMed] [Google Scholar]
- Burska A, Roget K, Blits M, Soto Gomez L, van de Loo F, Hazelwood LD, Verweij CL, Rowe A, Goulielmos GN, van Baarsen LG, Ponchel F. Gene expression analysis in RA: towards personalized medicine. Pharmacogenomics J. 2014;14:93–106. doi: 10.1038/tpj.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J. The functional role of long non-coding RNAs and epigenetics. BIological Procedures Online. 2014;16:1. doi: 10.1186/1480-9222-16-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Patterson Nick, Reich David. Population differentiation as a test for selective sweeps. Genome research. 2010;20:393–402. doi: 10.1101/gr.100545.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Jin P, Qin ZS. DIVAN: accurate identification of non-coding disease-specific risk variants using multi-omics profiles. Genome biology. 2016;17:252. doi: 10.1186/s13059-016-1112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi KR. The dark side of the human genome. Nature. 2016;538:275–277. doi: 10.1038/538275a. [DOI] [PubMed] [Google Scholar]
- Chinwalla AT, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Cieply B, Carstens Russ P. Functional roles of alternative splicing factors in human disease. Wiley Interdisciplinary Reviews: RNA. 2015;6:311–326. doi: 10.1002/wrna.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee SRS, Berman B, Coetzee G, Noushmehr H. FunciSNP: An R/Bioconductor Tool Integrating Functional Non-coding Datasets with Genetic Association Studies to Identify Candidate Regulatory SNPs. Nucleic Acids Research. 2012 doi: 10.1093/nar/gks542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium MGS. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Davydov EDLG, Sirota Marina, Cooper Gregory M, Sidow Arend, Batzoglou Serafim. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++ PLOS Computational Biology. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon M, Shin JW, Carninci P. Paradigm shifts in genomics through the FANTOM projects. Mamm Genome. 2015;26:1–12. doi: 10.1007/s00335-015-9593-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl A, Boyle AP. Deciphering ENCODE. Trends Genet. 2016;32:238–249. doi: 10.1016/j.tig.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Dunham I, Kulesha E, Iotchkova V, Morganella S, Birney E. FORGE: A tool to discover cell specific enrichments of GWAS associated SNPs in regulatory regions. 2014 Preprint at http://biorxiv.org/content/early/2014/12/20/013045.
- Edwards S, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eric L, Greer YS. Histone methylation: a dynamic mark in health, disease and inheritance. Nature Review Genetics. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlaino M, Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. An integrative approach to predicting the functional effects of small indels in non-coding regions of the human genome. BMC bioinformatics. 2017;18:442. doi: 10.1186/s12859-017-1862-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi MLD, Mohammad-Noori M, Beer MA. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol. 2014a;10 doi: 10.1371/journal.pcbi.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi MLD, Mohammad-Noori M, Beer MA. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol. 2014b;10:e1003711. doi: 10.1371/journal.pcbi.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RAWG, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:492–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez AMV, Reva B, Ritchie GR, Creixell P, Karchin R, Vazquez M, Fink JL, Kassahn KS, Pearson JV, Bader GD, Boutros PC, Muthuswamy L, Ouellette BF, Reimand J, Linding R, Shibata T, Valencia A, Butler A, Dronov S, Flicek P, Shannon NB, Carter H, Ding L, Sander C, Stuart JM, Stein LD, Lopez-Bigas N, International Cancer Genome Consortium Mutation Pathways and Consequences Subgroup of the Bioinformatics Analyses Working Group Computational approaches to identify functional genetic variants in cancer genomes. Nat Methods. 2013;10:723–729. doi: 10.1038/nmeth.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grarup N, et al. The diabetogenic VPS13C/C2CD4A/C2CD4B rs7172432 variant impairs glucose-stimulated insulin response in 5,722 non-diabetic Danish individuals. Diabetologia. 2011;54:789–794. doi: 10.1007/s00125-010-2031-2. [DOI] [PubMed] [Google Scholar]
- Graur D. An upper limit on the functional fraction of the human genome. Genome biology and evolution. 2017;9:1880–1885. doi: 10.1093/gbe/evx121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubert FZJ, Kasowski M, Ursu O, Spacek DV, Martin AR, Greenside P, Srivas R, Phanstiel DH, Pekowska A, Heidari N, Euskirchen G, Huber W, Pritchard JK, Bustamante CD, Steinmetz LM, Kundaje A, Snyder M. Genetic Control of Chromatin States in Humans Involves Local and Distal Chromosomal Interactions. Cell. 2015;162:1051–1065. doi: 10.1016/j.cell.2015.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundberg EME, Sandling JK, Hedman AK, Keildson S, Buli A, Busche S, Yuan W, Nisbet J, Sekowska M, Wilk A, Barrett A, Small KS, Ge B, Caron M, Shin SY, Multiple Tissue Expression Resource Consortium. Lathrop M, Dermitzakis ET, McCarthy MI, Spector TD, Bell JT, Deloukas P. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet. 2013;93:876–890. doi: 10.1016/j.ajhg.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulko BHM, Gronau I, Siepel A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat Genetics. 2015;47:276–283. doi: 10.1038/ng.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RM, et al. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell. 2017;170:522–533.e515. doi: 10.1016/j.cell.2017.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy Diane RC, Loscalzo Joseph. Epigenetic Modifications: Basic Mechanisms and Role in Cardiovascular Disease. NIH. 2011;123:2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidari N, Phanstiel DH, He C, Grubert F, Jahanbani F, Kasowski M, Zhang MQ, Snyder MP. Genomewide map of regulatory interactions in the human genome. Genome Research. 2014;24:1905–1917. doi: 10.1101/gr.176586.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger KE, Weir Bruce S. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nature Reviews Genetics. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins R, Repo S, Barsky D, Andreoletti G, Moult J, Brenner SE. Reports from CAGI: The Critical Assessment of Genome Interpretation. Human Mutation. 2017;38:1039–1041. doi: 10.1002/humu.23290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-F, Gulko B, Siepel A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nature genetics. 2017;49:618–624. doi: 10.1038/ng.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, Higgs DR. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genetics. 2014;46:205–212. doi: 10.1038/ng.2871. [DOI] [PubMed] [Google Scholar]
- Ionita-Laza IMK, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nature genetics. 2016;48:20. doi: 10.1038/ng.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennifer Harrow AF, Gonzalez Jose M, Tapanari Electra, et al. GENCODE: The reference human genome annotation for ENCODE project. Genome Research. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski MFG, Heffelfinger Christopher, Hariharan Manoj, Asabere Akwasi, Waszak Sebastian M, Habegger Lukas, Rozowsky Joel, Shi Minyi, Urban Alexander E, Hong Mi-Young, Karczewski Konrad J, Huber Wolfgang, Weissman Sherman M, Gerstein Mark B, Korbel Jan O, Snyder Michael. Variation in Transcription Factor Binding Among Humans. Science. 2010;328:232–235. doi: 10.1126/science.1183621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Research. 2016;26:990–999. doi: 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana EFY, Chakravarty D, Demichelis F, Rubin MA, Gerstein M. Role of non-coding sequence variants in cancer. Nat Rev Genet. 2016;17:93–108. doi: 10.1038/nrg.2015.17. [DOI] [PubMed] [Google Scholar]
- Khurana EFY, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, Das J, Abyzov A, Balasubramanian S, Beal K, Chakravarty D, Challis D, Chen Y, Clarke D, Clarke L, Cunningham F, Evani US, Flicek P, Fragoza R, Garrison E, Gibbs R, Gümüs ZH, Herrero J, Kitabayashi N, Kong Y, Lage K, Liluashvili V, Lipkin SM, MacArthur DG, Marth G, Muzny D, Pers TH, Ritchie GR, Rosenfeld JA, Sisu C, Wei X, Wilson M, Xue Y, Yu F, 1000 Genomes Project Consortium. Dermitzakis ET, Yu H, Rubin MA, Tyler-Smith C, Gerstein M. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science. 2013:342. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen HWS, Gschwind AR, Raghav SK, Witwicki RM, Orioli A, Migliavacca E, Wiederkehr M, Gutierrez-Arcelus M, Panousis NI, Yurovsky A. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science. 2013;342:744–747. doi: 10.1126/science.1242463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher MWD, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014;46:5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landers JE, et al. Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences. 2009;106:9004–9009. doi: 10.1073/pnas.0812937106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521:436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- Lee D, Gorkin DUBM, Strober BJ, Asoni AL, McCallion AS, Beer MA. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet. 2015:47. doi: 10.1038/ng.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice LAHS, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar GHR, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- Li MJWJ. Current trend of annotating single nucleotide variation in humans–A case study on SNVrap. Methods. 2015;79–80:32–40. doi: 10.1016/j.ymeth.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Li MJ, et al. cepip: context-dependent epigenomic weighting for prioritization of regulatory variants and disease-associated genes. Genome biology. 2017;18:52. doi: 10.1186/s13059-017-1177-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MJ, et al. Predicting regulatory variants with composite statistic. Bioinformatics. 2016;32:2729–2736. doi: 10.1093/bioinformatics/btw288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh KWC, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human non-synonymous and splice site SNVs. Hum Mutation. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyuan Guo YD, Chang Suhua, Zhang Kunlin, Wang Jing. rSNPBase: a database for curated regulatory SNPs. Nucleic Acids Research. 2013;42:d1033–1039. doi: 10.1093/nar/gkt1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizio MJH, Shimoji Hisashi, Severin Jessica, Kasukawa Takeya, Sahin Serkan, Abugessaisa Imad, Fukuda Shiro, Hori Fumi, Ishikawa-Kato Sachi, Mungall Christopher J, Arner Erik, Baillie J Kenneth, Bertin Nicolas, Bono Hidemasa, de Hoon Michiel, Diehl Alexander D, Dimont Emmanuel, Freeman Tom C, Fujieda Kaori, Hide Winston, Kaliyaperumal Rajaram, Katayama Toshiaki, Lassmann Timo, Meehan Terrence F, Nishikata Koro, Ono Hiromasa, Rehli Michael, Sandelin Albin, Schultes Erik A, ’t Hoen Peter AC, Tatum Zuotian, Thompson Mark, Toyoda Tetsuro, Wright Derek W, Daub Carsten O, Itoh Masayoshi, Carninci Piero, Hayashizaki Yoshihide, Forrest Alistair RR, Kawaji Hideya, the FANTOM consortium Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biology. 2014;16:1. doi: 10.1186/s13059-014-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas Ward, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Research. 2012;40:d930–934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maria Gutierrez-Arcelus HO, Lappalainen Tuuli, Montgomery Stephen B, Buil Alfonso, Yurovsky Alisa, Bryois Julien, Padioleau Ismael, Romano Luciana, Planchon Alexandra, Falconnet Emilie, Bielser Deborah, Gagnebin Maryline, Giger Thomas, Borel Christelle, Letourneau Audrey, Makrythanasis Periklis, Guipponi Michel, Gehrig Corinne, Antonarakis StylianosE, Dermitzakis EmmanouilT. Tissue-Specific Effects of Genetic and Epigenetic Variation on Gene Regulation and Splicing. PLOS Genetics. 2015;11:e1004958. doi: 10.1371/journal.pgen.1004958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxim Barenboim TM. ChroMoS: an integrated web tool for SNP classification, prioritization and functional interpretation. Bioinformatics. 2013;29:2197–2198. doi: 10.1093/bioinformatics/btt356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G, vd G B, Degner JF, Cain CE, Banovich NE, Raj A, Lewellen N, Myrthil M, Gilad Y, Pritchard JK. Identification of genetic variants that affect histone modifications in human cells. Science. 2013a;342:747–749. doi: 10.1126/science.1242429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G, vd G B, Degner JF, Cain CE, Banovich NE, Raj A, Lewellen N, Myrthil M, Gilad Y, Pritchard JK. Identification of genetic variants that affect histone modifications in human cells. Science. 2013b;342:747–749. doi: 10.1126/science.1242429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer T, D ME, M JS. Long non-coding RNAs: insights into functions. Nature Review Genetics. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Michael KK, Leung HYX, Lee Leo J, Frey Brendan J. Deep learning of the tissue-regulated splicing code. Bioinformatics. 2014;30:121–129. doi: 10.1093/bioinformatics/btu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monlong J, Calvo M, Ferreira PG, Guigo R. Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat Commun. 2014;5:4698. doi: 10.1038/ncomms5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgane Thomas-Chollier AH, Heinig Matthias, O’Keeffe Sean, Masri Nassim El, Roider Helge G, Manke Thomas, Vingron Martin. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nature Protocols. 2011;6:1860–1869. doi: 10.1038/nprot.2011.409. [DOI] [PubMed] [Google Scholar]
- Li Mulin Jun, Wang Lily Yan, Xia Zhengyuan, Sham Pak Chung, Wang Junwen. GWAS3D: detecting human regulatory variants by integrative analysis of genome-wide associations, chromosome interactions and histone modifications. Nucleic Acids Research. 2013:gkt456. doi: 10.1093/nar/gkt456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Mulin Jun, S PC, Wang Junwen. Genetic variant representation, annotation and prioritization in the post-GWAS era. Cell Research. 2012;22:1505–1508. doi: 10.1038/cr.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nainar S, Marshall PR, Tyler CR, Spitale RC, Bredy TW. Evolving insights into RNA modifications and their functional diversity in the brain. Nature Neuroscience. 2016;19:1292–1298. doi: 10.1038/nn.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair P, Vihinen M. A Benchmark Database for Variations. Human Mutation. 2013;34:42–49. doi: 10.1002/humu.22204. [DOI] [PubMed] [Google Scholar]
- Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium. O’Dushlaine C, Rossin L, Lee PH, Duncan L, Parikshak NN, Newhouse S, Ripke S, Neale BM, Holmans Peter A, Breen Gerome. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18:199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts LAsKH, Keildson Sarah, Knights Andrew J, Abreu-Goodger Cei, van de Bunt Martijn, Guerra-Assunc José Afonso, Bartonicek Nenad, van Dongen Stijn, Reedik Mägi JN, Barrett MR, Nica Alexandra C, Quail Michael A, Small Kerrin S, Glass Daniel, Enright Anton J, Winn John, MuTHER Consortium. Deloukas Panos, Dermitzakis Emmanouil T, McCarthy Mark I, Spector Timothy D, Durbin Richard, L CM. Extent, Causes, and Consequences of Small RNA Expression Variation in Human Adipose Tissue. POS Genetics. 2012;8:e1002704. doi: 10.1371/journal.pgen.1002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, Tsankov A, Shang L, Krumholz K, Jagadeesan P, Woodard CM, Sun B, Vilboux T, Zimmer M, Forero E, Moroziewicz DN, Martinez H, Malicdan MC, Weiss KA, Vensand LB, Dusenberry CR, Polus H, Sy KT, Kahler DJ, Gahl WA, Solomon SL, Chang S, Meissner A, Eggan K, Noggle SA. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods. 2015;12:885–892. doi: 10.1038/nmeth.3507. [DOI] [PubMed] [Google Scholar]
- Peterson TA, Mort M, Cooper DN, Radivojac P, Kann MG, Mooney SD. Regulatory Single-Nucleotide Variant Predictor Increases Predictive Performance of Functional Regulatory Variants. Human Mutation. 2016;37:1137–1143. doi: 10.1002/humu.23049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petretto E, Mangion J, Dickens NJ, Cook SA, Kumaran MK, Lu H, Fischer J, Maatz H, Kren V, Pravenec M, Hubner N, Aitman TJ. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet. 2006;2:e172. doi: 10.1371/journal.pgen.0020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popadin KMG-A, Dermitzakis Emmanouil T, Antonaraki Stylianos E. Genetic and Epigenetic Regulation of Human lincRNA Gene Expression. The American Journal of Human Genetics. 2013;93:1015–1026. doi: 10.1016/j.ajhg.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D, Xie X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Research. 2016:44. doi: 10.1093/nar/gkw226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang DCY, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:3. doi: 10.1093/bioinformatics/btu703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rands CM, Meader S, Ponting CP, Lunter G. 8.2% of the human genome is constrained: variation in rates of turnover across functional element classes in the human lineage. PLoS genetics. 2014;10:e1004525. doi: 10.1371/journal.pgen.1004525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie GRDI, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11:6. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MF, Shihab HA, Gaunt TR, Campbell C. CScape: a tool for predicting oncogenic single-point mutations in the cancer genome. Scientific Reports. 2017a;7:11597. doi: 10.1038/s41598-017-11746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. 2017b doi: 10.1093/bioinformatics/btx536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan NM, Morris SW, Porteous DJ, Taylor MS, Evans KL. SuRFing the genomics wave: an R package for prioritising SNPs by functionality. Genome medicine. 2014;6:79. doi: 10.1186/s13073-014-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Ripke S, Neale BM, Corvin A, Walters JT, Farh KH, Holmans PA, Lee P, Bulik-Sullivan B, Collier DA, Huang H, Pers TH, Agartz I, Agerbo E, Albus M, Alexander M, Amin F, Bacanu SA, Begemann M, Belliveau RA, Jr, Bene J, Bergen SE, Bevilacqua E, Bigdeli TB, Black DW, Bruggeman R, Buccola NG, Buckner RL, Byerley W, Cahn W, Cai G, Campion D, Cantor RM, Carr VJ, Carrera N, Catts SV, Chambert KD, Chan RC, Chen RY, Chen EY, Cheng W, Cheung EF, Chong SA, Cloninger CR, Cohen D, Cohen N, Cormican P, Craddock N, Crowley JJ, Curtis D, Davidson M, Davis KL, Degenhardt F, Del Favero J, Demontis D, Dikeos D, Dinan T, Djurovic S, Donohoe G, Drapeau E, Duan J, Dudbridge F, Durmishi N, Eichhammer P, Eriksson J, Escott-Price V, Essioux L, Fanous AH, Farrell MS, Frank J, Franke L, Freedman R, Freimer NB, Friedl M, Friedman JI, Fromer M, Genovese G, Georgieva L, Giegling I, Giusti-Rodríguez P, Godard S, Goldstein JI, Golimbet V, Gopal S, Gratten J, de Haan L, Hammer C, Hamshere ML, Hansen M, Hansen T, Haroutunian V, Hartmann AM, Henskens FA, Herms S, Hirschhorn JN, Hoffmann P, Hofman A, Hollegaard MV, Hougaard DM, Ikeda M, Joa I, Julià A, Kahn RS, Kalaydjieva L, Karachanak-Yankova S, Karjalainen J, Kavanagh D, Keller MC, Kennedy JL, Khrunin A, Kim Y, Klovins J, Knowles JA, Konte B, Kucinskas V, Ausrele Kucinskiene Z, Kuzelova-Ptackova H, Kähler AK, Laurent C, Keong JL, Lee SH, Legge SE, Lerer B, Li M, Li T, Liang KY, Lieberman J, Limborska S, Loughland CM, Lubinski J, Lönnqvist J, Macek M, Jr, Magnusson PK, Maher BS, Maier W, Mallet J, Marsal S, Mattheisen M, Mattingsdal M, McCarley RW, McDonald C, McIntosh AM, Meier S, Meijer CJ, Melegh B, Melle I, Mesholam-Gately RI, Metspalu A, Michie PT, Milani L, Milanova V, Mokrab Y, Morris DW, Mors O, Murphy KC, Murray RM, Myin-Germeys I, Müller-Myhsok B, Nelis M, Nenadic I, Nertney DA, Nestadt G, Nicodemus KK, Nikitina-Zake L, Nisenbaum L, Nordin A, O’Callaghan E, O’Dushlaine C, O’Neill FA, Oh SY, Olincy A, Olsen L, Van Os J, Pantelis C, Papadimitriou GN, Papiol S, Parkhomenko E, Pato MT, Paunio T, Pejovic-Milovancevic M, Perkins DO, Pietiläinen O, Pimm J, Pocklington AJ, Powell J, Price A, Pulver AE, Purcell SM, Quested D, Rasmussen HB, Reichenberg A, Reimers MA, Richards AL, Roffman JL, Roussos P, Ruderfer DM, Salomaa V, Sanders AR, Schall U, Schubert CR, Schulze TG, Schwab SG, Scolnick EM, Scott RJ, Seidman LJ, Shi J, Sigurdsson E, Silagadze T, Silverman JM, Sim K, Slominsky P, Smoller JW, So HC, Spencer CA, Stahl EA, Stefansson H, Steinberg S, Stogmann E, Straub RE, Strengman E, Strohmaier J, Stroup TS, Subramaniam M, Suvisaari J, Svrakic DM, Szatkiewicz JP, Söderman E, Thirumalai S, Toncheva D, Tosato S, Veijola J, Waddington J, Walsh D, Wang D, Wang Q, Webb BT, Weiser M, Wildenauer DB, Williams NM, Williams S, Witt SH, Wolen AR, Wong EH, Wormley BK, Xi HS, Zai CC, Zheng X, Zimprich F, Wray NR, Stefansson K, Visscher PM, Adolfsson R, Andreassen OA, Blackwood DH, Bramon E, Buxbaum JD, Børglum AD, Cichon S, Darvasi A, Domenici E, Ehrenreich H, Esko T, Gejman PV, Gill M, Gurling H, Hultman CM, Iwata N, Jablensky AV, Jönsson EG, Kendler KS, Kirov G, Knight J, Lencz T, Levinson DF, Li QS, Liu J, Malhotra AK, McCarroll SA, McQuillin A, Moran JL, Mortensen PB, Mowry BJ, Nöthen MM, Ophoff RA, Owen MJ, Palotie A, Pato CN, Petryshen TL, Posthuma D, Rietschel M, Riley BP, Rujescu D, Sham PC, Sklar P, St Clair D, Weinberger DR, Wendland JR, Werge T, Daly MJ, Sullivan PF, O’Donovan MC. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Odom DT. Five vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EMZJ, Zhou W, Chen J, Mohlke KL, Chen YE, Willer CJ. GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics. 2015;31:2601–2606. doi: 10.1093/bioinformatics/btv201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian M, Waszak OD, Gschwind Andreas R, Kilpinen Helena, Raghav Sunil K, Witwicki Robert M, Orioli Andrea, Wiederkehr Michael, Panousis Nikolaos I, Yurovsky Alisa, Romano-Palumbo Luciana, Planchon Alexandra, Bielser Deborah, Padioleau Ismael, Udin Gilles, Thurnheer Sarah, Hacker David, Hernandez Nouria, Reymond Alexandre, Deplancke Bart, Dermitzakis Emmanouil T. Population Variation and Genetic Control of Modular Chromatin Architecture in Humans. Cell. 2015;162:1039–1050. doi: 10.1016/j.cell.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nature Reviews Genetics. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day INM, Campbell C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31:1536–1543. doi: 10.1093/bioinformatics/btv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel AGB, Pedersen Jakob S, Hinrichs Angie S, Hou Minmei, Kate Rosenbloom HC, Spieth John, Hillier LaDeana W, Stephen Richards GMW, Wilson Richard K, Gibbs Richard A, Kent W James, M W, Haussler David. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Research. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon G, Coetzee GAC, Hazelett Dennis J. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015:btv470. doi: 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Schubach M, Jacobsen JO, Köhler S, Zemojtel T, Spielmann M, Jäger M, Hochheiser H, Washington NL, McMurry JA, Haendel MA, Mungall CJ, Lewis SE, Groza T, Valentini G, Robinson PN. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am J Hum Genet. 2016;99:595–606. doi: 10.1016/j.ajhg.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PDBE, Howells K, Phillips A, Mort M, Cooper DN. Human Gene Mutation Database: towards a comprehensive central mutation database. Journal of medical genetics. 2008;45:124–126. doi: 10.1136/jmg.2007.055210. [DOI] [PubMed] [Google Scholar]
- Taher L, Narlikar L, Ovcharenko I. Identification and computational analysis of gene regulatory elements. Cold Spring Harb Protoc. 2015;1:24–51. doi: 10.1101/pdb.top083642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata A, Ionita-Laza I, Gogos JA, Xu B, Karayiorgou M. De novo synonymous mutations in regulatory elements contribute to the genetic etiology of autism and schizophrenia. Neuron. 2016;89:940–947. doi: 10.1016/j.neuron.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taudt A, C-T M, J F. Genetic sources of population epigenomic variation. Nature Reviews Genetics. 2016;17:319–332. doi: 10.1038/nrg.2016.45. [DOI] [PubMed] [Google Scholar]
- The GTEX Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, Raychaudhuri S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature Genetics. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velde KJ, et al. GAVIN: Gene-Aware Variant INterpretation for medical sequencing Genome biology. 2017;18:6. doi: 10.1186/s13059-016-1141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genomics. 2012;13:S2. doi: 10.1186/1471-2164-13-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KYWY, Liu Bo, Sanyal Amartya, Corces-Zimmerman Ryan, Chen Yong, Bryan R, Protacio Lajoie Angeline, Flynn Ryan A, Gupta Rajnish A, Wysocka Joanna, Lei Ming, Dekker Job, Helms Ill A, Chang Howard Y. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waszak SMDO, Gschwind AR, Kilpinen H, Raghav SK, Witwicki RM, Orioli A, Wiederkehr M, Panousis NI, Yurovsky A, Romano-Palumbo L, Planchon A, Bielser D, Padioleau I, Udin G, Thurnheer S, Hacker D, Hernandez N, Reymond A, Deplancke B, Dermitzakis ET. Population Variation and Genetic Control of Modular Chromatin Architecture in Humans. Cell. 2015;162:1039–1050. doi: 10.1016/j.cell.2015.08.001. [DOI] [PubMed] [Google Scholar]
- West A, Gaszner M, Felsenfeld G. Insulators: many functions, many mechanisms. Genes & Dev. 2003;16:271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nature reviews Genetics. 2011;13:59–69. doi: 10.1038/nrg3095. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, et al. A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nature genetics. 2010;42:864–868. doi: 10.1038/ng.660. [DOI] [PubMed] [Google Scholar]
- Yang TT, et al. Role of transcription factor NFAT in glucose and insulin homeostasis. Molecular and cellular biology. 2006;26:7372–7387. doi: 10.1128/MCB.00580-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunfei Guo DVC, Wang Kai. Enlight: web-based integration of GWAS results with biological annotations. Bioinformatics. 2014;31:275–276. doi: 10.1093/bioinformatics/btu639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Lupski JR. Non-coding genetic variants in human disease. Hum Mol Genet. 2015;24:R102–R110. doi: 10.1093/hmg/ddv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Zhang Y, Lange K, Fan R. A cross-population extended haplotype-based homozygosity score test to detect positive selection in genome-wide scans. Statistics and its interface. 2011;4:51. doi: 10.4310/SII.2011.v4.n1.a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JTO. Predicting effects of noncoding variants with deep learning-based sequence model. Nature methods. 2015;12:931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.