

Abstract
The increasing global prevalence of obesity and its associated disorders point to an urgent need for the development of novel and effective therapeutic strategies that induce healthy weight loss. Obesity is characterized by hyperleptinemia and central leptin resistance. In an attempt to identify compounds that could reverse leptin resistance and thus promote weight loss, we analyzed a library of small molecules with mRNA expression profiles similar to that of celastrol, a naturally-occurring compound we previously identified as a leptin sensitizer. By this process we identified another natural compound, withaferin A, that also acts as a leptin sensitizer. We found that withaferin A treatment of diet-induced obese mice resulted in a 20-25% reduction of body weight, while also decreasing obesity-associated abnormalities including hepatic steatosis. Withaferin A marginally affects the body weight of ob/ob and db/db mice, which are both deficient in leptin signaling. In addition, withaferin A, unlike celastrol, has beneficial effects on glucose metabolism independently from its leptin-sensitizing effect. Our results show that the metabolic abnormalities of diet-induced obesity can be mitigated by sensitizing animals to endogenous leptin, and indicate that withaferin A is a potential leptin sensitizer with additional anti-diabetic actions.
INTRODUCTION
There has been a progressive increase in the global rate of obesity over the past two decades, and it is predicted that approximately half of the US population will be obese by the year 2030 (ref. 1). Obesity confers a significant risk for associated disorders such as insulin resistance and type 2 diabetes, atherosclerosis, cardiovascular disease and cancer2,3. Despite enormous efforts in academia and industry to develop anti-obesity medications, the drugs that have reached market thus far have had only marginal effects (in the range of 3-10%) on body weight, and most have been withdrawn from the market due to their side effects4. Therefore, there is an urgent need for safe and effective medical treatments for obesity.
Leptin is a protein hormone involved in the regulation of a plethora of physiological parameters, including food intake, energy expenditure, reproduction, immune function, and glucose homeostasis5-8. The discovery of leptin more than two decades ago raised much hope that an effective treatment had been found for obesity9; however, except for the very rare cases of obesity caused by genetic deficiency of leptin, treatment with this hormone alone does not reduce the body weight of the majority of obese individuals who are hyperleptinemic10-13.
A number of pathologies have been proposed to reduce leptin signaling, and thus to induce leptin resistance, during the progression of obesity14-22. However, a complete understanding of the molecular mechanisms that underlie these pathologies is missing. One such mechanism is endoplasmic reticulum (ER) stress. The ER is an intracellular organelle that is responsible for the synthesis of membrane and secretory proteins, as well as cholesterol and lipids. Perturbations in endoplasmic reticulum homeostasis lead to ER stress, which activates a complex signaling cascade called the unfolded protein response (UPR)23-25. UPR activation maintains or re-establishes ER homeostasis as a cellular defense mechanism, and basal activity of this signaling cascade is required for normal physiological processes23-25. Despite the beneficial role of the acute UPR signaling in maintaining cellular homeostasis, prolonged ER stress contributes to the pathogenesis of many diseases including type 2 diabetes, cancer, atherosclerosis, as well as inflammatory and neurodegenerative diseases24-27.
Recently, we and others have documented that increased ER stress in hypothalamic neurons plays a key role in the development of leptin resistance and consequently obesity28-30. Induction of ER stress alone is sufficient to trigger leptin resistance in otherwise leptin-sensitive lean animals, and alleviation of this stress in obese mice, by agents such as 4-phenyl butyrate (4-PBA) or Tauroursodeoxycholic acid (TUDCA), can potentiate the weight-reducing and anorectic effect of exogenously administered leptin28. The spliced form of X-box binding protein-1 (XBP1s) is one of the cardinal regulators of ER homeostasis31,32, and we and others have previously reported a positive role of XBP1s in the regulation of leptin signaling28,33.
Based on the above lines of evidence, we searched the Broad Institute Connectivity Map (CMAP) database34 for small molecules with gene expression signatures that are highly similar to the expression profiles of tissues that overexpress XBP1s, and of tissues obtained from mice treated with 4-PBA and TUDCA35. We identified celastrol as a very potent leptin sensitizer that reduces food intake and body weight in diet-induced obese (DIO) mice, but has little effect on lean, ob/ob or db/db mice35.
Celastrol was obtained from combined analysis of different CMAP outputs, by using six different gene expression signatures from livers and hypothalamus, which show reduced ER stress and leptin resistance35. Thus, we reasoned that the gene expression profile of celastrol itself would serve as a consolidated signature to search compounds that act as leptin sensitizers and also validate our strategy for discovering new leptin sensitizers.
By using this approach we identified withaferin A with a similar gene expression profile with that of celastrol. Withaferin A is a steroidal lactone that belongs to the withanolide family of compounds that are isolated from the leaves, berries and roots of the medicinal plant Withania somnifera (also known as winter cherry or ashwagandha)36. Withaferin A is one of more than 50 chemicals identified in the Withania somnifera extract36. The extract of Withania somnifera has been used in traditional Indian medicine for centuries to treat tumors, stress, anxiety, aging, inflammation and autoimmune diseases37,38
RESULTS
Identification of withaferin A as a leptin sensitizer
We hypothesized that compounds with gene expression signatures similar to that of celastrol, which is a strong leptin sensitizer35, will have similar effects on leptin sensitization. To test this hypothesis, we created a celastrol gene expression signature from the microarrays of mouse embryonic fibroblasts (MEFs) treated with vehicle or celastrol. We chose the 20 genes that were most highly upregulated, and another 20 genes that were most downregulated by celastrol treatment (Fig. 1a,b), and then selected available human probes matching these mouse genes (Supplementary Fig. 1a). This human probe combination was used for a query in CMAP as the gene expression signature of celastrol (see Online Methods section for detailed information about CMAP). Certain conditions such as ER stress can have distinctive gene expression profiles for the same phenotype: reduced ER stress can have both increased and reduced chaperone expression profiles35. Therefore, we used the absolute enrichment score as the metric from CMAP analysis results as we did during the discovery of celastrol and identified withaferin A as one of three top-ranking chemical compounds, the first of which was celastrol itself (Fig. 1c). CMAP scored withaferin A as the second closest molecule to celastrol when the enrichment score itself was used (Supplementary Fig. 1b). To check whether gene expression profiles obtained in different settings for celastrol and withaferin A also show the same relationship, we obtained microarray results for celastrol and withaferin A from the CMAP database, and again observed a high level of similarity in the heatmap analysis between the first 50 genes that were upregulated or downregulated (Fig. 1d).
Figure 1.
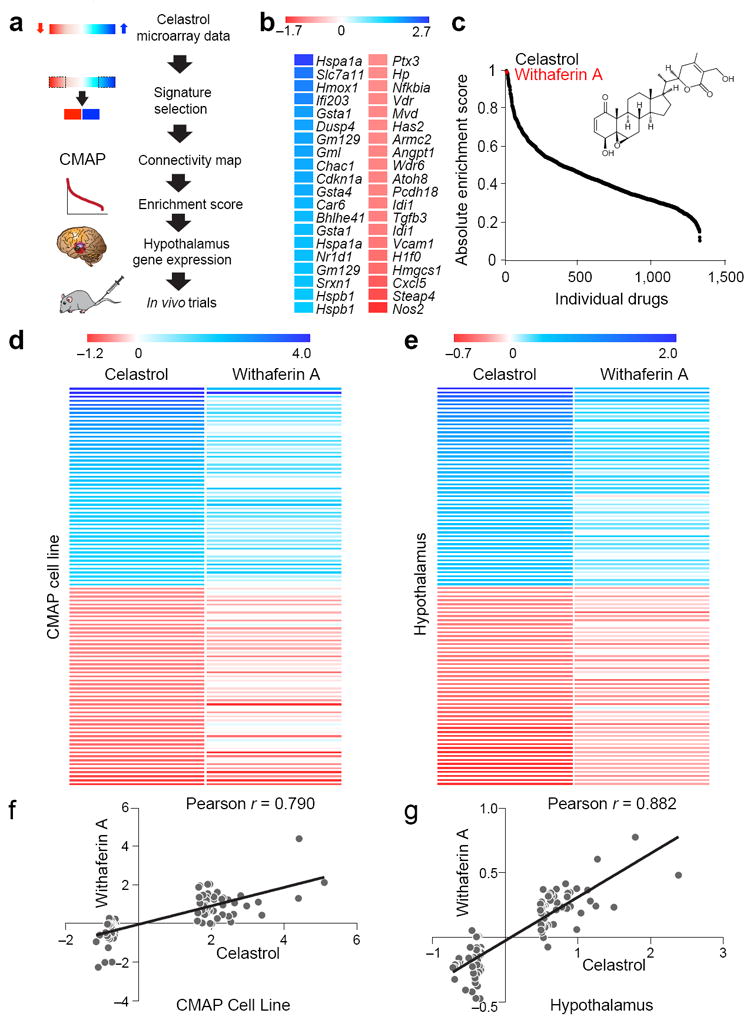
Identification of withaferin A with similar gene expression profile as celastrol. (a) Summary flow chart showing the identification of withaferin A as a leptin sensitizer and anti-obesity candidate. (b) Heat map representing the celastrol-regulated genes (20 most upregulated, blue; 20 most downregulated, red) obtained from MEFs treated with vehicle or celastrol (250 nM, 7 h). See also Supplementary Fig. 1a. (c) Distribution of absolute enrichment scores of the individual compounds in the CMAP database obtained using celastrol gene expression signature (Supplementary Fig. 1a). Also indicated is the chemical structure of withaferin A. (d) The heat maps generated from the CMAP database representing the celastrol regulated genes (50 most upregulated, blue; 50 most downregulated, red) and the corresponding changes in the same genes induced by withaferin A. (e) The heat maps representing the most upregulated (blue) and the most downregulated (red) 50 genes in the hypothalamus of celastrol-treated DIO mice (100 μg/kg, 4 d) and their corresponding gene expression changes in the hypothalamus of withaferin A-treated DIO mice (2 mg/kg, 4 d). The color scales in b,d,e represent the celastrol- and withaferin A-induced logarithmic fold changes in gene expressions. (f,g) Distribution of the celastrol- versus withaferin A-induced logarithmic fold changes in the expression of the genes in d,e, respectively. Line of best fit, and Pearson product-moment correlation coefficients (r) are shown.
Considering the possible effects of leptin sensitizers on the hypothalamus, we also investigated whether celastrol and withaferin A create similar gene expression profiles in the hypothalamus. Analysis of microarray results showed a similarity between the hypothalamic gene expression profiles of celastrol and withaferin A-treated mice in the heatmap analysis (Fig. 1e). Heatmap analysis of a microarray result would only provide qualitative measures. To be able to quantitatively analyze the similarity between the hypothalamic gene expression signatures of celastrol and withaferin A, and also compare this analysis with the results obtained from cell line experiments in CMAP, we first quantified the degree of correlation between gene expression signatures of celastrol and withaferin A from the CMAP-cell line in Fig. 1d by using Pearson product-moment correlation analysis. Indeed, we found a high degree of correlation with an r value of 0.79 (Fig. 1f). This analysis was consistent with the CMAP analysis, which showed a high degree correlation between the expression profiles of celastrol and withaferin A. Next, we used Pearson’s correlation analysis for hypothalamic gene expression profiles of these two molecules in Fig. 1e. We found that the degree of correlation was even higher between the hypothalamus gene expression signatures of celastrol and withaferin A, with an r value of 0.882 (Fig. 1g). These results provide strong support that withaferin A creates similar alterations in the hypothalamic gene expression profile of DIO mice compared to those after celastrol treatment. Based on the similarity between the gene expression profiles of celastrol and withaferin A, we tested the hypothesis that withaferin A would induce physiological responses similar to those of celastrol by acting as a leptin sensitizer.
Withaferin A reduces bodyweight of DIO mice
To assess whether withaferin A acts as a leptin sensitizer, and leads to weight loss, DIO mice were treated either with vehicle or withaferin A. The vehicle-treated group retained a stable body weight throughout the study (Fig. 2a,b). However, treatment of DIO mice with withaferin A led to a highly significant decrease in body weight during the treatment period (P < 0.001) (Fig. 2a), equivalent to a total weight loss of 22.8% (Fig. 2b).
Figure 2.
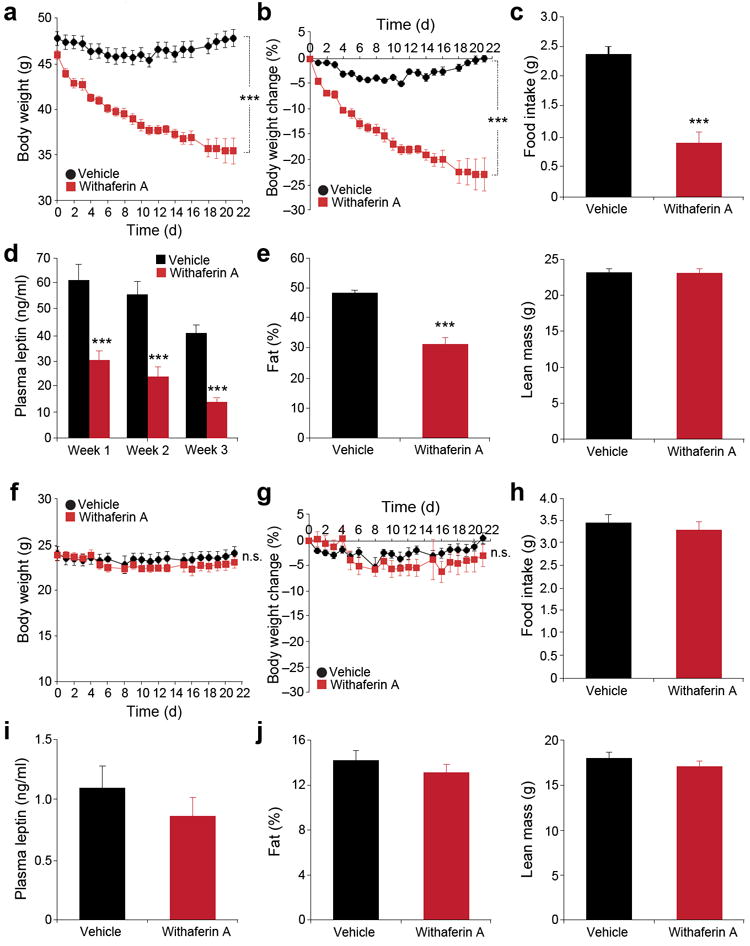
Withaferin A reduces body weight and food intake of diet-induced obese mice but not that of lean mice. (a–e) DIO mice received vehicle or withaferin A (1.25 mg/kg) for 21 d. (a) Body weight and (b) percent change in body weight (n = 9, vehicle; n = 8, withaferin A). (c) Daily food intake during the first week treatment. The experiments in a–c were repeated in five cohorts (total n = 42, vehicle; n = 40, withaferin A). (d) Plasma leptin concentrations of DIO mice after 1, 2, and 3 weeks of treatment (n = 10 per group). (e) Fat percentage (left panel) and lean mass (right panel) after 3-week treatment (n = 9, vehicle; n = 8, withaferin A), which were repeated in five cohorts (total n = 46, vehicle; n = 44, withaferin A). (f–j) Lean mice received vehicle or withaferin A (1.25 mg/kg) for 21 d. (f) Body weight and (g) percent change in body weight (n = 9, vehicle; n = 10, withaferin A). (h) Daily food intake during the first week treatment. The experiments in f–h were repeated in three cohorts (total n = 21 per group). (i) Circulating leptin concentrations after 3-week treatment (n = 9, vehicle; n = 10, withaferin A), which were repeated in three cohorts (total n = 19, vehicle; n = 18, withaferin A). (j) Fat percentage (left) and lean mass (right) of lean mice after 3-week treatment (n = 9, vehicle; n = 10, withaferin A). Values are averages ± s.e.m. P values are determined by two-way ANOVA (a,b,f,g) or Student’s t test (c,d,e,h,i,j). ***P < 0.001. n.s.; not significant.
One of the most pronounced effects of leptin treatment is the suppression of food intake. Withaferin A-treated mice also consumed significantly lower amount of food (62%), compared to vehicle group (P < 0.001) (Fig. 2c and Supplementary Fig. 2a,b). Consistent with increased leptin sensitivity, circulating leptin concentrations in the withaferin A-treated group were significantly lower than vehicle group (Fig. 2d). Next, we used dual-energy X-ray absorptiometry (DEXA) scans to determine fat and muscle mass and found that the withaferin A group showed significantly lower fat mass by 35% (P < 0.001) compared to vehicle group, without changing the total lean mass (Fig. 2e).
To address the possibility that the reduction in food intake and the subsequent weight loss derives from a toxic effect of the treatment, we reasoned that toxicity should be observed in both obese and lean animals, whereas a leptin sensitizer should reduce body weight and food intake preferentially in DIO mice. Neither vehicle treatment nor withaferin A treatment altered the body weight of lean mice (Fig. 2f,g). Food intake of lean mice was also not affected by withaferin A (Fig. 2h), and we observed no significant differences in plasma leptin between the groups (Fig. 2i). DEXA scans revealed that withaferin A administration did not change the fat or lean mass of lean mice (Fig. 2j). However, when we administered withaferin A to wild type mice which were heavier (~30 g) and were kept on chow diet, withaferin A administration led to a significant decrease in body weight and also lower food intake compared with vehicle treatment (Supplementary Fig. 2c–e), indicating that the effect of withaferin A on bodyweight loss is not specifically due to HFD-feeding, but related to the absolute body weight of the mice.
Withaferin A minimally affects ob/ob and db/db mice
Next, we administered withaferin A to db/db and ob/ob mice to test for the dependency of the effects of the compound on leptin signaling as both these strains lack this pathway (the former due to absence of the receptor and the latter due to the absence of the hormone). Administration of withaferin A to male db/db mice did not produce body weight reduction, while the vehicle-treated group gained body weight during the experimental period (Fig. 3a). Db/db mice treated with withaferin A gained a slight amount of body weight relative to the pre-treatment period, but the vehicle-treated group gained more weight compared to the withaferin A-treated group (14% versus 4% increase in body weight, vehicle versus withaferin A, P < 0.001) (Fig. 3b). These findings indicate that withaferin A’s influence on bodyweight in db/db mice is minimal compared to that in DIO mice (Figs. 2a and 3a). We observed a slightly (but not significantly) lower amount of food consumption from withaferin A-treated db/db mice compared to the vehicle-treatment group (Fig 3c) and no difference in leptin concentrations between the two groups (Fig. 3d). Withaferin A administration also did not significantly alter the fat or lean mass of db/db mice compared to vehicle treatment (Fig. 3e). The same withaferin A treatment in leptin-deficient ob/ob mice led to a small but insignificant decrease in body weight (Fig. 3f), whereas vehicle-treated ob/ob mice displayed a slight gain in body weight during the treatment period (Fig. 3f,g). However, at the end of trial, analysis of percent changes in the body weight between vehicle and withaferin A-treated ob/ob mice resulted in a significant difference (Fig. 3g). There was no significant difference in absolute body weight between two groups at the end of treatment (Fig. 3f). Food intake (Fig. 3h) and fat mass (Fig. 3i) in withaferin A-treated ob/ob mice were slightly (but significantly) lower compared to vehicle group, but there was no difference in lean mass between the two groups (Fig. 3i); the total fat mass reduction was much smaller relative to that seen in DIO mice (35% versus 7% reduction in fat, DIO versus ob/ob mice) (Figs. 2e and 3i).
Figure 3.
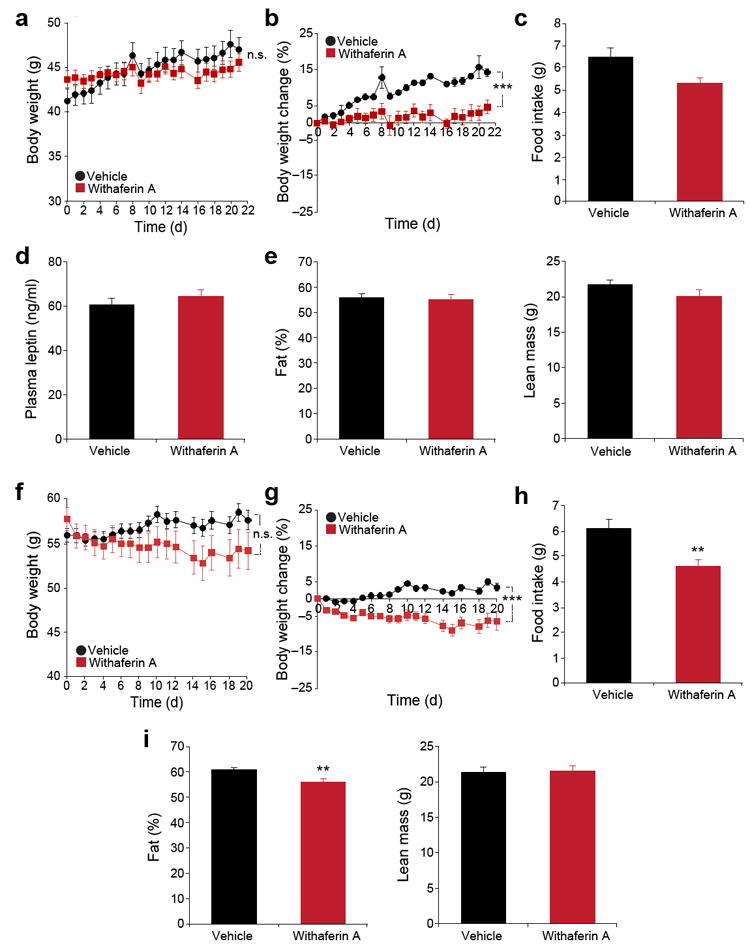
Leptin signaling-deficient mice are resistant to the weight reducing effect of withaferin A. (a–e) db/db mice received vehicle or withaferin A (1.25 mg/kg) for 21 d. (a) Body weight and (b) percent change in bodyweight during the treatment (n = 10, vehicle; n = 8, withaferin A). The experiments in a,b were repeated in seven cohorts (total n = 53, vehicle; n = 47, withaferin A). (c) Daily food intake during the first week treatment. (d) Plasma leptin concentrations after 3-week treatment period (n = 10, vehicle; n = 7, withaferin A). (e) Fat percentage (left) and lean mass (right) after 21-d treatment (n = 10, vehicle; n = 9, withaferin A). (f–i) ob/ob mice received vehicle or withaferin A (1.25 mg/kg) for 20 d. (f) Body weight and (g) percent change in bodyweight (n = 10 per group). The experiments in f,g were repeated in three cohorts (total n = 29, vehicle; n = 22, withaferin A). (h) Daily food intake during the first week treatment. (i) Fat percentage (left) and lean mass (right) of ob/ob mice after 20 d of treatment (n = 10 per group). Values are averages ± s.e.m. P values are determined two-way ANOVA (a,b,f,g) or by Student’s t test (c,d,e,h,i). ** P < 0.01, *** P < 0.001. n.s.; not significant.
Withaferin A increases the potency of leptin
While lean mice are sensitive to acute administration of leptin39, it is known that DIO mice do not respond to exogenous leptin treatment due to leptin resistance40,41. To provide further support for the leptin-sensitizing role of withaferin A, we first tested the potency of leptin on the regulation of food intake and bodyweight in leptin-resistant DIO mice, with or without withaferin A treatment. First, we administered either vehicle or withaferin A to DIO mice and then injected saline or leptin on the second day of vehicle or withaferin A treatment. The group treated with vehicle plus leptin (Veh + Lep) showed no significant change in food consumption compared with vehicle plus saline (Veh + Sal) group (Supplementary Fig. 3a). However, withaferin A treatment alone (Wit A + Sal) led to significantly lower food intake (42%) compared with Veh+Lep group (P < 0.01). Despite already reduced food consumption, leptin injection after withaferin A treatment (Wit A + Lep) further lowered food intake by 65% relative to the Veh + Sal or 71% relative to the Veh + Lep group (P < 0.001) (Supplementary Fig. 3a). Also, parallel to the alteration of food intake, body weight was significantly lower in the Wit A + Lep group, compared to all other three cohorts (Supplementary Fig. 3b). Leptin caused no significant changes in the bodyweight of the Veh + Lep group compared to the Veh + Sal group (Supplementary Fig. 3b).
Ob/ob mice are sensitive to administration of exogenous leptin39,42,43. However, we chose a low dose of leptin, which is minimally effective in the ob/ob mice, in order to investigate whether withaferin A can also increase sensitivity of ob/ob mice to very low doses of leptin. Ob/ob mice were pre-treated with vehicle or withaferin A. Following withaferin A treatment, we divided each set of mice into two subgroups, and further treated these groups with saline or leptin. The low dose of leptin did not lead to significant loss of bodyweight, but did block further weight gain when compared with Veh + Sal group (Supplementary Fig. 3c). Withaferin A treatment alone did not cause any weight loss. However, the Wit A + Lep group lost significantly more weight than Veh + Sal or Wit A + Sal groups (Supplementary Fig. 3c). Leptin treatment in the vehicle group led to a 29% lower food intake compared to that for the vehicle plus saline group (Supplementary Fig. 3d); however, the same dose of leptin in the withaferin A-treated group led to a 40% lower food consumption when compared to the withaferin A plus saline group (Supplementary Fig. 3d).
The hypothalamic sites of action of celastrol and withaferin A
We have previously shown that celastrol increases the sensitivity of the leptin receptor signaling system in the whole hypothalamus35. However, we have not previously determined which nuclei in the hypothalamus were affected by celastrol treatment. Arcuate nucleus (ARC), ventromedial hypothalamus (VMH) and dorsomedial hypothalamus (DMH) have been suggested as critical sites for leptin action44-46. Thus, we first determined the status of STAT3Tyr705 phosphorylation in ARC, VMH and DMH of DIO mice by using immunohistofluorescence (IHF) analysis, with or without celastrol treatment. DIO mice administered vehicle or celastrol were subsequently received saline or a bolus dose of leptin. Afterwards, total fluorescence intensities (TFI, hereafter intensities) and total cell numbers (TCN, hereafter cell numbers) of p-STAT3Tyr705-positive cells in ARC, VMH and DMH were determined for each mouse, and experiments from two independent cohorts were combined for final analysis.
Celastrol alone (Cel + Sal) led to significantly greater p-STAT3Tyr705-positive cell numbers in the DMH compared to the vehicle group (Veh + Sal) (P < 0.001) (Supplementary Fig. 4a,b). Leptin treatment alone (Veh + Lep) also led to larger p-STAT3Tyr705-positive cell numbers compared to Veh + Sal (Supplementary Fig. 4a,b), while celastrol plus leptin treatment (Cel + Lep) resulted in greater p-STAT3Tyr705-positive cell numbers compared to Veh + Lep (Supplementary Fig. 4a,b). In the VMH, Cel + Sal mice show significantly larger p-STAT3Tyr705-positive cell numbers compared to the Veh + Sal group (P < 0.05) (Supplementary Fig. 4c,d), and Cel + Lep administration produced greater p-STAT3Tyr705-positive cell numbers compared to the Veh + Lep group (Supplementary Fig. 4c,d). In the ARC, celastrol treatment led to greater p-STAT3Tyr705-positive cell numbers compared to the Veh + Sal group, while Cel + Lep did not show any significant difference in p-STAT3Tyr705-positive cell numbers compared to Veh + Lep (Supplementary Fig. 5e,f). We detected significantly greater p-STAT3Tyr705 intensities only in Cel + Lep mice, when compared to the Veh + Sal group (Supplementary Fig. 4a–f). Taken together, our data show that celastrol potentiates leptin signaling mainly in the DMH and VMH.
We then investigated how STAT3Tyr705 phosphorylation is modulated by withaferin A treatment in the whole hypothalamus. DIO mice first received a single dose of vehicle or withaferin A, and 15 h later, mice were administered with either saline or leptin. Leptin injection in vehicle-treated obese mice did not produce a greater extent of STAT3Tyr705 phosphorylation (Fig. 4a). However, a single injection of withaferin A led to significantly higher level of basal STAT3Tyr705 phosphorylation when compared to Veh + Sal and Veh + Lep groups. Administration of leptin after injection of withaferin A created a much greater degree of STAT3Tyr705 phosphorylation when compared to all other groups (Fig. 4a).
Figure 4.
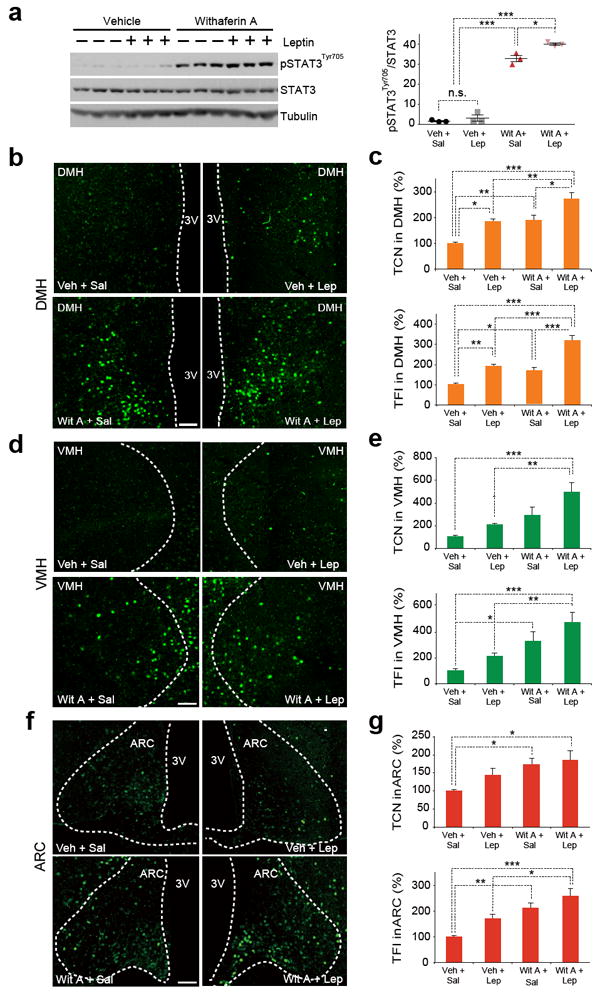
Withaferin A increases leptin sensitivity in the hypothalamus of DIO mice. (a) Representative immunoblots of p-STAT3Tyr705, total STAT3 and tubulin from whole hypothalamus (left), and the quantified ratio between p-STAT3Tyr705 and total STAT3 (right). DIO mice received vehicle or withaferin A (5 mg/kg, 15 h), and then were injected with saline or leptin (1 mg/kg, 30 min). The experiments in a,b were repeated three times with similar outcomes (total n = 9, Veh + Sal; n = 10, Veh + Lep; n = 9, Wit A + Sal; n = 10, Wit A + Lep). (b–g) DIO mice were administered with vehicle or withaferin A (2 mg/kg) for 3 d and subsequently received saline or leptin (1 mg/kg). The hypothalamic samples were analyzed by immunohistofluorescence staining using p-STAT3Tyr705 specific antibodies. (b,d,f) Representative images of p-STAT3Tyr705 positive cells from (b) dorsomedial hypothalamus (DMH), (d) ventromedial hypothalamus (VMH) and (f) arcuate nucleus (ARC) (total numbers of analyzed images are provided in Online Methods). (c,e,g) The quantified results of total p-STAT3Tyr705 positive cell numbers (TCN, top panel) and fluorescence intensities (TFI, bottom panel) of the groups in b,d,f. Bar graphs in c,e,g represent the average of three experiments (total n = 12 mice per group). Values are averages ± s.e.m. P values are determined by one-way ANOVA with Bonferroni (a) or Tukey post hoc test (c,e,g). See Online Methods for the details of statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001. n.s.; not significant. Scale bars, 100 μm.
We next analyzed STAT3Tyr705 phosphorylation levels in DMH, VMH and ARC by IHF staining approach. In the DMH, mice treated with withaferin A alone (Wit A + Sal) showed significantly higher levels of both p-STAT3Tyr705-positive cell numbers and intensities compared with Veh + Sal group (P < 0.01) (Fig. 4b,c), while withaferin A plus leptin treatment (Wit A + Lep) produced higher p-STAT3Tyr705-positive cell numbers and intensities when compared to the Veh + Lep group (Fig. 4b,c). In the VMH, Wit A + Sal treatment led to significantly greater p-STAT3Tyr705 intensities compared with Veh + Sal (P < 0.05) (Fig. 4d,e), while both p-STAT3Tyr705-positive cell numbers and intensities of Wit A + Lep mice were significantly greater when compared to those of Veh + Lep group (P < 0.01) (Fig. 4d,e). In the ARC, Wit A + Sal mice showed markedly higher levels of both p-STAT3Tyr705-positive cell numbers and intensities when compared with Veh + Sal group (Fig. 4f,g); in addition, Wit A + Lep treatment produced significantly higher p-STAT3Tyr705 intensities in ARC than Veh + Lep (P < 0.05) (Fig. 4f,g). In sum, withaferin A, similar to celastrol, also sensitizes leptin receptor signaling significantly in the DMH and VMH.
Next we analyzed gene expression levels of agouti-related peptide (Agrp), neuropeptide Y (Npy), pro-opiomelanocortin (Pomc) and suppressor of cytokine signaling 3 (Socs3). Withaferin A treatment led to significantly higher Agrp and Socs3 expression and significantly lower Pomc expression without leading to a change in NPY expression levels (Fig. 5a).
Figure 5.
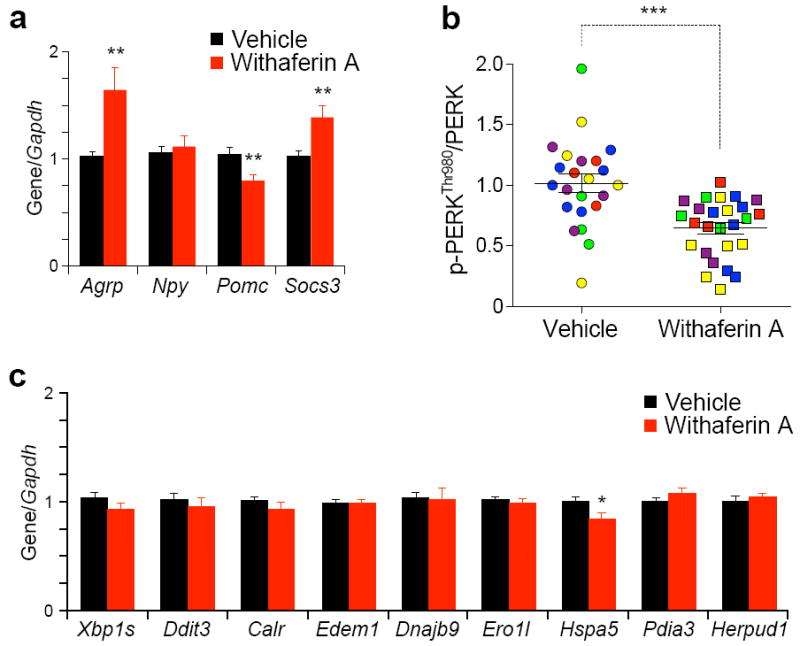
Withaferin A reduces ER stress. (a–c) DIO mice received vehicle or withaferin A (2 mg/kg) for 3 d. (a) Agrp, Npy, Pomc and Socs3 mRNA expression in the hypothalamus. (b) The quantified ratio of the signals between p-PERKThr980 and total PERK of the hypothalamic samples from five independent experiments in Supplementary Fig. 5c (total n = 23, vehicle; n = 26, withaferin A). (c) mRNA expression levels of spliced Xbp1s, Ddit3 and endoplasmic reticulum chaperones in the hypothalamus. Bar graphs in a,c represent the average of three independent experiments (total n = 16 per group). Values are represented as mean ± s.e.m. P values were determined by Student’s t test (a,c) or two-way ANOVA (b). *P < 0.05, **P < 0.01, ***P < 0.001. See also Supplementary Fig. 5.
Withaferin A reduces ER stress
We first treated the Tuberous sclerosis complex 2 (TSC2) knockout (Tsc2−/−) MEFs, which are characterized by intrinsically elevated ER stress47, with increasing doses of withaferin A and documented that withaferin A greatly reduced PERKThr980 phosphorylation, which indicates that withaferin A reduces ER stress; results of four independent experiments are presented in Supplementary Fig. 5a,b. We then investigated the status of ER stress in the hypothalamus of withaferin A-treated mice. Five independent cohorts of mice were used in this experiment (Supplementary Fig. 5c) and combined analysis was performed. Results from these experiments documented that withaferin A treatment led to less PERKThr980 phosphorylation in the hypothalamus, in a highly significant manner (Fig. 5b) indicating that withaferin A alleviates hypothalamic ER stress. We next investigated the transcript levels of Xbp1s, C/EBP homologous protein (Ddit3) and various other ER chaperones in the whole hypothalamus of mice treated with vehicle or withaferin A. Most of the expression levels of ER chaperones were unaltered during withaferin A treatment, except glucose-regulated protein 78 (Hspa5), of which expression level was significantly lower in the withaferin A treatment when compared with the vehicle-treated group (Fig. 5c).
Withaferin A has been proposed as a heat shock protein 90 (Hsp90) inhibitor48. Hsp70 expression has been used as an indicator of Hsp90 activity and Hsp70 levels were reported to be increased in the conditions that Hsp90 activity is inhibited49. Therefore, we measured Hsp70 protein amount in the hypothalamus from DIO mice treated vehicle or withaferin A. As shown in Supplementary Fig. 5d, withaferin A administration did not alter Hsp70 protein level in the hypothalamus, despite that the same dose of withaferin A effectively suppressed food intake of DIO mice in the same experiment (data not shown), indicating that withaferin A affects food intake and body weight of mice independent of Hsp90 inhibition in the hypothalamus.
Furthermore, withaferin A has been suggested to be an inhibitor of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway50. We thus investigated whether withaferin A treatment inhibits NF-κB activity in the hypothalamus. Analysis of NF-κB target genes showed that most of the NF-κB target genes were unaltered with withaferin A treatment (Supplementary Fig. 5e), indicating that withaferin A action does not rely on inhibition of NF-κB in the hypothalamus at the doses administered in our study.
Withaferin A blocks a reduction in energy expenditure
Food restriction or starvation reduces basal metabolic energy expenditure41,51. However, despite its anorexigenic ability, leptin does not lead to a reduction in energy expenditure41. To investigate the metabolic changes induced by withaferin A, we undertook metabolic chamber experiments and determined the values for energy expenditure, for physical activity levels, and for the respiratory exchange ratio (RER) at the earlier times of treatment where leptin action is at the highest level in the DIO mice. Withaferin A treatment in DIO mice led to a significant decrease in food intake (data not shown). But despite this reduction, no significant differences in energy expenditure were seen between the vehicle- and withaferin A-treated DIO mice (Fig. 6a). However, significantly lower RER values in the withaferin A-treated mice, compared to the vehicle-treated control group, was noted both in night and light cycles (Fig. 6b), suggesting that the withaferin A treatment increased the utilization of fat as an energy source. Analysis of the activity of the mice treated with withaferin A showed significantly less movement during the dark cycle compared to vehicle treatment, but no alteration during the light cycle (Fig. 6c).
Figure 6.
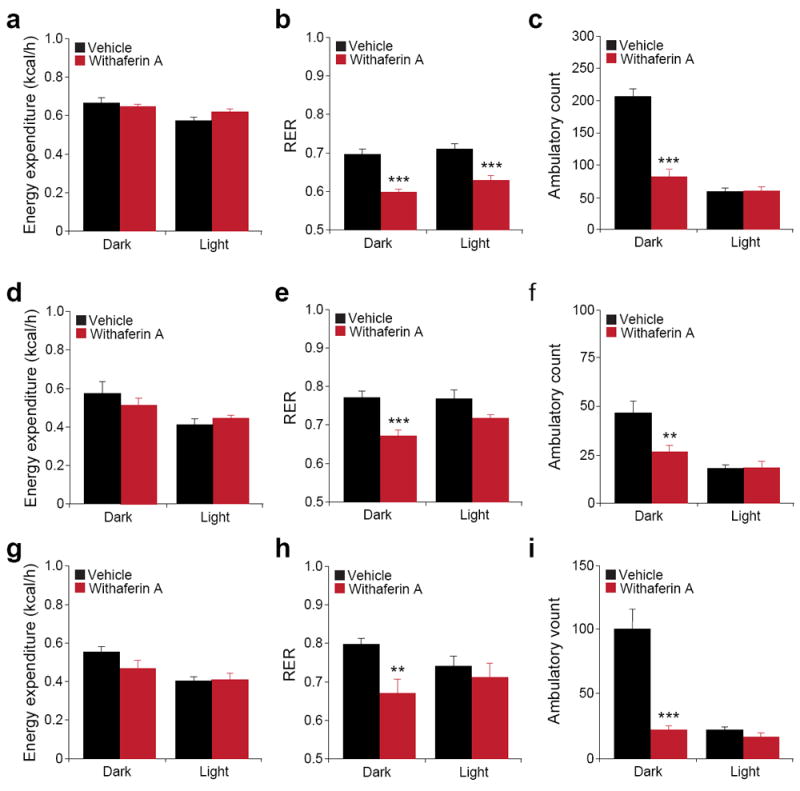
Withaferin A’s beneficial effect on metabolic homeostasis. Individually caged (a–c) DIO (total n = 16 per group from two cohorts), (d–e) ob/ob (n = 5 per group) and (g–i) db/db mice (n = 6 per group) were placed in metabolic cages and received vehicle or withaferin A (1.5 mg/kg) for 3 d. Energy expenditure (kcal/h) of (a) DIO, (d) ob/ob and (g) db/db mice. Respiratory exchange ratios (RER) (VCO2/VO2) of (b) DIO, (e) ob/ob and (h) db/db mice. Ambulatory physical activity of (c) DIO, (f) ob/ob and (i) db/db mice. Bar graphs represent average of two dark (24–36 h and 48–60 h) and two light cycles (12–24 h and 36–48 h). Results in a–c are the average of two independent cohorts. Values are averages ± s.e.m. P values are determined by Student’s t test (a–i). **P < 0.01, ***P < 0.001.
We also analyzed metabolic parameters of ob/ob and db/db mice. Withaferin A treatment did not create any significant changes in energy expenditure of ob/ob mice (Fig. 6d); however withaferin A treatment in ob/ob mice led to significantly lower RERs and physical activity compared to the vehicle treatment in only the dark cycle (Fig. 6e,f). Analysis of metabolic parameters in the db/db mice revealed similar regulations with withaferin A treatment (Fig. 6g–i).
Withaferin A treatment loses its potency to reduce body weight after 3 weeks of treatment (Fig. 2a,b), in accordance with amelioration of hyperleptinemia (Fig. 2d); this result led us to reason that the effect of withaferin A on metabolic parameters in DIO mice should also diminish with decreasing leptin levels. We thus repeated the metabolic chamber experiments after weight loss had occurred and bodyweight had stabilized. At the end of 18 d treatment, the withaferin A-treated group was significantly leaner than the vehicle group – mice in this group were consuming similar amount of food with vehicle group and yet not gaining further weight. We saw no significant differences in energy expenditure between the vehicle and withaferin A-treated groups (Supplementary Fig. 6a). RER values returned back to control levels and no differences were noted in physical activity (Supplementary Fig. 6b,c). Analyses of energy expenditure, RER values and activity also yielded no differences between vehicle- or withaferin A-treated ob/ob (Supplementary Fig. 6d–f) or db/db (Supplementary Fig. 6g–i) mice, despite continuing presence of obesity in these groups.
We next investigated whether withaferin A-treatment and the consequent weight loss have positive effects on the liver and on other metabolic parameters. Analysis of liver sections after 21 d of withaferin A treatment showed that the hepatic steatosis was completely resolved in withaferin A-treated mice (Supplementary Fig. 7a). No difference was seen in the fat content of livers of withaferin A-treated obese mice and of vehicle-treated lean control mice after the treatment period. Plasma alanine transaminase (ALT) and aspartate transaminase (AST) concentrations were significantly lower in withaferin A-treated mice compared to vehicle group (Supplementary Fig. 7b,c), indicating an improved status of liver function. Blood cholesterol concentrations were also significantly lower in withaferin A group compared with control (Supplementary Fig. 7d). To rule out the possibility that the reduced food intake and decreased body weight by withaferin A treatment resulted from increased thyroid activity, we measured serum triiodothyronine (T3) concentrations and found that T3 concentrations were actually lower in withaferin A-treated mice compared to control, indicating the mice were not in a possible thyrotoxicosis (Supplementary Fig. 7e).
Withaferin A improves glucose homeostasis of obese mice
We next analyzed the effect of withaferin A on glucose metabolism. A glucose tolerance test (GTT) revealed significantly faster disposal of glucose from the circulation in the withaferin A-treated DIO mice (Supplementary Fig. 8a). An insulin tolerance test (ITT) also showed a significantly greater responsiveness to insulin in the withaferin A-treated DIO mice compared to the vehicle group (Supplementary Fig. 8a). We also found that blood glucose and insulin concentrations were lower in the withaferin A-treated DIO mice compared to vehicle treatment (Supplementary Fig. 8b).
We next studied the effect of withaferin A on glucose homeostasis in lean mice. Performance of GTT showed no difference in glucose tolerance under withaferin A treatment in lean mice (Supplementary Fig. 8c), as did the ITT (Supplementary Fig. 8c), with no change in blood glucose and insulin concentrations (Supplementary Fig. 8d). However, analysis of glucose homeostasis revealed a significant effect in db/db and ob/ob mouse models; as shown in Supplementary Fig. 8e, clearance of glucose from the circulation was significantly faster in withaferin A-treated db/db compared with vehicle group. There was no difference in response to insulin during ITT between the two groups (Supplementary Fig. 8e). Blood glucose concentrations were significantly lower in the withaferin A-treated db/db mice compared with vehicle group (Supplementary Fig. 8f), without any significant difference in circulating insulin concentrations (Supplementary Fig. 8f). Similar results were obtained for withaferin A-treated ob/ob mice in comparison to their controls (Supplementary Fig. 8g,h). Taken together, our results indicate that, in addition to being an anti-obesity agent, withaferin A also has anti-diabetic effects that are independent of its anorexigenic capabilities.
DISCUSSION
More than two decades have passed since the historic discovery of leptin by Friedman and coworkers9, but no viable leptin-centric treatment for obesity has been developed to date. Soon after the initial publications on leptin, it was suggested that obesity is a condition of leptin-resistance10-12. Over the course of the last twenty years, hopes for a leptin-oriented treatment of obesity have progressively diminished, as many attempts at re-sensitizing the brains of obese individuals to endogenous leptin failed. These unsuccessful efforts to increase leptin sensitivity and utilize the hyperleptinemic state of obesity to treat the condition also contributed substantially to the debate on whether or not leptin resistance actually exists6,52. Our findings on celastrol and now on withaferin A potentially change the landscape – they not only provide strong support for the notion of leptin resistance, but more importantly they also show that the hyperleptinemic state of obesity can be leveraged for therapeutic purposes by increasing leptin sensitivity.
In contrast to the major focus of drug discovery - which has been targeting a single molecule in a specific pathway (in this case this would be the ER stress-signaling pathway) – here we have employed a different approach. We analyzed the outputs of various interventions that had positive effects on different aspects of the ER system. Celastrol was discoverable from reverse analysis, combination, and ultimately absolute value-scoring algorithms of six different microarrays that were obtained from a variety of inputs leading to outputs that improved ER homeostasis. A major challenge in validating such a methodology was to prove that a compound that emerged from these studies serves as a condensed platform for the discovery of new drugs. Indeed, our findings on celastrol led us to discover withaferin A, and thereby we also documented that this system, and the discovery of celastrol, were not just coincidental.
Treatment of HFD-fed obese and hyperleptinemic mice with withaferin A led to a robust reduction in food intake as well as in body weight. Both changes are dependent on high levels of circulating leptin. As the leptin levels gradually decrease over the treatment period, the effect of withaferin A also gradually diminishes. Also, no withaferin A-induced changes in food consumption or in body weight were observed in lean mice, which have low levels of circulating leptin. However, withaferin A led to small but significant changes in body weight (percent changes from the initial point) of ob/ob and db/db mice, probably due to the transient decrease in food intake of these mice during the early treatment period. In both mice, the effect on food intake was minimal when compared to the reduction that is seen in DIO model. Several possibilities come to mind with regard to why withaferin A treatment might have this effect on db/db and ob/ob mice but not in lean mice. First, in addition to leptin sensitization, withaferin A may also be sensitizing the central nervous system to another cytokine/factor that only weakly affects the regulation of food intake and is upregulated in obese but not in lean mice. Second, withaferin A may be sensitizing the brain to another hormone/cytokine that is upregulated in obesity, and is part of a signaling pathway that has significant crosstalk with the leptin receptor signaling system. The latter possibility is supported by our observation that the withaferin A-associated reduction in food intake is slightly greater in ob/ob mice than in db/db mice; ob/ob mice have functional leptin receptor signaling but lack leptin, whereas db/db mice have high levels of circulating leptin, but no functional leptin receptor signaling system. Further investigation is needed to reveal the presence and identity of such potential factors. But even if they exist, their contribution to the regulation of food intake and energy balance is likely to be minimal compared to that of leptin, as indicated by the differences between the responses of DIO mice and ob/ob or db/db mice to withaferin A. Furthermore, the effects of withaferin A on ob/ob or db/db are transient and disappear despite continuation of obesity, which provides further support that the effects of withaferin A on food intake at the doses that we used are due to increased leptin sensitivity.
Withaferin A also has a more pronounced effect on blood glucose levels in ob/ob and db/db mice than does celastrol. One explanation for this may be that, in obesity models, withaferin A has a slightly higher influence on food intake and body weight of these models, which might drive the improvement in glucose homeostasis. However, as discussed above, the alterations in food intake and body weight of withaferin A-treated ob/ob and db/db mice were minimal. Thus, these results suggest that the positive effect of withaferin A on glucose metabolism is not merely a secondary response to weight loss, but that withaferin A also could be positively influencing anti-diabetic pathways in a leptin-independent manner. This action of withaferin A is different than celastrol’s effects on glucose metabolism in ob/ob and db/db mice; at the doses celastrol showed its strong leptin sensitizer effects, unlike withaferin A, it was unable to reduce the blood glucose levels in the ob/ob and db/db mouse models.
Organisms respond to fasting or calorie restriction by decreasing their basal energy expenditure, in order to compensate for decreased food intake and preserve energy. However, it is believed that leptin-induced anorexia or reduced calorie consumption does not lead to lower energy expenditure. Indeed, as for celastrol, withaferin A treatment did not lead to reduction of energy expenditure in DIO mice. Collectively, results obtained from treatments with withaferin A or with celastrol indicate that establishing leptin sensitivity within the context of a previous leptin-resistant condition might not increase energy expenditure to a level that is higher than in the leptin resistant state. However, increasing leptin sensitivity with withaferin A or with celastrol does block the reduction of energy expenditure that is otherwise seen in calorie-restricted mice41.
Further evidence for the effects of withaferin A as a leptin sensitizer derives from experiments in which we analyzed the physiological and biochemical responses to acute leptin treatment in the presence or absence of withaferin A treatment. Immunohistofluorescence analysis further documented that acute leptin administration in withaferin A-treated mice, compared to the vehicle-treated group, leads to a robustly greater presence of STAT3Tyr705 phosphorylation in the hypothalamus, particularly in the DMH and VMH, as is similarly observed with celastrol treatment.
We identified withaferin A based on the similarity of its gene expression signature to that of celastrol. Whether the mechanisms for creation of the gene expression profiles of celastrol and withaferin A are the same is not known. However, considering that withaferin A’s leptin-independent effects on glucose homeostasis in ob/ob and db/db mice are not observed with use of celastrol, it is possible that the two compounds use different pathways to create the same outcome in gene expression profile, or that they use a common pathway but withaferin A also independently activates another anti-diabetic pathway.
We show that withaferin A treatment leads to a reduction in ER stress without affecting the expression levels of most of the chaperones, and ER stress is known to create leptin resistance28. A recent report indicated that celastrol’s anti-obesity effect could be due to peripheral actions of celastrol through activation of HSF1-PGC1α axis53. While celastrol could be also activating the HSF1-PGC1α axis, we believe that this effect is not the main effect by which celastrol suppresses appetite and reduces body weight, because we see a very mild effect in ob/ob or db/db mice at the doses that we used for celastrol. Withaferin A, which has a similar gene expression profile to that of celastrol, also exerts its effects mainly on DIO mice and not on ob/ob and db/db mice. In this sense, it will be highly unlikely that the anti-obesity effect of withaferin A would be mediated through activation of pathways including HSF1-PGC1α axis in the periphery.
We identified withaferin A, another potent anti-obesity agent, without targeting a specific molecule but rather using the similarity in the gene expression signature of celastrol and withaferin A. We believe this methodology is a more powerful tool than screening for molecules for single targets. Indeed, more than 15 years of efforts spent on identifying single molecular targets that cause leptin resistance have not produced effective treatments for obesity. Our results indicate that improving the ER system, reducing ER stress, and using the outcomes of gene expression profiling will be an important future strategy for further discovery of anti-obesity drugs.
Finally, Withania somnifera extracts, which also contain withaferin A, have been used in humans for centuries37,38, and the leptin system is strongly preserved among mammals including mice and humans54. Considering all of this information, we believe that translating the use of withaferin A for the treatment of obesity in humans holds great promise for the future.
Online Methods
Reagents
Phospho-PERKThr980 (Clone C33E10, cat. 3192), PERK (Clone 16F8, cat. 3179), phospho-STAT3Tyr705 (Cat. 9131; Clone D3A7, Cat. 9145), STAT3 (Clone 79D7, cat. 4904), α-tubulin (Clone 11H10, cat. 2125) and Hsp70 (Cat. 4872)-specific antibodies were purchased from Cell Signaling Technology (Beverley, MA). β-Actin (Clone C4, cat. sc-47778) and Hsp90 (Clone F-8, cat. sc-13119)-specific antibodies, and horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). BM chemiluminescence western blotting substrate (POD) kit was from Roche Diagnostics, Inc. (Indianapolis, IN). TRIzol reagent was from Invitrogen (Carlsbad, CA). iScript cDNA synthesis kit was purchased from Bio-Rad (Hercules, CA). RNeasy MinElute Cleanup Kit was from Qiagen (Valencia, CA). Mouse recombinant leptin was from R&D Technologies (North Kingstown, RI), and recombinant human insulin was from Eli Lilly and Company (Indianapolis, IN). Mouse leptin ELISA kit and ultra-sensitive mouse insulin enzyme-linked immunosorbent assay
(ELISA) kits were from Crystal Chem, Inc. (Downers Grove, IL). Alanine Transaminase (ALT) color endpoint assay kit and Aspartate Transaminase (AST) color endpoint assay kit were from Bio Scientific (Austin, TX). Wako Cholesterol E assay kit was from Wako Diagnostics (Richmond, VA). Taqman® primers, probes, Taqman® universal PCR master mix, SYBR Green Super Mix, fetal bovine serum and Dulbecco’s Modified Eagle Medium (DMEM, with 25mM glucose) were purchased from Life Technologies. 10% buffered formaline phosphate, and dimethyl sulfoxide (DMSO) was from Fischer BioReagents (Fremont, CA). Superfrost Plus microscopy slides and micro cover glasses were purchased from VWR (Radnor, PA). TSC+/+ and TSC−/− MEFs are kindly provided by D. Kwiatkowski (Dana-Farber Cancer Institute)55 and not tested for mycoplasma contamination. Withaferin A was purchased from ChromaDex (Irvine, CA) and celastrol was from BOC Sciences (Shirley, NY). We followed the instruction of antibody manufacturers to dilute antibodies in our experiments and any deviation is stated in online methods.
Animals
The Animal Care and Use Committee at Boston Children’s Hospital has approved all animal experiments conducted in this study. Males of C57BL/6J, ob/ob or db/db mice were obtained from Jackson Laboratories. C57BL/6J male mice were placed on high fat diet (HFD, 45 kcal% from fat) at the age of 3 or 7 weeks and maintained on the same diet for 16-20 weeks. HFD was purchased from Research Diets (New Brunswick, NJ). 8- or 9-week-old ob/ob, db/db, and lean mice were kept on normal chow diet (NCD, 13.5% calories from fat) that was purchased from Lab Diet (St Louis, MO). Mice were housed in a 12-h dark/light cycle with the dark cycle encompassing 7 PM to 7 AM and had free access to food and water.
Microarray from mouse embryonic fibroblasts
We plated mouse embryonic fibroblasts (MEFs) on six 10-cm culture dishes in DMEM containing 10% fetal bovine serum (FBS), and incubated them at 37 °C with 5% CO2. After 16 h, we removed the cell medium and washed the cells twice with DMEM containing 1% FBS. We then added 10 ml of 1% FBS DMEM with DMSO or celastrol (250 nM). After 7 h of incubation, we removed the medium from the plates, and snap froze the culture dishes in liquid nitrogen. For RNA extraction, the plates were placed on ice for 15 s. We then added 1 ml of TRIzol reagent to the plates, and incubated at room temperature for 15 min. The cells in TRIzol were collected in 1.5 ml tubes. We then added 200 μl of chloroform, followed by vortexing for 20 s. The tubes were centrifuged at 4 °C for 15 min at 13,400 × g. We transferred the clear top phase to new vials containing 400 μl isopropanol. After 10 min of incubation at room temperature, the tubes were centrifuged at 4 °C for 12 min at 13,400 × g. RNA pellets were washed with 75% ethanol twice. RNA pellets were air dried for 10 min at room temperature and dissolved in 30 μl of RNase free water. We further purified the RNAs using RNeasy MinElute Cleanup Kit according to the manufacturer’s protocols. 1 μg of total RNA was used for microarray analysis using Affymetrix Mouse Gene 1.0 ST arrays.
Microarray from mouse hypothalamus
We treated 12 DIO mice that were fed on HFD for 20 weeks with vehicle for four consecutive days. After this acclimation period, we treated each group of four DIO mice with vehicle, celastrol (100 μg/kg) or withaferin A (2 mg/kg) 30 min prior to dark cycle by intraperitoneal injection for 4 d. After 14 h of the last injection, we extracted hypothalami and froze them in liquid nitrogen. We kept the hypothalamus at −80 °C until later RNA extraction. On the day of RNA extraction, we added 1 ml of TRIzol to each hypothalamus sample. The tissues were homogenized in TissueLyser II (Qiagen, Valencia, CA). We performed the hypothalamic RNA purification and microarray analysis as described in the “microarray from mouse embryonic fibroblasts“ section.
Identifying celastrol-like leptin sensitizer using connectivity map (CMAP)
As explained previously34,35, CMAP is a database with a collection of gene expression dataset from cultured human cell lines treated with various small molecules. Users can search CMAP database with two lists of genes (referred as “signatures” and obtained from any experimental conditions), one in which genes are upregulated, and the other in which genes are downregulated. CMAP reports enrichment scores (which lie between −1 and 1) of all the drugs based on relative correlations between query signature and reference gene expression profiles of individual drugs in the CMAP database. Higher positive scores for a given signature indicate that the experimental results of the particular drug treatment in CMAP show a gene expression profile similar to the signature that is provided by the user.
Administration of withaferin A
We carried out intraperitoneal injections 90 min prior to the dark cycle. Mice were acclimated for 4 d by daily intraperitoneal injection of vehicle (DMSO) before the start of withaferin A injection. Withaferin A was dissolved in DMSO (25 μl), and administered to the mice intraperitoneally once a day. We injected corresponding vehicle groups intraperitoneally with a total volume of 25 μl DMSO once a day. Daily food intake (Figures 2c,h and 3c,h) is an average of 3-d food intake during the first week of treatment. Any deviation of the procedure is stated in corresponding section of online methods or in figure legends.
Immunohistofluorescence staining of phospho-STAT3Tyr705 in the hypothalamus
For celastrol treatment (Supplementary Fig. 4), DIO mice were acclimated with 25 μl of DMSO by intraperitoneal injection 60 min prior to dark cycle for 4 d. Then, mice were divided into two groups, and one group was intraperitoneally injected with 25 μl of vehicle and the other with celastrol (100 μg/kg) for 3 d. 14 h after the third injection, each group of mice was injected one more time with vehicle or celastrol (200 μg/kg). 6 h later, each group was divided into two sub-groups and each sub-group was intraperitoneally administered with either 100 μl of saline or leptin (1 mg/kg in 100 μl of saline). After 40 min of leptin or saline administration, the brain was fixated with 4% paraformaldehyde (PFA) through cardiac perfusion. Following overnight fixation in 4% PFA, the brains were incubated sequentially with 20% sucrose and 30% sucrose for 2 d, and frozen in Tissue-Tek OCT compound (Sakura Finetek; Torrance, CA) in dry ice. A total of 48 sections (30 μm/section, from bregma −0.9 to −2.3), including the whole arcuate nucleus (ARC), ventromedial hypothalamus (VMH) and dorsomedial hypothalamus (DMH) from each mouse, were collected using cryostat (Leica). Half of sections (24 sections) of each brain were subjected to p-STAT3Tyr705 staining. The floating sections were washed with phosphate buffered saline (PBS: 3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, 135 mM NaCl, pH 7.4) for 5 min three times at room temperature and then sequentially incubated with 0.3% H2O2-1% NaOH for 20 min, 0.3% glycine for 10 min, and 0.03% sodium dodecyl sulfate for 10 min. Following 1 h incubation with blocking buffer (3% normal goat serum, 0.3% Triton X-100, 0.02% NaN3 in PBS), the sections were incubated with p-STAT3Tyr705 antibody (1:3,000 in blocking buffer; Clone D3A7, cat. 9145, Cell Signaling) for 2 d at 4 °C. After washing for 5 min with PBST (0.05% Tween 20 in PBS) three times, the sections were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG for 1 h at room temperature. After three additional washes with PBST, the sections were placed in microscopy glasses and covered with cover glasses, and then subjected to image analysis. The image from the whole hypothalamic area of each section was acquired with a ZEISS 710 confocal microscope under 20 X objective (tile scan, resolution: 512 × 512 pixels). The sections without detectable p-STAT3Tyr705 signal were not subjected to the image processing. Therefore, total 154 images for Veh + Sal, 166 images for Veh + Lep, 167 images for Wit A + Sal and 167 images for Wit A + Lep group were analyzed for Fig. 4b–g. Total 93 images for Veh + Sal, 95 images for Veh + Lep, 98 images for Cel + Sal and 97 images for Cel + Lep group were used for the analysis of Supplementary Fig. 4. The p-STAT3Tyr705-positive cell numbers and fluorescence intensities representing p-STAT3Tyr705 were blindly analyzed by using ImageJ software with the option of analysis particle, which determined area of measurement and calculated mean grey value, particle numbers and integrated fluorescence densities. The same setting was applied for all image analysis. The sum of cell numbers (p-STAT3Tyr705 positive total cell number, TCN) and fluorescence intensities (p-STAT3Tyr705 positive total fluorescence intensity) from sections of ARC, VMH and DMH of each brain were calculated and presented as the percentage to control group (vehicle plus saline treated group).
For withaferin A treatment (Fig. 4b–g), DIO mice were acclimated with vehicle 60 min prior to dark cycle for 4 d. Then, mice were divided into two groups receiving either vehicle or withaferin A (2 mg/kg) for 3 d. 14 h after the final injection, each group of mice was further divided into two sub-groups, and one sub-group was intraperitoneally administered with 100 μl of saline and the other sub-group was received with leptin (1 mg/kg in 100 μl of saline). After 40 min of leptin or saline injection, the brain was fixated and samples were analyzed as described above.
Leptin administration and food intake/body weight measurements
We administered vehicle or withaferin A (1.5 mg/kg) intraperitoneally once a day for 2 d 90 min prior to dark cycle to DIO mice. 1 h after the second injection, we further divided the vehicle and withaferin A-injected groups into two subgroups. Each subgroup of mice received either saline or leptin (1 mg/kg, intraperitoneally). Food intake and body weight changes were measured 15 h after saline/leptin administrations (Supplementary Fig. 3a,b).
Leptin administration to ob/ob mice
For experiments in Supplementary Fig. 3c,d, we treated 8-week-old male ob/ob mice with either vehicle or withaferin A (1 mg/kg) for five consecutive days after 4-d acclimation with vehicle. On the sixth day, we divided each group of mice into two subgroups and injected each subgroup with either saline or leptin (0.1 mg/kg) together with vehicle or withaferin A for 2 more weeks. Therefore, we had four groups: vehicle plus saline, vehicle plus leptin, withaferin A plus saline, and withaferin A plus leptin treatment. We recorded food intake and body weights of each mouse daily. Percent suppression in food intake for vehicle and withaferin A groups during the second week of leptin treatment is calculated as follows: 100 × (Food intake of leptin group)/(Food intake of saline group).
Metabolic cage measurements
For metabolic chamber measurements, we used three different mouse models: male DIO mice that were fed on HFD for 16 weeks, and 8-week-old male ob/ob and db/db on regular chow diet. We caged mice individually for at least a week prior to the metabolic chamber experiments. We used Columbus Instruments Comprehensive Lab Animal Monitoring System (CLAMS) for metabolic measurements. Mice were pre-treated with vehicle (DMSO) by intraperitoneal injection for 4 d (acclimation period), and placed into the metabolic chambers on the fifth day. We then injected them with either vehicle or withaferin A (1.5 mg/kg) for three consecutive days in the metabolic cages. Mice had free access to food and water. Energy expenditure (EE) and respiratory exchange ratio (RER) were measured by indirect calorimetry. We measured volumetric rate of oxygen consumption (VO2) and carbon dioxide production (VCO2) by CLAMS, and EE and RER are calculated according to the following formulas:
Metabolic chambers had infrared light sources and optical sensors to detect beam breaks. Total number of beam breaks per mouse was interpreted as physical activity.
For metabolic chamber measurements obtained after chronic withaferin A administration, we individually caged DIO, ob/ob, and db/db mice and treated them with vehicle by intraperitoneal injection for 4 d. We then started administering withaferin A (2 mg/kg) or vehicle for the next 18 d. Subsequently, we placed the mice into metabolic chambers and continued the daily intraperitoneal injections of vehicle and withaferin A for three additional days.
Total protein extraction from hypothalamus and western blotting
For the analysis of hypothalamic STAT3Tyr705 phosphorylation by western blotting (Fig. 4a), DIO mice were acclimated with vehicle for 4 d and then received either vehicle or withaferin A (5 mg/kg, intraperitoneally) 30 min prior to dark cycle. After 15 h of vehicle or withaferin A injection, mice received a single injection of either saline or leptin (1 mg/kg, intraperitoneally). After 40 min, we excised hypothalami and flash froze them in liquid nitrogen. For the analysis of PERKThr980 phosphorylation and Hsp70 levels (Fig. 5b and Supplementary Fig. 5c,d), DIO mice previously acclimated for 1 week were administered intraperitoneally with either vehicle or withaferin A (2 mg/kg) 3 h prior to dark cycle for 3 d. 6 h after the third injection of vehicle or withaferin A, the hypothalamus was collected and subjected to immunoblot analysis.
Hypothalamic tissues were homogenized with a bench-top TissueLyser II in ice-cold tissue lysis buffer (25 mM Tris-HCl, pH 7.4; 100 mM NaF; 50 mM Na4P2O7; 10 mM Na3VO4; 10 mM EGTA; 10 mM EDTA; 1% NP-40; supplemented with phosphatase and protease inhibitors). After homogenization, lysates were rotated for 1 h at 4 °C and then subjected to centrifugation at 13,400 × g for 10 min at 4 °C. Protein concentration of the supernatant was quantified using a Protein Assay Kit (Bio-Rad). Protein samples were mixed with 5 × Laemmli buffer and boiled at 95 °C for 5 min before loading on sodium dodecyl sulfate poly acrylamide gels (SDS-PAGE). After electrophoresis, we transferred the proteins onto PVDF membranes at 4 °C, 100 V for 2 h, and blocked the membranes in TBST (Tris Buffered Saline with Tween-20; 10 mM Tris-base, 150 mM NaCl, 0.1% Tween-20, pH 7.4) with 10% blocking reagent. We incubated the membranes with primary antibodies overnight in TBST containing 10% blocking reagent. After primary incubation, we washed the membranes three times for 20 min with TBST. We then incubated the membranes with secondary antibodies in TBST with 10% blocking reagent for 1 h at room temperature. After washing the membrane three times in TBST, we developed the membranes using a chemiluminescence assay system and quantified band intensities using ImageJ (NIH). We followed the instruction of antibody manufacturers to dilute antibodies in our western blotting.
RNA extraction, cDNA Synthesis and quantitative real-time PCR
DIO mice acclimated with DMSO for 4 d received DMSO or withaferin A (2 mg/kg) intraperitoneally 3 h before dark cycle for 3 d. The hypothalamus was removed 6 h after the final injections. Total RNA for quantitative real time-PCR (qPCR) was extracted from the hypothalamus with TRIzol lysis reagent following manufacturer’s protocol. Complementary DNA (cDNA) was synthesized with 1 μg of total RNA using iScript cDNA synthesis kit (Bio-Rad) with the following conditions: 25 °C for 5 min, 42 °C for 30 min, and 85 °C for 5 min. QPCR was conducted by Taqman® or SYBR-based qPCR assay on QuantStudio™ 6 Flex Real-Time PCR system (Life Technologies). The relative expression of genes of interest was calculated by comparative Ct method, and Gapdh (for Taqman® assay) or Rn18s (for SYBR green-based qPCR) was used as an endogenous control. Taqman® primers and probes were obtained from Life Technologies and their assay identification (ID) numbers are as follows:
Spliced XBP1 (Xbp1s) (assay ID: Mm03464496_m1), Socs3 (Mm00545913_s1), Gapdh (Mm99999915_g1), Ero1l (Mm00469296_m1), Pomc (Mm00435874_m1), Npy (Mm03048253_m1), Agrp (Mm00475829_g1), Calr (Mm00482936_m1), Edem1 (Mm00551797_m1), Dnajb9 (Mm01622956_s1), Hspa5 (Mm00517690_g1), Pdia3 (Mm00433130_m1), Herpud1 (Mm00445600_m1).
The primers for SYBR green-based qPCR were synthesized by Integrated DNA Technologies (IDT) and their sequences were as follows:
Bcl2l1 forward: 5’-GACAAGGAGATGCAGGTATTGG-3’
Bcl2l1 reverse: 5’-TCCCGTAGAGATCCACAAAAGT-3’
Ccl3 forward: 5’- TTCTCTGTACCATGACACTCTGC-3’
Ccl3 reverse: 5’- CGTGGAATCTTCCGGCTGTAG-3’
Ccl4 forward: 5’-TTCCTGCTGTTTCTCTTACACCT-3’
Ccl4 reverse: 5’-CTGTCTGCCTCTTTTGGTCAG-3’
Ccl5 forward: 5’-GCTGCTTTGCCTACCTCTCC-3’
Ccl5 reverse: 5’-TCGAGTGACAAACACGACTGC-3’
Ccl7 forward: 5’-GCTGCTTTCAGCATCCAAGTG-3’
Ccl7 reverse: 5’-CCAGGGACACCGACTACTG-3’
Ccl12 forward: 5’- ATTTCCACACTTCTATGCCTCCT-3’
Ccl12 reverse: 5’- ATCCAGTATGGTCCTGAAGATCA-3’
Cd14 forward: 5’- CTCTGTCCTTAAAGCGGCTTAC-3’
Cd14 reverse: 5’- GTTGCGGAGGTTCAAGATGTT-3’
Cxcl9 forward: 5’- TCCTTTTGGGCATCATCTTCC-3’
Cxcl9 reverse: 5’- TTTGTAGTGGATCGTGCCTCG-3’
Cxcl10 forward: 5’-CCAAGTGCTGCCGTCATTTTC-3’
Cxcl10 reverse: 5’-GGCTCGCAGGGATGATTTCAA-3’
Hgf forward: 5’-ATGTGGGGGACCAAACTTCTG-3’
Hgf reverse: 5’-GGATGGCGACATGAAGCAG-3’
Nos2 forward: 5’- GTTCTCAGCCCAACAATACAAGA-3’
Nos2 reverse: 5’- GTGGACGGGTCGATGTCAC-3’
Sod2 forward: 5’- CAGACCTGCCTTACGACTATGG-3’
Sod2 reverse: 5’- CTCGGTGGCGTTGAGATTGTT-3’
Blood collection
We collected blood from the tail vein with heparinized capillary tubes, transferred the blood to ice-cold 1.5 ml tubes, and centrifuged them at 2,000 × g for 20 min at 4 °C. We transferred the plasma portions to new vials and stored them at −80 °C until further analysis.
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
For GTT, mice were fasted for 15 h and dextrose (1 g/kg for lean and DIO mice, 0.5 g/kg for ob/ob and db/db mice) was administrated intraperitoneally. Blood glucose levels were measured from the tail before dextrose administration and 15, 30, 60, 90 and 120 min after the administration.
For ITT, mice were fasted for 6 h starting at 8 AM until 2 PM. Recombinant human insulin (1 IU/kg for lean and DIO mice, 2 IU/kg for ob/ob and db/db mice) was administered intraperitoneally. Blood glucose levels were measured from the tail before insulin administration and 15, 30, 60, 90 and 120 min after the administration.
Hormone and metabolite measurements from mouse plasma
We measured plasma leptin, insulin, cholesterol, alanine transaminase (ALT), and aspartate transaminase (AST) using the corresponding ELISA or assay kits according to the manufacturer’s instructions. We used 5 μl of plasma sample of lean mice, and 5 μl of five times diluted plasma sample from DIO and db/db mice for leptin ELISA. We used 5 μl of plasma from lean, DIO, ob/ob, or db/db mice for insulin ELISA and AST assay, 10 μl for ALT assay and 3 μl for cholesterol assay.
Triiodothyronine (T3) radioimmunoassay
Serum total T3 was measured as previously described using a modified Coat-a-Count radioimmunoassay (Siemens) and T3 charcoal uptake to correct for serum binding56,57.
Hematoxylin and eosin (H&E) staining
We treated DIO mice or lean mice with vehicle or withaferin A (1.25 mg/kg) for 3 weeks. At the end of the treatment period, we dissected liver tissue and stored it in 10% buffered formalin phosphate. Paraffin embedded liver sections were H&E stained.
Statistical analysis
All data are presented as mean ± s.e.m. Statistical significance is measured with Prism version 6 (GraphPad, La Jolla, CA) or SPSS version 23 software (IBM, Armonk, NY) via the Student’s t-test (two-tailed), one-way or two-way ANOVA as indicated in figure legends. P values smaller than 0.05 were considered significant. Numbers of cohorts and n values for each experiment were indicated in figure legends. Dead or sick mice before the end of experiments or statistical outliers (judged by Grubb’s outlier test) were excluded in the final analysis. No blinding or randomization was used. No statistical method was used to predetermine sample size and sample size was determined based on previous experiments and literatures. The variance was similar in the groups being compared.
For statistical analysis of the experimental results in Fig. 4 and Supplementary Figs 3 and 4, we used one-way ANOVA because of the following reasons. Withaferin A and celastrol require leptin to increase leptin receptor signaling and anti-obesity effects. However, withaferin A or celastrol treatment alone does demonstrate anti-obesity effect in DIO mice due to high level of circulating endogenous leptin, and their anti-obesity effect is further increased by acute exogenous leptin administration. Thus, we are interested in single factor effect of the drug treatment (withaferin A or celastrol) under the same level of leptin (either within saline group with similar level of endogenous leptin or within the leptin group with the similar level of endogenous plus exogenous leptin), and used one-way ANOVA with post hoc test (Fig. 4 and Supplementary Figs 3 and 4).
Supplementary Material
Acknowledgments
We greatly appreciate the time of M. White for critical reading of our manuscript and for his contributions and suggestions. We thank S. Cabi and I. Cakir for their initial experimental contributions to this work. We also thank to S. Mert and B. Akosman for their help on some of the experiments, and S. Huang and M. Mulcahey-Maynard for measurement of serum T3 levels. This work was mainly supported by the funds provided to U. Ozcan from Department of Medicine, Boston Children’s Hospital, and also by grant R01DK098496 (U.O.) from the National Institutes of Health, American Diabetes Association Career Development grant #7-09-CD-10 (U.O.) and support from Fidelity Biosciences Research Initiative.
Footnotes
AUTHOR CONTRIBUTIONS
U.O. came up with the idea to investigate compounds that have similar gene expression to celastrol by using CMAP. U.O. directed this and subsequent analyses and picked withaferin A as a candidate for the treatment of type 2 diabetes and obesity. J.Lee, J.Liu, X.F., M.A.S.H., P.M., D.I. and J.W.C. performed experiments under the direction of U.O. U.O., J.Lee, J.Liu and X.F. analyzed data. U.O. wrote the manuscript, and J.Lee, J.Liu, X.F. were involved with the writing and the preparation of the manuscript.
COMPETING FINANCIAL INTERESTS
U. Ozcan is a scientific founder, shareholder, and scientific advisory board and board of directors member of ERX Pharmaceuticals Inc.
References
- 1.Wang Y, Beydoun MA, Liang L, Caballero B, Kumanyika SK. Will all Americans become overweight or obese? estimating the progression and cost of the US obesity epidemic. Obesity. 2008;16:2323–2330. doi: 10.1038/oby.2008.351. [DOI] [PubMed] [Google Scholar]
- 2.Olshansky SJ, et al. A potential decline in life expectancy in the United States in the 21st century. The New England journal of medicine. 2005;352:1138–1145. doi: 10.1056/NEJMsr043743. [DOI] [PubMed] [Google Scholar]
- 3.Berrington de Gonzalez A, et al. Body-mass index and mortality among 1.46 million white adults. The New England journal of medicine. 2010;363:2211–2219. doi: 10.1056/NEJMoa1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodgers RJ, Tschop MH, Wilding JP. Anti-obesity drugs: past, present and future. Disease models & mechanisms. 2012;5:621–626. doi: 10.1242/dmm.009621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalamaga M, et al. Leptin at the intersection of neuroendocrinology and metabolism: current evidence and therapeutic perspectives. Cell metabolism. 2013;18:29–42. doi: 10.1016/j.cmet.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 6.Myers MG, Jr, et al. Challenges and opportunities of defining clinical leptin resistance. Cell metabolism. 2012;15:150–156. doi: 10.1016/j.cmet.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dietrich MO, Horvath TL. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends in neurosciences. 2013;36:65–73. doi: 10.1016/j.tins.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Friedman JM. Modern science versus the stigma of obesity. Nature medicine. 2004;10:563–569. doi: 10.1038/nm0604-563. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 10.Frederich RC, et al. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nature medicine. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 11.Considine RV, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. The New England journal of medicine. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 12.Maffei M, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature medicine. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 13.Heymsfield SB, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 14.Mori H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nature medicine. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 15.Howard JK, et al. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nature medicine. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 16.Loh K, et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell metabolism. 2011;14:684–699. doi: 10.1016/j.cmet.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horvath TL, et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14875–14880. doi: 10.1073/pnas.1004282107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneeberger M, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155:172–187. doi: 10.1016/j.cell.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dietrich MO, Liu ZW, Horvath TL. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell. 2013;155:188–199. doi: 10.1016/j.cell.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diano S, et al. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nature medicine. 2011;17:1121–1127. doi: 10.1038/nm.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long L, Toda C, Jeong JK, Horvath TL, Diano S. PPARgamma ablation sensitizes proopiomelanocortin neurons to leptin during high-fat feeding. The Journal of clinical investigation. 2014;124:4017–4027. doi: 10.1172/JCI76220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren D, Li M, Duan C, Rui L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell metabolism. 2005;2:95–104. doi: 10.1016/j.cmet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews Molecular cell biology. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 24.Park SW, Ozcan U. Potential for therapeutic manipulation of the UPR in disease. Seminars in immunopathology. 2013;35:351–373. doi: 10.1007/s00281-013-0370-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. The Journal of biological chemistry. 2014;289:1203–1211. doi: 10.1074/jbc.R113.534743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 27.Ozcan U, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozcan L, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell metabolism. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 29.Ramirez S, Claret M. Hypothalamic ER stress: A bridge between leptin resistance and obesity. FEBS Lett. 2015;589:1678–1687. doi: 10.1016/j.febslet.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 30.Won JC, et al. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity. 2009;17:1861–1865. doi: 10.1038/oby.2009.194. [DOI] [PubMed] [Google Scholar]
- 31.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 32.Calfon M, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 33.Williams KW, et al. Xbp1s in Pomc Neurons Connects ER Stress with Energy Balance and Glucose Homeostasis. Cell metabolism. 2014 doi: 10.1016/j.cmet.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamb J, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mirjalili MH, Moyano E, Bonfill M, Cusido RM, Palazon J. Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules. 2009;14:2373–2393. doi: 10.3390/molecules14072373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winters M. Ancient medicine, modern use: Withania somnifera and its potential role in integrative oncology. Alternative medicine review : a journal of clinical therapeutic. 2006;11:269–277. [PubMed] [Google Scholar]
- 38.Mishra LC, Singh BB, Dagenais S. Scientific basis for the therapeutic use of Withania somnifera (ashwagandha): a review. Alternative medicine review : a journal of clinical therapeutic. 2000;5:334–346. [PubMed] [Google Scholar]
- 39.Halaas JL, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 40.Van Heek M, et al. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. The Journal of clinical investigation. 1997;99:385–390. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halaas JL, et al. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pelleymounter MA, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 43.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. The Journal of clinical investigation. 1996;98:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fei H, et al. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:7001–7005. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hakansson ML, Brown H, Ghilardi N, Skoda RC, Meister B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1998;18:559–572. doi: 10.1523/JNEUROSCI.18-01-00559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozcan U, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Molecular cell. 2008;29:541–551. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu Y, et al. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochemical pharmacology. 2010;79:542–551. doi: 10.1016/j.bcp.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neckers L. Heat shock protein 90: the cancer chaperone. Journal of biosciences. 2007;32:517–530. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 50.Kaileh M, et al. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. The Journal of biological chemistry. 2007;282:4253–4264. doi: 10.1074/jbc.M606728200. [DOI] [PubMed] [Google Scholar]
- 51.Harris RB, Kelso EW, Flatt WP, Bartness TJ, Grill HJ. Energy expenditure and body composition of chronically maintained decerebrate rats in the fed and fasted condition. Endocrinology. 2006;147:1365–1376. doi: 10.1210/en.2005-1156. [DOI] [PubMed] [Google Scholar]
- 52.Arch JR, Stock MJ, Trayhurn P. Leptin resistance in obese humans: does it exist and what does it mean? International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity. 1998;22:1159–1163. doi: 10.1038/sj.ijo.0800779. [DOI] [PubMed] [Google Scholar]
- 53.Ma X, et al. Celastrol Protects against Obesity and Metabolic Dysfunction through Activation of a HSF1-PGC1alpha Transcriptional Axis. Cell metabolism. 2015;22:695–708. doi: 10.1016/j.cmet.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Denver RJ, Bonett RM, Boorse GC. Evolution of leptin structure and function. Neuroendocrinology. 2011;94:21–38. doi: 10.1159/000328435. [DOI] [PubMed] [Google Scholar]
- 55.Kwiatkowski DJ, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 56.Zavacki AM, et al. Type 1 iodothyronine deiodinase is a sensitive marker of peripheral thyroid status in the mouse. Endocrinology. 2005;146:1568–1575. doi: 10.1210/en.2004-1392. [DOI] [PubMed] [Google Scholar]
- 57.Kaplan MM. Subcellular alterations causing reduced hepatic thyroxine-5’-monodeiodinase activity in fasted fats. Endocrinology. 1979;104:58–64. doi: 10.1210/endo-104-1-58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.