

Abstract
Purpose of review
Long non-coding RNAs (lncRNAs) have emerged as powerful regulators of nearly all biological processes. Their cell-type and tissue-specific expression in health and disease provides new avenues for diagnosis and therapy. This review highlights the role of lncRNAs that are involved in cardiovascular disease (CVD) with a special focus on cell types involved in cardiac injury and remodelling, vascular injury, angiogenesis, inflammation, and lipid metabolism.
Recent findings
Almost 98% of the genome does not encode for proteins. LncRNAs are among the most abundant type of RNA in the non-coding genome. Accumulating studies have uncovered novel lncRNA-mediated regulation of CVD-associated genes, signalling pathways, and pathophysiological responses. Targeting lncRNAs in vivo using short antisense oligonucleotides or by gene editing has provided important insights into disease pathogenesis through epigenetic, transcriptional, or translational mechanisms. Although cross-species conservation still remains a major obstacle, there is increasing appreciation that altered expression of lncRNAs associates with stage-specific CVD pathologies and in human patient cohorts, providing new opportunities for diagnosis and therapy.
Summary
A better understanding of lncRNAs will not only fundamentally improve our understanding of key signaling pathways in CVD, but also aid in the development of effective new therapies and RNA-based biomarkers.
Keywords: lncRNAs, non-coding RNA, cardiovascular disease
Introduction
Despite recent advances, cardiovascular disease (CVD) is the leading cause of death worldwide. Major mechanistic gaps in the understanding of signaling pathways involved in tissue homeostasis and pathophysiological conditions remain and contribute to the lack of targeted therapeutics in the field of CVD. The traditional view on non-coding RNA (ncRNA) as “junk of the genome” has been replaced by the appreciation that these transcripts may influence disease-associated genes and contribute to disease pathogenesis. Recent advances in next generation sequencing, especially RNA sequencing (RNA-Seq), has enabled research on a new class of ncRNAs termed long non-coding (lncRNAs). These non-coding sequences are defined as being longer than 200 nucleotides in length, yet exhibit mRNA-like features such as being 5’-capped, spliced, and polyadenylated. Because of their complex tertiary structure, lncRNAs may interact with RNA, DNA, or protein to control transcriptional, epigenetic, or translational responses. Extensive research in nearly every disease model has highlighted to date their important role in regulating gene expression and key signaling pathways. Although a fast growing number of studies have begun to elucidate our understanding of their function, for the vast majority, the precise function remains poorly understood, particularly in CVD. Herein, we will highlight examples of lncRNAs (Figure 1) with significant translational value in CVD and future directions.
Figure 1.
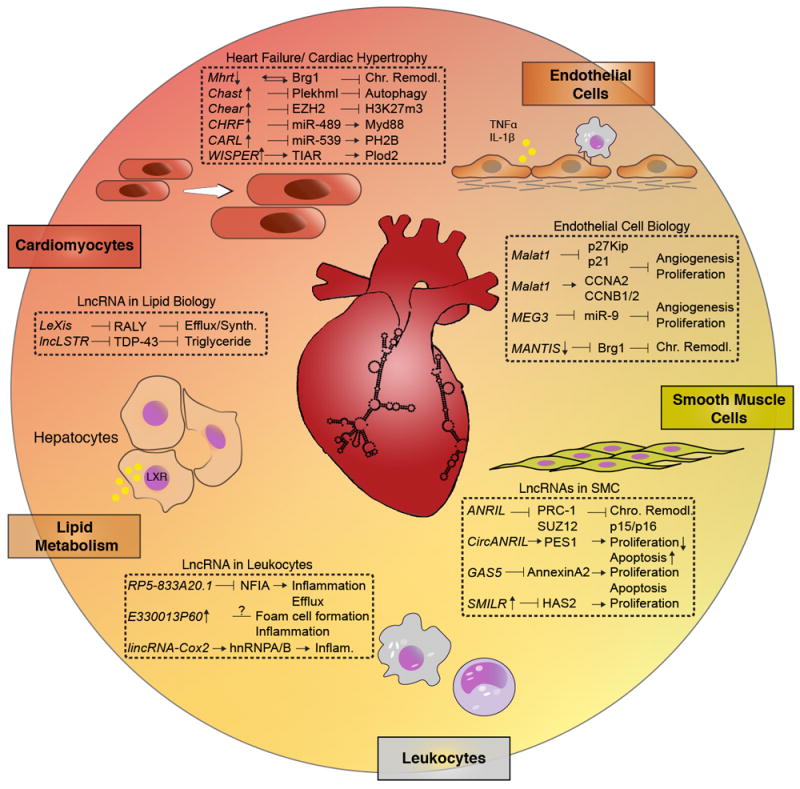
Selection of lncRNAs involved in cardiac injury and remodelling, endothelial cell biology and angiogenesis, vascular smooth muscle cell proliferation, leukocytes and inflammation, and lipid metabolism.
LncRNAs in Cariomyocytes
Cardiac hypertrophy is characterized by thickening of the ventricle wall, subsequently reducing the contractile capability of the heart to pump blood. Prolonged hypertrophy of cardiomyocytes leads to heart failure [1]. In a transverse aortic constriction (TAC)-mice model, Han et al. [2] have identified a cluster of lncRNAs, which are antisense to myosin heavy chain 7 (Myh7) loci. Although myosin heavy-chain associated RNA transcript (Mhrt) loci lies antisense to Myh7, it’s regulatory role could not be simply explained by RNA:RNA interference. Interestingly, Mhrt binds to Brg1, an important member of the chromatin repressor complex [2]. Brg1 has a specific dual-binding feature for either Mhrt or chromatinized DNA, enabling competitive inhibition by which Mhrt prevents Brg1 from binding to its genomic DNA targets, thus blocking its chromatin remodelling function. Furthermore, as a consequence of pressure-induced myocardial injury, Brg1 represses Mhrt expression, providing a Mhrt-Brg1 feedback circuit. Han et al. [2] demonstrated that Mhrt protected the heart from excessive hypertrophy and heart failure by restoring Mhrt expression in transgenic mice. Since the human Mhrt is also repressed in various types of myopathic hearts, this lncRNA-chromatin feedback circuit may be critical for protecting the heart in response to injury [2].
In a whole-genome lncRNA profiling study of TAC-operated mice, Viereck et al. [3] identified a RNA transcript named cardiac hypertrophy-associated (Chast) being highly expressed in cardiomyocytes from hypertrophic hearts. Similarly, human Chast homolog expression was induced in hypertrophic heart tissue from aortic stenosis patients. Silencing of Chast by GapmeRs prevented pathological cardiac remodelling in mice; vice versa progression of hypertrophy was achieved by viral-based overexpression of Chast in vitro and in vivo. Mechanistically, Chast overlaps with Plekhml, which is a regulatory factor for autophagy [4]. Plekhml expression inversely correlated with Chast and silencing of Plekhml mimicked the Chast overexpression phenotype, thereby blocking autophagy of cardiomyocytes and promoting cardiac hypertrophy [3].
More recently, a lncRNA named cardiac-hypertrophy-associated epigenetic regulator (Chaer) was found to be highly induced in hearts of mice with pressure-overload-induced heart failure. Chaer is specifically expressed in cardiomyocytes, but not in fibroblasts. Hypertrophy as well as TAC-induced cardiac fibrosis was significantly attenuated in Chaer-KO mice, whereas no changes on histology could be observed under normal conditions. Mechanistically, Chear binds transiently to PRC2 through EZH2-binding in the onset of hypertrophy, thereby limiting PRC2-mediated H3K27m3 promoter methylation of cardiac hypertrophy-associated genes. Gain-of-function of Chaer mimicked the hypertrophic phenotype in human pluripotent stem cells derived from cardiomyocytes, suggesting cross-species conservation of its function [5].
Wang et al. [6] identified a different lncRNA termed cardiac hypertrophy-related factor (CHRF) being highly induced in cardiomyocytes of mice treated with Ang-II. Furthermore, CHRF expression was induced in heart tissue of patients with heart failure. Mechanistically, CHRF acts as a sponge for miR-489, preventing its target Myd88 from degradation [6]. Similar to CHRF, the cardiac apoptosis-related lncRNA (CARL) has been reported to act as a molecular sponge for miR-539, thereby competitively binding with PHB2. PHB2 is important for mitochondrial function and regulation of apoptosis [7]. Hence, CARL suppresses myocytes from undergoing mitochondrial fission and apoptosis, thereby preventing myocardial infarction injury-induced cardiac remodelling [8].
Finally, a cardiac fibroblast-enriched lncRNA named WISPER (WIsp2 SuPer-Enhancer associated RNA) was found to regulate cardiac fibrosis. Silencing of WISPER in vivo using short antisense oligonucleotides (ASO) attenuated MI-induced fibrosis and remodelling by injection pre- or post-injury. Moreover, WISPER expression was significantly increased in cardiac tissues from patients with aortic stenosis [9].
LncRNAs in endothelial cells
The endothelium has an important role in vascular homeostasis. Numerous factors may damage endothelial structure and function, including physical and biochemical injury and immune-mediated damage, resulting in endothelial dysfunction, an early predictor of atherosclerosis. Emerging studies have revealed important implications of lncRNAs in endothelial cell (EC) physiology and their regulatory roles in endothelial dysfunction, suggesting that their therapeutic regulation could be relevant to CVD.
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is a lncRNA highly expressed in different types of ECs in both the macro- and microvasculature and has been implicated in controlling EC inflammation and angiogenesis [10-12]. For example, MALAT1 expression is profoundly increased by hypoxia, high glucose, and oxidative stress. Silencing of MALAT1 reduced EC proliferation by inhibiting cell cycle progression, decreasing the number of cells in S-phase under basal or hypoxic conditions, and after VEGF-stimulation. Mechanistically, MALAT1 silencing reduced the S-phase cyclins CCNB1, CCNB2, and CCNA2, while increasing the cell cycle inhibitory genes p21 and p27Kip1 [10]. Similar results were observed by Liu. et al. [12] where MALAT1 knockdown using GapmeRs reduced cell viability of retinal ECs, while MALAT1 overexpression had the opposite effect. Interestingly, MALAT1 knockdown decreased phosphorylated p38 levels in retinal ECs, while p38 MAPK inhibition strongly blocked the effect of MALAT1-induced proliferation of retinal ECs, suggesting that MALAT1 regulates the hyper-proliferation of retinal ECs through p38 MAPK signaling. In addition, MALAT1 silencing decreased glucose-induced up-regulation of inflammatory mediators IL-6 and TNFα through activation of SAA3 in ECs [11]. Consistent with these in vitro observations, intraocular injection of MALAT1 shRNA in streptozotocin-induced diabetic rats decreased the number of apoptotic retinal cells, alleviated vascular leakage induced by hyperglycemia, and significantly reduced retinal inflammation [12]. Under non-diabetic conditions, pharmacological inhibition of MALAT1 in mice undergoing hindlimb ischemia reduced blood flow recovery and capillary density, verifying studies that silencing of MALAT1 profoundly impairs EC proliferation, and leads to a block in vessel outgrowth in vitro and in vivo [1]. Phenotypically, MALAT1-KO mice showed a delayed vessel extension in the retina and a reduction of the vessel density when compared with wild-type littermates. Moreover, the number of proliferating EC, as identified by phosphohistone H3 staining, was significantly reduced in MALAT1-KO mice [10]. While the molecular mechanisms mediating MALAT1’s angiogenic effects has not been clarified, it may serve as an endogenous sponge to reduce the microRNA (miRNA) miR-26b and induce the expression of the autophagy-related gene ULK2 and in turn EC survival [13]. Because the related family member miR-26a harbors a near identical seed sequence to miR-26b, a known anti-angiogenic miRNA in post-MI repair and diabetic wound healing [14,15], future studies will be of interest to assess if MALAT1 may mediate angiogenic effects via interactions with this miRNA. Collectively, these studies highlight an important role for MALAT1 in regulating both vascular inflammation and angiogenesis.
Maternally expressed gene 3 (MEG3) is a lncRNA with regulatory roles in both angiogenesis and diabetes-related microvascular dysfunction [16]. MEG3 expression was significantly reduced in the retinas of STZ-induced diabetic mice and in vitro in retinal ECs upon oxidative stress and high glucose stimulation. Knockdown of MEG3 in vivo aggravates retinal vessel dysfunction, as observed by serious capillary degeneration, inflammation, and increased microvascular leakage. Knockdown of MEG3 in retinal ECs increased their proliferation, migration, and tube formation capacity, through a mechanism mediated by the activation of the PI3K/Akt signaling pathway. MEG3 exerts its function in part by negatively regulating miR-9, hence acting as a miRNA sponge in vascular ECs, where it regulates angiogenesis and proliferation [17]. Furthermore, MEG3 expression is increased in senescent human ECs and MEG3 silencing rescued age-induced impairment of angiogenesis in cultured ECs. Moreover, in mice undergoing hindlimb ischemia, MEG3 inhibition augmented blood flow recovery [18]. Finally, Meg3-KO mice showed increased expression of vascular endothelial growth factor pathway genes and increased cortical microvessel density in Meg3-null embryos, suggesting the important role of MEG3 in vascularization and angiogenesis [19].
Another lncRNA strongly regulated in ECs is MANTIS (lncRNA n342419), a nuclear localized lncRNA controlled by the histone demethylase JARID1B, suggesting a chromatin regulatory function [20]. MANTIS was reduced in patients with idiopathic pulmonary arterial hypertension (IPAH) and in a rat IPAH disease model, whereas it was induced in ECs isolated from human glioblastoma patients as well as in carotid arteries of Macaca fascicularis subjected to atherosclerosis regression diet. Functional silencing of MANTIS by CRISPR/Cas9-mediated deletion or oligonucleotide delivery (siRNAs or GapmeRs) inhibited angiogenic sprouting, tube formation, and migration of ECs in vitro and in vivo in mice injected with matrigel-embedded HUVECs. Mechanistically, MANTIS interacts with Brg1, the catalytic subunit of the SWI/SNF chromatin remodelling complex and regulates SOX18, SMAD6 and COUP-TFII, which are all implicated in angiogenesis modulation.
LncRNAs in smooth muscle cells
In atherogenesis, ECs, platelets, and inflammatory leukocytes release mediators, such as cytokines and growth factors that will induce changes of VSMCs from the contractile, quiescent state to the active synthetic state, inducing VSMC proliferation and migration, and extracellular matrix (ECM) protein deposition. In this context, recent studies have shown that several lncRNAs play an important role in regulating the VSMC proliferation and migration, modulating neointimal formation, and atherosclerosis progression.
The 9p21.3 risk locus, identified in several genome-wide association studies (GWAS) for coronary artery disease (CAD) susceptibility, is adjacent to the last exons of the antisense ncRNA in the INK4 locus (ANRIL) and encompasses multiple single nucleotide polymorphisms (SNPs). This increased risk is independent of all known CAD risk factors, which led to the idea that this locus may induce a different biological pathway relevant for atherosclerosis development [21]. Interestingly, silencing ANRIL at different target exons (exon1/19) in human aortic VSMCs induced regulatory effects on different genes, suggesting that different splicing variants of ANRIL might play distinct roles in cell physiology [22]. Consistent with this, two ANRIL transcripts (EU741058 and NR_003529) were found significantly increased in human atherosclerotic plaque tissues and peripheral blood mononuclear cells from CAD patients as compared to healthy subjects, while another ANRIL transcript, DQ485454, remained unaffected [23].
Mechanistically, Yap et al. showed in chromatin fractions that ANRIL directly binds to CBX7, a polycomb repression complex-1 (PRC-1) component [24], suggesting that ANRIL may affect chromatin activity. Independently, Kotake et al. [25] showed that ANRIL associates with SUZ12, a component of PRC-2. Inhibition of ANRIL disrupts the binding of PRC-1 and PRC-2 on the INK4 locus, increasing the mRNA levels of p15 and p16, two of the genes encoded by the INK4 locus and limiting cellular life span [24-26]. Taken together, these data suggest that ANRIL binds to and recruits PRC-1 and -2 to the INK4 locus, leading to the repression of p15 and p16 transcription.
Adding another layer of complexity, a recent study revealed that circularization of ANRIL activates a different cellular mechanism, conferring atheroprotection by controlling rRNA maturation and modulating pathways of atherogenesis [27]. CircANRIL binds to PES1, an essential 60S-preribosomal assembly factor, impairing pre-rRNA processing and ribosome biogenesis in VSMC and macrophages. As a consequence, circANRIL induces nucleolar stress and p53 activation, resulting in the inhibition of proliferation and induction of apoptosis, as observed for the linearANRIL. Future studies will be informative to assess the functionality of circANRIL in relevant atherosclerotic models.
Growth arrest–specific 5 (GAS5) is a lncRNA that plays important roles in several biological processes including apoptosis, cell proliferation, and cell growth arrest [28,29] and it was recently observed to regulate vascular remodelling in hypertension [30]. GAS5 knockdown aggravated the microvascular dysfunction in a hypertension rat model, as shown by increased capillary leakage and retinal neovascularization. Moreover, GAS5 silencing regulated VSMC phenotypic conversion, ECs activation and proliferation, and EC-VSMC communication in vitro and in vivo. In response to oxidative stress, GAS5 knockdown partially reversed the H2O2-induced reduction of VSMC and EC viability and reversed apoptosis in response to hypoxia stress. GAS5 knockdown also accelerated VSMC proliferation and migration, and decreased the expression of contractile marker proteins including α-smooth muscle actin and calponin, by the β-catenin signaling pathway [30]. Similar results were observed in another study, where GAS5 regulated the VSMC proliferation and migration through AnnexinA2, a Ca2+-dependent RNA-binding protein [31]. GAS5 expression was reduced In response to isoproterenol-induced cardiac fibrosis in rats, and in cardiac fibroblasts treated with TGF-b1. Overexpression of GAS5 inhibited the proliferation of cardiac fibroblasts by decreasing the expression of miR-21 and indirectly regulating one of its targets, PTEN, suggesting its importance of GAS5 in another CVD-associated disease [32].
Another lncRNA highly expressed in SMC after interleukin-1α and PDGF stimulation is the smooth muscle–induced lncRNA enhances replication (SMILR). Following stimulation, SMILR expression was increased in both the cytoplasm and nucleus, and was detected in conditioned media. Furthermore, knockdown of SMILR significantly reduced cell proliferation and the expression of the nearby gene HAS2 [24]. However, no change in the expression of HAS1 and HAS3 isoforms was observed, or for HAS2-AS1 lncRNA or the ZHX2 gene, indicating that the SMILR knockdown effect is specific to HAS2. Increased expression of SMILR was observed in human samples from patients with unstable atherosclerotic plaques and in plasma from patients with high plasma C-reactive protein levels, suggesting SMILR may be a driver of VSMC proliferation, with potential implications in atherogenesis [33].
LncRNAs in lipid biology
Disordered lipid metabolism is one of the pathological processes contributing to the onset and progression of atherosclerosis [34]. Emerging studies have indicated that lncRNAs may serve as key regulators of lipid homeostasis. For example, the lncRNA named LeXis, was recently identified to regulate liver X receptors (LXRs)-mediated cholesterol synthesis. Sallam et al [35] demonstrated that LeXis promotes cholesterol efflux and inhibition of cholesterol biosynthesis by binding to a heterogeneous ribonucleoprotein called RALY. The binding of LeXis to RALY affected its ability to interact with DNA, which in turn prevented cholesterol synthesis via transcriptional control of a subset of genes. As expected, total serum cholesterol was reduced in mice upon adenoviral administration of LeXis [35]. In addition to LeXis, a liver-enriched lncRNA termed lncLSTR was found to regulate systemic triglyceride (TG) metabolism in vivo. Knockdown of lncLSTR using adenovirus reduced plasma TG levels both in lncLSTR-KO and ApoE-/- mice, without changes in total cholesterol levels. Mechanistically, lncLSTR interacted with TDP-43 and altered FXR activity, which subsequently altered a gene cascade involved in TG clearance [36]. The translational value of human of LeXis (which does not have a human lncRNA homologue) and lncLSTR remains unknown.
LncRNAs in monocytes and macrophages
A hallmark for the initial stage of atherosclerosis is the recruitment of leukocytes to activated ECs. After migration into the intima and engulfment of modified lipoprotein particles, monocytes become lipid-laden macrophages, known as foam cells [37,38]. In the core region of a atherosclerotic plaque are a variety of immune cells present to orchestrate a series of inflammatory steps [39-41]. Although our understanding of the role of lncRNAs in regulating inflammatory cells in this multistep process of lesion progression is nascent, Hu et al. [42] identified increased expression of lncRNA RP5-833A20.1 in human foam cells. Overexpression of RP5-833A20.1 in THP-1 macrophages reduced cholesterol efflux and increased inflammatory cytokines, including IL-1β, IL-6, and TNFα. Mechanistically, RP5-833A20.1 decreased the expression of NFIA by inducing miR-382-5p expression, which adds a new mechanism of lncRNA-miRNA interaction. However, the specific mechanism of how RP5-833A20.1 regulates miR-382-5p expression for macrophage foam formation will require further investigation [42].
Reddy et al. [43] identified lncRNA E330013P60 by comparing the expression pattern of bone marrow derived macrophage (BMDM) in diabetic db/db mice and non-diabetic db/+ mice. Diabetes induced E330013P60 expression and promoted the polarization of macrophages into a more pro-inflammatory phenotype. E330013P60 loci overlays with MiR143HG, which has been described in cardiac lineage development, known as CARMEN [44]. LncRNA E330013P06 overexpression in macrophages induced the expression of pro-inflammatory genes and promoted foam cell formation [43]. The biological relevance for this lncRNA in vivo remains poorly understood.
By RNA-Seq studies of mouse BMDMs stimulated with the a synthetic Tlr2 ligand, Carpenter et al. [45] identified lincRNA-Cox2. In loss-of-function studies they demonstrated that lincRNA-Cox2 regulates various immune genes (e.g. Ccl5, IL-6) [45]. A another study found that in intestinal epithelial cells, lincRNA-Cox2 was induced by TNFα through activation of the NF-κB signaling pathway and knockdown of lincRNA-Cox2 enhanced the transcription of Il12b induced by TNFα [46]. In a recent study, lincRNA-Cox2 was found to act as a coactivator of NF-κB for the transcription of late inflammatory genes through modulating SWI/SNF-mediated chromatin remodeling in macrophages in response to LPS stimulation [47]. Although lincRNA-Cox2 has extensively been described to function as a regulator of inflammatory processes, its role in CVD requires further investigation.
Conclusion and Future directions
Accumulating studies demonstrate that lncRNAs hold promise as important regulators to alter key biological processes for CVD susceptibility, progression, and regeneration in a cell-type and/or tissue-specific manner (Figure 1).
Diagnostically, lncRNA provide an interesting class as biomarkers, since their expression tracks with pathophysiological processes. Moreover, lncRNA-associated loci implicated in regions that may bear relevance for CVD susceptibility are of tremendous interest, for example ANRIL and the chromosome 9p21 CAD locus [48,49]. In this manner, lncRNAs provide a new powerful class of regulatory factors that may uncover how SNPs in the non-coding genome may influence CVD susceptibility.
The translational value of more than the estimated 50,000 human lncRNAs has yet to be shown [50]. This may be due to: i) over-reliance of lncRNA biology in mouse studies because of poor cross-species conservation on the level of sequence identity. However, tissue-specific lncRNA expression shows strong conservation across species and often the genomic locus (“neighborhood”) is well-conserved [51]; ii) little is known about the precise expression of lncRNA in a cell-type specific manner in human patients over the onset of a given disease; and iii) delivery of inhibitors to lncRNAs, especially in a cell-type or tissue specific manner, remains challenging. Nucleic acid-based therapeutics constitutes the next frontier in CVD management [52]. Accumulating studies utilize nucleic acid-based therapeutics, i.e. ASO that are chemically modified to increase specificity and resistance towards enzymatic degradation. Targeting lncRNAs using ASOs may hold therapeutic potential as analogous ASO-based strategies have been used for FDA-approved drugs, such as mipomersen that targets apolipoprotein B in homozygous familial hypercholesterolemia.
Taken together, because lncRNAs exhibit tremendous effects on cell-type specific cellular function, pathophysiological signaling pathways, and animal models of disease, targeting lncRNAs or harnessing their diagnostic potential may provide a unique opportunity to exploit the non-coding genome to fine-tune cardiovascular tissue homeostasis in health and disease.
Key Points.
LncRNAs are among the most abundant type of non-coding RNAs and control expression of disease-associated genes important to CVD pathogenesis
Examples of lncRNAs that modulate cardiomyocyte physiology and left ventricular remodeling include: Mhrt, Chast, Chear, CHRF, CARL, and WISPER.
MALAT1, MEG3, and MANTIS regulate endothelial cell homeostasis, whereas ANRIL, GAS5, and SMILR control vascular smooth muscle cell phenotypic responses.
LeXis and lncLSTR modulate lipid biology and RP5-833A20.1, E330013P06, and lincRNA-Cox2 regulate leukocyte inflammatory signaling pathways with potential therapeutic applications in atherosclerosis
Targeting lncRNAs using chemically modified oligonucleotides may provide a novel therapeutic strategy for controlling pathogenesis in a range of CV disease states
Acknowledgments
Financial support and sponsorship:
This work was supported by the National Institutes of Health (HL115141, HL117994, HL134849, and GM115605 to M.W.F.), the Arthur K. Watson Charitable Trust (to M.W.F.), and the Dr. Ralph & Marian Falk Medical Research Trust (to M.W.F.).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest.
References
- 1.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.Han P, Li W, Lin C-H, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin C-Y, Lin C-J, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M, Dittrich M, Maetzig T, Zimmer K, Remke J, et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. 2016;8:326ra22–326ra22. doi: 10.1126/scitranslmed.aaf1475. ** Example of a deleterious lncRNA (Chast) that is expressed highly in cardiomyocytes from hypertrophic hearts in mice and heart tissue from aortic stenosis patients. Silencing of Chast by GapmeRs prevented pathological cardiac remodelling in mice providing a potential therapeutic target. [DOI] [PubMed] [Google Scholar]
- 4.McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Molecular cell. 2015;57:39–54. doi: 10.1016/j.molcel.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Zhang X-J, Ji Y-X, Zhang P, Deng K-Q, Gong J, Ren S, Wang X, Chen I, Wang H, et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. 2016;22:1131–1139. doi: 10.1038/nm.4179. ** The lncRNA Chaer is specifically expressed in cardiomyocytes, but not in fibroblasts. Chaer-KO mice exhibited reduced hypertrophy and fibrosis in response to TAC-induced myocardial injury. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K, Liu F, Zhou L-Y, Long B, Yuan S-M, Wang Y, Liu C-Y, Sun T, Zhang X-J, Li P-F. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 7.Osman C, Merkwirth C, Langer T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J Cell Sci. 2009;122:3823–3830. doi: 10.1242/jcs.037655. [DOI] [PubMed] [Google Scholar]
- 8.Wang K, Long B, Zhou LY, Liu F, Zhou QY. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation - ProQuest. Nature. 2014 doi: 10.1038/ncomms4596. no volume. [DOI] [PubMed] [Google Scholar]
- 9.Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting C-C, Alexanian M, Maric D, Maison D, Nemir M, Young RA, et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aai9118. ** Example of a cardiac fibroblast-enriched lncRNA named WISPER that was found to regulate cardiac fibrosis. Silencing of WISPER in vivo attenuated MI-induced fibrosis and remodelling Moreover, WISPER expression was significantly increased in cardiac tissues from patients with aortic stenosis suggesting a potential role in human fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michalik KM, You X, Manavski Y, Doddaballapur A, Zörnig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389–1397. doi: 10.1161/CIRCRESAHA.114.303265. [DOI] [PubMed] [Google Scholar]
- 11.Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med. 2015;19:1418–1425. doi: 10.1111/jcmm.12576. * Example of lncRNA regulating endothelial inflammation. MALAT1 silencing decreased glucose-induction of inflammatory mediators IL-6 and TNFα through activation of SAA3 in ECs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J-Y, Yao J, Li X-M, Song Y-C, Wang X-Q, Li Y-J, Yan B, Jiang Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014;5:e1506. doi: 10.1038/cddis.2014.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z, Li J, Tang N. Long noncoding RNA Malat1 is a potent autophagy inducer protecting brain microvascular endothelial cells against oxygen-glucose deprivation/reoxygenation-induced injury by sponging miR-26b and upregulating ULK2 expression. Neuroscience. 2017;354:1–10. doi: 10.1016/j.neuroscience.2017.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Icli B, Wara AKM, Moslehi J, Sun X, Plovie E, Cahill M, Marchini JF, Schissler A, Padera RF, Shi J, et al. MicroRNA-26a regulates pathological and physiological angiogenesis by targeting BMP/SMAD1 signaling. Circ Res. 2013;113:1231–1241. doi: 10.1161/CIRCRESAHA.113.301780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Icli B, Nabzdyk CS, Lujan-Hernandez J, Cahill M, Auster ME, Wara AKM, Sun X, Ozdemir D, Giatsidis G, Orgill DP, et al. Regulation of impaired angiogenesis in diabetic dermal wound healing by microRNA-26a. J Mol Cell Cardiol. 2016;91:151–159. doi: 10.1016/j.yjmcc.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu G-Z, Tian W, Fu H-T, Li C-P, Liu B. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem Biophys Res Commun. 2016;471:135–141. doi: 10.1016/j.bbrc.2016.01.164. [DOI] [PubMed] [Google Scholar]
- 17.He C, Yang W, Yang J, Ding J, Li S, Wu H, Zhou F, Jiang Y, Teng L, Yang J. Long Noncoding RNA MEG3 Negatively Regulates Proliferation and Angiogenesis in Vascular Endothelial Cells. DNA Cell Biol. 2017;36:475–481. doi: 10.1089/dna.2017.3682. [DOI] [PubMed] [Google Scholar]
- 18.Boon RA, Hofmann P, Michalik KM, Lozano-Vidal N, Berghäuser D, Fischer A, Knau A, Jaé N, Schürmann C, Dimmeler S. Long Noncoding RNA Meg3 Controls Endothelial Cell Aging and Function: Implications for Regenerative Angiogenesis. J Am Coll Cardiol. 2016;68:2589–2591. doi: 10.1016/j.jacc.2016.09.949. [DOI] [PubMed] [Google Scholar]
- 19.Gordon FE, Nutt CL, Cheunsuchon P, Nakayama Y, Provencher KA, Rice KA, Zhou Y, Zhang X, Klibanski A. Increased expression of angiogenic genes in the brains of mouse meg3-null embryos. Endocrinology. 2010;151:2443–2452. doi: 10.1210/en.2009-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leisegang MS, Fork C, Josipovic I, Richter F, Preussner J, Hu J, Miller MJ, Epah JN, Hofmann P, Günther S, et al. Long Noncoding RNA MANTIS Facilitates Endothelial Angiogenic Function. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.116.026991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarinova O, Stewart AFR, Roberts R, Wells G, Lau P, Naing T, Buerki C, McLean BW, Cook RC, Parker JS, et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. 2009;29:1671–1677. doi: 10.1161/ATVBAHA.109.189522. [DOI] [PubMed] [Google Scholar]
- 22.Congrains A, Kamide K, Oguro R, Yasuda O, Miyata K, Yamamoto E, Kawai T, Kusunoki H, Yamamoto H, Takeya Y, et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis. 2012;220:449–455. doi: 10.1016/j.atherosclerosis.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Holdt LM, Beutner F, Scholz M, Gielen S, Gäbel G, Bergert H, Schuler G, Thiery J, Teupser D. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol. 2010;30:620–627. doi: 10.1161/ATVBAHA.109.196832. [DOI] [PubMed] [Google Scholar]
- 24.Yap KL, Li S, Muñoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou M-M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Molecular cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011;30:1956–1962. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan G, Mathur R, Hu X, Liu Y, Zhang X, Peng G, Lu X. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell Signal. 2013;25:1086–1095. doi: 10.1016/j.cellsig.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, Kohlmaier A, Herbst A, Northoff BH, Nicolaou A, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7:12429. doi: 10.1038/ncomms12429. * Example of a circular non-coding RNA (CircANRIL) that binds to PES1, an essential 60S-preribosomal assembly factor, and in turn impairs pre-rRNA processing and ribosome biogenesis in VSMC and macrophages. Consequently, circANRIL induces nucleolar stress and p53 activation, resulting in the inhibition of proliferation and induction of apoptosis, as observed for the linear ANRIL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3:ra8–ra8. doi: 10.1126/scisignal.2000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams GT, Mourtada-Maarabouni M, Farzaneh F. A critical role for non-coding RNA GAS5 in growth arrest and rapamycin inhibition in human T-lymphocytes. Biochem Soc Trans. 2011;39:482–486. doi: 10.1042/BST0390482. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y-N-Z, Shan K, Yao M-D, Yao J, Wang J-J, Li X, Liu B, Zhang Y-Y, Ji Y, Jiang Q, et al. Long Noncoding RNA-GAS5: A Novel Regulator of Hypertension-Induced Vascular Remodeling. Hypertension. 2016;68:736–748. doi: 10.1161/HYPERTENSIONAHA.116.07259. [DOI] [PubMed] [Google Scholar]
- 31.Li L, Li X, The E, Wang L-J, Yuan T-Y, Wang S-Y, Feng J, Wang J, Liu Y, Wu Y-H, et al. Low expression of lncRNA-GAS5 is implicated in human primary varicose great saphenous veins. PLoS ONE. 2015;10:e0120550. doi: 10.1371/journal.pone.0120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao H, Zhang J-G, Qin R-H, Dai C, Shi P, Yang J-J, Deng Z-Y, Shi K-H. LncRNA GAS5 controls cardiac fibroblast activation and fibrosis by targeting miR-21 via PTEN/MMP-2 signaling pathway. Toxicology. 2017;386:11–18. doi: 10.1016/j.tox.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 33.Ballantyne MD, Pinel K, Dakin R, Vesey AT, Diver L, Mackenzie R, Garcia R, Welsh P, Sattar N, Hamilton G, et al. Smooth Muscle Enriched Long Noncoding RNA (SMILR) Regulates Cell Proliferation. Circulation. 2016;133:2050–2065. doi: 10.1161/CIRCULATIONAHA.115.021019. * Example of a VSMC-enriched lncRNA that controls proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 35.Sallam T, Jones MC, Gilliland T, Zhang L, Wu X, Eskin A, Sandhu J, Casero D, Vallim TQ de A, Hong C, et al. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature. 2016;534:124–128. doi: 10.1038/nature17674. ** Example of a lncRNA that regulates lipid metabolism in response to LXR activation in mice. There is no human homologue of this lncRNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li P, Ruan X, Yang L, Kiesewetter K, Zhao Y, Luo H, Chen Y, Gucek M, Zhu J, Cao H. A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice. Cell Metab. 2015;21:455–467. doi: 10.1016/j.cmet.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 38.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 39.Bobryshev YV, Lord RS. S-100 positive cells in human arterial intima and in atherosclerotic lesions. Cardiovasc Res. 1995;29:689–696. [PubMed] [Google Scholar]
- 40.Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–138. doi: 10.1161/01.atv.6.2.131. [DOI] [PubMed] [Google Scholar]
- 41.Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995;92:1084–1088. doi: 10.1161/01.cir.92.5.1084. [DOI] [PubMed] [Google Scholar]
- 42.Hu Y-W, Zhao J-Y, Li S-F, Huang J-L, Qiu Y-R, Ma X, Wu S-G, Chen Z-P, Hu Y-R, Yang J-Y, et al. RP5-833A20.1/miR-382-5p/NFIA-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis and inflammatory reaction. Arterioscler Thromb Vasc Biol. 2015;35:87–101. doi: 10.1161/ATVBAHA.114.304296. [DOI] [PubMed] [Google Scholar]
- 43.Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, Bhatt K, Leung A, Wu X, Putta S, et al. Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. Diabetes. 2014;63:4249–4261. doi: 10.2337/db14-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ounzain S, Micheletti R, Arnan C, Plaisance I, Cecchi D, Schroen B, Reverter F, Alexanian M, Gonzales C, Ng SY, et al. CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J Mol Cell Cardiol. 2015;89:98–112. doi: 10.1016/j.yjmcc.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 45.Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science (New York, N Y) 2013;341:789–792. doi: 10.1126/science.1240925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tong Q, Gong A-Y, Zhang X-T, Lin C, Ma S, Chen J, Hu G, Chen X-M. LincRNA-Cox2 modulates TNF-α-induced transcription of Il12b gene in intestinal epithelial cells through regulation of Mi-2/NuRD-mediated epigenetic histone modifications. FASEB J. 2016;30:1187–1197. doi: 10.1096/fj.15-279166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu G, Gong A-Y, Wang Y, Ma S, Chen X, Chen J, Su C-J, Shibata A, Strauss-Soukup JK, Drescher KM, et al. LincRNA-Cox2 Promotes Late Inflammatory Gene Transcription in Macrophages through Modulating SWI/SNF-Mediated Chromatin Remodeling. Journal of immunology (Baltimore, Md : 1950) 2016;196:2799–2808. doi: 10.4049/jimmunol.1502146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, et al. Genomewide Association Analysis of Coronary Artery Disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nitsche A, Rose D, Fasold M, Reiche K, Stadler PF. Comparison of splice sites reveals that long noncoding RNAs are evolutionarily well conserved. RNA. 2015;21:801–812. doi: 10.1261/rna.046342.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Washietl S, Kellis M, Garber M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014;24:616–628. doi: 10.1101/gr.165035.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haemmig S, Feinberg MW. Viewpoints Targeting LncRNAs in Cardiovascular Disease: Options and Expeditions. Circ Res. 2016 doi: 10.1161/CIRCRESAHA. [DOI] [PMC free article] [PubMed] [Google Scholar]