

Abstract
Loss-of-function mutations of the protein kinase PERK (EIF2AK3) in humans and mice cause permanent neonatal diabetes and severe proinsulin aggregation in the endoplasmic reticulum (ER), highlighting the essential role of PERK in insulin production in pancreatic β cells. As PERK is generally known as a translational regulator of the unfolded protein response (UPR), the underlying cause of these β cell defects has often been attributed to derepression of proinsulin synthesis, resulting in proinsulin overload in the ER. Using high-resolution imaging and standard protein fractionation and immunological methods we have examined the PERK-dependent phenotype more closely. We found that whereas proinsulin aggregation requires new protein synthesis, global protein and proinsulin synthesis are down-regulated in PERK-inhibited cells, strongly arguing against proinsulin overproduction being the root cause of their aberrant ER phenotype. Furthermore, we show that PERK regulates proinsulin proteostasis by modulating ER chaperones, including BiP and ERp72. Transgenic overexpression of BiP and BiP knockdown (KD) both promoted proinsulin aggregation, whereas ERp72 overexpression and knockdown rescued it. These findings underscore the importance of ER chaperones working in concert to achieve control of insulin production and identify a role for PERK in maintaining a functional balance among these chaperones.
Keywords: chaperone, endoplasmic reticulum (ER), insulin secretion, intracellular trafficking, protein aggregation, BiP, PERK, beta cells
Introduction
Minor changes in circulating levels of insulin can elicit a major perturbance of glucose homeostasis. Consequently, insulin synthesis and secretion are finely controlled within pancreatic β cells (1). The discovery of monogenic forms of permanent neonatal diabetes and Type 2 β cell decompensation have highlighted the importance and vulnerability of post-translational proinsulin processing and forward trafficking out of the endoplasmic reticulum (ER)5 (2). Once thought to be housekeeping functions, proinsulin folding, ER-associated degradation (ERAD)-mediated quality/quantity control, and anterograde trafficking are regulated as a function of the metabolic state and demand for insulin (2, 3). These steps ensure that β cells can rapidly mobilize and secrete insulin while maintaining sufficient intracellular stores for later secretion events.
PERK (EIF2AK3) is one of a small number of genes essential for maintaining glucose homeostasis. As such, PERK deficiency results in permanent neonatal diabetes in humans and mice (4, 5). In humans, PERK deficiency leads to Wolcott-Rallison syndrome (WRS), which, in addition to permanent neonatal diabetes, causes exocrine pancreas atrophy, growth retardation, and osteopenia (6). Perk knock-out (KO) mice exhibit the same set of anomalies as are seen in human WRS patients (7, 8) and have served as the model system to study the molecular and cellular basis of neonatal diabetes as well as the normal functions of PERK. We previously showed that insulin insufficiency in Perk KO mice was specifically caused by the absence of PERK function in β cells and is associated with an aberrant accumulation of proinsulin within the ER, denoted as the Impacted-ER phenotype (9–12). Mice carrying a targeted mutation of eIF2α Ser51, the primary substrate of PERK kinase activity, also exhibit the Impacted-ER phenotype (13), providing further evidence that PERK is essential for normal β cell function and insulin production.
Two alternative hypotheses have dominated the subsequent inquiry into the molecular mechanisms that govern PERK function in β cells. The first was based on early observations of chemically induced ER stress in cultured cells. During ER stress, PERK is activated to phosphorylate its major substrate, eIF2α, leading to depressed global translation initiation and increased expression of transcription factors that function to either relieve the stress or, failing that, induce apoptosis. Thus, it was initially postulated that the proinsulin overload within the ER lumen associated with PERK and eIF2α mutations was caused by derepression of protein synthesis (7, 14, 15).
Upon further investigation into the cellular outcomes of PERK ablation in mice and cultured cells, an alternative hypothesis began to emerge. PERK deficiency was found to lead to defects in ERAD and anterograde proinsulin trafficking that were correlated with changes in expression of BiP, ERp72, and other ER chaperones (10). These ER chaperones are essential for coordinating proinsulin folding, disulfide bond formation, and protein quality control (2, 16–22). Furthermore, repeated experiments in cultured cells and isolated islets found no evidence of global protein or proinsulin over-synthesis when PERK was ablated (9–12, 23, 24). Although these findings did not support the ER stress-based explanation of PERK function, they did not directly refute the protein overload hypothesis.
The recent availability of a highly specific inhibitor of PERK kinase activity, GSK2606414 (PERKi) (25, 26), provided a means to study the consequences of acute and controlled ablation of PERK in β cells. Using this inhibitor, Harding and co-workers (27) reported a modest derepression of proinsulin synthesis in rat islets and the appearance of proinsulin in high-molecular weight (HMW) aggregates in cultured β cells. They interpreted these correlative findings as additional evidence for the proinsulin over-synthesis model of PERK function in β cells. In parallel, we discovered that within seconds to minutes, PERKi treatment perturbs calcium fluxes, which are critical for glucose-stimulated insulin secretion. Moreover, we found that within 24 h, PERKi induces an Impacted-ER phenotype that is indistinguishable from what is seen in genetic models of PERK ablation, indicating that the phenotype is a generalized feature of PERK ablation in β cells (11).
The goal of the work presented herein is to directly test the two competing hypotheses for how the absence of PERK function leads to β cell dysfunction and the Impacted-ER phenotype. Consistent with our previous finding in genetic models of PERK ablation we do not observe increases in global protein or proinsulin synthesis in a population of PERKi-treated cells. On the contrary, we clearly demonstrate that both are significantly reduced, specifically in Impacted-ER cells, thereby ruling out the ER stress-based proinsulin over-synthesis hypothesis. Instead, we uncover compelling evidence for the alternative hypothesis that loss of functional coordination among key PERK-dependent ER chaperones is primarily responsible for β cell dysfunctions and diabetes in Perk mutant mice and humans afflicted with WRS.
Results
β cells from WRS patients display a proinsulin Impacted-ER phenotype
Proinsulin abnormally accumulates in the ER of β cells in rodent models of PERK ablation (Fig. 1, A–D, and Fig. S1, A and B), and this is purported to be the major reason for insulin insufficiency and diabetes in Perk KO mice (9–12). Because the diabetes in Perk KO mice is similar to the diabetes in WRS-afflicted humans, we asked if they share the same subcellular β cell defects. As was previously shown (28), the number of insulin-positive β cells was greatly diminished in pancreata from human WRS. Furthermore, human WRS islets also contained Impacted-ER cells that were indistinguishable from those seen in Perk KO islets (Fig. 1, E and F). More detailed examination (Fig. 1, G–I) of PERK-inhibited β cells in culture identified intermediate cells in which proinsulin could be detected both within and just outside of its normal Golgi localization (Fig. 1H). “Super” Impacted-ER cells in which proinsulin was dispersed throughout the cell body, reaching the periphery, were also observed (Fig. 1I). Additionally, the structure of the Golgi appears less organized, but not wholly lost, in the super Impacted-ER cells. This observation is consistent with previous data that showed mixing of the ER and Golgi compartments when PERK function was ablated genetically (10).
Figure 1.

The Impacted-ER phenotype correlates with proinsulin aggregation and can also be found in WRS patients. A–F, immunodetection of insulin (green), proinsulin (red), and DAPI (blue) in wildtype mouse pancreata (A), DMSO-treated INS1 832/13 cells (C), and normal human pancreata (E) shows proinsulin in perinuclear patches associated with the Golgi and insulin dispersed in secretory granules throughout the cytoplasm. The Impacted-ER phenotype in PKO mouse pancreata (B), PERKi-treated INS1 832/13 cells (D), and human pancreata from a 3-month-old WRS patient (F) as characterized by distended ER and an aberrant accumulation of proinsulin. Insets in A and B are enlarged ×30 to show detail. White arrowheads indicate representative cells exhibiting the Impacted-ER phenotype. Scale bar = 30 μm. G–I, immunodetection of proinsulin (red), a Golgi-specific marker, RCAS1 (green), and DAPI (blue) in INS1-832/13 cells treated for 24 h with 1 μm PERKi or an equal volume of DMSO. Cells were imaged on a high-resolution microscope to show punctate proinsulin staining, which largely overlaps the Golgi area in control cells and then progressively spreads throughout the cytoplasmic area as the ER becomes distended and the cell becomes impacted under PERKi conditions. Scale bar = 10 μm. J, INS1 832/13 cells treated for 24 h with 1 μm PERKi and HMW aggregates were isolated and then electrophoresed under non-reducing (NR) and reducing (R) conditions. Immunoreactive NR proinsulin (ProI.) aggregates have dramatically retarded electrophoretic mobility and can only migrate a short distance into the gel. K, INS1 832/13 cells treated for 0, 4, 8, and 24 h with 1 μm PERKi. HMW protein aggregates were analyzed as described in J. A representative blot is shown with quantification of the NR samples representing n = 5 per treatment presented as mean ± S.D. with the input data points shown as black dots. Statistical significance is calculated relative to the PERKi 0 h control; *, p ≤ 0.05; **, p < 0.01. L, INS1 832/13 cells were treated for 6, 8, 12, 18, and 24 h with 1 μm PERKi. Proinsulin and DAPI were detected by immunocytochemistry, and Impacted-ER cells were counted as a frequency of total cells. Quantification represents n = 4 per treatment. Different letters indicate statistically significant difference. M, INS1 832/13 cells treated for 0, 4, 8, and 24 h with 1 μm PERKi or for 2 h with 1 μm thapsigargin (Tg) as a positive control of ER stress. A representative blot is shown with quantification of n = 4 per treatment. Different letters indicate statistically significant difference.
The Impacted-ER phenotype is correlated with proinsulin aggregation in β cells
The appearance of proinsulin as insoluble, HMW aggregates in PERKi-treated cells (27) in culture (Fig. 1D) suggested a biochemical basis for the Impacted-ER phenotype. A previously described ultracentrifugation method (27) was employed to determine whether aggregate formation is temporally correlated with the Impacted-ER phenotype in INS1 832/13 cells treated with PERKi. Proinsulin aggregates were detectable at 4 h and continued to increase through 24 h post-treatment with PERKi (Fig. 1, J and K). The timing of this aggregation coincided with the appearance of the Impacted-ER phenotype cytologically (Fig. 1L). Importantly, eIF2α-P levels at these time points were not elevated in contrast to levels seen in cells exposed to the ER stress-inducing agent thapsigargin, indicating the absence of a canonical ER stress response (Fig. 1M).
Proinsulin aggregation is independent of derepressed protein synthesis but requires new protein synthesis
It was previously reported by Harding and co-workers (27) that PERKi modestly increases proinsulin synthesis in rat whole pancreatic islets, which was correlated with the formation of HMW proinsulin aggregates in cultured mouse MIN6 β cells. However, when we repeated these experiments using rat INS1 832/13 β cells, we found no significant change in proinsulin (Fig. 2, A and B) or global protein synthesis over time (Fig. 2, C and D), whereas both were significantly reduced at all time points by treatment with the global protein synthesis inhibitor anisomycin (AN).
Figure 2.

Proinsulin aggregation occurs independently of derepressed protein synthesis. A, INS1 832/13 cells treated for 1 or 12 h with 1 μm PERKi and 50 nm AN. Shown are representative autoradiographs of metabolically labeled proinsulin immunopurified from cell lysates pulse-labeled with [35S]methionine/cysteine for the last 30 min. Quantification was carried out using the ImageQuant software on specialized image files generated using the Storage Phosphor setting on the Typhoon PhosphorImager (GE Healthcare) and represents n = 3 per treatment. Statistical significance is calculated relative to the DMSO control; ***, p < 0.001. B, INS1 832/13 cells treated for 24 h with 1 μm PERKi and 15 nm AN. Shown are representative autoradiographs as described in A. Quantification was carried out as described for A and represents n = 8 per treatment. Statistical significance is calculated relative to the DMSO control; **, p ≤ 0.01; ***, p < 0.001. C, INS1 832/13 cells treated for 1 h with 1 μm PERKi, 10 or 50 nm AN. Shown are representative autoradiographs of metabolically labeled total cell lysates pulse-labeled with [35S]methionine/cysteine for the last 30 min. The entire molecular weight range of each lane was quantified by ImageQuant as described for A. The graph represents n = 3 per treatment. Statistical significance is calculated relative to the DMSO control; ***, p < 0.001. D, INS1 832/13 cells treated for 12 h with 1 μm PERKi, 10 or 50 nm AN. Shown are representative autoradiographs as described in C. Quantification represents n = 3 per treatment. Statistical significance is calculated relative to the DMSO control; ***, p < 0.001. E, INS1 832/13 cells treated for 12 h with 1 μm PERKi. The methionine analog HPG (50 μm) was added 1 h before harvest. Newly synthesized proteins were quantified by fluorescence detection of the incorporated derivatized methionine using ImageJ software as described under “Experimental procedures.” Quantification represents n = 4 per treatment. Statistical significance was calculated relative to the DMSO control. F, INS1 832/13 cells treated for 4, 8, 12, and 24 h with 1 μm PERKi. HPG was added for the last hour as described in A. Intensity of staining was measured for Impacted and non-impacted cells based on proinsulin staining. Quantification represents n = 8 per treatment. Statistical significance was calculated relative to the non-impacted control; *, p ≤ 0.05; **, p < 0.01. G–N, immunodetection of DAPI (blue), proinsulin (red), and newly synthesized protein (green) in DMSO-treated INS1 832/13 cells (B–E) and 4-h PERKi-treated INS1 832/13 cells (F-I). White arrowheads indicate impacted cells. Scale bar = 20 μm.
Impacted-ER cells typically comprise less than 20% of the total cell population in PERKi-treated cells. Therefore, our inability to detect PERKi-induced changes in proinsulin or total protein synthesis may have been due to the overwhelming excess of unaffected β cells that obscured changes in the comparatively minor population of Impacted-ER cells. To assess the status of protein synthesis only in Impacted-ER cells, a single cell-based fluorescence assay was employed, which incorporates an alkyne-modified analog of methionine, which allows for fluorescence detection of cells that are active for protein synthesis. When fluorescence intensities of total populations of INS1 832/13 cells were compared, no significant differences in total protein synthesis were found in cells treated with PERKi compared with the controls (Fig. 2E). However, when Impacted-ER and non-Impacted-ER cells within the same PERKi-treated population were compared, a reduction in total protein synthesis by ∼50% for Impacted-ER compared with non-Impacted-ER cells was observed at all time points tested (Fig. 2, F–N). A significant reduction was also seen in MIN6 cells (Fig. S2A). These results strongly argue against total protein or proinsulin over-synthesis as being the cause of ER and proinsulin dysfunctions seen in PERK-ablated β cells.
In normal β-cells there is a low steady-state level of proinsulin in the ER due to rapid transit to the Golgi (9, 10, 12, 29), and therefore it is unlikely that the amount of proinsulin in the ER at initiation of PERK inhibition is sufficient to cause the massive aggregation of proinsulin in the ER several hours later. Thus, we speculated that aggregate formation requires ongoing new proinsulin synthesis. To test this, β cells were co-treated with PERKi and a moderately high dose (50 nm) of AN, which reduced proinsulin synthesis by 40% after 1 h and 65% after 12 h (Fig. 2A). As expected, this level of reduction in proinsulin synthesis was correlated with an almost complete elimination of the Impacted-ER phenotype at 24 h (Fig. 3A). However, a lower dose of AN (15 nm), which achieved a 25% reduction in proinsulin synthesis after 24 h (Fig. 2B), was permissive for proinsulin aggregation and the Impacted-ER phenotype, albeit to a lesser degree than PERKi treatment alone (Fig. 3, B and C). To determine whether prior reduction of proinsulin synthesis predisposes cells to aggregate formation in response to PERKi, cells were pre-treated with AN for 15 min before adding PERKi. Under these conditions, the Impacted-ER phenotype was still readily observable after 24 h of PERK inhibition, albeit to a slightly lesser extent than with co-treatment (Fig. 3D).
Figure 3.
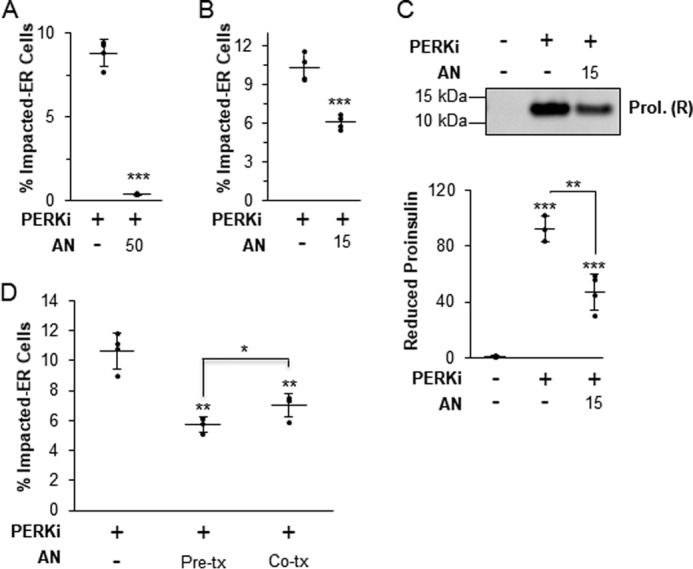
Protein aggregation requires new protein synthesis. A, INS1 832/13 cells treated for 24 h with 1 μm PERKi and 50 nm AN. Quantification represents n = 4 per treatment. Statistical significance is calculated relative to the PERKi only treatment; ***, p < 0.001. B, INS1 832/13 cells treated for 24 h with 1 μm PERKi and 15 nm AN. Quantification represents n = 8 per treatment. Statistical significance is calculated relative to the PERKi only treatment; ***, p < 0.001. C, INS1 832/13 cells treated for 24 h with 1 μm PERKi and 15 nm AN. HMW protein aggregates were pelleted and electrophoresed under reducing conditions. A representative blot is shown with quantification representing n = 4 per treatment. Statistical significance is calculated relative to the DMSO control or as indicated; **, p ≤ 0.01; ***, p < 0.001. D, INS1 832/13 cells treated for 24 h with 1 μm PERKi and 15 nm anisomycin was added either 15 min prior to addition of PERKi (Pre-tx) or at the same time as the PERKi (Co-tx). Quantification represents n = 4 per treatment. Statistical significance is calculated relative to the PERKi only treatment or as indicated; *, p ≤ 0.05; **, p < 0.01.
To summarize, PERK inhibition still leads to proinsulin aggregation even when protein synthesis is modestly reduced, however, some level of ongoing synthesis is needed to provide proinsulin to the accumulating aggregates once their formation has been initiated by loss of PERK function. Collectively, these findings strongly argue that the root cause of the Impacted-ER phenotype in PERK-ablated cells is not ER client protein over-synthesis.
PERK mediates expression of ER chaperones
Previously, we showed that BiP and ERp72 expression in Perk-deficient mice and cultured β cells were substantially elevated compared with other ER trafficking-related proteins (9, 10). Similarly, in INS1 832/13 cells treated for 24 h with PERKi we observed an increase in Erp72 and Bip mRNAs (Fig. 4A). BiP protein levels were modestly elevated after a 24-h PERKi treatment, with the most significant increase occurring between 8 and 24 h of PERKi exposure (Fig. 4B). Thus, up-regulation of this critical chaperone is delayed with respect to the onset of proinsulin aggregate formation.
Figure 4.
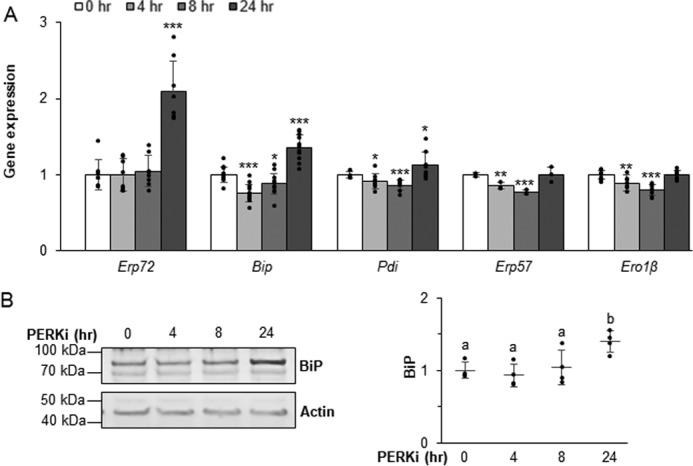
PERK mediates expression of ER chaperones. A and B, mRNA (A) and protein (B) expression levels quantified in INS1 832/13 cells treated with 1 μm PERKi. Quantification represents n = 8 per treatment. Statistical significance is calculated relative to 0 h control; *, p ≤ 0.05; ***, p < 0.001 (A). Representative Western blots are shown; different letters indicate statistically significant difference (B).
In both INS1 832/13 and MIN6 PERKi-treated cells, the transcript for the chaperone Pdi, which is also known to promote proinsulin folding (9, 20, 30–32), is modestly increased. However, this increase was not as large as was seen for Bip and Erp72 mRNAs (Fig. 4A and Fig. S3). ERp72 protein has been shown to be significantly elevated in neonatal Perk KO islets and modestly so in β cells expressing a dominant-negative Perk transgene (9). In cultured β cells treated with PERKi, however, ERp72 protein levels were inconsistently elevated (not shown). Protein level changes for ERO1β or two of the other PDI family of chaperones, PDI and ERp57, were not observed. However, we previously demonstrated that the redox state of both ERp72 and ERp57 are significantly altered in models of genetic Perk ablation (9), which suggests that coordinated chaperone function is achieved as much by maintaining physiological foldase activity as it is by the overall amount of the chaperone proteins.
BiP co-localizes and co-immunoprecipitates with proinsulin in PERK-ablated cells
BiP is normally dispersed throughout the ER and therefore occupies most of the cell body in wildtype β cells from mice, and humans, whereas proinsulin almost exclusively localizes to the Golgi (Fig. 5, A–C, G–I, and M–O). By contrast, in all queried models of PERK ablation, including Perk KO β cells (Fig. 5, D–F), PERKi-treated INS1 832/13 cells (Fig. 5, J–L), and pancreata from WRS patients (Fig. 5, P–R), BiP and proinsulin aberrantly co-localize within an Impacted-ER (Fig. 5F, L, and R, and Fig. S4, A–J). High proinsulin protein levels are correlated with up-regulation of BiP, specifically in Perk KO β cells displaying an Impacted-ER (12). To examine this further, we assessed BiP expression levels over increasing PERKi treatment times and found that 80–90% of the cells overexpressing BiP co-localized with Impacted-ER. This correlation showed no significant variation over the 6 to 24 h time course of PERKi treatment (Fig. 5S). These results confirm our previous finding that elevated BiP levels correlate with the appearance of the Impacted-ER phenotype under these conditions. There is also an enhanced interaction between BiP and proinsulin by co-immunoprecipitation that was absent in vehicle-treated controls (Fig. 5T). The very low frequency of Impacted-ER β cells within the first 6 h of PERK inhibition (Fig. 1L) precluded our ability to adequately assess the degree of association between BiP and proinsulin by co-immunoprecipitation or the HMW assay at early time points. However, given that by 6 h ∼85% of Impacted-ER cells also over-express BiP (Fig. 5S), an abnormally enhanced interaction between BiP and proinsulin that occurs early and becomes overt by 24 h seems highly likely.
Figure 5.

BiP co-localizes and co-immunoprecipitates with proinsulin in PERK-ablated cells. A–R, immunodetection of BiP (green) and proinsulin (red) in wildtype mouse pancreata (A–C), DMSO-treated INS1 832/13 cells (G–I), and normal human pancreata (M–O) shows proinsulin in perinuclear patches associated with the Golgi and BiP is dispersed throughout the ER. In PKO mouse pancreata (D–F), PERKi-treated INS1 832/13 cells (J–L), and human pancreata from a 3-month-old WRS patient (P–R), both proinsulin and BiP are overexpressed and dispersed throughout the abnormally distended ER. Scale bar = 25 μm for A–F and 30 μm for G–R. S, INS1 832/13 cells treated for 6, 8, 12, 18, and 24 h with 1 μm PERKi. Proinsulin, BiP, and DAPI were detected by immunocytochemistry. Cells overexpressing BiP were readily scored as having intense BiP staining throughout the ER. Quantification represents n = 4 per treatment. T, INS1 832/13 cells treated for 24 h with 1 μm PERKi and then immunoprecipitated (IP) with BiP antibody. Representative image of IP/Western blot is shown with quantification representing n = 3 per treatment. Statistical significance is calculated relative to the DMSO control; *, p ≤ 0.05; **, p < 0.01; WB, Western blot.
BiP functions within an optimal range to promote proper proinsulin trafficking
If the functional coordination of ER chaperones is the critical downstream effector of PERK in β cells, then manipulation of their levels and/or activity should alter PERKi-induced proinsulin aggregation. This should be especially true for BiP, which, in addition to its vital role in proinsulin folding, is also a key regulator of PERK that responds to physiological calcium fluxes in response to the major insulin secretagogue glucose (17, 33, 35). Adenoviral overexpression of BiP (Adv-BiP) alone at both low (500) and high (3000) multiplicity of infection (m.o.i.) (Fig. 6, A and B) caused an accumulation of proinsulin in the ER indistinguishable from the Impacted-ER phenotype (Fig. 6C versus Fig. 5, J–L) but at a lower frequency than what is observed in PERKi-treated cells (Fig. 6D). BiP overexpression was monitored at the single-cell level by GFP (green fluorescent protein) fluorescence in infected cells as the shuttle vector used to produce the adenovirus had a humanized recombinant GFP (22). BiP overexpression did not alter global protein or proinsulin synthesis (Fig. S5A). When BiP was overexpressed to higher levels (3000 m.o.i.), modest increases in UPR markers (Fig. S5B) were seen. However, these effects were absent at lower levels of overexpression (500 m.o.i.; Fig. S5B), which still caused proinsulin aggregation (Fig. 6, C and D).
Figure 6.

The balance of BiP is important in properly regulating proinsulin trafficking. A and B, INS1 832/13 cells infected with a BiP-overexpressing adenovirus or control adenovirus overexpressing GFP at 500 (A) or 3000 (B) m.o.i. prior to treatment for 24 h with 500 nm PERKi. A representative blot is shown with quantification representing n = 4 per treatment. Statistical significance was calculated relative to the Adv-GFP/DMSO control; *, p ≤ 0.05; ***, p < 0.001. C, immunostaining of INS1 832/13 cells infected with AdV-BiP (500 MOI) using proinsulin (red) and DAPI (blue). The GFP image shows that the cells with Impacted-ER are infected with the BiP-overexpressing adenovirus. Scale bar = 30 μm. D, INS1 832/13 cells infected with AdV-BiP as described in A and B followed by treatment for 24 h with 500 nm PERKi. Quantification represents n = 4 per treatment. Different letters indicate statistically significant difference. E, INS1 832/13 cells transfected with BiP siRNAs or a non-targeting negative control siRNA. A representative blot is shown with quantification representing n = 4 per treatment. Statistical significance is calculated relative to the negative siRNA control; **, p < 0.01. F, INS1 832/13 cells treated for 24 h with 500 nm PERKi and treated for the final 1 h with 0.5 μg/ml of Subtilase cytotoxin (SubAB) or the inactive mutant SubAA272B (Mut). A representative blot is shown with quantification representing n = 4 per treatment. Different letters indicate statistically significant differences. G, INS1 832/13 cells transfected with BiP or negative control siRNAs or treated with SubAB or the inactivated mutant in combination with PERKi treatment as was done in E and F. Quantification represents n = 4 per treatment. Statistical significance is calculated relative to the negative siRNA control or the SubAA272B inactive mutant treated with PERKi; ***, p < 0.001. H, INS1 832/13 cells were transfected with BiP or negative control siRNAs and treated for the final 24 h with 1 μm PERKi. HMW protein aggregates were pelleted and electrophoresed as described in the legend to Fig. 1J. Representative blots are shown with quantification representing n = 4 per treatment. Different letters indicate statistically significant differences. I, INS1 832/13 cells were transfected for 48 h with V5-tagged wild-type proinsulin–KDEL (INS+-KDEL). Twenty-four hours after transfection, cells were then infected with a BiP-overexpressing adenovirus or control adenovirus overexpressing GFP at 500 m.o.i. prior to treatment for 24 h with 1 μm PERKi. Representative blots are shown with quantification representing n = 4 per treatment. Different letters indicate statistically significant differences. J, INS1 832/13 cells were transfected for 48 h with V5-tagged wildtype proinsulin-KDEL (INS+-KDEL). Cells were treated for the final 24 h with 1 μm PERKi and the final 1 h with 0.5 μg/ml of SubAB or the inactive mutant SubAA272B (Mut). Representative blots are shown. Different letters indicate statistically significant differences.
Because BiP overexpression alone caused an Impacted-ER phenotype, we hypothesized that BiP knockdown would have the opposite effect and rescue the phenotype in PERKi-treated cells. Two methods for reducing the amount of active BiP were used: siRNA-mediated KD and subtilase cytotoxin (SubAB), which specifically and acutely cleaves and inactivates BiP (36). Unexpectedly, we found that reducing BiP by 50% in PERK-inhibited cells (Fig. 6, E and F) more than doubled the frequency of cells with Impacted-ER (Fig. 6G) and significantly increased proinsulin aggregation (Fig. 6H). Acutely reducing BiP using SubAB increased the percentage of cells with Impacted-ER in a time-dependent manner (Fig. S6, A and B).
It was previously shown that genetic ablation of PERK in AD293 cells causes impaired ERAD of exogenously expressed proinsulin (10). To determine whether the same holds true for β cells and if BiP plays a role, similar experiments in INS1 832/13 cells were performed. Cells were transfected with a recombinant proinsulin that harbors a KDEL ER-retention signal as well as a V5 epitope tag that allows for simplified detection on immunoblots (10). At 48 h post-transfection, the cells were infected with AdV-BiP or the AdV-GFP control and PERKi was also added. At 24 h post-infection/PERKi treatment, cells were harvested and both BiP and V5-tagged proinsulin were quantified. Only trace amounts of V5-tagged proinsulin were detected in untreated AdV-GFP controls cells (Fig. 6I). Under these conditions the KDEL ER-retention signal impedes anterograde trafficking of proinsulin out of the ER, which stimulates its removal by ERAD (10). The addition of PERKi led to an over 20-fold increase in immunodetectable V5-proinsulin, which suggests impaired degradation, consistent with our previous findings in AD293 cells genetically ablated for PERK. In BiP overexpressing β cells, there was a modest but significant elevation of V5-proinsulin, suggesting that an imbalance of active BiP can, on its own, disrupt degradation of proinsulin by ERAD. When PERKi was added to cells overexpressing BiP, this effect was greatly amplified, but was not significantly different from what was observed in similarly treated AdV-GFP control cells. When SubAB toxin was then used to carry out BiP knockdown in this system it too dramatically reduced ERAD proteolysis of proinsulin both in the presence and absence of PERK inhibition (Fig. 6J). Collectively, these results confirm the importance of BiP for normal proinsulin folding and forward trafficking. However, they are inconsistent with the widely accepted view that protein mis-folding and its resultant negative cellular and physiological consequences can be overcome by simply increasing chaperone protein levels (37).
Knockdown of ATF6 worsens PERKi-induced proinsulin aggregation by simultaneously increasing proinsulin synthesis and reducing BiP expression
Expression of ER chaperones, including BiP, is tightly regulated in all cell types, in part, by binding of the transcription factors ATF6 and XBP1 to ER stress elements (ERSE) upstream of their promotors. Recently, ATF6 has emerged as the predominant ERSE-specific transcriptional regulator of BiP in β cells (38). The ability of the ER chaperone transcriptional regulators to dynamically modulate ER chaperone expression is critical to the proper function of highly secretory cell types, such as plasma or β cells, as they attempt to meet the ever-changing demands for their secretory products under normal physiological conditions. The regulation of ER chaperone expression by ATF6 and XBP1 has been best characterized in the context of ER stress (39–43); however, they have also been shown to play important roles in basal chaperone expression in the absence of ER protein overload (38, 39). Basal expression of activated ATF6 is an important regulator of BiP and proinsulin synthesis in islets and isolated β cells and is essential for their normal function and survival (38). Upon activation by either ER stress or other physiological stimuli, ER-localized ATF6 translocates to the Golgi where it is proteolytically cleaved to release a transcriptionally active ATF6 p50 domain, which then enters the nucleus and stimulates the transcription of target genes, including Bip (43, 44). ATF6 exists in two isoforms, Atf6α and Atf6β, and deletion of both isoforms results in embryonic lethality, indicating that ATF6 has an essential cellular function (44, 45). Volchuk and co-workers (38) demonstrated that knockdown of ATF6α in β cells reduces BiP expression while simultaneously increasing the expression of insulin via the Ins2 gene. Importantly, they were unable to detect any evidence of ER stress in ATF6 KD cells.
The hypothesis that the coordination of functional PERK-dependent ER chaperones governs proinsulin forward trafficking predicts that the combined effects of reduced BiP expression and increased proinsulin production seen in ATF6 KD cells should lead to proinsulin folding and trafficking problems even when PERK remains functional. To test this, β cells were subjected to siRNA-mediated ATF6 KD for a total of 48 h with PERKi or a vehicle control introduced during the last 24 h of knockdown. Cells were then monitored for any indications of ER stress or proinsulin trafficking defects. An increase in the spliced form of Xbp1 was observed in PERK-ablated cells irrespective of ATF6 KD. Sec61a, which has been shown to be specifically up-regulated by the IRE1 arm of the ER stress response (46), remained unchanged, as did the known ER stress indicators, Chop and Gadd34 (Fig. 7A). This absence of ER stress is consistent with previous findings in all models of PERK ablation (9, 23, 24), as well as for knockdown of ATF6 in cultured β cells (38). As was also previously shown (38), ATF6α KD on its own reduced both ATF6α and BiP (mRNA and protein) expression compared with the negative siRNA control (Fig. 7A), and there was a small but significant increase in proinsulin protein (Fig. 7B). When the subcellular location of proinsulin after 48 h of ATF6 KD was examined, a small but significant percentage of cells exhibiting an Impacted-ER phenotype (Fig. 7C) were observed. Although we were unable to see Impacted-ER cells by 28 h of ATF6 KD (Fig. 7D), we did detect a significant increase in total intracellular proinsulin within the PERKi-treated population (data not shown; p value< 0.05), suggesting that the cells were progressing toward an overtly Impacted-ER phenotype.
Figure 7.

ATF6 knockdown alters BiP and proinsulin levels under nonstress conditions. A and B, mRNA (A) and protein (B) expression levels quantified in INS1 832/13 cells transfected with ATF6 siRNAs for 48 h and treated with 1 μm PERKi for the last 24 h prior to harvest. Representative Western blots are shown (B). Quantification represents n = 6 per treatment. Different letters indicate statistically significant difference. C, INS1 832/13 cells were transfected with ATF6 siRNAs for 48 h and 1 μm PERKi was added for the last 24 h. Quantification represents n = 8 per treatment. D and E, INS1 832/13 cells were transfected with ATF6 siRNAs for 28 h and 1 μm PERKi was added for the last 4 h prior to harvest. Quantification represents n = 4 per treatment. Different letters indicate statistically significant differences.
The consequences of adding PERKi to cells with reduced ATF6 expression provide additional clues as to the mechanisms in which proper proinsulin trafficking is dependent on BiP function. When PERKi was included during the last 24 of the 48 h of ATF6 KD, the levels of BiP mRNA (Fig. 7A) and protein (Fig. 7B) were partially restored, and the amount of intracellular proinsulin was dramatically higher (Fig. 7B). If restoration of BiP protein levels was the critical determinant for rescuing chaperone function, then the most likely outcome of this observed partial recovery of BiP expression would be amelioration of proinsulin accumulation and the Impacted-ER phenotype. Instead, we found just the opposite: the frequency of Impacted-ER β cells was higher in ATF6 KD cells treated with PERKi for 24 h (Fig. 7C). Initially, we were somewhat surprised to find that long-term PERK ablation restored BiP expression levels although the expression of its dominant transcriptional activator, ATF6, continued to be significantly knocked down. However, at earlier time points, when proinsulin aggregation is already present (Fig. 7D), we found no increase in Bip expression in response to PERKi irrespective of ATF6 KD (Fig. 7E). The increased expression of BiP that occurs several hours later is most likely due to proteolytic cleavage and activation of residual ATF6, which is known to occur after long-term PERK ablation due to mixing of the ER and Golgi compartments (9, 47).
Other ER chaperones play a role in maintaining proper proinsulin folding
Because other ER chaperones play supporting roles in BiP-dependent proinsulin folding and processing, it was important to ask if altering their expression might affect the formation of the Impacted-ER phenotype. ERp72 seemed to most warrant further investigation because it is the most highly expressed chaperone at the mRNA level in all established models of PERK inhibition and also co-localizes with proinsulin in Impacted-ER β cells (9). Although both overexpressing and reducing functional BiP exacerbated the negative effects of PERKi, manipulation of ERp72 expression had the opposite effect. Adenoviral overexpression (Fig. 8A) and siRNA-mediated KD (Fig. 8C) of ERp72 both partially rescued the Impacted-ER phenotype in PERKi-treated cells (Fig. 8, B and D). However, when KD of BiP and ERp72 were combined, the effect of the BiP KD was dominant (Fig. 8E).
Figure 8.

The balance of other ER chaperones is also important in properly regulating proinsulin trafficking. A, INS1 832/13 cells infected with an ERp72-overexpressing adenovirus or control adenovirus overexpressing GFP at 1000 m.o.i. A representative blot is shown with quantification representing n = 3 per treatment. Statistical significance was calculated relative to the AdV-GFP control; **, p < 0.01. B, INS1 832/13 cells infected with AdV-ERp72 as described in A followed by treatment for 24 h with 1 μm PERKi. Quantification represents n = 4 per treatment. Statistical significance is calculated relative to AdV-GFP/PERKi; ***, p < 0.001. C, INS1 832/13 cells transfected with ERp72 siRNAs. A representative blot is shown with quantification representing n = 4 per treatment. Statistical significance is calculated relative to the negative siRNA control; **, p < 0.01. D, INS1 832/13 cells were transfected with siRNAs as described in C, and 1 μm PERKi was added for the last 24 h prior to harvest. Quantification represents n = 8 per treatment. Statistical significance is calculated relative to the negative siRNA treated with PERKi; ***, p < 0.001. E, INS1 832/13 cells were transfected with BiP and ERp72 siRNAs, and 1 μm PERKi was added for the last 24 h prior to harvest. Quantification represents n = 4 per treatment. Different letters indicate statistically significant differences. F, INS1 832/13 cells transfected with PDI or negative control siRNA. A representative blot is shown with quantification representing n = 4 per treatment. Statistical significance is calculated relative to the negative siRNA control; *, p ≤ 0.05. G, INS1 832/13 cells were transfected with siRNA as described in F, and 1 μm PERKi was added for the last 24 h prior to harvest. Quantification represents n = 4 per treatment. Statistical significance is calculated relative to the negative siRNA treated with PERKi; ***, p < 0.001. H, INS1 832/13 cells treated for 24 h with 1 μm PERKi and then immunoprecipitated (IP) with PDI antibody. Representative image of IP/Western blot (WB) is shown with quantification representing n = 3 per treatment. Statistical significance is calculated relative to the input or IP controls; **, p ≤ 0.01; ***, p < 0.001. I, INS1 832/13 cells were transfected with ATF6 siRNAs for 48 h, mRNA quantification represents n = 3 per treatment. Statistical significance is calculated relative to the negative siRNA control; ***, p < 0.001. J, INS1 832/13 cells infected with a DNAJC3-overexpressing adenovirus or control adenovirus overexpressing GFP at 1000 m.o.i. followed by treatment for 24 h with 1 μm PERKi. Quantification represents n = 4 per treatment. Statistical significance is calculated relative to AdV-GFP/PERKi; ***, p < 0.001. K, INS1 832/13 cells infected with AdV-DNAJC3 as described in J. A representative blot is shown with quantification representing n = 3 per treatment. Statistical significance was calculated relative to the AdV-GFP control; **, p < 0.01; ***, p < 0.001.
The mRNA encoding the ER chaperone Pdi was modestly up-regulated by PERKi treatment (Fig. 4A). As with Erp72, siRNA KD of PDI by over 80% (p value <0.05; Fig. 8F) reduced the frequency of Impacted-ER (Fig. 8G). PERKi treatment also led to enhanced interaction of PDI and proinsulin (Fig. 8H). DNAJC3, a co-chaperone of BiP that promotes its ATPase activity (48), is highly expressed in β cells (49). DNAJC3 deficiency also leads to postnatal diabetes due to reduced insulin production (49). Although siRNA-mediated knockdown of Dnajc3 resulted in a 60% reduction in mRNA levels (Fig. 8I), there was no reduction in immunodetectable DNAJC3 protein nor was there any change in the frequency of Impacted-ER cells (not shown). Adenoviral overexpression of DNAJC3 resulted in a 6-fold increase in DNAJC3 protein levels, regardless of PERKi treatment (p value <0.001), which was correlated with a decrease in total cellular proinsulin and a significant reduction in Impacted-ER cell frequency (Fig. 8, J–K). Taken together, these results support our assertion that proinsulin trafficking requires a functional balance among the PERK-dependent ER chaperone and co-chaperones that mediate folding, quality control, and anterograde trafficking.
Discussion
The elucidation of the UPR pathway in cell culture for the three major UPR regulators (PERK, IRE1, and ATF6) formed the basis to the hypothesis that whole organism defects would be due to a loss of UPR regulation. Thus, the accumulation of proinsulin in the ER of Perk KO β cells was attributed to derepressed protein synthesis and protein overload within the ER (7, 14, 15). However, the growing number of studies using genetic and pharmacological ablation of PERK in β cells consistently found evidence for aberrant proinsulin aggregation in the ER, induction of chaperone expression, and defects in ERAD and trafficking in the absence of an ER stress response (9, 11, 12, 47). Understanding the physiological function of PERK is particularly important to the study of a large number of protein aggregation diseases for which PERK has been implicated, including the diabetes and osteopenia associated with WRS. Using a highly specific and potent inhibitor of PERK (PERKi) to acutely block PERK activity, we were able to directly test the hypothesis that the Impacted-ER arises by derepression of protein synthesis (27). We found that PERKi treatment of cultured β cells consistently causes proinsulin aggregation while reducing proinsulin and protein synthesis in Impacted-ER cells, establishing that proinsulin over-synthesis is not responsible for the Impacted-ER phenotype. Although proinsulin aggregation in PERK-inhibited cells is not caused by protein over-synthesis, it is the result of accumulating a large amount of newly synthesized proinsulin within the ER lumen over several hours.
The widely held perception that increasing ER chaperone levels promotes protein folding and trafficking was largely based on studies of ER stress. By contrast, we found that BiP overexpression resulted in proinsulin aggregation within the ER that mimicked the Impacted-ER phenotype, demonstrating that more chaperone activity is not necessarily better. Reducing BiP levels in the presence of PERKi exacerbated proinsulin aggregation resulting in the most extreme form we have ever seen. Thus, too much or too little of BiP results in proinsulin trafficking dysfunctions. BiP overexpression is known to have differential effects on trafficking of ER client proteins, causing the ER retention of some whereas hastening anterograde trafficking of others (50, 51). BiP can precipitate with misfolded forms of proinsulin (15, 52–54) and has enhanced interaction with ER-accumulated proinsulin in eIF2α Ser51 mutant mice (15). Likewise, an enhanced association of BiP and proinsulin was observed in PERKi-treated cells. We surmise that BiP temporarily restrains ER client protein trafficking as a function of their sequence and/or structure and the physiological conditions within the ER. We found comparatively less proinsulin aggregation when BiP is overexpressed compared with PERK inhibition, suggesting that changes in other ER chaperones other than BiP are also important. Indeed, altering the expression of ERp72, PDI, and DNAJC3 all had an effect on Impacted-ER frequency. Finally, if BiP levels are restored late in the development of the Impacted-ER phenotype, the proinsulin aggregates cannot be untangled and forward trafficking was restored as seen in cells treated with PERKi after 24 h of ATF6 KD.
The greater importance of a functional chaperone balance, as opposed to overall amount, implies strongly that PERK primarily regulates BiP activity, which has been shown to occur via changes in post-translational modification (17, 55–60). Furthermore, BiP is a major ER calcium-binding ATPase, and both ATP and calcium modulate its chaperone activity (17, 48, 61–63). ATP and calcium are, in turn, dynamically regulated in β cells by glucose levels within a normal physiological range (64). We previously showed that PERK acutely regulates cytoplasmic and ER calcium and that PERK activity is regulated inversely by the level of cytoplasmic and ER calcium as a function of glucose or other insulin secretagogues (11). Thus, PERK may first regulate BiP chaperone activity through regulating calcium fluxes, which impact proinsulin folding and trafficking in response to changes in metabolic demand for insulin. Recent studies by van Vliet et al. (65) suggest that PERK regulation of calcium dynamics may be mediated through interaction between the ER and plasma membranes.
By 4 h of PERK inhibition, HMW proinsulin aggregates and the Impacted-ER phenotype can be detected. However, an increase in BiP protein levels does not appear until 24 h, which coincides with the frequency of Impacted-ER β cells reaching its maximum. So, even though artificially elevating BiP can cause proinsulin aggregation, BiP overexpression does not appear to initiate proinsulin aggregation in PERK-inhibited cells. We postulate that the defect in proinsulin trafficking seen in PERK-inhibited cells is triggered by acute disruption of PERK-dependent ER calcium homeostasis, which negatively impacts BiP chaperone activity. The cell, having sensed the loss of this critical chaperone function, then elicits a downstream adaptive response by increasing the expression of BiP through an as yet unidentified mechanism. This response is, however, ineffectual.
Based upon our previous findings using genetic ablation of Perk (9), we assumed that the transcriptional aspect of this mechanism would involve up-regulation of ATF6, as it is the predominant transcriptional regulator of BiP in β cells (38). However, no overall increase in Atf6 mRNA with PERKi treatment was observed, and our attempts to detect the endogenous, active, cleaved form of ATF6 were hindered by the lack of available antibodies. The influence of PERK inhibition in the context of ATF6 KD provides further support to our contention that regulation of BiP activity by PERK controls the early events involving proinsulin folding and forward trafficking. The recently discovered inverse relationship between BiP and proinsulin expression in response to ATF6 KD in unstressed β cells (38, 66) prompted us to ask if these conditions might give rise to proinsulin aggregates. When this was tested, we found evidence for progression toward Impacted-ER by 28 h of ATF6 KD and a low frequency of Impacted-ER cells at 48 h thereby confirming that the combination of reduced BiP and increased proinsulin seen under conditions of ATF6 KD are permissive for proinsulin aggregation. We surmise that ATF6 KD pretreatment magnifies the effects of subsequent PERKi on proinsulin aggregation by reducing the amount of functional BiP whereas simultaneously supplying more, newly synthesized proinsulin for the growing aggregates. The increase in BiP expression in the presence of ATF6 KD after long-term exposure to PERKi is most likely attributable to induced cleavage and activation of the residual endogenous ATF6 protein, which is known to occur due to mixing of the ER and Golgi compartments that occurs after several hours of PERK ablation (9, 10). One possibility for why the delayed increase in BiP protein failed to reverse PERKi-induced proinsulin aggregation is that the partial restoration of chaperone capacity occurred too late in the aggregation process. However, it is also possible that the proinsulin aggregates persisted because the newly synthesized BiP was rendered functionally inactive by the continued presence of the inhibitor.
The active, spliced form of Xbp1 also stimulates Bip transcription during ER stress, and we observed an increased formation of active Xbp1 in PERKi-treated cells with or without ATF6 KD. However, PERKi treatment did not up-regulate the downstream transcriptional ER-stress Xbp-1 targets, Chop and Gadd34, and the PERKi/ATF6 KD combination had no significant impact on Bip mRNA levels compared with ATF6 KD alone. These results are consistent with previous reports (38) and suggest that XBP1 plays a relatively minor role compared with ATF6 in regulating BiP expression in β cells under non-stressed conditions.
The precise role of PERK's major substrate, eIF2α, in ER chaperone regulation is presently unclear. Guanabenz, which inhibits GADD34 and eIF2α de-phosphorylation during ER stress, dramatically reduced the Impacted-ER phenotype and HMW proinsulin aggregation in β cells with minimal impact on global protein synthesis (Fig. S7, A–C) (67). However, its purported effects on stabilizing the phosphorylated form of eIF2α were only seen in the MIN6 β cell line, whereas in the more physiologically competent INS1 832/13β cell line, guanabenz treatment modestly reduced eIF2α-P levels irrespective of PERK inhibition (data not shown). However, guanabenz is also an α agonist of the α2-adrenergic receptor, which impacts intracellular calcium uptake (68). Therefore, guanabenz might fully or partially restore nascent chaperone function in the presence of PERK inhibition thereby promoting forward trafficking by a mechanism that is independent of the PERK–eIF2α pathway. We have shown that PERK, and calcineurin, acutely regulate ER calcium uptake in β cells via store-operated calcium channels (11) and perhaps it is this pathway that is bypassed by the application of guanabenz to PERK-inhibited β cells.
Numerous reports highlight the importance of substrate phosphorylation by PERK for up-regulating ER chaperones expression in a plethora of tissue and cell types undergoing ER stress (10, 69, 70). Furthermore, eIF2α Ser51–Ala homozygous mutant mice are diabetic at birth and have Impacted-ER β cells emphasizing the importance of PERK and eIF2α in maintaining normal β cell function (14). Likewise, we have shown in our previous work and herein that reducing eIF2α-P levels via PERK ablation causes ER dysfunction and misregulation of ER chaperones. However, the sequence and timing of these events are inconsistent with a mechanism that is initiated by global protein or proinsulin over-synthesis or a canonical ER stress response. Alterations in calcium dynamics in response to PERK inhibition in β cells (11) occurs within seconds and HMW proinsulin aggregates can be detected as early as 4 h. The rapidity of both of these responses precludes them as being initiated by adaptive effects on translation via eIF2α. This suggests that the role of eIF2α, whereas important, is to act downstream of the immediate effects of PERK ablation within the ER, presumably through translational regulation of adaptive functions that attempt to restore proper proinsulin folding and trafficking.
To summarize, within seconds to minutes of PERK ablation, ER calcium dynamics are perturbed (11), and we hypothesize that this negatively impacts the activity and/or cooperativity of the resident ER chaperones. Absent the participation of the necessary foldases, inappropriate disulfide bonds form between newly synthesized proinsulin monomers resulting in an accumulation of HMW proinsulin aggregates that are detectable within 4 h of PERK inhibition. The appearance of these aggregates is temporally correlated with the appearance of an expanded and distended ER and a partial merging of the ER and Golgi, leading to cleavage of ATF6 and a progressive inability to clear the defective proinsulin cargo due to ERAD defects (10). As the β cell senses that its ER folding capacity is compromised, it attempts to restore proper function by up-regulating ER chaperone transcription. In the case of BiP and other ERSE-regulated ER chaperones, newly cleaved, active ATF6α, and perhaps spliced XBP1, are likely to be key transcriptional regulators of this adaptive response, with eIF2α playing a role in modulating the subsequent translation of the chaperones and perhaps other key adaptive proteins.
Experimental procedures
Genetic strains
Perk KO strains (8) were congenic for C57BL/6J or 129SvEvTac or were of mixed genetic background. All animal studies were approved by the Penn State Institutional Animal Care and Use Committee (IACUC).
Cell culture
Rat INS1 832/13 (Dr. Christopher Newgard, Duke University) and mouse MIN6 (Dr. Jun-Ichi Miyazaki, Osaka University, Japan) β cells were cultured in supplemented RPMI with 11 mm glucose and Dulbecco's modified Eagle's medium with 25 mm glucose, respectively, as previously described (9).
Adenoviral infection
AdV-GFP and AdV-BiP (Allen Volchuk, University of Toronto) (22) as well as AdV-ERp72 and AdV-DNAJC3 (Vector BioLabs) were added to cells for 2 h. Infection time was increased to 8 h for AdV-BiP at 500 m.o.i. The cells were washed once with phosphate-buffered saline (PBS), fresh medium was added, and the cells incubated for a total of 26 h post-infection with additional treatments occurring at 2 h post-infection as indicated.
Plasmid and siRNA transfection
The wildtype proinsulin–KDEL construct with a V5 epitope-tag (INS+-KDEL) was previously described (10). The INS+-KDEL was transfected using Lipofectamine 3000 reagent (Invitrogen). Cells were transfected with BiP siRNAs (20 nm), ERp72 siRNAs (10 nm), PDI siRNA (10 nm), ATF6 siRNAs (20 nm), DNAJC3 siRNAs (40 nm), or a non-targeting negative siRNA control (Invitrogen) using Lipofectamine RNAiMAX reagent (Invitrogen). Media was replaced 6 h post-transfection to minimize cytotoxic effects of DNA–lipid complexes, and cells were incubated for a total of 48 h, unless otherwise noted. This timing allowed for sufficient protein knockdown to occur prior to subsequent treatment with PERKi for up to 24 h.
Reagents
GSK2606414 PERK inhibitor (PERKi) (Jeffrey Axten and Rakesh Kumar, GlaxoSmithKline) and anisomycin (Sigma) were prepared as stock solutions in dimethyl sulfoxide. Guanabenz (Sigma) was prepared in water. Subtilase cytotoxin (SubAB) and the inactive mutant (SubAA272B) were purified from recombinant Escherichia coli as described previously (34, 71).
Immunohistochemistry
Whole pancreata were harvested as previously described (10). MIN6 and INS1 832/13 cells were immunostained using primary antibodies for proinsulin (Beta Cell Biology Consortium and Hytest), insulin (Abcam), and BiP (Santa Cruz) and appropriate secondary antibodies were conjugated with Alexa Fluor 488 or 546 dye (Molecular Probes) as previously described (11). Slides were mounted using anti-fade reagent with DAPI (Invitrogen) to label nuclei. Fluorescence images were captured with a Nikon Eclipse E1000 and Image-Pro Plus (Phase 3 Imaging Systems, GE Healthcare, Inc.). Impacted-ER cells were quantified as previously described (10). Super resolution images of INS1 832/13 cells grown on coverslips were immunostained using primary antibodies for proinsulin (Beta Cell Biology Consortium and Hytest), BiP (Santa Cruz), and the Golgi-specific marker, RCAS1 (Cell Signaling), followed by appropriate secondary antibodies conjugated with Alexa Fluor 488, 546, or 647 dye (Molecular Probes). Slides were mounted using prolong gold reagent with DAPI (Invitrogen) to label nuclei. Slides were allowed to cure for at least 48 h before fluorescence images were captured with a Nikon N-SIM.
Proinsulin and total protein synthesis
Proinsulin synthesis was measured by labeling with 200 μCi/ml of EXPRESS35S label (Met/Cys) (PerkinElmer Life Sciences) for 30 min in methionine/cysteine-free media. Cells were lysed in cold lysis buffer: Triton X-100 buffer (20 mm HEPES, pH 7.5, 150 mm NaCl, 1% Triton X-100, 10% glycerol, 1 mm EDTA, 1:100 protease inhibitor mixture (Sigma)). Lysates were cleared by centrifugation at 20,000 × g for 15 min at 4 °C. Immunoprecipitation of proinsulin was performed on cleared lysates of equal protein concentration as determined by Coomassie Brilliant Blue (Sigma) staining on a fraction of the lysates that were electrophoretically separated by standard SDS-PAGE. To the remaining lysates Protein A/G PLUS-Agarose (Santa Cruz) and a mixture of anti-insulin SC9168 (Santa Cruz Biotechnology) and anti-insulin I2018 (Sigma) antisera were added. After overnight incubation at 4 °C, the samples were centrifuged at low speed to pellet the bound material. Pellets were washed three times with cold lysis buffer and then boiled in sample buffer under reducing conditions to release the bound proteins from the Protein A/G-agarose beads. The precipitated proteins were separated on 10% BisTris pre-cast polyacrylamide gels (ThermoFisher Scientific), and the gels were fixed and dried. Incorporation was quantified using the Typhoon FLA 7000 (GE Healthcare) and ImageQuant software. Detection and quantification of newly synthesized global protein within individual cells was done using the Click-iT® HPG AlexaFluor® Protein Synthesis Assay Kit following the manufacturer's protocol with minor modifications. Briefly, cells were preincubated with DMSO or PERKi for a specified amount of time followed by co-incubation with the methionine analog l-homoproparargylglycine (HPG) and drug for 60–90 min in methionine-free media. Cells were then fixed in 10% formalin, permeablized in 0.5% Triton X-100 in PBS, and then detected using the Click-iT® reaction mixture. Incorporated HPG causes an alkyne-modified protein, which is detected with Alexa Fluor® 488. Quantification was based on fluorescence intensity levels measured with ImageJ software.
Immunoblot analysis and immunoprecipitation
Cells were lysed directly by boiling in 2× SDS sample buffer prior to electrophoresis on 4–12% BisTris pre-cast polyacrylamide gels (ThermoFisher Scientific). Appropriate horseradish peroxidase-conjugated secondary antibodies (Sigma) were used, and ECL-2 fluorescence (ThermoFisher Scientific) was detected using the Typhoon FLA 7000 (GE Healthcare) and quantified using ImageQuant software. Samples were normalized to the Actin signal of the same sample as a loading control. HMW proinsulin aggregates were isolated using a modification of the method previously described (27). Further details can be found in the supporting Experimental procedures. Primary antibodies used included: eIF2α-P (Invitrogen), eIF2α (gift from S. Kimball), puromycin (Millipore), C-peptide (BioVision), α-tubulin (Sigma), actin (Sigma), ERp72 (Stressgen and Santa Cruz), PDI (Enzo Life Sciences), P58IPK/DNAJC3 (Cell Signaling), and BiP (Santa Cruz). Proinsulin was also detected by blotting with SC-9168 (Santa Cruz) anti-insulin antibody. Immunoprecipitation of BiP and PDI were performed using anti-BiP (Santa Cruz) or anti-PDI (Enzo Life Sciences) and Protein A/G PLUS-Agarose (Santa Cruz).
RNA isolation and gene expression
Total RNA was extracted using the illustra RNAspin Mini Kit (GE Healthcare). Reverse transcription was performed using qScript cDNA supermix (Quanta). Quantitative mRNA measurement was carried out using a quantitative PCR core kit for SYBR Green I (Quanta) with the StepOnePlus detection system (Applied Biosystems). Gene expression levels were normalized to Gapdh and/or Actin levels of the same sample. See supporting Experimental Procedures for mouse and rat primer sequences.
Statistical analysis
All numerical data were represented as mean ± S.D. with the input data points shown as black dots. Statistical significance was determined using Student's t test.
Author contributions
Conceptualization, C. R. S., R. W., J. H., B. C. M., D. R.C.; methodology, C. R. S., R. W., J. H., R. A. B., B. C. M., D. R. C.; validation, C. R. S., R. W., J. H., R. A. B., B. C. M., D. R. C.; formal analysis, C. R. S., R. W., J. H., R. A. B.; investigation, C. R. S., R. W., J. H., S. C. B., R. A. B., B. C. M.; resources, J. C. P., A. W. P., S. C-F., M. N.; writing/original draft, C. R. S., R. W.; writing/review and editing, C. R. S., B. C. M., R. A. B., D. R. C.; visualization? C. R. S.; funding acquisition, C. R. S., B. C. M., and D. R. C.
Supplementary Material
Acknowledgments
We thank Dr. Jeff Axten and Dr. Rakesh Kumar for providing the GSK2606414 PERK inhibitor and discussions about the effects of PERK inhibition. We thank Dr. Christopher Newgard, Dr. Scott Kimball, Dr. Jun-Ichi Miyazaki, Dr. Allen Volchuk, and Dr. Anne Bertolotti for providing experimental materials.
Note added in proof
In the version of this article that was published as a Paper in Press on February 14,2018, Fig. 1, A and B, was missing insets and in Fig. 3, panel D was missing. This error has now been corrected.
This work was supported by National Institutes of Health Grant R01 DK88140 and a Huck Graduate Research Dissertation Award (to C. R. S.) from the Huck Institutes of the Life Sciences at Penn State University. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7, Tables S1 and S2, and “Experimental procedures”.
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- WRS
- Wolcott-Rallison syndrome
- HMW
- high-molecular weight
- AN
- anisomycin
- GFP
- green fluorescent protein
- UPR
- unfolded protein response
- ERSE
- ER stress element
- PDI
- protein-disulfide isomerase
- KD
- knockdown
- eIF
- eukaryote initiation factor
- PERKi
- PERK inhibitor
- SubAB
- subtilase cytotoxin
- DAPI
- 4′,6-diamidino-2-phenylindole
- m.o.i.
- multiplicity of infection
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HPG
- l-homoproparargylglycine
- Adv-BiP
- adenoviral overexpression of BiP.
References
- 1. Alarcón C., Wicksteed B., and Rhodes C. J. (2002) Regulation of the production and secretion of insulin. Av. Diabetol. 18, 168–174 [Google Scholar]
- 2. Weiss M. A. (2009) Proinsulin and the genetics of diabetes mellitus. J. Biol. Chem. 284, 19159–19163 10.1074/jbc.R109.009936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagamatsu S., Bolaffi J. L., and Grodsky G. M. (1987) Direct effects of glucose on proinsulin synthesis and processing during desensitization. Endocrinology 120, 1225–1231 10.1210/endo-120-4-1225 [DOI] [PubMed] [Google Scholar]
- 4. Greeley S. A., Naylor R. N., Philipson L. H., and Bell G. I. (2011) Neonatal diabetes: an expanding list of genes allows for improved diagnosis and treatment. Curr. Diab. Rep. 11, 519–532 10.1007/s11892-011-0234-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kanakatti Shankar R., Pihoker C., Dolan L. M., Standiford D., Badaru A., Dabelea D., Rodriguez B., Black M. H., Imperatore G., Hattersley A., Ellard S., Gilliam L. K., and SEARCH for Diabetes in Youth Study Group. (2013) Permanent neonatal diabetes mellitus: prevalence and genetic diagnosis in the SEARCH for Diabetes in Youth Study. Pediatr Diabetes 14, 174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Delépine M., Nicolino M., Barrett T., Golamaully M., Lathrop G. M., and Julier C. (2000) EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 25, 406–409 10.1038/78085 [DOI] [PubMed] [Google Scholar]
- 7. Harding H. P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D. D., and Ron D. (2001) Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 7, 1153–1163 10.1016/S1097-2765(01)00264-7 [DOI] [PubMed] [Google Scholar]
- 8. Zhang P., McGrath B., Li S., Frank A., Zambito F., Reinert J., Gannon M., Ma K., McNaughton K., and Cavener D. R. (2002) The PERK eukaryotic initiation factor 2 kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 22, 3864–3874 10.1128/MCB.22.11.3864-3874.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feng D., Wei J., Gupta S., McGrath B. C., and Cavener D. R. (2009) Acute ablation of PERK results in ER dysfunctions followed by reduced insulin secretion and cell proliferation. BMC Cell Biol. 10, 61 10.1186/1471-2121-10-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gupta S., McGrath B., and Cavener D. R. (2010) PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 59, 1937–1947 10.2337/db09-1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang R., McGrath B. C., Kopp R. F., Roe M. W., Tang X., Chen G., and Cavener D. R. (2013) Insulin secretion and Ca2+ dynamics in beta-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. J. Biol. Chem. 288, 33824–33836 10.1074/jbc.M113.503664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang W., Feng D., Li Y., Iida K., McGrath B., and Cavener D. R. (2006) PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 4, 491–497 10.1016/j.cmet.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 13. Back S. H., Scheuner D., Han J., Song B., Ribick M., Wang J., Gildersleeve R. D., Pennathur S., and Kaufman R. J. (2009) Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 10, 13–26 10.1016/j.cmet.2009.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scheuner D., Song B., McEwen E., Liu C., Laybutt R., Gillespie P., Saunders T., Bonner-Weir S., and Kaufman R. J. (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176 10.1016/S1097-2765(01)00265-9 [DOI] [PubMed] [Google Scholar]
- 15. Scheuner D., Vander Mierde D., Song B., Flamez D., Creemers J. W., Tsukamoto K., Ribick M., Schuit F. C., and Kaufman R. J. (2005) Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat. Med. 11, 757–764 10.1038/nm1259 [DOI] [PubMed] [Google Scholar]
- 16. Forster M. L., Sivick K., Park Y. N., Arvan P., Lencer W. I., and Tsai B. (2006) Protein disulfide isomerase-like proteins play opposing roles during retrotranslocation. J. Cell Biol. 173, 853–859 10.1083/jcb.200602046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hendershot L. M. (2004) The ER chaperone BiP is a master regulator of ER function. Mt. Sinai J. Med. 71, 289–297 [PubMed] [Google Scholar]
- 18. Kim M. K., Kim H. S., Lee I. K., and Park K. G. (2012) Endoplasmic reticulum stress and insulin biosynthesis: a review. Exp. Diabetes Res. 2012, 509437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ni M., and Lee A. S. (2007) ER chaperones in mammalian development and human diseases. FEBS Lett. 581, 3641–3651 10.1016/j.febslet.2007.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rajpal G., Schuiki I., Liu M., Volchuk A., and Arvan P. (2012) Action of protein disulfide isomerase on proinsulin exit from endoplasmic reticulum of pancreatic beta-cells. J. Biol. Chem. 287, 43–47 10.1074/jbc.C111.279927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ron D., and Harding H. P. (2012) Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harb. Perspect. Biol. 4, a013177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang L., Lai E., Teodoro T., and Volchuk A. (2009) GRP78, but not protein-disulfide isomerase, partially reverses hyperglycemia-induced inhibition of insulin synthesis and secretion in pancreatic β-cells. J. Biol. Chem. 284, 5289–5298 10.1074/jbc.M805477200 [DOI] [PubMed] [Google Scholar]
- 23. Iida K., Li Y., McGrath B. C., Frank A., and Cavener D. R. (2007) PERK eIF2α kinase is required to regulate the viability of the exocrine pancreas in mice. BMC Cell Biol. 8, 38 10.1186/1471-2121-8-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei J., Sheng X., Feng D., McGrath B., and Cavener D. R. (2008) PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. J. Cell. Physiol. 217, 693–707 10.1002/jcp.21543 [DOI] [PubMed] [Google Scholar]
- 25. Atkins C., Liu Q., Minthorn E., Zhang S. Y., Figueroa D. J., Moss K., Stanley T. B., Sanders B., Goetz A., Gaul N., Choudhry A. E., Alsaid H., Jucker B. M., Axten J. M., and Kumar R. (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002 10.1158/0008-5472.CAN-12-3109 [DOI] [PubMed] [Google Scholar]
- 26. Axten J. M., Medina J. R., Feng Y., Shu A., Romeril S. P., Grant S. W., Li W. H., Heerding D. A., Minthorn E., Mencken T., Atkins C., Liu Q., Rabindran S., Kumar R., Hong X., et al. (2012) Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207 10.1021/jm300713s [DOI] [PubMed] [Google Scholar]
- 27. Harding H. P., Zyryanova A. F., and Ron D. (2012) Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J. Biol. Chem. 287, 44338–44344 10.1074/jbc.M112.428987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Collardeau-Frachon S., Vasiljevic A., Jouvet A., Bouvier R., Senée V., and Nicolino M. (2015) Microscopic and ultrastructural features in Wolcott-Rallison syndrome, a permanent neonatal diabetes mellitus: about two autopsy cases. Pediatr. Diabetes 16, 510–520 10.1111/pedi.12201 [DOI] [PubMed] [Google Scholar]
- 29. Haataja L., Snapp E., Wright J., Liu M., Hardy A. B., Wheeler M. B., Markwardt M. L., Rizzo M., and Arvan P. (2013) Proinsulin intermolecular interactions during secretory trafficking in pancreatic beta cells. J. Biol. Chem. 288, 1896–1906 10.1074/jbc.M112.420018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu M., Hodish I., Haataja L., Lara-Lemus R., Rajpal G., Wright J., and Arvan P. (2010) Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol. Metab. 21, 652–659 10.1016/j.tem.2010.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Jeso B., Park Y. N., Ulianich L., Treglia A. S., Urbanas M. L., High S., and Arvan P. (2005) Mixed-disulfide folding intermediates between thyroglobulin and endoplasmic reticulum resident oxidoreductases ERp57 and protein disulfide isomerase. Mol. Cell. Biol. 25, 9793–9805 10.1128/MCB.25.22.9793-9805.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zito E., Chin K. T., Blais J., Harding H. P., and Ron D. (2010) ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 188, 821–832 10.1083/jcb.200911086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bertolotti A., Zhang Y., Hendershot L. M., Harding H. P., and Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 10.1038/35014014 [DOI] [PubMed] [Google Scholar]
- 34. Talbot U. M., Paton J. C., and Paton A. W. (2005) Protective immunization of mice with an active-site mutant of subtilase cytotoxin of Shiga toxin-producing Escherichia coli. Infect. Immun. 73, 4432–4436 10.1128/IAI.73.7.4432-4436.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Resendez E. Jr., Attenello J. W., Grafsky A., Chang C. S., and Lee A. S. (1985) Calcium ionophore A23187 induces expression of glucose-regulated genes and their heterologous fusion genes. Mol. Cell. Biol. 5, 1212–1219 10.1128/MCB.5.6.1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paton A. W., Beddoe T., Thorpe C. M., Whisstock J. C., Wilce M. C., Rossjohn J., Talbot U. M., and Paton J. C. (2006) AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443, 548–552 10.1038/nature05124 [DOI] [PubMed] [Google Scholar]
- 37. Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., and Hartl F. U. (2013) Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 10.1146/annurev-biochem-060208-092442 [DOI] [PubMed] [Google Scholar]
- 38. Teodoro T., Odisho T., Sidorova E., and Volchuk A. (2012) Pancreatic beta-cells depend on basal expression of active ATF6α-p50 for cell survival even under nonstress conditions. Am. J. Physiol. Cell Physiol 302, C992–C1003 10.1152/ajpcell.00160.2011 [DOI] [PubMed] [Google Scholar]
- 39. Acosta-Alvear D., Zhou Y., Blais A., Tsikitis M., Lents N. H., Arias C., Lennon C. J., Kluger Y., and Dynlacht B. D. (2007) XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66 10.1016/j.molcel.2007.06.011 [DOI] [PubMed] [Google Scholar]
- 40. Lee A. H., Iwakoshi N. N., and Glimcher L. H. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 10.1128/MCB.23.21.7448-7459.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamamoto K., Suzuki N., Wada T., Okada T., Yoshida H., Kaufman R. J., and Mori K. (2008) Human HRD1 promoter carries a functional unfolded protein response element to which XBP1 but not ATF6 directly binds. J. Biochem. 144, 477–486 10.1093/jb/mvn091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamamoto K., Yoshida H., Kokame K., Kaufman R. J., and Mori K. (2004) Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 136, 343–350 10.1093/jb/mvh122 [DOI] [PubMed] [Google Scholar]
- 43. Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., and Mori K. (2000) ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755–6767 10.1128/MCB.20.18.6755-6767.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamamoto K., Sato T., Matsui T., Sato M., Okada T., Yoshida H., Harada A., and Mori K. (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 13, 365–376 10.1016/j.devcel.2007.07.018 [DOI] [PubMed] [Google Scholar]
- 45. Wu J., Rutkowski D. T., Dubois M., Swathirajan J., Saunders T., Wang J., Song B., Yau G. D., and Kaufman R. J. (2007) ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364 10.1016/j.devcel.2007.07.005 [DOI] [PubMed] [Google Scholar]
- 46. Adamson B., Norman T. M., Jost M., Cho M. Y., Nuñez J. K., Chen Y., Villalta J. E., Gilbert L. A., Horlbeck M. A., Hein M. Y., Pak R. A., Gray A. N., Gross C. A., Dixit A., Parnas O., Regev A., and Weissman J. S. (2016) A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21 10.1016/j.cell.2016.11.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gupta S., McGrath B., and Cavener D. R. (2009) PERK regulates the proliferation and development of insulin-secreting beta-cell tumors in the endocrine pancreas of mice. PLoS ONE 4, e8008 10.1371/journal.pone.0008008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Petrova K., Oyadomari S., Hendershot L. M., and Ron D. (2008) Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 27, 2862–2872 10.1038/emboj.2008.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ladiges W. C., Knoblaugh S. E., Morton J. F., Korth M. J., Sopher B. L., Baskin C. R., MacAuley A., Goodman A. G., LeBoeuf R. C., and Katze M. G. (2005) Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 54, 1074–1081 10.2337/diabetes.54.4.1074 [DOI] [PubMed] [Google Scholar]
- 50. Dorner A. J., Wasley L. C., and Kaufman R. J. (1992) Overexpression of GRP78 mitigates stress induction of glucose regulated proteins and blocks secretion of selective proteins in Chinese hamster ovary cells. EMBO J. 11, 1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Watson L. M., Chan A. K., Berry L. R., Li J., Sood S. K., Dickhout J. G., Xu L., Werstuck G. H., Bajzar L., Klamut H. J., and Austin R. C. (2003) Overexpression of the 78-kDa glucose-regulated protein/immunoglobulin-binding protein (GRP78/BiP) inhibits tissue factor procoagulant activity. J. Biol. Chem. 278, 17438–17447 10.1074/jbc.M301006200 [DOI] [PubMed] [Google Scholar]
- 52. Liu M., Li Y., Cavener D., and Arvan P. (2005) Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J. Biol. Chem. 280, 13209–13212 10.1074/jbc.C400475200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tiwari A., Schuiki I., Zhang L., Allister E. M., Wheeler M. B., and Volchuk A. (2013) SDF2L1 interacts with the ER-associated degradation machinery and retards the degradation of mutant proinsulin in pancreatic beta-cells. J. Cell Sci. 126, 1962–1968 10.1242/jcs.117374 [DOI] [PubMed] [Google Scholar]
- 54. Wang J., Takeuchi T., Tanaka S., Kubo S. K., Kayo T., Lu D., Takata K., Koizumi A., and Izumi T. (1999) A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Invest. 103, 27–37 10.1172/JCI4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aprile F. A., Dhulesia A., Stengel F., Roodveldt C., Benesch J. L., Tortora P., Robinson C. V., Salvatella X., Dobson C. M., and Cremades N. (2013) Hsp70 oligomerization is mediated by an interaction between the interdomain linker and the substrate-binding domain. PLoS ONE 8, e67961 10.1371/journal.pone.0067961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chambers J. E., Petrova K., Tomba G., Vendruscolo M., and Ron D. (2012) ADP ribosylation adapts an ER chaperone response to short-term fluctuations in unfolded protein load. J. Cell Biol. 198, 371–385 10.1083/jcb.201202005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fabrizio G., Di Paola S., Stilla A., Giannotta M., Ruggiero C., Menzel S., Koch-Nolte F., Sallese M., and Di Girolamo M. (2015) ARTC1-mediated ADP-ribosylation of GRP78/BiP: a new player in endoplasmic-reticulum stress responses. Cell. Mol. Life Sci. 72, 1209–1225 10.1007/s00018-014-1745-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Freiden P. J., Gaut J. R., and Hendershot L. M. (1992) Interconversion of three differentially modified and assembled forms of BiP. EMBO J. 11, 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gething M.-J. (1999) Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 10, 465–472 [DOI] [PubMed] [Google Scholar]
- 60. Laitusis A. L., Brostrom M. A., and Brostrom C. O. (1999) The dynamic role of GRP78/BiP in the coordination of mRNA translation with protein processing. J. Biol. Chem. 274, 486–493 10.1074/jbc.274.1.486 [DOI] [PubMed] [Google Scholar]
- 61. Lièvremont J. P., Rizzuto R., Hendershot L., and Meldolesi J. (1997) BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J. Biol. Chem. 272, 30873–30879 10.1074/jbc.272.49.30873 [DOI] [PubMed] [Google Scholar]
- 62. Munro S., and Pelham H. R. (1986) An Hsp70-like protein in the ER: identity with the 78 kDa glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 46, 291–300 10.1016/0092-8674(86)90746-4 [DOI] [PubMed] [Google Scholar]
- 63. Preissler S., Chambers J. E., Crespillo-Casado A., Avezov E., Miranda E., Perez J., Hendershot L. M., Harding H. P., and Ron D. (2015) Physiological modulation of BiP activity by trans-promoter engagement of the interdomain linker. Elife 4, e08961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vishnu N., Jadoon Khan M., Karsten F., Groschner L. N., Waldeck-Weiermair M., Rost R., Hallström S., Imamura H., Graier W. F., and Malli R. (2014) ATP increases within the lumen of the endoplasmic reticulum upon intracellular Ca2+ release. Mol. Biol. Cell 25, 368–379 10.1091/mbc.E13-07-0433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Vliet A. R., Giordano F., Gerlo S., Segura I., Van Eygen S., Molenberghs G., Rocha S., Houcine A., Derua R., Verfaillie T., Vangindertael J., De Keersmaecker H., Waelkens E., Tavernier J., Hofkens J., et al. (2017) The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol. Cell 65, 885–899.e6 10.1016/j.molcel.2017.01.020 [DOI] [PubMed] [Google Scholar]
- 66. Seo H. Y., Kim Y. D., Lee K. M., Min A. K., Kim M. K., Kim H. S., Won K. C., Park J. Y., Lee K. U., Choi H. S., Park K. G., and Lee I. K. (2008) Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology 149, 3832–3841 10.1210/en.2008-0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsaytler P., Harding H. P., Ron D., and Bertolotti A. (2011) Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94 10.1126/science.1201396 [DOI] [PubMed] [Google Scholar]
- 68. Norez C., Vandebrouck C., Antigny F., Dannhoffer L., Blondel M., and Becq F. (2008) Guanabenz, an α2-selective adrenergic agonist, activates Ca2+-dependent chloride currents in cystic fibrosis human airway epithelial cells. Eur. J. Pharmacol. 592, 33–40 10.1016/j.ejphar.2008.06.103 [DOI] [PubMed] [Google Scholar]
- 69. Teske B. F., Wek S. A., Bunpo P., Cundiff J. K., McClintick J. N., Anthony T. G., and Wek R. C. (2011) The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 22, 4390–4405 10.1091/mbc.E11-06-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wek R. C., and Cavener D. R. (2007) Translational control and the unfolded protein response. Antioxid. Redox Signal. 9, 2357–2371 10.1089/ars.2007.1764 [DOI] [PubMed] [Google Scholar]
- 71. Paton A. W., Srimanote P., Talbot U. M., Wang H., and Paton J. C. (2004) A new family of potent AB(5) cytotoxins produced by Shiga toxigenic Escherichia coli. J. Exp. Med. 200, 35–46 10.1084/jem.20040392 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.