

Abstract
Fatty acid–binding proteins (FABPs) are intracellular lipid carriers that regulate inflammation, and pharmacological inhibition of FABP5 reduces inflammation and pain. The mechanism(s) underlying the anti-inflammatory effects associated with FABP5 inhibition is poorly understood. Herein, we identify a novel mechanism through which FABP5 modulates inflammation. In mice, intraplantar injection of carrageenan induces acute inflammation that is accompanied by edema, enhanced pain sensitivity, and elevations in proinflammatory cytokines and prostaglandin E2 (PGE2). Inhibition of FABP5 reduced pain, edema, cytokine, and PGE2 levels. PGE2 is a major eicosanoid that enhances pain in the setting of inflammation, and we focused on the mechanism(s) through which FABP5 modulates PGE2 production. Cyclooxygenase 2 (COX-2) and microsomal prostaglandin E synthase 1 (mPGES-1) are enzymes up-regulated at the site of inflammation and account for the bulk of PGE2 biosynthesis. Pharmacological or genetic FABP5 inhibition suppressed the induction of mPGES-1 but not COX-2 in carrageenan-injected paws, which occurred predominantly in macrophages. The cytokine interleukin 1β (IL-1β) is a major inducer of mPGES-1 during inflammation. Using A549 cells that express FABP5, IL-1β stimulation up-regulated mPGES-1 expression, and mPGES-1 induction was attenuated in A549 cells bearing a knockdown of FABP5. IL-1β up-regulates mPGES-1 via NF-κB, which activates the mPGES-1 promoter. Knockdown of FABP5 reduced the activation and nuclear translocation of NF-κB and attenuated mPGES-1 promoter activity. Deletion of NF-κB–binding sites within the mPGES-1 promoter abrogated the ability of FABP5 to inhibit mPGES-1 promoter activation. Collectively, these results position FABP5 as a novel regulator of mPGES-1 induction and PGE2 biosynthesis during inflammation.
Keywords: fatty acid binding protein, prostaglandin, inflammation, pain, NF-κB, FABP, mPGES-1
Introduction
Pain is a frequent reason for seeking medical care, and approximately 30% of older adults experience chronic pain (1–3). Nonsteroidal anti-inflammatory drugs (NSAIDs)3 and opioids are mainstay treatments for chronic pain. However, chronic NSAID use is associated with gastrointestinal hemorrhage, and even acute NSAID use increases the incidence of myocardial infarctions (4, 5). Chronic opioid use is associated with significant addiction and overdose liability (6–8). Consequently, there is a need to develop novel efficacious non-opioid analgesics.
Fatty acid–binding proteins (FABPs) are intracellular carriers for fatty acids and related bioactive lipids, such as the endocannabinoid anandamide (9–11). In addition to the cytosolic transport of lipids, FABPs deliver ligands to the nucleus wherein they activate nuclear receptors (10, 12–14). Ten FABP isoforms are expressed in mammals, and we previously demonstrated that pharmacological inhibition of FABP5 produces analgesic effects by enhancing endocannabinoid signaling (15–18). We also demonstrated that FABP5 is expressed in nociceptors, primary sensory neurons that transmit noxious information from peripheral tissues to the dorsal horn of the spinal cord (18–20). In addition to nociceptors, FABP5 is found in cells of the innate immune system, primarily macrophages and dendritic cells (21, 22), raising the possibility that FABP5 may also regulate pain indirectly by suppressing the production of proinflammatory mediators. In addition to FABP5, macrophages and dendritic cells also express FABP4, which can likewise regulate inflammation (23).
Tissue inflammation leads to the production of a variety of mediators that activate and/or sensitize nociceptors, resulting in enhanced nociceptor activation and an exacerbation of pain termed hyperalgesia (24, 25). Proinflammatory cytokines and the eicosanoid prostaglandin E2 (PGE2) are important mediators of inflammatory hyperalgesia (24). PGE2 is synthesized from arachidonic acid through the sequential actions of cyclooxygenase 1 or cyclooxygenase 2 enzymes (COX-1 and COX-2, respectively) followed by microsomal prostaglandin E synthase 1 (mPGES-1) (Fig. 1). During inflammation, COX-2 becomes highly induced, leading to enhanced PGE2 biosynthesis (26). Although there are three enzymes that can catalyze the biosynthesis of PGE2 (Fig. 1), mPGES-1 appears to be the principal isoform that contributes to PGE2 biosynthesis in the setting of inflammation and pain (26, 27). mPGES-1 knockout (KO) mice exhibit attenuated inflammatory responses and reduced pain sensitivity (27), positioning this enzyme as a major regulator of PGE2 biosynthesis. In the current study, we explored the mechanism(s) through which FABP5 regulates inflammation. Our findings uncover a novel role for FABP5 as an essential regulator of mPGES-1 induction and PGE2 biosynthesis. These effects are unique to FABP5 because mPGES-1 induction is unaltered in FABP4 KO mice, highlighting a unique feature of FABP5 in controlling inflammation, thus positioning FABP5 as a modulator of mPGES-1 and PGE2 associated pain and inflammation.
Figure 1.

Biosynthetic pathway for PGE2. Arachidonic acid is converted into prostaglandin H2 by COX-1 and COX-2 enzymes. Prostaglandin H2 is subsequently converted into PGE2 by mPGES-1, mPGES-2, and cytosolic prostaglandin E synthase (cPGES) enzymes. COX-2 and mPGES-1 are the enzymes that contribute to the bulk of PGE2 biosynthesis during inflammation.
Results
Effect of FABP4 and FABP5 inhibition upon inflammation and pain
Administration of carrageenan into the hind paw of WT mice results in the rapid development of edema and hyperalgesia (Fig. 2, A and B). We previously demonstrated that FABP5 inhibitors produce antinociceptive (i.e. pain-reducing) and anti-inflammatory effects in this model (15, 16). Consistent with this, carrageenan-associated edema was significantly reduced in FABP5 KO mice (Fig. 2A). Carrageenan administration also induces hyperalgesia, which manifests as reduced hind paw withdrawal latencies in response to a thermal stimulus (Fig. 2B). Compared with WT mice injected with carrageenan, thermal withdrawal thresholds were significantly higher in FABP5 KO mice, indicative of an antinociceptive phenotype (Fig. 2B). In contrast, FABP4 KO mice displayed normal development of edema and hyperalgesia (Fig. 2, A and B).
Figure 2.

FABP5 inhibition produces anti-inflammatory and antinociceptive effects. A, WT, FABP4 KO, and FABP5 KO mice received intraplantar injections of carrageenan, and paw edema was measured 4 h later. The change in paw edema from baseline is indicated. *, p < 0.05 versus WT mice (n = 6). B, thermal withdrawal latencies in WT, FABP4 KO, and FABP5 KO mice before and 4 h after intraplantar injection of carrageenan. *, p < 0.05 versus WT mice after carrageenan injection (n = 9). C, CGRP release in sections of lumbar spinal cords obtained from carrageenan- and saline-injected WT mice. Mice received an intraplantar injection of carrageenan or saline, and CGRP release was measured 4 h later in spinal sections obtained from the ipsilateral (carrageenan-injected) or contralateral (saline-injected) side. **, p < 0.01 (n = 6). D, capsaicin evoked CGRP release in lumbar spinal slices from WT and FABP5 KO. Spinal sections were obtained as in C. CGRP release was quantified in ipsilateral (carrageenan-injected) slices before and after treatment with capsaicin (1 μm, 30 min). *, p < 0.05; **, p < 0.01; ***, p < 0.001 (n = 6). E, TNFα, IL-1β, and IL-6 levels in paws at baseline and 4 h after carrageenan injection. Mice were injected with vehicle or SBFI26 (20 mg/kg, i.p.) 30 min before carrageenan injection. Cytokine levels were normalized to baseline levels in WT mice. *, p < 0.05 versus carrageenan-injected WT mice (n = 6). F, PGE2 levels in paws at baseline and 4 h after carrageenan injection in WT mice treated with vehicle or 20 mg/kg SBFI26 and in FABP5 KO mice. **, p < 0.01 versus carrageenan injected WT mice (n = 6). Error bars represent S.E.
To confirm the behavioral data, we examined the release of calcitonin gene–related peptide (CGRP) from lumbar spinal cord sections of WT and FABP5 KO mice that received an intraplantar injection of saline or carrageenan. CGRP is released from nociceptors into the dorsal horn of the spinal cord, and its release is enhanced in sensitized nociceptors, such as during inflammatory hyperalgesia in rodents and humans (28, 29). We injected mice with carrageenan and collected lumbar spinal cords, which were bisected along the midline to yield ipsilateral (carrageenan-injected) and contralateral (saline-injected) sections. Similar levels of CGRP release were observed in contralateral sections of WT and FABP5 KO mice (Fig. 2C). Compared with the contralateral sections, spinal CGRP release was elevated in ipsilateral spinal sections of WT but not FABP5 KO mice (Fig. 2C). Importantly, compared with ipsilateral spinal sections obtained from WT mice, CGRP release was significantly lower in ipsilateral sections obtained from FABP5 KO mice (Fig. 2C), indicative of reduced sensitization of nociceptors after inflammation in FABP5 KO mice. We recently reported that FABP5 is expressed in nociceptors that coexpress CGRP and the transient receptor potential vanilloid-1 (TRPV1) ion channel (18). TRPV1 mediates thermal hyperalgesia and is activated by the exogenous ligand capsaicin, and its activation enhances CGRP release (28, 30, 31). To determine whether FABP5 deletion alters capsaicin-evoked CGRP release, we incubated ipsilateral spinal cord sections with 1 μm capsaicin for 30 min and quantified CGRP release. Capsaicin induced a significant increase in spinal CGRP release in WT sections and a smaller but significant increase in sections from FABP5 KO mice (Fig. 2D). These data indicate that FABP5 inhibition reduces nociceptor sensitization after inflammation, consistent with the behavioral phenotypes of FABP5 KO mice.
Effect of FABP5 inhibition upon cytokines and PGE2
Peripheral inflammation induces the recruitment of innate immune cells, predominantly neutrophils and macrophages, which secrete proinflammatory cytokines that sensitize nociceptors (32, 33). As expected, carrageenan administration elevated the levels of tumor necrosis factor α (TNFα), interleukin 1β (IL-1β), and interleukin 6 (IL-6) in the paws of WT mice (Fig. 2E). Compared with WT mice, levels of these cytokines were significantly lower in paws of WT mice treated with the FABP5 inhibitor SBFI26 (15) and in FABP5 KO mice. Of note, IL-1β contributes to inflammatory pain by up-regulating the expression of COX-2 and mPGES-1 and enhancing inflammatory PGE2 biosynthesis (34–36). To determine whether FABP5 inhibition reduces PGE2 levels in inflamed paws, we quantified PGE2 in paws of WT, SBFI26-treated, and FABP5 KO mice. As expected, PGE2 was elevated in the paws of WT mice treated with carrageenan (Fig. 2F). PGE2 levels were significantly reduced in WT mice treated with SBFI26 and in FABP5 KO mice (Fig. 2F), confirming that FABP5 regulates PGE2 biosynthesis during inflammation. We examined the expression of FABP5 in cross-sections of mouse paws before and after carrageenan injection using a validated FABP5 antibody (18). At baseline, FABP5 staining was predominantly observed in the epidermis and in resident macrophages (Fig. 3). Carrageenan administration resulted in the recruitment of FABP5-expressing macrophages to the site of inflammation (Fig. 3). Collectively, these results indicate that FABP5 regulates PGE2 biosynthesis in inflamed tissue, possibly within macrophages.
Figure 3.
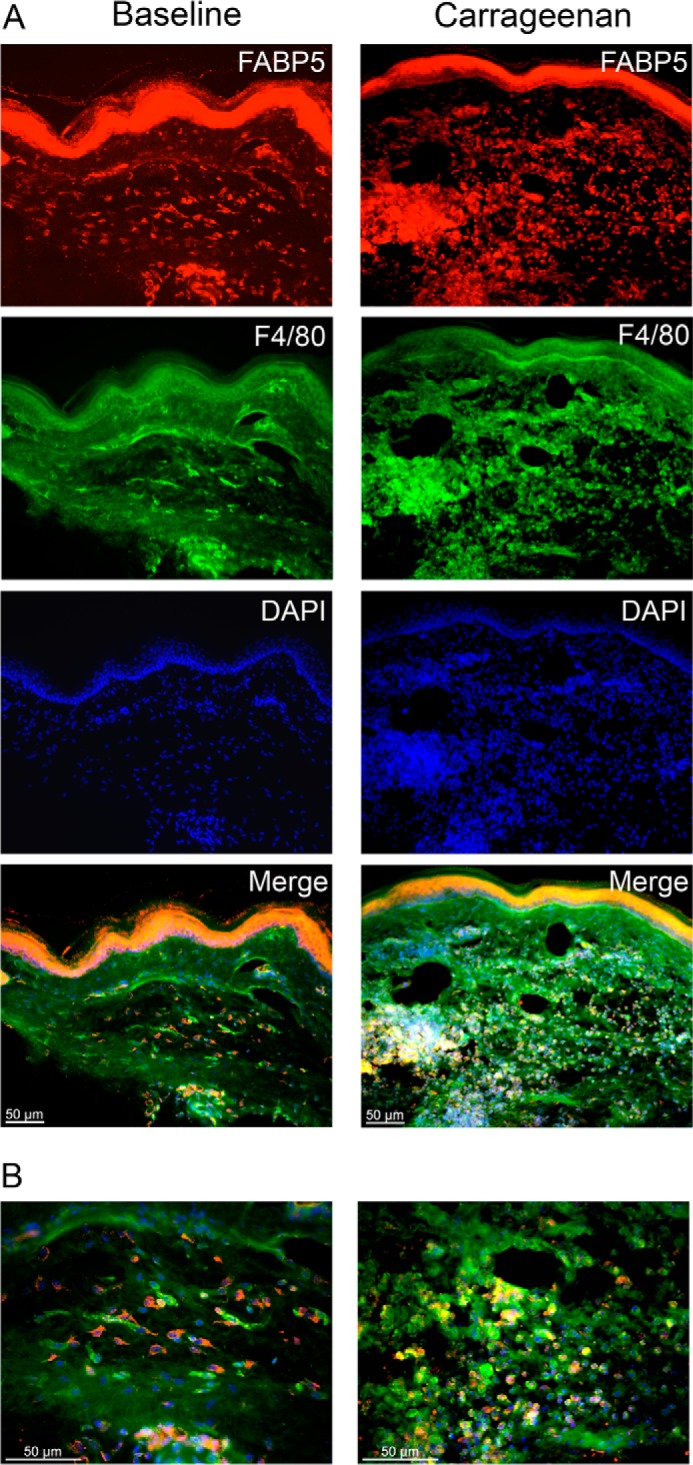
FABP5 expression in paws after inflammation. A, immunolocalization of FABP5 in sections of paw tissue before and after intraplantar injection of carrageenan. Left panels, FABP5 was predominantly expressed in the epidermis of control paws with some expression in F4/80+ macrophages. Right panels, carrageenan injection resulted in the recruitment of FABP5-expressing F4/80+ macrophages to the site of inflammation. B, higher magnification merged images of those shown in A demonstrating robust expression of FABP5 in F4/80+ macrophages in carrageenan-injected paws.
FABP5 is a cytosolic transport protein that binds to arachidonic acid, the precursor for PGE2 (37). Therefore, it is possible that FABP5 may regulate PGE2 biosynthesis in immune cells by shuttling arachidonic acid to COX-2, which is localized on the endoplasmic reticulum. To test this experimentally, human THP-1 monocytic cells, which express FABP5 but not other FABP isoforms (38), were differentiated into macrophages using phorbol myristate acetate and subsequently activated with lipopolysaccharide (LPS) to ensure robust PGE2 biosynthesis as described (39). Treatment with LPS for 24 h was required to induce COX-2 expression (Fig. 4A). After activation with LPS for 24 h, the cells were washed and incubated for 2 h in the presence or absence of SBFI26 to determine whether acute FABP5 inhibition in fully activated cells can reduce PGE2 production. Indeed, FABP5 inhibition modestly but significantly suppressed PGE2 levels in activated THP-1 cells (Fig. 4B). Treatment of cells with the nonselective COX-1/2 inhibitor diclofenac robustly inhibited PGE2 production (Fig. 4B). To rule out the possibility that SBFI26 directly inhibits COX enzymes, we confirmed that SBFI26 did not inhibit the activity of purified COX-1 or COX-2 (Fig. 4C). To differentiate the effects of SBFI26 on arachidonic acid transport from direct effects on COX enzymes, we lysed cells and isolated membrane fractions from activated THP-1 cells. The membranes were incubated with arachidonic acid to stimulate PGE2 biosynthesis (Fig. 4D). Coincubation of membranes with arachidonic acid in the presence of diclofenac significantly reduced PGE2 production (Fig. 4D), confirming the presence of COX-mediated PGE2 biosynthesis. In contrast, SBFI26 did not affect PGE2 production, confirming that it does not inhibit enzymes that mediate PGE2 biosynthesis. These data are consistent with a model wherein FABP5 may transport arachidonic acid toward PGE2 biosynthesis, but this is unlikely to fully account for the robust reduction in PGE2 levels observed in paws after inflammation.
Figure 4.

Effect of FABP5 inhibition upon PGE2 levels in activated THP-1 cells. A, Western blot of COX-2 induction in THP-1 cells after incubation with 5 μg/ml LPS for 2 or 24 h. B, PGE2 levels in THP-1 cells before and after activation with LPS. The cells were incubated with vehicle (PBS; control) or 5 μg/ml LPS for 24 h to induce PGE2 biosynthesis. The cells were subsequently washed and incubated with SBFI26, diclofenac, or vehicle (0.1% DMSO) for 2 h, and PGE2 levels were quantified in the media. *, p < 0.05; **, p < 0.01 versus LPS-activated vehicle-treated cells (n = 6). C, effect of SBFI26 upon purified COX-1 and COX-2 activity (n = 4). D, PGE2 biosynthesis in membrane fractions of control and LPS-activated THP-1 cells. Membranes were isolated 24 h after LPS stimulation and incubated with 1 μm arachidonic acid in the presence or absence of SBFI26 or diclofenac. **, p < 0.01 versus LPS-activated vehicle-treated membranes (n = 6). Error bars represent S.E.
FABP5 regulates mPGES-1 induction
As noted above, paw inflammation induces COX-2 and mPGES-1 expression, raising the possibility that FABP5 may regulate PGE2 by modulating the induction of these enzymes. We first examined the effect of FABP5 inhibition upon the induction of COX-2. Carrageenan administration enhanced COX-2 expression in paws, which was unaffected by FABP5 deletion (Fig. 5A). In contrast, carrageenan-induced up-regulation of mPGES-1 was highly suppressed in FABP5 KO mice (Fig. 5B). The induction of mPGES-1 in FABP4 KO mice was comparable with that observed in WT mice (Fig. 5B), indicating that the regulation of mPGES-1 expression during inflammation is a unique feature of FABP5. A similar profile was observed in WT mice treated with SBFI26, confirming that acute pharmacological and genetic FABP5 inhibition produces similar effects upon mPGES-1 induction (Fig. 5C). To confirm this further, we examined the expression and localization of mPGES-1 in carrageenan-injected paws. At baseline, very low levels of mPGES-1 immunoreactivity were observed in paws (Fig. 5, D and E). After carrageenan injection, elevated mPGES-1 expression was observed in paws of WT mice that colocalized to a large extent with the macrophage marker F4/80 (Fig. 5D). Weaker staining was also observed in the epidermis and eccrine sweat glands. Carrageenan administration failed to induce mPGES-1 expression in paws of FABP5 KO mice (Fig. 5D), confirming the Western blot results.
Figure 5.

Effect of FABP5 inhibition upon COX-2 and mPGES-1 induction in carrageenan-injected paws. A, left, COX-2 expression in hind paws of WT and FABP5 KO mice injected with saline or carrageenan. The paws were harvested 24 h after carrageenan administration. Right, quantification of COX-2 and GAPDH intensity ratios. COX-2 levels are reported as the ratio of COX-2/GAPDH signal intensity (n = 6). B, left, mPGES-1 induction in hind paws of WT, FABP4 KO, and FABP5 KO mice injected with carrageenan. Right, quantification of mPGES-1 signal intensities. *, p < 0.05 versus carrageenan-injected WT mice (n = 6). C, left, induction of mPGES-1 in carrageenan-injected hind paws of WT mice receiving an intraperitoneal injection of vehicle or 20 mg/kg SBFI26. Right, quantification of mPGES-1 and GAPDH signal intensities. **, p < 0.01 versus carrageenan-injected mice (n = 6). D, immunofluorescence of mPGES-1 and F4/80+ macrophage expression in the paws of WT and FABP5 KO mice. Note the low mPGES-1 expression at baseline and its increase primarily in F4/80+ macrophages after carrageenan injection in WT mice. In FABP5 KO mice, carrageenan injection did not lead to an increase in mPGES-1 expression. E, higher magnification merged images of those shown in D. Error bars represent S.E.
We previously demonstrated that FABP5 inhibition produces analgesic effects by enhancing activation of cannabinoid receptors and peroxisome proliferator–activated receptor α (PPARα) by their endogenous ligands (15–18). To determine whether FABP5 inhibition suppresses mPGES-1 induction through activation of these receptors, we treated FABP5 KO mice with the cannabinoid receptor 1 and 2 antagonists AM251 and AM630 (3 mg/kg, i.p.), respectively, or with the PPARα antagonist GW6471 (4 mg/kg, i.p.) prior to carrageenan injection. Cannabinoid and PPARα receptor antagonism did not alter mPGES-1 induction in FABP5 KO mice (Fig. 6, A and B). Previous work has demonstrated that cells of the innate immune system express the related receptor PPARγ, whose activation can reduce mPGES-1 induction (40). Treatment of FABP5 KO mice with the PPARγ antagonist GW9662 (2 mg/kg, i.p.) likewise did not affect mPGES-1 induction (Fig. 6, B and C). We corroborated the Western blot results by quantifying tissue PGE2, which demonstrated similarly suppressed PGE2 levels in FABP5 KO mice treated with vehicle or the receptor antagonists (Fig. 6D).
Figure 6.

Involvement of cannabinoid and PPAR receptors in mPGES-1 induction. A, induction of mPGES-1 in WT and FABP5 KO mouse hind paws after injection with carrageenan. Mice were administered vehicle, the cannabinoid receptor 1 and 2 antagonists AM251 and AM630 (3 mg/kg, i.p.), or the PPARα antagonist GW6471 (4 mg/kg, i.p.). Paws were harvested 24 h after carrageenan injection. B, quantification of mPGES-1/GAPDH band intensities of Western blots described in A and C. *, p < 0.05 versus carrageenan-injected WT mice (n = 6–8). C, induction of mPGES-1 in paws of WT and FABP5 KO mice treated with vehicle or the PPARγ antagonist GW9662 (2 mg/kg, i.p.). D, PGE2 levels in hind paws of WT and FABP5 KO mice administered AM251/AM630, GW6471, or GW9662. PGE2 levels were quantified at baseline and 24 h after carrageenan injection. *, p < 0.05; **, p < 0.01 versus carrageenan-injected WT mice (n = 6). Error bars represent S.E.
FABP5 induces mPGES-1 via NF-κB
IL-1β is elevated during inflammation, and our data demonstrate that FABP5 inhibition reduces IL-1β levels in carrageenan-injected paws (Fig. 2E). IL-1β is also a major inducer of mPGES-1 expression (34–36), indicating that FABP5 inhibition may curtail mPGES-1 induction indirectly by suppressing IL-1β release. It is also possible that FABP5 may directly regulate mPGES-1 induction in cells activated by IL-1β. To test this, we used A549 cells, which up-regulate mPGES-1 in response to IL-1β stimulation (34). Importantly, A549 cells express FABP5 but do not express other FABP isoforms, thus providing an ideal system to examine the contribution of FABP5 to mPGES-1 induction (38). We confirmed that treatment of A549 cells with IL-1β (1 ng/ml) results in the up-regulation of mPGES-1 (Fig. 7A). A549 cells bearing a knockdown of FABP5 were generated, and we confirmed that cells expressing FABP5 shRNA exhibited attenuated mPGES-1 induction in response to IL-1β (Fig. 7, A and B).
Figure 7.

Effect of FABP5 inhibition upon mPGES-1 induction and NF-κB activity in A549 cells. A, left, induction of mPGES-1 and COX-2 in control and FABP5 shRNA–expressing A549 cells treated with 1 ng/ml IL-1β or vehicle. Right, quantification of Western blots. *, p < 0.05 versus IL-1β–treated control cells at the same time point (n = 6). B, left, knockdown of FABP5 by shRNA. Right, quantification of Western blots. *, p < 0.05 (n = 3). C, left, phosphorylated and total NF-κB in control and FABP5 shRNA–expressing A549 cells treated with 1 ng/ml IL-1β. Right, quantification of phosphorylated/total NF-κB. *, p < 0.05 versus IL-1β–treated control cells at the same time point (n = 6). D, left, Western blot of nuclear and cytoplasmic NF-κB in control and FABP5 shRNA–expressing A549 cells incubated with 1 ng/ml IL-1β or vehicle for 30 min. The cells underwent fractionation, and nuclear and cytosolic fractions were collected. The purity of the fractions was confirmed using GAPDH and histone H3 as cytosolic and nuclear markers, respectively. C, cytosolic fraction; N, nuclear fraction. Right, quantification of nuclear NF-κB/histone H3 levels. *, p < 0.05 versus IL-1β–treated control cells (n = 4). E, NF-κB reporter activity in control and FABP5 shRNA A549 cells expressing the NF-κB luciferase reporter construct. The cells were treated with 1 ng/ml IL-1β or vehicle for 6 h, and luciferase and β-galactosidase signals were quantified. Results are reported as luciferase/β-galactosidase signal and normalized to control cells. **, p < 0.01 versus IL-1β–treated control cells (n = 6). Error bars represent S.E.
It was previously reported that IL-1β stimulation activates NF-κB, which enhances mPGES-1 expression in multiple cell types, including A549 cells (34, 39). A549 cells treated with IL-1β exhibited robust NF-κB activation as evidenced by enhanced NF-κB phosphorylation, which was reduced in FABP5 knockdown cells (Fig. 7C). Upon activation, NF-κB rapidly translocates into the nucleus to stimulate gene transcription, including that of mPGES-1 (34, 39). To determine whether FABP5 inhibition modulates the nuclear entry of NF-κB, A549 cells were stimulated with IL-1β, and the nuclear and cytoplasmic fractions were isolated. The purity of the fractions was confirmed using GAPDH and histone H3 as cytoplasmic and nuclear markers, respectively (Fig. 7D). Prior to the addition of IL-1β, NF-κB was not detected in the nucleus (Fig. 7D). Upon stimulation with IL-1β, robust NF-κB immunoreactivity was detected in the nuclei of A549 cells (Fig. 7D). In contrast, nuclear NF-κB levels were significantly lower in IL-1β–treated A549 cells bearing a knockdown of FABP5 (Fig. 7D). To confirm reduced nuclear NF-κB activity, we examined NF-κB activity using a luciferase reporter containing NF-κB–binding sites upstream of the luciferase open reading frame. Binding of NF-κB to its binding sites stimulates luciferase expression. NF-κB reporter activity was elevated after IL-1β stimulation of A549 cells, and this was significantly lower in FABP5 shRNA cells, confirming reduced nuclear NF-κB activity upon FABP5 knockdown (Fig. 7E). In addition to mPGES-1, NF-κB mediates the induction of COX-2 in response to IL-1β (34, 35). We confirmed that IL-1β induced COX-2 expression in A549 cells, and this was reduced upon FABP5 knockdown (Fig. 7A).
To determine whether FABP5 inhibition reduces the activity of the mPGES-1 promoter, we examined mPGES-1 promoter activation in A549 cells expressing a reporter wherein luciferase expression is driven by the human mPGES-1 promoter (Fig. 8A) (41). Treatment of cells with IL-1β produced a significant increase in promoter activity (Fig. 8B). In FABP5 knockdown cells, baseline promoter activity was lower than in the corresponding control cells. Treatment of FABP5 knockdown cells with IL-1β produced a small increase in promoter activity (Fig. 8B), which was significantly lower than observed in IL-1β–treated control cells. To determine whether FABP5 regulates mPGES-1 promoter activity via NF-κB–mediated activation of the mPGES-1 promoter, we deleted the NF-κB–binding sites within the mPGES-1 promoter as described (Fig. 8A) (41). This promoter variant (termed ΔNF-κB) retains binding sites for the Egr-1 transcription factor that can induce mPGES-1 expression (41, 42). Compared with the WT promoter, the baseline activity of the ΔNF-κB promoter was significantly lower in both control and FABP5 shRNA cells and showed activity levels similar to the WT promoter when expressed in FABP5 shRNA cells (Fig. 8C). In control and FABP5 knockdown cells, addition of IL-1β produced small but significant increases in promoter activity that were comparable in magnitude (Fig. 8D), indicating that FABP5 knockdown does not affect the activity of the mPGES-1 promoter lacking the NF-κB–binding sites.
Figure 8.

FABP5 modulates mPGES-1 promoter activity in A549 cells. A, schematic of the mPGES-1 promoter luciferase constructs, which contain the human mPGES-1 promoter upstream of luciferase. The ΔNF-κB construct lacks base pairs −631 to −177 that contain the NF-κB–binding sites. B, WT mPGES-1 promoter activity in control and FABP5 shRNA A549 cells treated with 1 ng/ml IL-1β or vehicle for 6 h. Promoter activity is represented as luciferase/β-galactosidase and is normalized to control A549 cells. *, p < 0.05; ***, p < 0.001. ##, p < 0.01 versus control cells (n = 6). C, baseline promoter activity in control and FABP5 shRNA–expressing A549 cells transfected with the WT or ΔNF-κB constructs. **, p < 0.01 versus control cells expressing the WT construct (n = 6). D, promoter activity in control and FABP5 shRNA A549 cells expressing the ΔNF-κB promoter construct. The cells were treated with 1 ng/ml IL-1β or vehicle for 6 h, and luciferase and β-galactosidase activities were quantified. Data are normalized to control cells. *, p < 0.05 (n = 6). Error bars represent S.E.
Discussion
This study identifies FABP5 as a novel modulator of inflammatory mPGES-1 induction and PGE2 biosynthesis, thus positioning FABP5 inhibitors as potential anti-inflammatory and analgesic drugs (15, 16). Although FABP4 and FABP5 are coexpressed in innate immune cells and FABP4 inhibition can reduce the inflammatory output of macrophages (21, 23, 43), our data demonstrate that the modulation of inflammatory PGE2 biosynthesis is restricted to FABP5. Inflammation induces the release of cytokines and PGE2, which sensitize nociceptors and lead to the development of hyperalgesia (24). Consequently, inhibition of COX-1/2 or mPGES-1 and blocking proinflammatory cytokines produce analgesic effects (24, 27, 44). In this study, we demonstrated that FABP5 inhibition reduces tissue PGE2 and proinflammatory cytokine levels as well as paw edema and hyperalgesia after carrageenan administration. Consistent with the antinociceptive effects observed using behavioral approaches, FABP5 KO mice also exhibited attenuated spinal CGRP release, indicative of reduced nociceptor sensitization. This raises the possibility that the antinociceptive effects observed after FABP5 inhibition may stem at least in part from reduced inflammatory output at the site of inflammation, such as via suppressed PGE2 release. We recently demonstrated that FABP5 is abundantly expressed in nociceptors (18), and its inhibition within this site may contribute to the observed analgesic effects that are mediated by cannabinoid and PPARα receptors. The identification and functional characterization of the site(s) wherein FABP5 modulates nociception will require animal models bearing selective disruption of FABP5 in each of these cell populations.
Our data demonstrate that FABP5 regulates inflammatory PGE2 biosynthesis through two independent mechanisms (Fig. 9). In the first, FABP5 serves as an intracellular carrier for arachidonic acid, and we hypothesized that FABP5 may transport arachidonic acid to COX enzymes, which reside within the endoplasmic reticulum of cells (45, 46). Indeed, our data demonstrate that FABP5 inhibition in activated THP-1 cells reduced PGE2 biosynthesis, suggesting that arachidonic acid transport may be partially disrupted after FABP5 inhibition. The second and likely more important mechanism stems from our discovery that the induction of mPGES-1 at the site of inflammation is suppressed upon FABP5 deletion. To our surprise, cannabinoid and PPAR receptor activation did not account for the effects of FABP5 inhibition upon mPGES-1 expression. Instead, our data reveal that FABP5 modulates mPGES-1 through at least two complementary mechanisms. Proinflammatory cytokines, such as IL-1β, induce mPGES-1 expression during inflammation (34–36). Pharmacological and genetic FABP5 inhibition reduced tissue IL-1β levels (Fig. 2E), thus likely suppressing its ability to up-regulate mPGES-1. FABP5 can also regulate mPGES-1 expression downstream of IL-1β signaling. Specifically, FABP5 knockdown reduced mPGES-1 induction in cells treated with IL-1β, effects that were mediated via suppressed NF-κB activation of the mPGES-1 promoter. Down-regulation of NF-κB activity following FABP5 inhibition has also been reported in other settings (47, 48), consistent with our data. It is noteworthy that, in addition to mPGES-1, NF-κB up-regulates COX-2 in cells treated with IL-1β (34, 35), and we confirmed that knockdown of FABP5 suppressed COX-2 induction in A549 cells as well. In contrast to these results, FABP5 inhibition did not affect COX-2 expression in carrageenan-injected paws (Fig. 5). This indicates that additional pathways may regulate COX-2 induction and/or distinct cell types may up-regulate its expression in vivo, independently of FABP5.
Figure 9.
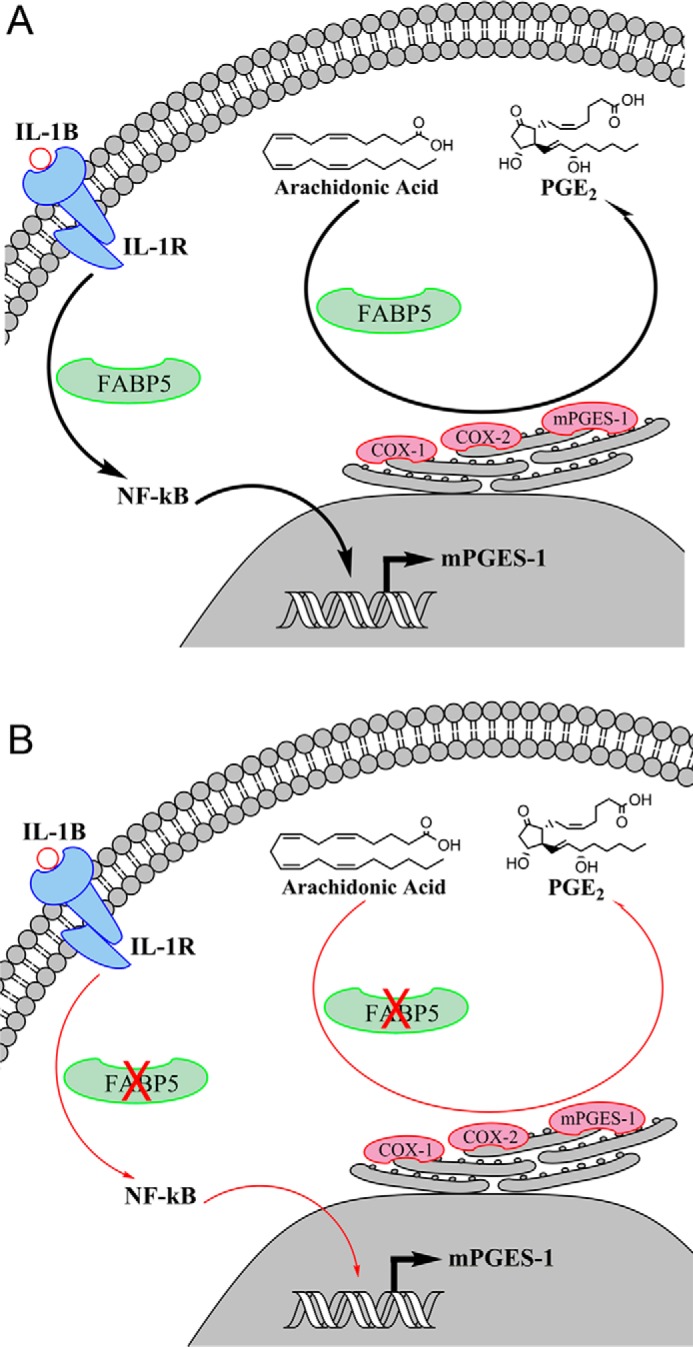
Schematic representation of the pathways through which FABP5 modulates PGE2 biosynthesis during inflammation. A, activation of the interleukin 1 receptor (IL-1R) by IL-1β triggers NF-κB phosphorylation and translocation to the nucleus wherein it binds to the mPGES-1 promoter and initiates mPGES-1 transcription. In parallel, FABP5 may transport arachidonic acid to COX-1/2 and mPGES-1, the main biosynthetic enzymes for PGE2, which reside on the endoplasmic reticulum. B, inhibition of FABP5 blunts NF-κB activation and nuclear translocation, resulting in attenuated induction of mPGES-1. FABP5 inhibition may additionally reduce arachidonic acid delivery to COX-1/2, resulting in suppression of PGE2 biosynthesis.
Carrageenan injection induces an influx of FABP5-expressing macrophages into the site of inflammation (Fig. 3). To our knowledge, the expression of mPGES-1 in paws after an inflammatory challenge has not been previously reported. Our histological results indicate that mPGES-1 is up-regulated predominantly in macrophages and to a lesser extent in the epidermis, which may account for the ability of macrophage- and skin-expressed FABP5 to modulate its expression during inflammation. As noted above, mPGES-1 is the principal enzyme that synthesizes PGE2 (26). Our data indicate that FABP5 deletion reduced paw PGE2 levels by ∼60%, which was accompanied by a near complete suppression of mPGES-1 induction (Figs. 2F and 5). This is consistent with previous reports demonstrating that selective inhibition of mPGES-1 reduces tissue PGE2 levels by ∼70% (49), indicating that other PGE2 biosynthetic enzymes may account for the remainder of PGE2 production.
In conclusion, the present results identify FABP5 as a novel modulator of inflammation and hyperalgesia. FABP5 is required for mPGES-1 induction and nociceptor sensitization during carrageenan-induced inflammation. Thus, in addition to cannabinoid and PPARα receptors (16, 17), suppression of PGE2 biosynthesis may contribute to the antinociceptive effects observed after FABP5 inhibition. These results, coupled with our previous findings that pharmacological FABP5 inhibition produces anti-inflammatory and antinociceptive effects, position FABP5 as an attractive target for the development of novel analgesics.
Experimental procedures
Chemicals
LPS (026:B6), phorbol myristate acetate, and λ-carrageenan were from Sigma; human IL-1β was from R&D Systems; and diclofenac, GW9662, GW6471, AM251, AM630, and arachidonic acid were from Cayman Chemical. SBFI26 was synthesized as described previously (18).
Constructs
The FABP5 shRNA, empty vector, and β-galactosidase constructs were described previously (10). The mPGES-1 promoter construct contains 631 base pairs of the mPGES-1 promoter inserted upstream of luciferase using XhoI and HindIII as described (41). To generate the ΔNF-κB construct lacking the NF-κB–binding sites (Δ−631 to −177), the mPGES-1-Luc construct was digested with SacI and religated as described (41). The NF-κB luciferase reporter construct was kindly provided by Dr. Laurie Krug (Stony Brook University) and was described previously (50). All constructs were validated by sequencing.
Cells
THP-1 and A549 cells were obtained from ATCC. THP-1 cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum and 20 μm β-mercaptoethanol. A549 cells were cultured in DMEM supplemented with 10% fetal bovine serum and 2 mm l-glutamine. HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin/streptomycin, 2 mm l-glutamine, and 1 mm sodium pyruvate in a humidified incubator containing 95% air and 5% CO2.
Lentiviral transduction
The shRNA targeting FABP5 was described previously (10). To obtain stable knockdown cells, shRNA or control plasmids were packaged into lentiviral particles in HEK293T cells and subsequently utilized to transduce A549 cells. The human FABP5 shRNA clone was cotransfected with third-generation lentiviral packaging plasmids pRSV-REV (catalog number 12253, Addgene, Cambridge, MA), pCMV-VSV-G (catalog number 8454, Addgene), and pCgpV (catalog number 320024, Cell Biolabs Inc., San Diego, CA) using GenJet Plus transfection reagent (SignaGen, Rockville, MD) according to the manufacturer's instructions. Forty-eight hours later, packaged lentivirus was collected and used to transduce A549 cells. Forty-eight hours after infection, 1 μg/ml puromycin (Fisher Scientific) was added to the culture medium to select for puromycin-resistant stably infected cells.
Animals
The experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Stony Brook University Institutional Animal Care and Use Committee (protocol number 277150). This study used male C57Bl/6 mice. FABP4 KO and FABP5 KO mice were kindly provided by Dr. Gokhan Hotamisligil. The mice were maintained on a 12:12-h light/dark cycle with ad libitum access to food and water.
Inflammation and hyperalgesia measurement
Baseline thermal withdrawal latencies were obtained using a Hargreaves plantar apparatus as described previously (15–18). Mice received an intraplantar injection of 1% λ-carrageenan into the hind paw and saline into the contralateral paw, and thermal withdrawal latencies were measured 4 h later. Edema was measured using a plethysmometer (Ugo Basile) in which fluid displacement by the hind paw before and after inflammation was measured. Edema is reported as the change in fluid displacement from baseline to the 4-h time point for the carrageenan-injected paw minus the change in fluid displacement for the saline-injected paw.
Histology
The protocol for immunofluorescence has been described recently (18). Briefly, mice were anesthetized and transcardially perfused first with saline and then with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Paws were immersed in fixative overnight at 4 °C and then transferred to 30% sucrose in 0.1 m phosphate buffer. Tissues were embedded and frozen using a liquid nitrogen–chilled isopentane bath. Cryostat sections 20 μm thick were thaw-mounted on slides, dried, and stored at −20 °C. For immunoprocessing, slides were thawed, immersed in 4% paraformaldehyde for 5 min, and rinsed in PBS three times. Following a 30-min block consisting of 10% normal donkey serum (Jackson ImmunoResearch Laboratories, catalog number 017-000-121) in PBS, slides were incubated for 24–48 h at 4 °C in a mixture of primary antibodies diluted in PBS containing 0.1% Tween 20 and 1% bovine serum albumin. Primary antisera used were rabbit anti-FABP5 (BioVendor, catalog number RD181060100) diluted 1:600, rabbit anti-mPGES-1 (Cayman Chemical, catalog number 160140) diluted 1:250, and rat anti-F4/80 (Abcam, catalog number ab6640) diluted 1:100. Following secondary antibody incubations, the slides were washed with PBS and immediately mounted with ProLong® Gold antifade mounting medium with DAPI (Life Technologies, catalog number P36931). Fluorescent immunoreactivity was observed with a Zeiss Axioplan 2 epifluorescence microscope. Images were captured using a Zeiss AxioCam HRm monochrome digital camera and AxioVision release 4.6 software. Images were only adjusted for brightness and contrast.
Western blotting
Western blotting was performed as described previously (18). The following antibodies were used: mPGES-1 (Abcam, catalog number ab180589), mPGES-1 (Cayman Chemical, catalog number 160140), COX-2 (Abcam, catalog number ab15191), FABP5 (BioVendor, catalog number RD181060100), NF-κB (Cell Signaling Technology, catalog number 8242), phospho-NF-κB (Cell Signaling Technology, catalog number 3033), GAPDH (Cell Signaling Technology, catalog number 5174), and histone H3 (Cell Signaling Technology, catalog number 4499). The following secondary antibody was used: goat anti-rabbit HRP (Life Technologies, catalog number A16104). The blots were imaged using a C-DiGit scanner, and band intensities were quantified. Data are reported as a ratio of target band intensity/GAPDH or histone H3 intensities.
CGRP release
Mice received an intraplantar injection of 1% λ-carrageenan into the hind paw and saline into the contralateral paw. Four hours later the mice were euthanized, and the lumbar spinal column was removed and submerged into cold EC1a solution (10 mm HEPES, 13 mm glucose, 151 mm NaCl, 2.5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, pH 7.4). Each hemisphere of the vertebrae was cut to expose the spinal cord. The lumbar enlargement was then cut out and bisected along the midline. The contralateral and ipsilateral spinal sections were subsequently incubated in EC1a buffer for 30 min at 37 °C to obtain baseline CGRP release. The sections were incubated with EC1a buffer containing 1 μm capsaicin for 30 min at 37 °C, and the media were collected. CGRP levels in the media were quantified using the CGRP ELISA kit (Betrin Pharma) according to the manufacturer's instructions.
Biochemical assays and cell fractionation
For the THP1 experiments, the cells were differentiated into macrophages by incubation with 100 ng/ml phorbol myristate acetate for 72 h followed by activation with 5 μg/ml LPS for 24 h. The media were subsequently removed, and the cells were incubated for 2 h in the presence or absence of SBFI26 or diclofenac. The media were subsequently collected, and PGE2 levels were quantified using the PGE2 monoclonal ELISA kit (Cayman Chemical) according to the manufacturer's instructions. To quantify PGE2 levels in paws, the paws were homogenized in ice-cold PBS, and tissue lysates were generated and used for quantification. COX-1 and COX-2 inhibition by SBFI26 or diclofenac was examined using COX inhibitor screening kits (Cayman Chemical). THP-1 membranes were isolated as we described previously (51). Fractionation of cells and isolation of nuclei and cytoplasmic fractions were performed using the NE-PER Nuclear and Cytoplasmic Extraction Reagent (Pierce) according to the manufacturer's instructions.
Promoter activity assays
For mPGES-1 promoter assays, A549 cells were cotransfected with a plasmid expressing luciferase under the control of the mPGES-1 promoter and a plasmid expressing β-galactosidase to control for transfection efficiency. To measure NF-κB activity, cells were transfected with the NK-kB luciferase plasmid alongside β-galactosidase. Twenty-four hours later, the cells were incubated with vehicle or 1 ng/ml IL-1β for 6 h in DMEM containing 0.1% fetal bovine serum. The cells were rinsed with PBS and lysed with Glo Lysis Buffer (Promega, catalog number E2661). The cells were subsequently incubated with luciferase substrate (Bright-Glo Luciferase Assay System, Promega, catalog number E2610), and luminescence was quantified using a F5 Filtermax multimode microplate reader (Molecular Devices, Sunnyvale, CA). β-Galactosidase 2× activity buffer was then added to all wells (β-Galactosidase Assay System, Promega, catalog number E2000). After a 30-min incubation at 37 °C, 1 m sodium carbonate was added to the wells, and absorbance was measured at 405 nm. Background signals in untransfected cells were subtracted from all samples. Luciferase activity is reported as a ratio of luminescence/β-galactosidase for each sample.
Statistical analysis
Data are presented as mean ± S.E. Student's t test or one-way or two-way analysis of variance followed by Dunnett or Tukey post hoc test was used to determine significances of differences between means as appropriate.
Author contributions
D. B. data curation; D. B., J. F., M. P., and M. K. formal analysis; D. B., M. P., and M. K. supervision; D. B., J. F., M. P. K., S. H. P., G. C., K. S., M. G., Y. L., M. W. E., N. S., and M. K. investigation; D. B., M. P. K., K. S., M. P., and M. K. methodology; D. B., J. F., I. O., M. P., and M. K. writing-review and editing; M. P. K., K. S., and M. G. visualization; S. Y. and I. O. resources; M. P. and M. K. conceptualization; M. P. and M. K. project administration; M. K. writing-original draft.
Acknowledgments
We thank Liqun Wang for help with molecular biology and members of our laboratories for helpful discussions. We also thank Sabine Grosch for providing the mPGES-1 luciferase construct.
This work was supported by National Institutes of Health Grants R01 DA035949 and DA035923 (to M. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NSAID
- nonsteroidal anti-inflammatory drug
- FABP
- fatty acid–binding protein
- PGE2
- prostaglandin E2
- mPGES
- microsomal prostaglandin E synthase
- COX
- cyclooxygenase
- CGRP
- calcitonin gene–related peptide
- PPAR
- peroxisome proliferator–activated receptor
- TRPV1
- transient receptor potential vanilloid-1.
References
- 1. Patel K. V., Guralnik J. M., Dansie E. J., and Turk D. C. (2013) Prevalence and impact of pain among older adults in the United States: findings from the 2011 National Health and Aging Trends Study. Pain 154, 2649–2657 10.1016/j.pain.2013.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johannes C. B., Le T. K., Zhou X., Johnston J. A., and Dworkin R. H. (2010) The prevalence of chronic pain in United States adults: results of an Internet-based survey. J. Pain 11, 1230–1239 10.1016/j.jpain.2010.07.002 [DOI] [PubMed] [Google Scholar]
- 3. Gaskin D. J., and Richard P. (2012) The economic costs of pain in the United States. J. Pain 13, 715–724 10.1016/j.jpain.2012.03.009 [DOI] [PubMed] [Google Scholar]
- 4. Bally M., Dendukuri N., Rich B., Nadeau L., Helin-Salmivaara A., Garbe E., and Brophy J. M. (2017) Risk of acute myocardial infarction with NSAIDs in real world use: Bayesian meta-analysis of individual patient data. BMJ 357, j1909 10.1136/bmj.j1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sostres C., Gargallo C. J., and Lanas A. (2013) Nonsteroidal anti-inflammatory drugs and upper and lower gastrointestinal mucosal damage. Arthritis Res. Ther. 15, Suppl. 3, S3 10.1186/ar4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Campbell G., Nielsen S., Larance B., Bruno R., Mattick R., Hall W., Lintzeris N., Cohen M., Smith K., and Degenhardt L. (2015) Pharmaceutical opioid use and dependence among people living with chronic pain: associations observed within the pain and opioids in treatment (POINT) cohort. Pain Med. 16, 1745–1758 10.1111/pme.12773 [DOI] [PubMed] [Google Scholar]
- 7. Vowles K. E., McEntee M. L., Julnes P. S., Frohe T., Ney J. P., and van der Goes D. N. (2015) Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 156, 569–576 10.1097/01.j.pain.0000460357.01998.f1 [DOI] [PubMed] [Google Scholar]
- 8. Palmer R. E., Carrell D. S., Cronkite D., Saunders K., Gross D. E., Masters E., Donevan S., Hylan T. R., and Von Kroff M. (2015) The prevalence of problem opioid use in patients receiving chronic opioid therapy: computer-assisted review of electronic health record clinical notes. Pain 156, 1208–1214 10.1097/j.pain.0000000000000145 [DOI] [PubMed] [Google Scholar]
- 9. Kaczocha M., Glaser S. T., and Deutsch D. G. (2009) Identification of intracellular carriers for the endocannabinoid anandamide. Proc. Natl. Acad. Sci. U.S.A. 106, 6375–6380 10.1073/pnas.0901515106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaczocha M., Vivieca S., Sun J., Glaser S. T., and Deutsch D. G. (2012) Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J. Biol. Chem. 287, 3415–3424 10.1074/jbc.M111.304907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Furuhashi M., and Hotamisligil G. S. (2008) Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 7, 489–503 10.1038/nrd2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Helledie T., Antonius M., Sorensen R. V., Hertzel A. V., Bernlohr D. A., Kølvraa S., Kristiansen K., and Mandrup S. (2000) Lipid-binding proteins modulate ligand-dependent trans-activation by peroxisome proliferator-activated receptors and localize to the nucleus as well as the cytoplasm. J. Lipid Res. 41, 1740–1751 [PubMed] [Google Scholar]
- 13. Schug T. T., Berry D. C., Shaw N. S., Travis S. N., and Noy N. (2007) Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 129, 723–733 10.1016/j.cell.2007.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan N. S., Shaw N. S., Vinckenbosch N., Liu P., Yasmin R., Desvergne B., Wahli W., and Noy N. (2002) Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 22, 5114–5127 10.1128/MCB.22.14.5114-5127.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berger W. T., Ralph B. P., Kaczocha M., Sun J., Balius T. E., Rizzo R. C., Haj-Dahmane S., Ojima I., and Deutsch D. G. (2012) Targeting fatty acid binding protein (FABP) anandamide transporters: a novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS One 7, e50968 10.1371/journal.pone.0050968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaczocha M., Rebecchi M. J., Ralph B. P., Teng Y. H., Berger W. T., Galbavy W., Elmes M. W., Glaser S. T., Wang L., Rizzo R. C., Deutsch D. G., and Ojima I. (2014) Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS One 9, e94200 10.1371/journal.pone.0094200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaczocha M., Glaser S. T., Maher T., Clavin B., Hamilton J., O'Rourke J., Rebecchi M., Puopolo M., Owada Y., and Thanos P. K. (2015) Fatty acid binding protein deletion suppresses inflammatory pain through endocannabinoid/N-acylethanolamine-dependent mechanisms. Mol. Pain 11, 52 10.1186/s12990-015-0056-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peng X., Studholme K., Kanjiya M. P., Luk J., Bogdan D., Elmes M. W., Carbonetti G., Tong S., Gary Teng Y. H., Rizzo R. C., Li H., Deutsch D. G., Ojima I., Rebecchi M. J., Puopolo M., et al. (2017) Fatty-acid-binding protein inhibition produces analgesic effects through peripheral and central mechanisms. Mol. Pain 13, 1744806917697007 10.1177/1744806917697007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Todd A. J. (2010) Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 11, 823–836 10.1038/nrn2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Latremoliere A., and Woolf C. J. (2009) Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain 10, 895–926 10.1016/j.jpain.2009.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reynolds J. M., Liu Q., Brittingham K. C., Liu Y., Gruenthal M., Gorgun C. Z., Hotamisligil G. S., Stout R. D., and Suttles J. (2007) Deficiency of fatty acid-binding proteins in mice confers protection from development of experimental autoimmune encephalomyelitis. J. Immunol. 179, 313–321 10.4049/jimmunol.179.1.313 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y., Li Q., Rao E., Sun Y., Grossmann M. E., Morris R. J., Cleary M. P., and Li B. (2015) Epidermal fatty acid binding protein promotes skin inflammation induced by high-fat diet. Immunity 42, 953–964 10.1016/j.immuni.2015.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hui X., Li H., Zhou Z., Lam K. S., Xiao Y., Wu D., Ding K., Wang Y., Vanhoutte P. M., and Xu A. (2010) Adipocyte fatty acid-binding protein modulates inflammatory responses in macrophages through a positive feedback loop involving c-Jun NH2-terminal kinases and activator protein-1. J. Biol. Chem. 285, 10273–10280 10.1074/jbc.M109.097907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Basbaum A. I., Bautista D. M., Scherrer G., and Julius D. (2009) Cellular and molecular mechanisms of pain. Cell 139, 267–284 10.1016/j.cell.2009.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen L., Yang G., and Grosser T. (2013) Prostanoids and inflammatory pain. Prostaglandins Other Lipid Mediat. 104–105, 58–66 10.1016/j.prostaglandins.2012.08.006 [DOI] [PubMed] [Google Scholar]
- 26. Guay J., Bateman K., Gordon R., Mancini J., and Riendeau D. (2004) Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J. Biol. Chem. 279, 24866–24872 10.1074/jbc.M403106200 [DOI] [PubMed] [Google Scholar]
- 27. Trebino C. E., Stock J. L., Gibbons C. P., Naiman B. M., Wachtmann T. S., Umland J. P., Pandher K., Lapointe J. M., Saha S., Roach M. L., Carter D., Thomas N. A., Durtschi B. A., McNeish J. D., Hambor J. E., et al. . P. (2003) Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. U.S.A. 100, 9044–9049 10.1073/pnas.1332766100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garry M. G., and Hargreaves K. M. (1992) Enhanced release of immunoreactive CGRP and substance P from spinal dorsal horn slices occurs during carrageenan inflammation. Brain Res. 582, 139–142 10.1016/0006-8993(92)90328-7 [DOI] [PubMed] [Google Scholar]
- 29. Schou W. S., Ashina S., Amin F. M., Goadsby P. J., and Ashina M. (2017) Calcitonin gene-related peptide and pain: a systematic review. J. Headache Pain 18, 34 10.1186/s10194-017-0741-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caterina M. J., Leffler A., Malmberg A. B., Martin W. J., Trafton J., Petersen-Zeitz K. R., Koltzenburg M., Basbaum A. I., and Julius D. (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313 10.1126/science.288.5464.306 [DOI] [PubMed] [Google Scholar]
- 31. Davis J. B., Gray J., Gunthorpe M. J., Hatcher J. P., Davey P. T., Overend P., Harries M. H., Latcham J., Clapham C., Atkinson K., Hughes S. A., Rance K., Grau E., Harper A. J., Pugh P. L., et al. (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187 10.1038/35012076 [DOI] [PubMed] [Google Scholar]
- 32. Zhang J. M., and An J. (2007) Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 45, 27–37 10.1097/AIA.0b013e318034194e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haraguchi K., Kawamoto A., Isami K., Maeda S., Kusano A., Asakura K., Shirakawa H., Mori Y., Nakagawa T., and Kaneko S. (2012) TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci. 32, 3931–3941 10.1523/JNEUROSCI.4703-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Catley M. C., Chivers J. E., Cambridge L. M., Holden N., Slater D. M., Staples K. J., Bergmann M. W., Loser P., Barnes P. J., and Newton R. (2003) IL-1β-dependent activation of NF-κB mediates PGE2 release via the expression of cyclooxygenase-2 and microsomal prostaglandin E synthase. FEBS Lett. 547, 75–79 10.1016/S0014-5793(03)00672-0 [DOI] [PubMed] [Google Scholar]
- 35. Han R., Tsui S., and Smith T. J. (2002) Up-regulation of prostaglandin E2 synthesis by interleukin-1β in human orbital fibroblasts involves coordinate induction of prostaglandin-endoperoxide H synthase-2 and glutathione-dependent prostaglandin E2 synthase expression. J. Biol. Chem. 277, 16355–16364 10.1074/jbc.M111246200 [DOI] [PubMed] [Google Scholar]
- 36. Wobst I., Schiffmann S., Birod K., Maier T. J., Schmidt R., Angioni C., Geisslinger G., and Grösch S. (2008) Dimethylcelecoxib inhibits prostaglandin E2 production. Biochem. Pharmacol. 76, 62–69 10.1016/j.bcp.2008.04.008 [DOI] [PubMed] [Google Scholar]
- 37. Kane C. D., Coe N. R., Vanlandingham B., Krieg P., and Bernlohr D. A. (1996) Expression, purification, and ligand-binding analysis of recombinant keratinocyte lipid-binding protein (MAL-1), an intracellular lipid-binding found overexpressed in neoplastic skin cells. Biochemistry 35, 2894–2900 10.1021/bi952476e [DOI] [PubMed] [Google Scholar]
- 38. Thul P. J., Åkesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H., Alm T., Asplund A., Björk L., Breckels L. M., Bäckström A., Danielsson F., Fagerberg L., Fall J., Gatto L., et al. (2017) A subcellular map of the human proteome. Science 356, eaal3321 10.1126/science.aal3321 [DOI] [PubMed] [Google Scholar]
- 39. Noma T., Takahashi-Yanaga F., Arioka M., Mori Y., and Sasaguri T. (2016) Inhibition of GSK-3 reduces prostaglandin E2 production by decreasing the expression levels of COX-2 and mPGES-1 in monocyte/macrophage lineage cells. Biochem. Pharmacol. 116, 120–129 10.1016/j.bcp.2016.07.014 [DOI] [PubMed] [Google Scholar]
- 40. Mendez M., and LaPointe M. C. (2003) PPARγ inhibition of cyclooxygenase-2, PGE2 synthase, and inducible nitric oxide synthase in cardiac myocytes. Hypertension 42, 844–850 10.1161/01.HYP.0000085332.69777.D1 [DOI] [PubMed] [Google Scholar]
- 41. Deckmann K., Rörsch F., Steri R., Schubert-Zsilavecz M., Geisslinger G., and Grösch S. (2010) Dimethylcelecoxib inhibits mPGES-1 promoter activity by influencing EGR1 and NF-κB. Biochem. Pharmacol. 80, 1365–1372 10.1016/j.bcp.2010.07.032 [DOI] [PubMed] [Google Scholar]
- 42. Naraba H., Yokoyama C., Tago N., Murakami M., Kudo I., Fueki M., Oh-Ishi S., and Tanabe T. (2002) Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J. Biol. Chem. 277, 28601–28608 10.1074/jbc.M203618200 [DOI] [PubMed] [Google Scholar]
- 43. Makowski L., Brittingham K. C., Reynolds J. M., Suttles J., and Hotamisligil G. S. (2005) The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IκB kinase activities. J. Biol. Chem. 280, 12888–12895 10.1074/jbc.M413788200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miller R. E., Miller R. J., and Malfait A. M. (2014) Osteoarthritis joint pain: the cytokine connection. Cytokine 70, 185–193 10.1016/j.cyto.2014.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Otto J. C., DeWitt D. L., and Smith W. L. (1993) N-Glycosylation of prostaglandin endoperoxide synthases-1 and -2 and their orientations in the endoplasmic reticulum. J. Biol. Chem. 268, 18234–18242 [PubMed] [Google Scholar]
- 46. Otto J. C., and Smith W. L. (1994) The orientation of prostaglandin endoperoxide synthases-1 and -2 in the endoplasmic reticulum. J. Biol. Chem. 269, 19868–19875 [PubMed] [Google Scholar]
- 47. Dallaglio K., Marconi A., Truzzi F., Lotti R., Palazzo E., Petrachi T., Saltari A., Coppini M., and Pincelli C. (2013) E-FABP induces differentiation in normal human keratinocytes and modulates the differentiation process in psoriatic keratinocytes in vitro. Exp. Dermatol. 22, 255–261 10.1111/exd.12111 [DOI] [PubMed] [Google Scholar]
- 48. Ogawa E., Owada Y., Ikawa S., Adachi Y., Egawa T., Nemoto K., Suzuki K., Hishinuma T., Kawashima H., Kondo H., Muto M., Aiba S., and Okuyama R. (2011) Epidermal FABP (FABP5) regulates keratinocyte differentiation by 13(S)-HODE-mediated activation of the NF-κB signaling pathway. J. Invest Dermatol 131, 604–612 10.1038/jid.2010.342 [DOI] [PubMed] [Google Scholar]
- 49. Leclerc P., Idborg H., Spahiu L., Larsson C., Nekhotiaeva N., Wannberg J., Stenberg P., Korotkova M., and Jakobsson P. J. (2013) Characterization of a human and murine mPGES-1 inhibitor and comparison to mPGES-1 genetic deletion in mouse models of inflammation. Prostaglandins Other Lipid Mediat. 107, 26–34 10.1016/j.prostaglandins.2013.09.001 [DOI] [PubMed] [Google Scholar]
- 50. Krug L. T., Moser J. M., Dickerson S. M., and Speck S. H. (2007) Inhibition of NF-κB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 3, e11 10.1371/journal.ppat.0030011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaczocha M., Glaser S. T., Chae J., Brown D. A., and Deutsch D. G. (2010) Lipid droplets are novel sites of N-acylethanolamine inactivation by fatty acid amide hydrolase-2. J. Biol. Chem. 285, 2796–2806 10.1074/jbc.M109.058461 [DOI] [PMC free article] [PubMed] [Google Scholar]