

Abstract
Background/Aim: Epithelial-mesenchymal transition (EMT) and cancer stem cells (CSC) are critically implicated in cancer metastasis and chemoresistance. Herein, we investigated integrin-linked kinase (ILK)’s role in human colon cancer (CRC) progression and chemoresistance in relation to EMT and CSC markers. Patients and Methods: Expression of ILK, EMT and CSC markers were evaluated by immunohistochemistry in 149 CRC samples. We also generated colon cancer cells resistant to 5-FU and oxaliplatin and studied the effect of ILK inhibition on drug response by MTT assay and on EMT and CSC markers’ expression. Results: ILK expression in human CRC correlates with EMT and CSC markers and is associated with metastasis and chemoresistance. ILK inhibition increases sensitivity of resistant cells to 5-FU and oxaliplatin and reduces the levels of EMT and CSC markers in 5-FU resistant cells. Conclusion: ILK overexpression in human CRC associates with EMT and CSC traits, contributing to tumor progression and chemoresistance.
Keywords: ILK, EMT, chemotherapy, chemoresistance, colorectal cancer, cancer stem cells
Colorectal cancer (CRC) is a leading cause of cancer-related mortality worldwide. Approximately 20% of patients have metastatic disease at the time of diagnosis. (1,2). 5-Fluorouracil (5-FU) based chemotherapy is the standard treatment for metastatic CRC (2). However, despite the treatment advances in recent years many patients do not respond to chemotherapy and their disease progresses, resulting in low overall survival rates (2).
Although several mechanisms are involved in cancer progression and chemotherapy resistance, recent evidence point to epithelial-mesenchymal transition (EMT) and cancer stem cells (CSCs) as having significant roles in these processes (3-6). EMT is a developmental program usurped by cancer cells in order to gain the ability to invade and metastasize (7) During EMT, epithelial cells undergo morphogenetic changes characterized by loss of cell-cell adhesion and apical-basolateral polarity, resulting in transition to migratory mesenchymal cells with invasive properties (7,8). EMT inducers, including Wnt/β-catenin and integrin signaling, trigger complex intracellular pathways converging to activation of transcription factors such as Snail, and ZEB; these act as master EMT regulators repressing E-cadherin expression and inducing expression of mesenchymal genes (8,9).
Intriguingly, EMT has been shown to not only drive tumor invasion and metastasis of carcinomas, but also generate cells with cancer stem cell (CSC) properties (10,11). An emerging body of evidence suggests that cancer initiation, progression and recurrence are mediated by a small population of tumor cells within tumors that harbor stem cell properties i.e. the ability to self-renew, differentiate and resist chemotherapy (12,13). Cells with CSC properties have been isolated from colorectal carcinomas based on markers such as CD44, Lgr5 and CD133 (14-18). Several studies thereafter have linked these CSC markers in human CRC with metastasis, chemotherapy resistance and poor prognosis (14,19-22). Therefore, targeting EMT signaling or CSC may provide a new path to CRC treatment (4,5,23).
Integrin-linked kinase (ILK) is a widely expressed serine/threonine protein kinase located in focal adhesions and plays a central role as a multifunctional effector of growth factor signalling and cell-matrix interactions (24,25). It has been reported to be critically involved in human carcinogenesis and represents a novel anti-cancer therapeutic target (24,25). ILK’s oncogenic functions are mediated by regulation of pathways involved in cell proliferation, survival, cell adhesion-migration and EMT, including the Wnt/β-catenin and PI3K/Akt pathways, as well as, the EMT regulators Snail and E-cadherin (24,25). Previous studies have shown that ILK is critically implicated in colon carcinogenesis and represents an important regulator of the Wnt/β-catenin pathway in this context (26-30). In accordance with these, we have previously demonstrated that ILK is involved in human colorectal cancer progression and correlates with down-regulation of E-cadherin and β-catenin activation (31). Interestingly, ILK has been implicated in mechanisms of drug resistance in cancer cells (32-35).
Considering ILK’s involvement in colon carcinogenesis and the evidence linking ILK to EMT and drug resistance in other cancers we assumed that ILK may significantly contribute to mechanisms of tumor progression and chemotherapy resistance in human CRC (24,25,32-35). In this respect, we evaluated the expression of ILK in relation to EMT and CSC markers, tumor progression and chemotherapy resistance in a series of human CRC specimens. We also generated human colon cancer cells resistant to 5-FU and oxaliplatin and examined the effects of pharmacological inhibition of ILK on drug response and on the expression of EMT and CSC markers.
Materials and Methods
Patient samples. The study included a total of 149 cases of primary CRC from patients that underwent surgery from 1999 to 2013. Formalin-fixed paraffin-embedded (FFPE) tissue samples were retrieved from the archives of the Departments of Pathology, “Agios Andreas” General Hospital and University Hospital of Patras, Greece. Ninety six of these tumors have been included in previous studies (31). Human tissue samples used in the study were paraffin blocks (redundant tissue) from the archives of the Departments of Pathology. The study was performed in accordance with the Helsinki declaration and was approved by the Committee on Research and Ethics and the Scientific Committee of the University Hospital of Patras, Greece. Histopathology of the tumors was revised by expert pathologists (VB, VZ) and clinical data were available from the Oncology Department, University Hospital of Patras, Greece. None of the patients had received chemo or radiotherapy prior to surgery. The mean age of patients was 69 (range=24-92) years; 89 (60%) were men and 60 (40%) women. Patients with metastatic disease (stage IV) (n=31) received 5-fluorouracil (5-FU)–based chemotherapy with FOLFOX (n=12), FOLFOX plus Bevacizumab (n=4), FOLFOX plus panitumumab (n=3), FOLFIRI (n=3), FOLFIRI plus Bevacizumab (n=5), FOLFIRI plus panitumumab or cetuximab (n=4). The overall response rate was evaluated over 4 to 6 courses of treatment according to the Response Evaluation Criteria in Solid Tumors (RECIST version 1.1). Seventeen patients with partial response, complete response or stable disease were defined as responders, whereas 14 patients with progressive disease were defined as non-responders. Clinicopathological characteristics of the tumors are shown in Table I. Staging and grading of tumors were performed according to the American Joint Committee on Cancer Criteria (AJCC)/TNM classification 8th edition and the WHO classification of tumours of the digestive system 4th edition respectively (36,37).
Table I. IHC expression of ILK, β-catenin and E-cadherin in human CRC in relation to clinicopathological parameters.

Statistical analysis were performed by Chi-square or Fisher’s exact test and p<0.05 was considered statistical significant. Depth of invasion (pT), lymph node metastasis (pN) and stage were determined based on AJCC, 8th edition (36).
Immunohistochemistry. A two-step immunoperoxidase method using secondary antibodies conjugated to peroxidase-labeled polymer (Envision detection kit, DAKO, Glostrup, Denmark) and DAB as the chromogen was performed as previously described (31,38). Staining for ILK, β-catenin and E-cadherin in this enriched cohort of tumors was performed as previously described (31). Primary antibodies used were rabbit anti-ILK (1:500, Upstate Biotechnology, Lake Placid, NY, USA), mouse anti-β-catenin (1:1.000, BD Biosciences, San Jose, CA, USA), mouse anti-E-cadherin (1:1000, BD Biosciences, San Jose, CA, USA), rabbit anti-ZEB (1:200, Sigma-Aldrich, St. Louis, MO, USA), mouse anti-Snail (1:40, Acris Antibodies GmbH, Herford, Germany), mouse anti-CD44 (1:80, Novocastra Laboratories Ltd, Newcastle, UK), rabbit anti-Lgr5 (1:2000, Sigma-Aldrich, St. Louis, MO, USA) and mouse anti-CD133 (1:80, EMD Millipore, Billerica, MA, USA). Appropriate negative controls (by omitting the primary antibody) and positive controls (basal cell carcinoma and hepatocellular carcinoma tissue samples) were performed in all cases. Evaluation of immunoreactivity in all cases was performed blinded to the case by an expert pathologist (VB). To obtain comparable results with our previous study, immunostaining for ILK, E-cadherin and β-catenin was scored on a 0-3 scale (0: negative, 1: weak, 2: moderate and 3 strong) as previously described (31), while stains for all other markers were evaluated using Histoscore (H-score) with values ranging from 0-300 as previously described (38).
Cell lines, cell culture and drugs. The human CRC cell lines HT29 and Caco-2 were purchased from the American Type Culture Collection (ATCC, University Boulevard Manassas, VA, USA). The Caco-2 cell line that differentiates spontaneously post-confluence into mature enterocytes was cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and all experiments were performed at sub-confluence (70-80% confluence) and at 0, 6, 10, 15 and 21 days post-confluence (39). HT29 cells were cultured in RPMI 1640 medium with 100 μg/ml penicillin G/streptomycin, 2.5 μg/ml amphotericin B, 50 μg/ml gentamycin and 10% fetal bovine serum (FBS). 5-FU and oxaliplatin were purchased from Teva Pharmaceuticals Hellas S.A. Resistant HT29 cells to chemotherapeutic drugs 5-FU (5-FUR) and oxaliplatin (OxalR) were generated in our lab according to the protocol described herein. Initially, the IC50 of 5-FU or oxaliplatin in parental HT29 cells was determined using MTT assay as described below. The parental HT29 cells were then cultured with 5-FU or oxaliplatin at a concentration equal to 1/5 of the IC50 value for two passages and the new IC50 of 5-FU or oxaliplatin was determined. Surviving cells were then exposed to a dose 2x 1/5 of IC50 for another two passages. The procedure was repeated until the final IC50 value was at least 5-fold greater than the IC50 value of the parental HT29 cells (40). For all experiments stably resistant 5-FUR or OxalR cells were continuously cultured in the presence of 5-FU or oxaliplatin at concentrations of 29 μΜ and 5 μΜ respectively. The specific ILK inhibitor QLT0267 was purchased from Dermira (Menlo Park, CA, USA) and was diluted in DMSO. For ILK inhibition in all indicated experiments HT29 5-FUR cells or OxalR were cultured for 48h with 25 μΜ or 1 μΜ of QLT0267 respectively. Control cells were cultured with DMSO only.
MTT assay. Sensitivity of HT29, 5-FUR and OxalR cells to 5-FU, oxaliplatin and QLT0267 was evaluated by the 3-[4,5-dimethylthiazol-2-yl]-2,5-dimethyltetrazolium bromide (MTT) assay. Briefly, cells were seeded in 24-well plates at a density of 1.5×104 cells/well and treated with a range of concentrations (from 0.1 μΜ to 100 μΜ) of 5-FU, oxaliplatin or QLT0267. Forty-eight hours later, MTT (AppliChem GmbH, Darmstadt, Germany) solution (5 mg/ml in PBS) was added at a 1:10 volume to each well and incubated for 2 h at 37˚C with CO2 levels at 5%. The medium was then removed and dark blue formazan crystals formed by live cells were dissolved by addition of 100 μl acidified isopropanol. The solution was transferred to 96-well plates and absorbance at a wavelength of 570 nm was measured with a microplate reader (Sunrise Tecan's Magellan™ 2, Männedorf, Switzerland) using 620 nm as the reference wavelength. The number of live cells was calculated by plotting growth curves on OriginPro 8 (OriginLab Corporation, Northampton, MA, USA) and using the built-in Dose Response algorithm. Results were expressed in terms of the concentration required to inhibit cell growth by 50% (IC50).
Immunoblotting. Cells were plated at Petri dishes. Protein was harvested from cells plated from 70% to 80% confluence. For ILK inhibition, 5-FUR and OxalR cells were treated with QLT0267 or DMSO as described above. Cells were lysed in lysis buffer (10 mM Tris–HCl pH 8, 140 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton, 1 mM sodium fluoride, 0.1% sodium deoxycholate, 0.5 mM Na-orthovanadate and 1% protease Inhibitor). Protein concentration was determined by the Bradford assay. Proteins were separated using SDS-PAGE on a 10% or 12% gel and transferred to a polyvinylidene difluoride membrane by electroblotting. Membranes were blocked for 2 h in TBS containing 0.05% Tween-20 and 5% non-fat dry milk and incubated overnight with the following primary antibodies: mouse anti ILK (1:1.000, BD Biosciences, San Jose, CA, USA), rabbit anti-Akt (1:1000, Cell Signaling, Leiden, The Netherlands), rabbit anti-p-AKT (1:500, Cell Signaling, Leiden, The Netherlands), mouse active-β-catenin (1:500, EMD Millipore, Billerica, MA, USA), mouse anti-β catenin (1:500, BD Biosciences, San Jose, CA, USA), mouse anti-E-cadherin (1:1,000, BD Biosciences, San Jose, CA, USA), mouse anti-Vimentin (1:500, Novocastra Laboratories Ltd, Newcastle, UK) mouse anti-Snail (1:500, EMD Millipore, Billerica, MA, USA), rabbit anti-Lgr5 (1:2.000, Sigma-Aldrich, St. Louis, MO, USA), mouse anti-villin (1:1,000, BD Biosciences, San Jose, CA, USA) and mouse anti-actin antibody (1:1,000, EMD Millipore, Billerica, MA, USA), or mouse anti-a-tubulin (1:20,000, Sigma-Aldrich, St. Louis, MO, USA) as a loading control for protein quantification. The following day, the membrane was incubated for 1 h with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:3,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The immunoreactive proteins were detected by a chemiluminescent detection system (Pierce, Waltham, MA, USA), according to the manufacturer’s instructions.
Immunoprecipitation and in vitro kinase assay. For protein purification, cells were incubated with lysis buffer (50 mM Hepes, PH 7.5), 150 mM NaCl, 1% NP-40, 0.5% DOC, 5 mM NaF, 1 mM sodium orthovanadate (Na3Vo4), 1 mM PMSF, 10 μg/ml leupeptin and 2.5 μg/ml aprotinin) for 30 min at 4˚C and supernatants were obtained by centrifugation at 13,000 rpm for 20 min at 4˚C. The protein concentration was measured using the Bradford assay. 250 μg of protein were pre-cleared by incubation with non-specific IgG and protein A Sepharose for 30 min at 4˚C and then centrifuged at 3,500 rpm for 2 min at 4˚C. Subsequently, equal supernatant volumes were incubated with 10 μg of mouse anti-ILK for 3 h at 4˚C and then A-sepharose was added followed by incubation for further 10 h. The immunoprecipitate was isolated by centrifugation at 3,500 rpm for 2 min at 4˚, washed twice with lysis buffer and kinase reaction buffer (50 mM Hepes PH 7.0, 10 mM MnCl2, 10 mM MgCl2, 2 mM NaF, 1 mM Na3VO4) and incubated for 20 min at 30˚C with 10 μg of Myelic Basic Protein (MBP) (Upstate Biotechnology, Lake Placid, NY, USA), the substrate of ILK, in kinase reaction buffer containing 10 μCi [γ-32P]ATP. The reaction was terminated by the addition of 2x SDS-PAGE sample buffer. The reaction products were visualized by polyacrylamide gel electrophoresis and autoradiography.
Immunofluorescence. Immunofluorescence was performed in HT29, 5-FUR and 5-FUR cells treated with QLT0267 (5-FUR+QLT) and grown on coverslips. Cells were fixed with Carson’s buffer solution for 10 min at room temperature and blocking was performed by treatment with 10% FBS, 3% BSA in PBS for 1 h at 37˚C, followed by incubation with the following primary antibodies: mouse anti-ILK (1:30, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-E-cadherin (1:1,000, BD Biosciences, San Jose, CA, USA), mouse anti-Vimentin (1:100, Novocastra Laboratories Ltd, Newcastle, UK), mouse anti-Snail (1:50, EMD Millipore, Billerica, MA, USA), rabbit anti-ZEB (1:100, Sigma-Aldrich, St. Louis, MO, USA), mouse anti-CD44 (1:50, Novocastra Laboratories Ltd, Newcastle, UK) and rabbit anti-Lgr5 (1:100, Sigma-Aldrich, St. Louis, MO, USA). Secondary antibodies used were Alexa 488-conjugated goat anti-rabbit IgG and Alexa 546-conjugated goat anti-mouse IgG (1:500, Invitrogen, Carlsbad, CA, USA). Nuclei were stained with Hoechst. For double immunofluorescence experiments in FFPE tissue samples, sections were first deparaffinized and rehydrated and then the same as the above protocol was applied. Primary antibodies used were mouse anti-ILK (1:30, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit anti-ZEB (1:100, Sigma-Aldrich, St. Louis, MO, USA) or rabbit anti-Lgr5 (1:100, Sigma-Aldrich, St. Louis, MO, USA).
Statistical analysis. All statistical analyses were performed with IBM SPSS v24 for Windows (SPSS Inc, Chicago, IL, USA) and p-values <0.05 were considered statistically significant. To test differences between groups of clinicopathological parameters (location, grade, depth of invasion, lymph node metastasis, stage, and response), non-parametric tests (Kruskal Wallis or Mann Whitney tests) for continuous variables and Chi-square or Fisher’s exact test for categorical variables (ILK, β-catenin and E-cadherin) were used. Differences in chemotherapy response and in expression of proteins among treatment subgroups based on the regimens used were evaluated with Fisher’s exact test and non-parametric Kruskal-Wallis test respectively. Correlations between markers’ expression were examined using Spearman’s rho. Prediction of lymph node (LN) metastasis and response to chemotherapy was evaluated by Multivariate Binary logistic regression analysis using the Enter method.
Results
ILK expression in human CRC is associated with tumor progression, EMT and CSC markers. In accordance to our previous study, in this enriched cohort of tumors (n=149), ILK was overexpressed in tumors compared to adjacent non-neoplastic colonic epithelium (28,31). Positive ILK expression with cytoplasmic or cytoplasmic and nuclear expression was found in 135/149 (90.6%) of cases (Figure 1). ILK expression correlated significantly with tumor progression parameters such as grade (Fisher’s exact test p=0.005), depth of invasion (pT) (Fisher’s exact test, p<0.001), lymph node (LN) metastasis (pN) (Fisher’s exact test, p<0.001) and TNM stage (Chi square, p<0.001) (Table I). Further supporting ILK’s implication in colorectal carcinogenesis, both expression and kinase activity of ILK were decreased upon differentiation of the human colon cancer Caco-2 cells to mature enterocytes (data not shown).
Figure 1. Immunohistochemical expression of ILK, EMT and CSC markers in human CRC. Representative cases of human CRC with strong expression of ILK (A), nuclear and cytoplasmic expression of β-catenin (B), loss of membranous expression of E-cadherin (C), strong nuclear expression of ZEB (D), strong expression (mainly cytoplasmic) of Snail (E), membranous expression of CD44 (F), strong expression (mainly cytoplasmic) of Lgr5 (G) and strong expression (mainly cytoplasmic) of CD133 (H). Bars correspond to 50 μm.

We next examined the expression of EMT markers β-catenin, E-cadherin, ZEB and Snail in human CRC. Adjacent non-neoplastic colonic epithelium showed strong membranous expression for E-cadherin and β-catenin and negative expression for EMT master regulators ZEB and Snail. In contrast, tumors showed nuclear expression of β-catenin in 138/149 (92.6%), decreased membranous E-cadherin expression (score <3) in 117/149 (78.5%), positive expression (nuclear) of ZEB in 118/149 (79.2%) with a mean H-score of 42.8±4.2 and positive expression (cytoplasmic and nuclear) of Snail in 80/92 (86.9%) cases with a mean H-score of 72.6±6.9 (Figure 1). This EMT expression profile was stronger in the invasive front of the tumors and tumors buds (data not shown). Significantly lower levels of E-cadherin (Fisher’s exact test, p=0.003) and higher expression levels of nuclear β-catenin (Fisher’s exact test, p=0.021) and ZEB (Mann Whitney p=0.011) were observed in high grade tumors and there was a statistical significant difference in the expression of all EMT markers examined in relation to depth of invasion (pT), LN metastasis (pN) and TNM stage (Tables I and II).
In addition, CSC markers CD44, Lgr5 and CD133 were overexpressed in CRC compared to adjacent non-neoplastic epithelium. In CRC, we found positive (membranous) expression of CD44 in 86/149 (57.7%) cases with a mean H-score of 36.3±6.9, positive (mainly cytoplasmic) expression of Lgr5 in 130/149 (87.2%) cases with a mean H-score of 64.5±5 and positive (mainly cytoplasmic) expression of CD133 in 68/92 (73.9%) cases with a mean H-score of 72.6±6.9 (Figure 1). Significantly higher expression levels of Lgr5 and CD133 were observed in high grade tumors and expression of all CSC markers were significantly higher with increased depth of invasion (pT), presence of LN metastasis (pN) and advanced stage (Table II).
Table II. IHC expression of EMT markers ZEB and Snail and CSC markers CD44, Lgr5 and CD133 in human CRC in relation to clinicopathological parameters.

Statistical analysis were performed by non-parametric tests and p<0.05 was considered statistical significant. Depth of invasion (pT), Lymph node metastasis (pN) and stage were determined based on AJCC, 8th edition (36).
Multivariate analysis also showed that among all the markers examined, ILK (B=1.44, p<0.001, OR=4.22), Snail (B=0.02, p=0.001, OR=1.02) and CD44 (B=0.02, p=0.036, OR=1.02) are independent predictors of lymph node metastasis in human CRC as shown in Table III.
Table III. Prediction of lymph node metastasis by multivariate regression analysis (Overall Model Fit: Chi Square= 56.765, df=5; p<0.001).
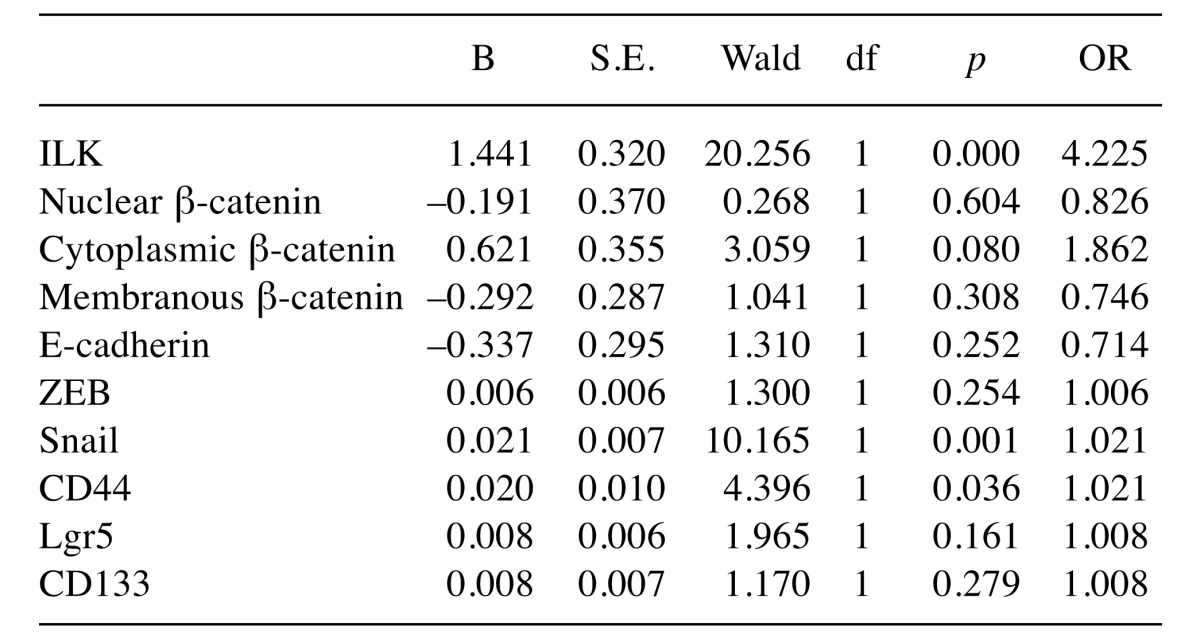
df: Degrees of freedom, p: significance, B: coefficient, S.E.: standard error, OR: odds ratio. p-Values <0.05 are considered significant.
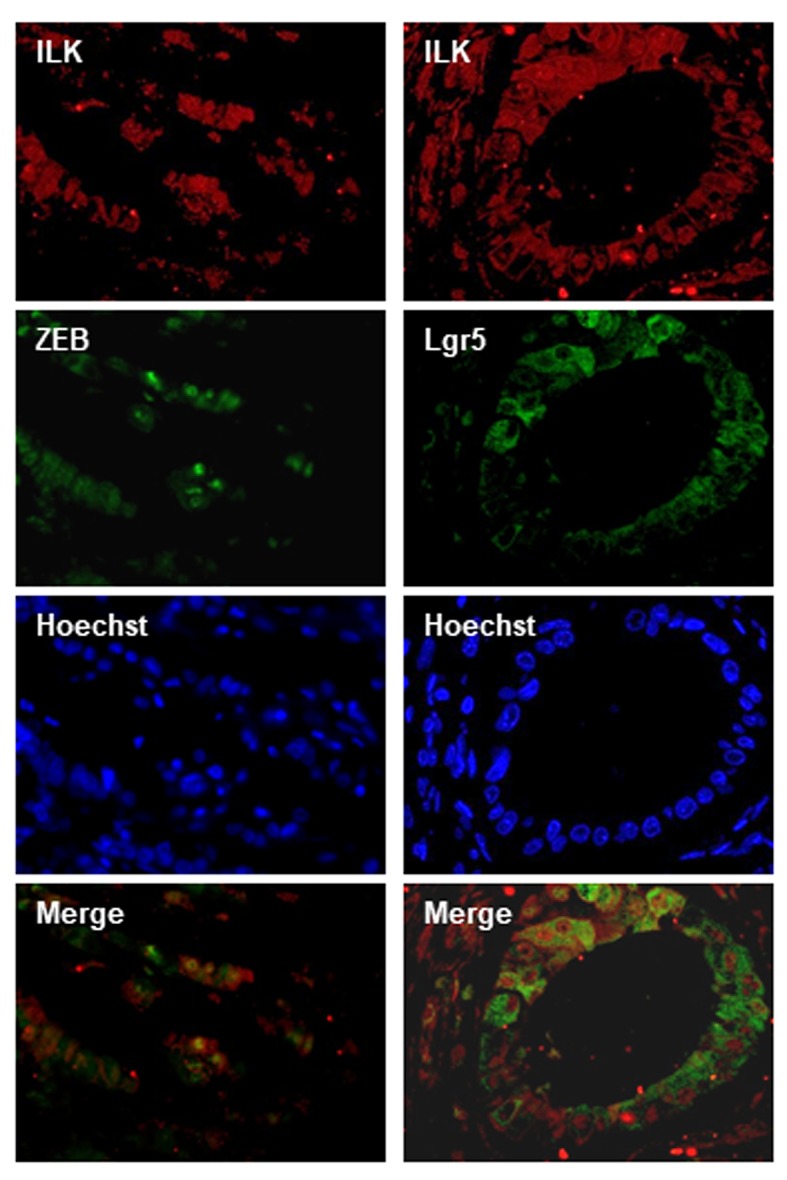
Notably, there was a significant correlation between EMT and CSCs markers as shown in Table IV. Immunohisto-chemical expression levels of ILK in human CRC significantly correlated with all the EMT markers examined including nuclear β-catenin, membranous E-cadherin, as well as ZEB and Snail. In addition, there was a significant positive correlation of ILK expression with the expression of all CSC markers i.e. CD44, Lgr5 and CD133 (Table IV). In line with these, double immunofluorescence experiments in human CRC showed co-localization of ILK with the EMT marker ZEB and the CSC marker Lgr5 in tumor cells (Figure 2).
Table IV. Correlations between expression of ILK, EMT and CSC markers in human CRC.

r: Correlation coefficient, p: significance (2-tailed), N: number. **Correlation is significant at the 0.01 level (2-tailed). *Correlation is significant at the 0.05 level (2-tailed).
Figure 2. ILK co-localizes with ZEB and Lgr5 in human CRC by double immunofluorescence. ILK expression is shown in red, ZEB and Lgr5 expression is shown in green. Nuclei are stained with Hoechst (blue). Merged photos of red and green (ILK-ZEB and ILK-Lgr5) show co-localization in tumor cells.
ILK overexpression in metastatic CRC is associated with resistance to chemotherapy. We next examined the expression of ILK, EMT and CSC markers in metastatic CRC in relation to response to chemotherapy. There was no significant differences in response between different treatment subgroups according to regimens used (Fisher’s exact test, p=0.915) and there was no significant differences in the expression of ILK, EMT and CSC markers between treatment subgroups (data not shown). Expression of ILK in tissue samples from metastatic CRC patients receiving 5-FU based chemotherapy was significantly higher in non-responders compared to responders (Fisher’s exact test p=0.006). Regarding EMT markers, non-responders showed lower levels of E-cadherin (Fisher’s exact test, p=0.048) and higher levels of ZEB (Mann-Whitney, p<0.001) compared to responders. Significantly higher expression of the CSC marker CD44 (Mann-Whitney, p=0.003) was also observed in non-responders to chemotherapy compared to responders. Results are shown in Tables I, II and Figure 3. Most importantly, expression of ILK (B=3.17, p=0.024, OR=23.7) and ZEB (B=0.208, p=0.049, OR=1.2) was shown by multivariate analysis to independently predict poor response of metastatic disease to chemotherapy (Table V).
Figure 3. Expression of ILK, E-cadherin, ZEB and CD44 in metastatic CRC is associated with chemotherapy response. Immunohistochemical expression of ILK (A, B), E-cadherin (C, D), ZEB (E, F) and CD44 (G, H) is shown in representative cases of responders (left column) and nonresponders (right column). Non-responders to chemotherapy show higher expression of ILK (B), ZEB (F) and CD44 (H) and decreased expression of E-cadherin (D) compared to responders. Bars correspond to 50 μm.

Table V. Prediction of response to 5-FU-based chemotherapy by regression analysis (Overall Model Fit: Chi Square= 31.5746; df=5; p<0.001).

df: Degrees of freedom, p: significance, B: coefficient, S.E.: standard error, OR: odds ratio. p-Values <0.05 are considered significant.
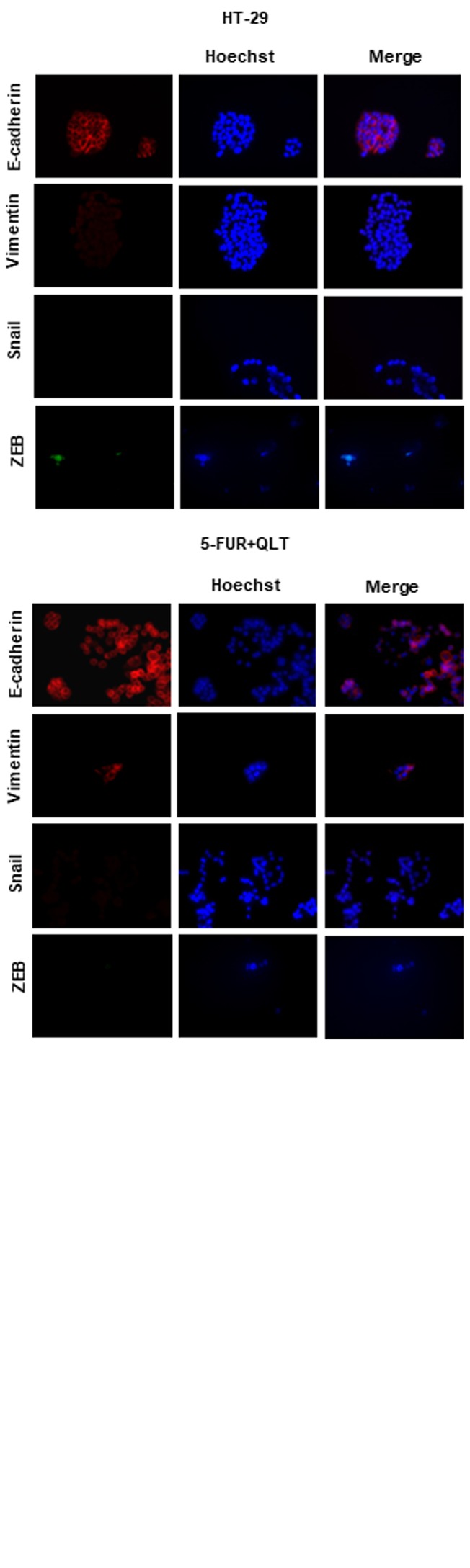
Pharmacologic inhibition of ILK reduces resistance to 5-FU and oxaliplatin and inhibits expression of EMT and CSC markers in 5-FU resistant colon cancer cells. We next generated cells resistant to 5-FU and oxaliplatin as described in materials and methods. The IC50 value of 5-FU in resistant cells (5-FUR) was 26 μΜ, as compared to 4.2 μΜ in the parental HT29 cell line. The IC50 value of oxaliplatin in resistant cells (OxalR) was 5.4 μΜ as compared to 0.9 μΜ in the parental HT29 cell line. The ILK specific inhibitor QLT0267 shows a 3 to 4-fold greater growth inhibition in 5-FUR cells (IC50 25 μΜ) and OxalR cells (IC50 35 μΜ), when compared to parental HT29 cells (IC50 >100 μΜ) as evaluated by the MTT assay (Figure 4A). Moreover, inhibition of ILK reduces acquired resistance to 5-FU and oxaliplatin, as the IC50 value of 5-FU is reduced from 26 μΜ to 10 μΜ and the IC50 value of oxaliplatin from 5.4 μΜ to 1.2 μΜ after pretreatment with QLT0267 for 48 h (Figure 4A). Protein levels of ILK and p-Akt (the main target of ILK kinase activity) are higher in 5FUR cells and OxalR cells compared to the parental HT29 cells by immunoblotting (Figure 4B). Also, consistent with the known effect of this specific ILK inhibitor on the levels of p-Akt, treatment of 5-FUR and OxalR cells with QLT0267 significantly reduces expression of p-Akt in resistant cells (Figure 4B) (33). In accordance with previous studies 5-FUR cells show morphologic changes by light microscopy consistent with EMT, as well as, an EMT and CSC related expression profile by immunoblotting i.e. decreased levels of the epithelial marker E-cadherin, increased expression of active β-catenin and mesenchymal markers vimentin and Snail and increased levels of the CSC marker Lgr5 (Figure 5) (41,42). However similar EMT changes were not observed in OxalR cells (data not shown). When examining the effect of ILK inhibition on EMT and CSC markers in 5-FUR cells, we observed up-regulation of E-cadherin and down-regulation of active β-catenin, vimentin, Snail and Lgr5 protein levels by immunoblotting in 5-FUR cells treated with QLT0267 (Figure 5). An EMT and CSC molecular profile in 5-FUR cells compared with the parental HT-29 and reversal of these changes upon inhibition of ILK with QLT0267 was also confirmed by immunofluorescence analysis (Figure 6).
Figure 4. Pharmacological inhibition of ILK in HT29 cells reduces acquired resistance to 5-FU and oxaliplatin and lowers levels of p-Akt. (A) Results from MTT assays. Logarithmic dose-response (growth) curves, with IC50 values indicated, showing the effect of QLT0267 on parental HT29 and resistant 5-FUR (i) or OxalR cells (ii), as well as the effect of 5-FU (iii) or oxaliplatin (iv) on resistant cells (5FUR and OxalR respectively) pretreated with QLT (5FURQLT and OxalRQLT respectively). QLT0267 results in 3- to 4-fold greater inhibition of cell proliferation of 5FUR (IC50=26 μΜ) (i) and OxalR cells (IC50=34 μΜ) (ii) compared to parental HT29 cells (IC50>100μΜ. Pre-treatment with QLT0267 increases sensitivity to growth inhibition by 5-FU and oxaliplatin. (B) Protein expression by immunoblotting of ILK and p-Akt in HT29 colon cancer cells resistant to 5-FU (5-FUR) and oxaliplatin (OxalR), as well as, in resistant cells treated with ILK inhibitor QLT0267 (5-FUR+QLT and OxalR+QLT). Representative results of three independent experiments are shown.

Figure 5. Pharmacological inhibition of ILK in HT29 cells resistant to 5-FU inhibits expression of EMT and CSC markers. (A) EMT-consistent phenotypic changes upon 5-FU resistance. Spindle-shape morphology and pseudopodia formation are noted in resistant to 5-FU (5-FUR), but not in parental HT29 cells. (B) Protein expression by immunoblotting of EMT markers (β-catenin, E-cadherin, Vimentin and Snail) and CSC markers (Lgr5) in HT29, 5-FUR cells and 5-FUR cells treated with QLT0267 (5-FUR+QLT). 5-FUR cells express higher protein levels of active β-catenin, lower levels of E-cadherin and higher levels of Vimentin, Snail and Lgr5 compared to parental HT29 cells, while QLT0267 treatment (5-FUR+QLT) reverses these effects. Representative results of three independent experiments are shown.

Figure 6. Immunofluorescence analysis of EMT markers in HT29, 5-FUR cells and 5-FUR cells pre-treated with QLT0267 (5-FUR+QLT). Decreased expression of E-cadherin and increased expression of vimentin, Snail and ZEB can be seen in 5-FU resistant cells (5-FUR) compared to HT29 cells while treatment of resistant cells with QLT0267 (5-FUR+QLT) results in increased expression of E-cadherin and decreased expression of Vimentin, Snail and ZEB. Nuclei are stained with Hoechstain. Representative results of three independent experiments are shown.
Discussion
CRC is a major cause of cancer-related morbidity and mortality worldwide (1). Chemotherapy is the standard treatment option for patients with metastatic unresectable disease (1,2). However, resistance to chemotherapeutic drugs prevents successful patient management indicating the need to identify the mechanisms involved. EMT and CSC properties are known to be critically implicated in cancer invasion, metastasis and drug-resistance (4,5,7,13,14). The present study addresses the role of ILK in tumor progression and chemotherapy resistance in human CRC in relation to EMT and CSC markers.
We showed that an EMT signature consisting of nuclear β-catenin, down-regulated E-cadherin and overexpression of ZEB and Snail is associated with invasion, metastasis and chemoresistance in human CRC. We also report for the first time that ZEB expression in our cohort of metastatic CRC is an independent predictor of poor response to chemotherapy and, in agreement with recent studies, we demonstrate that 5-FU resistant HT29 cells show features of EMT (41,42). Consistently, several lines of evidence indicate multiple interactions between E-cadherin, Wnt/β-catenin and the EMT key transcription factors Snail and ZEB inducing an invasive mesenchymal phenotype of epithelial cells in CRC (43-47). In addition, Snail and ZEB expression in human colon cancer specimens has been previously associated with tumor progression and/or poor prognosis (47-49) and only a few in vitro studies implicate these factors in cancer cell resistance to 5-FU (42,50). Our findings suggest that EMT is critically implicated in invasion, metastasis and chemotherapy resistance in human CRC and evaluation of EMT regulators, especially ZEB, in CRC specimens may have significant clinical relevance. However, considering the limitations of this study, such as the relatively small number of our metastatic CRC samples, further evaluation is needed.
Consistent with the significant role of CSCs in tumor progression and drug-resistance in cancer we demonstrate that CSC marker expression in human CRC specimens is associated with depth of tumor invasion, lymph node metastases, disease stage and resistance of metastatic disease to chemotherapy (4,5,12-14). Expression of CD44 was also shown to independently predict lymph node metastasis. In addition, 5-FU resistant colon cancer cells overexpress the CSC marker Lgr5 consistent with previous studies showing that chemoresistance in CRC is associated with stemness features (41,51). This is in agreement with previous studies that correlate CSC marker expression with tumor progression, poor prognosis and therapeutic resistance in human CRC (14,19-22,52). In further agreement with mounting evidence linking EMT to stemness we show a significant correlation between EMT and CSC marker expression in our samples (4,5,10,11,53). Congruently, ZEB and Snail have been shown to regulate stem cell properties of colon cancer cells (46,54,55). However, this is the first study, to the best of our knowledge, correlating a panel of EMT and CSC markers in tissue samples of CRC, supporting the notion that EMT in human CRC in vivo may generate cells with CSC properties promoting tumor progression and chemoresistance.
Interestingly, ILK overexpression in our cohort of human CRC is associated with tumor grade, invasion and metastasis as previously demonstrated and it is also an independent predictor of lymph node metastasis supporting a significant role of ILK in colorectal cancer progression (28,31). This is consistent with several previous studies showing that overexpression of ILK results in loss of cell-cell adhesion, anchorage-independent growth, induction of an invasive phenotype and tumorigenicity in nude mice (24,25). Notably, ILK expression in our samples is significantly associated with all EMT and CSM markers examined and also ILK co-localized with EMT and CSC markers in human colon cancer cells. Correlation of ILK with altered expression of E-cadherin and β-catenin has been previously demonstrated but this is the first report, to the best of our knowledge, linking ILK to Snail, ZEB and CSC markers in human CRC (31). These findings suggest that ILK overexpression may induce an EMT and CSC phenotype in human CRC in vivo and gain of this phenotype may, in part, account for the tumor-promoting role of ILK in CRC. In support of these findings, previous studies have implicated ILK as a critical regulator of EMT in cancer cells (29,56-58). Overexpression of ILK in epithelial cells induces an invasive mesenchymal phenotype concomitant with down-regulation of E-cadherin (29,56). Also, ILK is implicated in TGF-β and thymosin beta4 induced EMT in CRC, while inhibition of ILK in human colon carcinoma cells suppresses the EMT master regulator Snail (26,57,58). ILK is also known to regulate WNT/β-catenin in human CRC (26-30), a pathway with significant roles in proliferation and maintenance of both the normal stem cell compartment at the bottom of the colonic crypts, as well as cancer stem cells within the tumors (44). Moreover, it has been demonstrated that ILK regulates stem-cell features in breast cancer and chronic myeloid leukaemia and is required for the maintenance of intestinal stem cells in Drosophila (59-61).
We also showed that overexpression of ILK in our cohort is associated with poor response of metastatic disease to chemotherapy and along with the EMT marker ZEB, ILK expression is an independent predictor of chemotherapy response in metastatic CRC. Furthermore, we show for the first time that colon cancer cells resistant to 5-FU and oxaliplatin are more sensitive to ILK inhibition in vitro than non-resistant cells and ILK inhibitor QLT0267 increases the response of chemoresistant cells to treatment with 5-FU or oxaliplatin. In agreement with these, ILK has been implicated in chemotherapy resistance in glioma and lung cancer (34,35). Targeting ILK shows synergistic effects with chemotherapeutic drugs in animal models of breast and pancreatic cancer and also reduces multi-drug resistance in human gastric cancer cell lines (32,33,62,63). However, to the best of our knowledge, this is the first study linking ILK overexpression to mechanisms of chemoresistance in human colon cancer.
Further supporting the notion that ILK overexpression may attribute aggressive features and chemoresistance in human colon cancer through EMT and CSC related mechanisms, pharmacological inhibition of ILK in 5-FU resistant HT-29 cells significantly reverses the expression of EMT and CSC markers. Considering the importance of EMT and CSC features in cancer therapy resistance, ILK inhibition may prove of value as an EMT and CSC targeting treatment in the management of CRC. However additional studies are required to validate this hypothesis.
In conclusion, we provide evidence supporting the hypothesis that ILK overexpression in human CRC induces an EMT and CSC phenotype promoting invasion, metastasis and resistance to chemotherapy and targeting ILK may reverse these effects. However, further studies are required to evaluate ILK as a biomarker and therapeutic target in CRC.
Conflicts of Interest
The Authors declare that they have no conflict of interest.
Acknowledgements
The present work was funded by “K. Karatheodori 2015”, research fund, University of Patras and EOGE Oncology Research Fund, Greece.
References
- 1.Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 2.Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson A, Bodoky G, Ciardiello F, D'Hoore A, Diaz-Rubio E, Douillard JY, Ducreux M, Falcone A, Grothey A, Gruenberger T, Haustermans K, Heinemann V, Hoff P, Köhne CH, Labianca R, Laurent-Puig P, Ma B, Maughan T, Muro K, Normanno N, Österlund P, Oyen WJ, Papamichael D, Pentheroudakis G, Pfeiffer P, Price TJ, Punt C, Ricke J, Roth A, Salazar R, Scheithauer W, Schmoll HJ, Tabernero J, Taïeb J, Tejpar S, Wasan H, Yoshino T, Zaanan A, Arnold D. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–1422. doi: 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 3.Wenzel ES, Singh ATK. Cell-cycle checkpoints and aneuploidy on the path to cancer. In Vivo. 2018;32:1–5. doi: 10.21873/invivo.11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato R, Semba T, Saya H, Arima Y. Concise review: stem cells and epithelial-mesenchymal transition in cancer: biological impli-cations and therapeutic targets. Stem Cells. 2016;34:1997–2007. doi: 10.1002/stem.2406. [DOI] [PubMed] [Google Scholar]
- 6.Champeris Tsaniras S, Kanellakis N, Symeonidou IE, Nikolopoulou P, Lygerou Z, Taraviras S. Licensing of DNA replication, cancer, pluripotency and differentiation: an interlinked world. Semin Cell Dev Biol. 2014;30:174–180. doi: 10.1016/j.semcdb.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 8.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 9.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype. Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 10.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 12.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 13.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 14.Todaro M, Francipane MG, Medema JP, Stassi G. Colon cancer stem cells: promise of targeted therapy. Gastroenterology. 2010;138:2151–2162. doi: 10.1053/j.gastro.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 15.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, Shelton AA, Parmiani G, Castelli C, Clarke MF. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 17.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 18.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 19.Wahab SM, Islam F, Gopalan V, Lam AK. The Identifications and Clinical Implications of Cancer Stem Cells in Colorectal Cancer. Clin Colorectal Cancer. 2017;16:93–102. doi: 10.1016/j.clcc.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 20.Lugli A, Iezzi G, Hostettler I, Muraro MG, Mele V, Tornillo L, Carafa V, Spagnoli G, Terracciano L, Zlobec I. Prognostic impact of the expression of putative cancer stem cell markers CD133, CD166, CD44s, EpCAM, and ALDH1 in colorectal cancer. Br J Cancer. 2010;103:382–390. doi: 10.1038/sj.bjc.6605762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu HC, Liu YS, Tseng KC, Hsu CL, Liang Y, Yang TS, Chen JS, Tang RP, Chen SJ, Chen HC. Overexpression of Lgr5 correlates with resistance to 5-FU-based chemotherapy in colorectal cancer. Int J Colorectal Dis. 2013;28:1535–1546. doi: 10.1007/s00384-013-1721-x. [DOI] [PubMed] [Google Scholar]
- 22.Ong CW, Kim LG, Kong HH, Low LY, Iacopetta B, Soong R, Salto-Tellez M. CD133 expression predicts for non-response to chemotherapy in colorectal cancer. Mod Pathol. 2010;23:450–457. doi: 10.1038/modpathol.2009.181. [DOI] [PubMed] [Google Scholar]
- 23.Jung HJ. Chemical proteomic approaches targeting cancer stem cells: a review of current literature. Cancer Genomics Proteomics. 2017;14:315–332. doi: 10.21873/cgp.20042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- 25.McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase – essential roles in physiology and cancer biology. J Cell Sci. 2008;121:3121–3132. doi: 10.1242/jcs.017996. [DOI] [PubMed] [Google Scholar]
- 26.Tan C, Costello P, Sanghera J, Dominguez D, Baulida J, de Herreros AG, Dedhar S. Inhibition of integrin linked kinase (ILK) suppresses beta-catenin-Lef/Tcf-dependent transcription and expression of the E-cadherin repressor, snail, in APC-/- human colon carcinoma cells. Oncogene. 2001;20:133–140. doi: 10.1038/sj.onc.1204052. [DOI] [PubMed] [Google Scholar]
- 27.Marotta A, Tan C, Gray V, Malik S, Gallinger S, Sanghera J, Dupuis B, Owen D, Dedhar S, Salh B. Dysregulation of integrin-linked kinase (ILK) signaling in colonic polyposis. Oncogene. 2001;20:6250–6257. doi: 10.1038/sj.onc.1204791. [DOI] [PubMed] [Google Scholar]
- 28.Bravou V, Klironomos G, Papadaki E, Stefanou D, Varakis J. Integrin-linked kinase (ILK) expression in human colon cancer. Br J Cancer. 2003;89:2340–2341. doi: 10.1038/sj.bjc.6601482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novak A, Hsu SC, Leung-Hagesteijn C, Radeva G, Papkoff J, Montesano R, Roskelley C, Grosschedl R, Dedhar S. Cell adhesion and the integrin-linked kinase regulate the LEF-1 and beta-catenin signaling pathways. Proc Natl Acad Sci USA. 1998;95:4374–4379. doi: 10.1073/pnas.95.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oloumi A, McPhee T, Dedhar S. Regulation of E-cadherin expression and beta-catenin/Tcf transcriptional activity by the integrin-linked kinase. Biochim Biophys Acta. 2004;1691:1–15. doi: 10.1016/j.bbamcr.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Bravou V, Klironomos G, Papadaki E, Taraviras S, Varakis J. ILK over-expression in human colon cancer progression correlates with activation of beta-catenin, down-regulation of E-cadherin and activation of the Akt-FKHR pathway. J Pathol. 2006;208:91–99. doi: 10.1002/path.1860. [DOI] [PubMed] [Google Scholar]
- 32.Duxbury MS, Ito H, Benoit E, Waseem T, Ashley SW, Whang EE. RNA interference demonstrates a novel role for integrin-linked kinase as a determinant of pancreatic adenocarcinoma cell gemcitabine chemoresistance. Clin Cancer Res. 2005;11:3433–3438. doi: 10.1158/1078-0432.CCR-04-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalra J, Warburton C, Fang K, Edwards L, Daynard T, Waterhouse D, Dragowska W, Sutherland BW, Dedhar S, Gelmon K, Bally M. QLT0267, a small molecule inhibitor targeting integrin-linked kinase (ILK), and docetaxel can combine to produce synergistic interactions linked to enhanced cytotoxicity, reductions in P-AKT levels, altered F-actin architecture and improved treatment outcomes in an orthotopic breast cancer model. Breast Cancer Res. 2009;11:R25. doi: 10.1186/bcr2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia Z. Role of integrin-linked kinase in drug resistance of lung cancer. Onco Targets Ther. 2015;8:1561–1565. doi: 10.2147/OTT.S81447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang F, Wang B, Bao L, Zhao YS, Zhang SM, Zhang SQ. Overexpression of ILK promotes temozolomide resistance in glioma cells. Mol Med Rep. 2017;15:1297–1304. doi: 10.3892/mmr.2017.6157. [DOI] [PubMed] [Google Scholar]
- 36.Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR, Sullivan DC, Jessup JM, Brierley JD, Gaspar LE, Schilsky RL, Balch CM, Winchester DP, Asare EA, Madera M, Gress DM, Meyer LR. AJCC Cancer Staging Manual. 8th ed. New York, Springer. 2017 [Google Scholar]
- 37.Bosman FT, Carneiro F, Hruban RH, Theise ND. World Health Organization Classification of Tumours of the Digestive System. 4th edition. Lyon, IARC. 2010 [Google Scholar]
- 38.Karavias D, Maroulis I, Papadaki H, Gogos C, Kakkos S, Bravou V. Overexpression of CDT1 is a predictor of poor survival in patients with hepatocellular carcinoma. J Gastrointest Surg. 2016;20:568–579. doi: 10.1007/s11605-015-2960-7. [DOI] [PubMed] [Google Scholar]
- 39.Pinto M, Robine S, Appay MD, Appay MD, Robin S, Kedinger M, Triadon N, Dussanlxx E, Croix B, Zweibaum A. Enterocyte-like differentiation and polarization of the human colon carcinoma cell line Caco-2 in culture. Biol Cell. 1983;47:323–330. [Google Scholar]
- 40.Coley HM. Development of drug-resistant models. In: Cancer Cell Culture, Methods and Protocols. Langdom SP (ed) NJ, Totowa, Humana Press Inc. 2004:pp. 267–273. doi: 10.1385/1-59259-406-9:267. [DOI] [PubMed] [Google Scholar]
- 41.Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G, Samuel S, Kim MP, Lim SJ, Ellis LM. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–1957. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim AY, Kwak JH, Je NK, Lee YH, Jung YS. Epithelial-mesenchymal transition is associated with acquired resistance to 5-fluorocuracil in HT-29 colon cancer cells. Toxicol Res. 2015;31:151–156. doi: 10.5487/TR.2015.31.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 44.Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, Kirchner T. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005;179:56–65. doi: 10.1159/000084509. [DOI] [PubMed] [Google Scholar]
- 45.Sánchez-Tilló E, O de Barrios, Siles L, Cuatrecasas M, Castells A, Postigo A. β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA. 2011;108:19204–19209. doi: 10.1073/pnas.1108977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan F, Samuel S, Evans KW, Lu J, Xia L, Zhou Y, Sceusi E, Tozzi F, Ye XC, Mani SA, Ellis LM. Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012;1:5–16. doi: 10.1002/cam4.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kahlert C, Lahes S, Radhakrishnan P, Dutta S, Mogler C, Herpel E, Brand K, Steinert G, Schneider M, Mollenhauer M, Reissfelder C, Klupp F, Fritzmann J, Wunder C, Benner A, Kloor M, Huth C, Contin P, Ulrich A, Koch M, Weitz J. Overexpression of ZEB2 at the invasion front of colorectal cancer is an independent prognostic marker and regulates tumor invasion in vitro. Clin Cancer Res. 2011;17:7654–7663. doi: 10.1158/1078-0432.CCR-10-2816. [DOI] [PubMed] [Google Scholar]
- 48.Fan XJ, Wan XB, Yang ZL, Fu XH, Huang Y, Chen DK, Song SX, Liu Q, Xiao HY, Wang L, Wang JP. Snail promotes lymph node metastasis and Twist enhances tumor deposit formation through epithelial-mesenchymal transition in colorectal cancer. Hum Pathol. 2013;44:173–180. doi: 10.1016/j.humpath.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 49.Francí C, Gallén M, Alameda F, Baró T, Iglesias M, Virtanen I, García de Herreros A. Snail1 protein in the stroma as a new putative prognosis marker for colon tumours. PLoS One. 2009;4:5595. doi: 10.1371/journal.pone.0005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Findlay VJ, Wang C, Nogueira LM, Hurst K, Quirk D, Ethier SP, Staveley O'Carroll KF, Watson DK, Camp ER. SNAI2 modulates colorectal cancer 5-fluorouracil sensitivity through miR145 repression. Mol Cancer Ther. 2014;13:2713–2726. doi: 10.1158/1535-7163.MCT-14-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spelt L, Sasor A, Ansari D, Hilmersson KS, Andersson R. The prognostic role of cancer stem cell markers for long-term outcome after resection of colonic liver metastases. Anticancer Res. 2018;38:313–320. doi: 10.21873/anticanres.12224. [DOI] [PubMed] [Google Scholar]
- 53.Batsaikhan BE, Yoshikawa K, Kurita N, Iwata T, Takasu C, Kashihara H, Shimada M. Cyclopamine decreased the expression of sonic hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014;34:6339–6344. [PubMed] [Google Scholar]
- 54.Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, Teng HW, Yang SH, Lan YT, Chiou SH, Wang HW. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141:279–291. doi: 10.1053/j.gastro.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Wang L, Pappan L, Galliher-Beckley A, Shi J. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol Cancer. 2012;11:87. doi: 10.1186/1476-4598-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Somasiri A, Howarth A, Goswami D, Dedhar S, Roskelley CD. Overexpression of the integrin-linked kinase mesenchymally transforms mammary epithelial cells. J Cell Sci. 2001;114:1125–1136. doi: 10.1242/jcs.114.6.1125. [DOI] [PubMed] [Google Scholar]
- 57.Lee YI, Kwon YJ, Joo CK. Integrin-linked kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition. Biochem Biophys Res Commun. 2004;316:997–1001. doi: 10.1016/j.bbrc.2004.02.150. [DOI] [PubMed] [Google Scholar]
- 58.Huang HC, Hu CH, Tang MC, Wang WS, Chen PM, Su Y. Thymosin beta4 triggers an epithelial-mesenchymal transition in colorectal carcinoma by upregulating integrin-linked kinase. Oncogene. 2007;26:2781–2790. doi: 10.1038/sj.onc.1210078. [DOI] [PubMed] [Google Scholar]
- 59.Hsu EC, Kulp SK, Huang HL, Tu HJ, Salunke SB, Sullivan NJ, Sun D, Wicha MS, Shapiro CL, Chen CS. Function of integrin-linked kinase in modulating the stemness of IL-6-abundant breast cancer cells by regulating γ-secretase-mediated Notch1 activation in caveolae. Neoplasia. 2015;17:497–508. doi: 10.1016/j.neo.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rothe K, Babaian A, Nakamichi N, Chen M, Watanabe A, Mager D, Eaves CJ, Dedhar S, Jiang X. The focal adhesion complex Ilk-pinch-parvin is key to niche interactions and survival of quiescent stem cells in chronic myeloid leukemia in vitro and in vivo. Blood. 2016;128:936. [Google Scholar]
- 61.Lin G, Zhang X, Ren J, Pang Z, Wang C, Xu N, Xi R. Integrin signaling is required for maintenance and proliferation of intestinal stem cells in Drosophila. Dev Biol. 2013;377:177–187. doi: 10.1016/j.ydbio.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 62.Song W, Jiang R, Zhao CM. Role of integrin-linked kinase in multi-drug resistance of human gastric carcinoma SGC7901/ DDP cells. Asian Pac J Cancer Prev. 2012;13:5619–5625. doi: 10.7314/apjcp.2012.13.11.5619. [DOI] [PubMed] [Google Scholar]
- 63.Zalatnai A, Molnár J. Review. Molecular background of chemoresistance in pancreatic cancer. In Vivo. 2007;21:339–347. [PubMed] [Google Scholar]