

Abstract
Establishment of an in vivo small animal model of human tumor and human immune system interaction would enable preclinical investigations into the mechanisms underlying cancer immunotherapy. To this end, nonobese diabetic (NOD).Cg-PrkdcscidIL2rgtm1Wjl/Sz (null; NSG) mice were transplanted with human (h)CD34+ hematopoietic progenitor and stem cells, which leads to the development of human hematopoietic and immune systems [humanized NSG (HuNSG)]. HuNSG mice received human leukocyte antigen partially matched tumor implants from patient-derived xenografts [PDX; non–small cell lung cancer (NSCLC), sarcoma, bladder cancer, and triple-negative breast cancer (TNBC)] or from a TNBC cell line-derived xenograft (CDX). Tumor growth curves were similar in HuNSG compared with nonhuman immune-engrafted NSG mice. Treatment with pembrolizumab, which targets programmed cell death protein 1, produced significant growth inhibition in both CDX and PDX tumors in HuNSG but not in NSG mice. Finally, inhibition of tumor growth was dependent on hCD8+ T cells, as demonstrated by antibody-mediated depletion. Thus, tumor-bearing HuNSG mice may represent an important, new model for preclinical immunotherapy research.—Wang, M., Yao, L.-C., Cheng, M., Cai, D., Martinek, J., Pan, C.-X., Shi, W., Ma, A.-H., De Vere White, R. W., Airhart, S., Liu, E. T., Banchereau, J., Brehm, M. A., Greiner, D. L., Shultz, L. D., Palucka, K., Keck, J. G. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy.
Keywords: checkpoint inhibitor, mouse model, patient-derived xenograft, pembrolizumab
Translation of the discoveries from mouse models to clinical trials has been hindered by many genetic and biologic differences between humans and mice (1, 2). To overcome these constraints, humanized mice engrafted with a human immune system have been developed as an important tool for cancer research (3, 4). In conditioned nonobese diabetic (NOD).Cg-PrkdcscidIL2rgtm1Wjl/Sz (NSG) recipients, hematopoietic progenitor and stem cell (HPSC) engraftment leads to the generation of multiple lineages of hematopoietic and immune cells, including T cells, B cells, myeloid cells, and conventional, as well as plasmacytoid dendritic cells (5–7). Antigen-specific cytotoxic and IFN-γ-producing T-cell responses are detectable in humanized NSG (HuNSG) mice (3, 8, 9). Thus, HuNSG mice can develop partially functional human immune systems when engrafted with human HPSCs, suggesting that these HuNSG mice can be used as an important tool for immunotherapy in cancer research.
Immunotherapy has emerged as a promising cancer treatment via activation of the immune system and reduction of immune suppression within the tumor microenvironment [reviewed in refs. (10–12)]. Among the most efficient approaches for activation of anti-tumor immunity is the blockade of immune checkpoints. Under physiologic conditions, immune checkpoints are critical for maintaining self-tolerance and protecting peripheral tissues from collateral damage in response to infection (13). Tumors also use immune checkpoints to suppress anti-tumor immune responses. Blockade of checkpoint proteins, such as programmed cell death protein 1 (PD-1), has presented broad and diverse opportunities to enhance antitumor immunity with the potential to produce durable clinical responses [reviewed in refs. (14, 15)].
PD-1 is broadly expressed on activated CD4+, CD8+ T cells and CD4+ regulatory T (Treg) cells, as well as on B cells and NK cells (16, 17). PD-1 is also constitutively expressed on tumor-infiltrating lymphocytes (TILs) in a variety of tumor types (18), reflecting an exhausted T-cell status. PD-1 binds to 2 ligands: PD-1 ligand 1 (PD-L1; also known as B7-H1) and PD-L2 (B7-DC) (19–21). PD-L1 is broadly expressed on normal healthy tissues and malignant cells, whereas PD-L2 is expressed predominately by antigen-presenting cells (22). PD-L1 binding to PD-1 leads to inhibition of T-cell activation and effector function mediated by recruitment of tyrosine phosphatases to the immune synapse that disrupts T-cell receptor signaling (23). A large body of evidence has shown that PD-L1 expression is commonly upregulated in many different human cancer types, including melanoma, lung, and ovarian tumors (reviewed in refs. 14, 24).
Early-phase clinical trials investigating blockade of the PD-1/PD-L1 signaling pathway have shown positive clinical responses in some patients bearing melanoma, non–small cell lung cancer (NSCLC) and renal cell carcinoma tumors (25–27). Pembrolizumab, a highly selective humanized IgG4-κ mAb, has been the first U.S. Food and Drug Administration-approved anti-PD-1 therapy. The levels of PD-L1 expression in patient tumor samples correlate with higher response rates and a longer progression-free survival time (25, 28, 29). Thus, the expression levels of PD-L1 can identify those patients who are most likely to benefit from pembrolizumab. However, durable clinical responses have also been observed in patients considered to be negative for tumor PD-L1 expression (30), suggesting that additional mechanisms underlying PD-1/PD-L1 blockade therapy may be involved in mediating its therapeutic effects. Thus, it would be advantageous to establish an in vivo model system that would allow mechanistic studies regarding the mode of action of anti-PD-1 therapeutic agents.
Herein, we successfully established a humanized mouse model bearing human cancer cell line-derived xenograft (CDX) or patient-derived xenograft (PDX) tumors, the Onco-HuNSG model, using allogeneic but human leukocyte antigen (HLA) partially matched CD34+ HPSC donors and tumors. Onco-HuNSG mice might be useful in preclinical investigation of the efficacy of cancer immunotherapy.
MATERIALS AND METHODS
Mice
NSG mice were developed at The Jackson Laboratory (Sacramento, CA, USA) by backcrossing a complete null mutation at the Il2rg locus onto the NOD.Cg-Prkdcscid (NOD/SCID) strain (5, 31). HuNSG mice were generated as previously reported (31). In brief, human fetal liver CD34+-purified HPSCs were purchased from Stem Express (Folsm, CA, USA). HuNSG mice were generated by intravenous injection of 105 human CD34+ (hCD34+) HPSCs into 3-wk-old female NSG mice, 4 h post-140 cGy total body irradiation using the RS-2000 irradiator (Rad Source, Buford, GA, USA). The engraftment levels of hCD45+ cells were determined 12 wk post-HPSC transplantation by flow cytometric quantification of peripheral blood hCD45+ cells. HuNSG mice that had over 25% hCD45+ cells in the peripheral blood were considered as engrafted and humanized. HuNSG mice from different HPSC donors with different levels of engraftment were randomized into every treatment group in all of the experiments. Mice were maintained under defined flora with irradiated food at The Jackson Laboratory, according to guidelines established by the Institutional Animal Care and Use Committee.
CDX and PDX tumor explants
The MDA-MB-231 human triple-negative breast cancer (TNBC) cell line (ATCC HTB-26) was purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Leibovitz’s L-15 medium (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% heat-inactivated fetal bovine serum (GE Healthcare Life Sciences, HyClone Laboratories, Logan, UT, USA) and 1% penicillin-streptomycin (Thermo Fisher Scientific) at 37°C with 0% CO2. The MDA-MB-231 cell line was tested negative for gram-positive, gram-negative bacteria, and mycoplasma by PCR. Cell authentication was performed by Short Tandem Repeat Polymorphism DNA sequencing (SoftGenetics, State College, PA, USA). P5 MDA-MB-231 cells were used for tumor implantation. Patient tumor explants were obtained from surgical specimens of lung, breast, bladder, and sarcoma cancer from patients at the Davis Comprehensive Cancer Center, University of California, Davis (Davis, CA, USA). Written, informed consent was obtained from the patients before collection of specimens. PDX models were generated by implantation of PDX into NSG and HuNSG mice. In brief, patient-derived tumors were finely minced and loaded into 1-cc syringes with 14-gauge needles. Depending on the tumor model, 20–40 µl of homogenized tumor tissue was inoculated subcutaneously at the right flank of NSG mice while under anesthesia. The PDX tumors that were used in this study include NSCLC: LG0997, LG0978, LG1306, and LG1208; TNBC: BR1126 and BR0744; sarcoma: SA0209; and bladder: BL0293.
Tumor experiments
At least 12 wk post-human HPSC transplantation, 5 × 106 TNBC MDA-MB-231 cells, suspended in serum-free medium mixed with matrigel (Corning Life Sciences, Bedford, MA, USA), were injected into mammary fat pads in HuNSG mice. For PDX models, 20–40 µl of finely minced tumors was injected subcutaneously into the right flank of HuNSG mice. Treatment was started when the tumors reached 50–120 mm3 in volume. Vehicle control saline (Hospira, Austin, TX, USA) was injected intraperitoneally or intravenously every 5 d until the study end point. Pembrolizumab (anti-PD-1; Merck, Whitehouse Station, NJ, USA) was injected intraperitoneally or intravenously at 10 mg/kg for the first dose, followed by 5 mg/kg (i.p. or i.v.) dose every 5 d until the study end point. Bladder PDX tumor (BL0293) was injected with 10 mg/kg, i.p. pembrolizumab i.p., followed by 5 mg/kg, i.p. pembrolizumab, twice a week until the end of the study. Cisplatin (Teva Pharmachemie, Haarlem, The Netherlands) was injected intravenously at 2 mg/kg on d 0, 7, and 14. To deplete hCD8+ T cells in HuNSG mice, 200 µg anti-CD8 depletion mAb (clone RPA-T8; BioLegend, San Diego, CA, USA) was injected intraperitoneally into HuNSG mice, 1 d before pembrolizumab treatment and then followed by weekly intraperitoneal injections of anti-CD8 mAb for 4 wk. IgG1, κ isotype control mAb (clone MOPC-21) from BioLegend was injected intraperitoneally into control HuNSG mice on the same days as anti-CD8 depletion antibody. Tumors were measured by caliper every 3–4 d, and volumes (mm3) were calculated by (length × width)2/2.
Immunofluorescence microscopy
Tumors samples were frozen in optimum cutting temperature (Sakura Finetek, Torrance, CA, USA). Acetone-fixed cryosections (8 µm) were consecutively treated with 0.01% Triton X-100 for 15 min, 0.03% hyaluronidase for 15 min, FcR Block (Innovex Biosciences, Richmond, CA, USA) for 40 min, and Background Buster (Innovex Biosciences) for 20 min. The sections were then stained with primary antibodies diluted in PBS + 5% bovine serum albumin and 0.1% saponin for 1 h at room temperature, washed, and stained with the secondary antibodies at room temperature for 30 min. Nuclei were counterstained with DAPI (1 µg/ml) for 2 min. Primary antibodies included anti-hCD45 at 2 µg/ml (clone H130; BioLegend), FITC-conjugated anti-pan cytokeratin at 1:60 dilution (clone CK3-6H5; Miltenyi Biotec, Bergisch Gladbach, Germany), and AF647-conjugated anti-hCD8a at 1.25 µg/ml (clone RPA-T8; BioLegend). The secondary antibody included goat anti-mouse IgG1 AF568 at 1:2000 dilution (Thermo Fisher Scientific). For acquisition, data were acquired by sequential acquisition, and tile-scan imaging was performed on an SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany).
Antibodies and flow cytometry
Whole blood, spleen, and tumor tissues were processed for flow cytometry analysis. In brief, gentleMACS Octo Dissociator from Miltenyi Biotec was used for spleen and tumor-tissue dissociation, followed by filtering through 100 µm screens. After tissue dissociation, the cells were washed with PBS and incubated with the antibody panel for 15–20 min at room temperature. The red blood cells were lysed by Pharm Lyse (BD Biosciences, San Jose, CA, USA) for 8–12 min following antibody staining at room temperature. Live or dead cells were detected by LIVE/DEAD Fixable Red Dead Cell Stain Kit (Thermo Fisher Scientific). Samples were centrifuged and washed with PBS before suspension in PBS for data acquisition on BD FACSCanto II using FACSDiva software (BD Biosciences).
Fluorochrome–conjugated mAb to the following human antigens were used: CD45-V510 (clone HI30), CD45-phycoerythrin (PE; clone HI30), CD3-FITC (clone UCHT1), CD4-PE-cyanine 7 (clone SK3), CD8-allophycocyanin-cyanine 7 (clone SK1), CD19-allophycocyanin (clone HIB19), PD-1-PE (clone EH12.2H7), and PD-L1-V421 (clone 29E.2A3). All antibodies were purchased from BioLegend.
HLA genotyping of the McMaster Cohort
High-resolution HLA typing was performed by sequence-based typing (SBT) at the University of Oklahoma Health Sciences Center Clinical Laboratory Improvement Amendments/American Society for Histocompatibility & Immunogenetics-accredited HLA Typing Laboratory (Oklahoma City, OK, USA) using in-house methods. In brief, genomic DNA was extracted from HPSCs using a QIAamp DNA blood kit (Qiagen, Hilden, Germany), and a DNEasy Blood & Tissue Kit (Qiagen) was used to extract genomic DNA from tumor samples. After confirmation, the PCR product was purified using an ExoSAP-IT kit (USB) and was sequenced using BigDye Terminator v.3.1 (Thermo Fisher Scientific). Dye removal was conducted by ethanol precipitation. Sequencing reactions were performed on a 3730 capillary electrophoresis DNA sequencer (Thermo Fisher Scientific). Four-digit HLA types were determined using the HLA typing program Assign SBT (Olerup, West Chester, PA, USA).
Histocytometry
In situ quantitative analysis of breast cancer tissue was based on previously published methodology (32). In brief, a cryosection was stained by immunofluorescence for each breast cancer tissue to label nuclei and CD8. Whole-tissue scans were acquired using an SP8 confocal microscope (Leica Microsystems). Each scan was then analyzed by using the image analysis software Imaris (Bitplane, Zurich, Switzerland). With the use of the “spot” function in Imaris, the images were subdivided into individual cells, defined as having a nucleus diameter ≥6 µm. The accuracy of the segmentation was manually verified and adjusted if needed for each sample. Finally, for each generated spot, its x and y coordinates and the sum intensity values for all channels were exported into an fcs file to be visualized and quantified using FlowJo software (v.10; FlowJo, Ashland, OR, USA).
Statistical analysis
Statistical analyses were performed with GraphPad Prism 5 software, except a tumor growth curve comparison with 2 groups. Means ± sem are shown in all graphs. The nonparametric Mann-Whitney U test was applied to compare the results of cell numbers from different treatment groups. Tumor growth curves were analyzed by 2-way ANOVA, followed by Dunnett’s post-tests for experiment with 3 groups. For experiments with only 2 groups, tumor growth curves were analyzed by a Multivariate ANOVA test, followed by a univariate test with JMP 11 software. Differences of P < 0.05, P < 0.01, P < 0.001, and P < 0.0001 are considered statistically significant.
RESULTS
Onco-HuNSG mice support growth of partially HLA-matched human CDX and PDX tumors
The generation of Onco-HuNSG mice bearing CDX or PDX tumors is illustrated in Fig. 1A. We implanted the same PDX model in both Onco-HuNSG and NSG mice (Fig. 1B–D). HLA genotyping was performed for all PDX tumors, as well as human HPSC donors used in the Onco-HuNSG mice (Table 1). All 3 PDX models have partial HLA matches with their corresponding HPSC donors. The TNBC (BR0744) PDX tumor model grew slightly slower in Onco-HuNSG compared with NSG mice (Fig. 1B). This might be a result of multiple HLA class I and II loci matches between the TNBC (BR0744) PDX tumor and the CD34+ HPSC donor (Table 1), leading to stronger allogeneic immune responses against the tumor. The other 2 PDX models: NSCLC (LG0997P4; Fig. 1C) and sarcoma (SA0209P4; Fig. 1D) showed similar tumor growth rates between Onco-HuNSG and NSG mice. Our results showed that human PDX tumors can grow in Onco-HuNSG mice with partially HLA-matched allogeneic human immune systems.
Figure 1.

HuNSG mice support growth of partially HLA-matched human CDX and PDX tumors and immune cell populations in Onco-HuNSG mice. A) Experimental design for generating HuNSG mice bearing CDX or PDX tumors. B–D) Implantation of HLA partially matched PDX tumors BR0744P3 (B), LG0997P4 (C), and SA0209P4 (D) into NSG or HuNSG mice. PDX tumors were excised and implanted subcutaneously into NSG or HuNSG mice. Tumor measurements were calculated as (L × W × W)/2, where L is the length, and W is the width of the tumor. No statistical differences were observed in any tumor model between NSG and HuNSG mice. E) Flow cytometric analysis of hCD45+ cells in tumors from NSG or HuNSG mice. No hCD45+ cells were detected in tumors growing in NSG mice. F, G) At the study end point, hCD3, hCD4, hCD8, and hCD19 were stained for flow cytometric analysis to determine the populations of lymphocytes in tumor tissues (F) and peripheral blood (G) of 3 different PDX models. CD3+, CD4+, CD19+ cells are presented as percentage of cells within CD45+ cells. H) hCD45, hCD3, hCD4, and hCD8 were stained for flow cytometric analysis to determine numbers of TILs in MDA-MB-231 tumor-bearing HuNSG mice. Correlation was calculated by GraphPad Prism 5. **P < 0.01, ***P < 0.001.
TABLE 1.
HLA matching status between HPSC donors and tumors
| Tumor | CD34+ HPSC donors |
||
|---|---|---|---|
| 1 | 2 | 3 | |
| SA0209 | No HLA class I match; HLA class II: HLA-DRB3, DQB1 | ||
| BR0744 | HLA class I: HLA-A, -B, -C; HLA class II: HLA-DQB1, DQA1, DPB1, DPA1 | ||
| MDA-MB-231 | Responder: No HLA class I match; HLA class II: HLA-DRB1, DQB1, DQA1, DPA1 | ||
| BR1126 | Responder: HLA class I: HLA-C; HLA class II: HLA-DPA1 | Responder: HLA class I: HLA-A; HLA class II: HLA-DQA1, DPB1, DPA1 | Responder: HLA class I: HLA-C; HLA class II; DPA1 |
| LG1306 | Responder: No HLA class I match; HLA class II: HLA-DRB4, DQA1, DQB1 | Responder: no match | |
| LG0997 | HLA class I: HLA-A; HLA class II: HLA-DPA1 | ||
| LG1208 | Nonresponder: no HLA class I match; HLA class II: HLA-DRB1, DQB1, DQA1, DPB1, DPA1 | ||
| LG0978 | Nonresponder 7206: no HLA class I match; HLA class II: HLA-DRB3, DQA1, DPB1, DPA1 | Responder 7096: no HLA class I match; HLA class II: HLA-DPA1 | |
| BL0293 | Nonresponder 0912: no HLA class I match; HLA class II: HLA-DRB4, DPB1, DPA1 | Responder 6466: HLA class I: HLA-B, -C; HLA class II: DPB1, DPA1 | |
HLA matching between CDX/PDX tumor and CD34+ HPSC donors. HLA typing for all tumors and HPSC donors used was performed at the University of Oklahoma Health Sciences Center, as described in Materials and Methods. In brief, genomic DNA was extracted from HPSCs using a QIAamp DNA blood kit (Qiagen), and a DNEasy Blood & Tissue Kit (Qiagen) was used to extract genomic DNA from the tumor samples. Four-digit HLA types were determined using the HLA typing program Assign SBT (Olerup). The HLA matching loci between the CDX/PDX tumor and HPSC donors are shown.
Next, we used hCD45, a marker of human leukocytes, and flow cytometry to determine the degree of immune cell infiltration into the PDX tumors in Onco-HuNSG mice. As shown In Fig. 1E, all 3 PDX tumors engrafted in Onco-HuNSG mice displayed hCD45+ immune cell infiltrates, whereas no hCD45+ cells were detected in tumor-bearing NSG mice. This indicates that the human immune cells infiltrating the PDX tumors were derived from CD34+ donor HPSCs rather than from the surviving human immune cells of the original PDX tumors.
T-cell infiltration was detected using CD3+ staining in all 3 PDX tumors, whereas CD19+ B cells were only detectable in the TNBC (BR0744) tumor (Fig. 1F). The ratio of CD4+ and CD8+ T cells varied in different PDX tumors. The TNBC (BR0744) tumor showed significantly higher CD4+ T-cell infiltration (40.1 ± 7.4%) than CD8+ T-cell infiltration (15.2 ± 3.9%; P < 0.01). In contrast, CD8+ T-cell levels (57.5 ± 9.6%) were significantly higher than CD4+ T cells (24.6 ± 2.9%; P < 0.01) in the sarcoma (SA0209P4; Fig. 1F). The different CD4/CD8 T-cell ratios in different Onco-HuNSG tumor models reflect the heterogeneity of TIL density in patients with cancer [reviewed in Fridman et al. (33)]. Of note, these 3 PDX tumors in Onco-HuNSG mice had similar hCD45+ cells in blood circulation before tumor implantation. We also detected hCD33+ myeloid cells in both tumors (Supplemental Fig. S1A) and spleens (Supplemental Fig. S1B) of sarcoma (SA0209P4) and NSCLC (LG0997P4) Onco-HuNSG models. Analysis of the blood cells revealed that the sarcoma (SA0209P4) and the NSCLC (LG0997P4) Onco-HuNSG mice displayed more circulating CD4+ T cells than CD8+ T cells. Circulating B cells were also detected (Fig. 1G). Thus, the lymphocyte populations in both peripheral blood and tumor of Onco-HuNSG mice may vary, depending on the tumor model. In the TNBC cell line MDA-MB-231 CDX Onco-HuNSG model, the tumor volume was negatively correlated with the numbers of hCD45+, hCD4+, and hCD8+ T cells that infiltrated the tumor (Fig. 1H; P < 0.01 or P < 0.001), suggesting that the human T cells might be impeding tumor growth.
Onco-HuNSG mice responded to anti-PD-1 immunotherapy
Next, we determined whether anti-PD-1 pembrolizumab would show efficacy in Onco-HuNSG mice. The experimental protocol is outlined in Fig. 2A. We selected 1 TNBC CDX tumor model MDA-MB-231, 1 TNBC PDX tumor model BR1126, and 1 NSCLC tumor model LG1306 based on a high percentage of PD-L1+ cells in these tumors (Supplemental Table S1). When tumors reached 50–120 mm3 in volume, pembrolizumab was injected at 10 mg/kg i.p. for the first dose, followed by 5 mg/kg, i.p. dosage on d 5, 10, 15, 20, and 25.
Figure 2.

Pembrolizumab inhibited tumor growth in Onco-HuNSG mice. A) Experimental design and treatment schedule for anti-PD-1 and chemotherapy against CDX and PDX tumors. Treatments started when tumors reached 50–120 mm3 in volume. B, C) Animals were randomized into different experimental groups. Tumors from LG1306P5 (B) and MDA-MB-231 (C) Onco-HuNSG mice were processed into single-cell suspensions and stained, as described in Materials and Methods, for flow cytometric analysis. PD-1 was stained on both CD45− and CD45+ populations. D) MDA-MB-231 human breast cancer cells (5 × 106) in matrigel were injected into mammary fat pads of HuNSG mice. Vehicle control saline was injected intraperitoneally every 5 d for 25 d. Pembrolizumab was injected at 10 mg/kg on d 0, followed by 5 mg/kg on d 5, 10, 15, 20, and 25. E) TNBC PDX tumor BR1126P5 was trocared subcutaneously into HuNSG mice. Vehicle control saline was injected intraperitoneally every 5 d for 25 d. Cisplatin was injected at 2 mg/kg, i.v. on d 0, 7, and 14. Pembrolizumab was injected at 10 mg/kg, i.p. on d 0, followed injection at 5 mg/kg, i.p. on d 5, 10, 15, and 20. On d 21, both the cisplatin and pembrolizumab treatment groups have significantly smaller tumor size compared with the vehicle control group. F) NSCLC PDX LG1306P5 was trocared subcutaneously into HuNSG mice. Vehicle control saline and pembrolizumab (5 mg/kg) were injected intraperitoneally every 5 d for 25 d. For experiments with 2 groups, tumor growth response curves were analyzed by the Multivariate ANOVA test, followed by univariate test with JMP 11 software (D, F). For the experiment with 3 groups, tumor growth curves were analyzed by 2-way ANOVA, followed by Dunnett’s posttests using GraphPad Prism (E). *P < 0.05, **P < 0.01, ***P < 0.001.
To confirm that pembrolizumab did bind to its target PD-1 in our study, we quantified the percentage of PD-1+ cells in tumor tissues of NSCLC (LG1306) and TNBC (MDA-MB-231) Onco-HuNSG mice by flow cytometry. As shown in Fig. 2B, C, pembrolizumab treatment significantly decreased PD-1+ cell detection in hCD45+ cells in both tumor models (P < 0.01 and P < 0.0001). Percentages of PD-1+ cells in the hCD45− population in the TNBC (MDA-MB-231) Onco-HuNSG tumors were also significantly decreased (P < 0.0001), indicating that pembrolizumab binding to its target might compete with subsequent anti-PD-1 staining by antibody detection and prevented PD-1 detection by flow cytometry. In addition, the percentage of PD-L1+ cells in hCD45+ or hCD45− cell populations was not affected by pembrolizumab treatment (Supplemental Fig. S2).
We demonstrated that pembrolizumab treatment significantly delayed TNBC (MDA-MB-231) CDX growth compared with the vehicle control saline-treated Onco-HuNSG group (P < 0.001; Fig. 2D). Individual mouse responses to different treatments were also displayed in Supplemental Fig. S3A. Pembrolizumab also significantly inhibited tumor growth in both TNBC (BR1126P5) and NSCLC (LG1306P5) PDX Onco-HuNSG models (Fig. 2E, F). In the TNBC (BR1126P5) model, cisplatin was used as a positive control for tumor growth inhibition. Cisplatin is a chemotherapeutic drug that demonstrated efficacy in TNBC (BR1126P3) tumor growth inhibition in NSG mice (Supplemental Fig. S4). In the TNBC (BR1126P5) Onco-HuNSG mice, cisplatin treatment resulted in significant tumor growth inhibition as well (Fig. 2E; P < 0.001; Supplemental Fig. S3B). In the NSCLC (LG1306P5) Onco-HuNSG mice, pembrolizumab also significantly reduced tumor growth (Fig. 2F; P < 0.05).
HPSC donor variation to anti-PD-1 therapy in Onco-HuNSG mice
Pembrolizumab demonstrated a 30–35% durable response in patients with advanced melanoma, but not all patients responded to pembrolizumab treatment (28). The variable responses to anti-PD-1 therapy in patients might be a result of the interplay between the unique tumor microenvironment and the immune system in each patient. To test the impact of HPSC donor variability in Onco-HuNSG responses to anti-PD-1 therapy, we studied treatment responses in the same PDX tumor-bearing Onco-HuNSG mice generated from different HPSC donors. As shown in Fig. 3A, B and Supplemental Fig. S3C, D, in donor 6466-engrafted bladder PDX (BL0293P3) Onco-HuNSG mice, pembrolizumab significantly inhibited bladder (BL0293P3) tumor growth (P < 0.05), whereas in donor 0912-engrafted bladder PDX (BL0293P3) Onco-HuNSG mice did not respond to pembrolizumab treatment. In NSCLC (LG0978P5) Onco-HuNSG, tumor growth in donor 7096-engrafted Onco-HuNSG mice was significantly inhibited by pembrolizumab (Fig. 3C; P < 0.05; Supplemental Fig. S3E), but NSCLC (LG0978P5) growth was not affected by pembrolizumab treatment in donor 7206-engrafted Onco-HuNSG mice (Fig. 3D and Supplemental Fig. S3F), suggesting donor variation in response to anti-PD-1 therapy.
Figure 3.

Donor variations in response to pembrolizumab treatment in bladder (BL0293P3) and NSCLC (LG0978P5) Onco-HuNSG mice. A, B) Tumor growth curves of PDX bladder tumor BL0293P3 Onco-HuNSG in responder and nonresponder donors. The BL0293P3 tumor was implanted subcutaneously into HuNSG mice, established from 2 donor CD34+ HPSC sources: donor 6466 (A) and donor 0912 (B). Vehicle control saline was injected intraperitoneally twice a week until the end of the study. Pembrolizumab was injected 10 mg/kg, i.p. on d 0 followed by 5 mg/kg twice a week until the end of the study. C, D) Tumor growth curves of PDX NSCLC (LG0978P5) Onco-HuNSG in responder and nonresponder donors. The LG0978P5 tumor was trocared subcutaneously into HuNSG mice from 2 donor CD34+ HPSC sources: donor 7096 (C) and donor 7206 (D). Vehicle control saline was injected intraperitoneally every 5 d for 20 d on d 0, 5, 10, 15, and 20. Pembrolizumab was injected at 10 mg/kg, i.p. on d 0, followed by 5 mg/kg, i.p. injection on d 5, 10, 15, and 20. E, F) Whole blood from NSCLC (LG0978P5) Onco-HuNSG donor 7096 (E) and donor 7206 (F) mice was processed into single-cell suspension and stained for hCD45, hCD3, hCD4, and hCD8, as described in Materials and Methods, for flow cytometric analysis. Nonparametric Mann-Whitney U test was performed to compare the cell numbers in different treatment groups. *P < 0.05.
Next, we collected blood from the NSCLC (LG0978P5) Onco-HuNSG mice, and immune cell populations were analyzed by flow cytometry. We observed that CD4+ and CD8+ T cells were significantly increased in Onco-HuNSG mice engrafted with the responder donor 7096 in NSCLC (LG0978P5) Onco-HuNSG mice (Fig. 3E). In contrast, CD45+ leukocytes and CD4+ and CD8+ T cells from the pembrolizumab group all remained at the levels similar to the saline-treated control group in Onco-HuNSG mice established using the nonresponder donor 7206 (Fig. 3F). We also analyzed the TILs from both donors in the NSCLC (LG0978P5) lung model by flow cytometry, but the percentage of TILs in this model was barely detectable above background levels (<1%; data not shown).
To investigate the factors contributing to the successful response of Onco-HuNSG mice to anti-PD-1 therapy, we hypothesized that the natural level of PD-1 expression on CD45+ leukocytes might influence the efficacy of pembrolizumab. In all of the tumor models that were tested, including NSCLC (LG1306, LG1208, LG0978), TNBC (MDA-MB-231), and sarcoma (SA0209), we did not find such correlation (data not shown). Therefore, the prevalence of PD-1+ leukocytes within tumor does not determine the tumor response of Onco-HuNSG mice to pembrolizumab. In addition, we also compared the pretreatment T-cell levels in different tumor models. Although the levels of human T cells vary from donor to donor, successful responses to pembrolizumab treatment were observed in Onco-HuNSG mice that have 10–23% of pretreatment T-cell levels in blood circulation (data not shown).
Next, we typed HLA from all HPSC donors used in this study and compared to the tumor donor HLA (Table 1). We found that in all Onco-HuNSG models used, most of the HPSC donors and tumors were partially matched for several major HLA class I and II loci (Table 1). In the bladder PDX (BL0293P3) Onco-HuNSG mice, both the responder and the nonresponder HPSC donors had multiple HLA class II loci matched with the bladder PDX tumor, but only the responder donor had 2 HLA class I loci matched with the bladder PDX tumor (Table 1). In contrast, the responder donor in the lung (LG0978P5) Onco-HuNSG mice had no major HLA class I match with the lung (LG0978P5) PDX tumor (Table 1). In the NSCLC (LG0978P5) Onco-HuNSG model, the nonresponder donor (donor 7206) had 4 HLA class II matches but no major HLA class I match with the PDX tumor, which was similar to the matching status of “donor 1” in the TNBC (MDA-MB-231) model. However, donor 1 used in the TNBC (MDA-MB-231) Onco-HuNSG model did respond to anti-PD-1 therapy (Fig. 2D). In the TNBC (BR1126P5) Onco-HuNSG model, all 3 HPSC donors were partially matched with the TNBC (BR1126P5) PDX tumor at both HLA class I and class II loci, and all 3 HPSC donors responded to pembrolizumab treatment (Supplemental Fig. S5). Therefore, we did not discover any HLA matching pattern in major loci that can determine the response rates of human HPSC donors to anti-PD-1 therapy.
Immune cell infiltration in tumors from Onco-HuNSG mice treated with anti-PD-1 therapy
We examined immune cell populations in NSCLC (LG1306) and TNBC (MDA-MB-231) Onco-HuNSG mice following pembrolizumab treatment. Over 50% of the leukocytes in peripheral blood were hCD45+ in both NSCLC (LG1306) and TNBC (MDA-MB-231) Onco-HuNSG mice with no significant differences in percentages of hCD45+ cells between mice that received pembrolizumab or saline control treatment (Fig. 4A, B). We found that in peripheral blood, pembrolizumab treatment increased numbers of both CD4+ and CD8+ T cells in NSCLC (LG1306) Onco-HuNSG mice (Fig. 4A) but decreased numbers of both T-cell populations in TNBC (MDA-MB-231) Onco-HuNSG mice (Fig. 4B). The increase of CD4+ and CD8+ T-cell numbers in peripheral blood of pembrolizumab-treated NSCLC (LG1306) Onco-HuNSG mice is consistent with the aforementioned responder donor-engrafted lung PDX NSCLC (LG0978) Onco-HuNSG mice (Fig. 3E). Therefore, the composition of immune cell populations postpembrolizumab treatment may be tumor model dependent.
Figure 4.

Human immune cell populations in peripheral blood and tumors of the NSCLC (LG1306) and TNBC (MDA-MB-231) Onco-HuNSG model. Whole blood from LG1306 (A) or MDA-MB-231 (B) Onco-HuNSG mice was processed into single-cell suspensions and stained, as described in Materials and Methods, for flow cytometric analysis. Tumor tissues from LG1306 (C) or MDA-MB-231 (D) Onco-HuNSG mice were processed into single-cell suspensions and stained, as described in Materials and Methods, for flow cytometric analysis. The percentage of hCD3+CD4+ T cells, hCD3+CD8+ T cells, and hCD19+ B cells is shown within CD45+ cells. *P < 0.05; **P < 0.01.
In the tumor tissues of Onco-HuNSG mice, no significant differences in numbers of TILs were observed in either group in the NSCLC (LG1306) model (Fig. 4C), whereas in the TNBC (MDA-MB-231) tumor model, pembrolizumab decreased the frequency of both CD45+ and CD3+CD4+ T-cell populations (Fig. 4D; P < 0.05). B cells were not detected in the tumor tissues in either of these tumor models (data not shown).
To determine the location of TILs, in situ immunofluorescence microscopy analysis of NSCLC (LG1306) tumors was performed (Fig. 5A, B). Our results showed low numbers of CD8+ T cells present in both tumor margin (stromal tumor edge) and inside tumor parenchyma (tumor center) in saline-treated vehicle (Fig. 5A) and pembrolizumab (Fig. 5B) groups. However, in the TNBC (MDA-MD-231) Onco-HuNSG tumor model (Fig. 5C, D), we found large numbers of CD8+ T cells scattered around the tumor margin in the saline-treated vehicle group (Fig. 5C), whereas most CD8+ T cells were located within the tumor parenchyma in pembrolizumab group (Fig. 5D). Further quantification analysis using histocytometry was also conducted to show the drastic difference between T cells located in the tumor center vs. the tumor parenchyma (Supplemental Fig. S6). Our data also indicate that the number of tumor-infiltrating CD8+ T cells was quantitatively higher in the TNBC (MDA-MB-231) tumor than in the NSCLC (LG1306) tumor, which was consistent with our flow cytometry analysis of the tumor tissues from these 2 models (Fig. 4C, D).
Figure 5.

In situ characterization of CD8+ T-cell infiltration into the tumor. A, B) Tumor tissues from LG1306 were processed for histologic analysis and triple stained with DAPI (blue), hCD45 (yellow), and hCD8 (red). Representative tumor samples are shown from LG1306 treated with vehicle control (A) or pembrolizumab (B). CD8+ T cells were observed in both the tumor margin and center. C, D) Tumor tissues from MDA-MB-231 were processed for histologic analysis and stained with DAPI (blue) and hCD8 (red). Representative tumor samples are shown from MDA-MB-231 treated with vehicle control (C) or pembrolizumab (D). More CD8+ T cells were observed in the tumor center than the tumor margin in the pembrolizumab-treated tumor.
Tumor response to anti-PD-1 therapy in Onco-HuNSG mice is mediated by hCD8+ T cells
To evaluate the role of human immune cells in tumors after pembrolizumab treatment, we compared the tumor growth rates of the PDX lung model NSCLC (LG1306P5) in Onco-HuNSG and NSG mice (Fig. 6A, B). In Onco-HuNSG mice, 11 out of 13 mice responded to pembrolizumab treatment (Fig. 6A). In contrast, in NSG mice, pembrolizumab treatment resulted in a similar tumor growth rate as saline-treated vehicle control mice (Fig. 6B), indicating that the efficacy of pembrolizumab is dependent on the presence of human immune cells. Indeed, tumor regression in patients after pembrolizumab treatment requires pre-existing CD8+ T cells that are negatively regulated by PD-1/PD-L1-mediated adaptive-immune resistance (34). To define the role of hCD8+ T cells in the response to pembrolizumab in Onco-HuNSG mice, we depleted hCD8+ T cells by anti-hCD8 mAb treatment before pembrolizumab treatment. Our data revealed in TNBC (MDA-MB-231) Onco-HuNSG mice that the depletion of hCD8+ T cells results in the abrogation of the efficacy of pembrolizumab (Fig. 6C; P < 0.001). Therefore, hCD8+ T cells are required for anti-PD-1 therapy efficacy against the TNBC MDA-MB-231 tumor in Onco-HuNSG mice.
Figure 6.
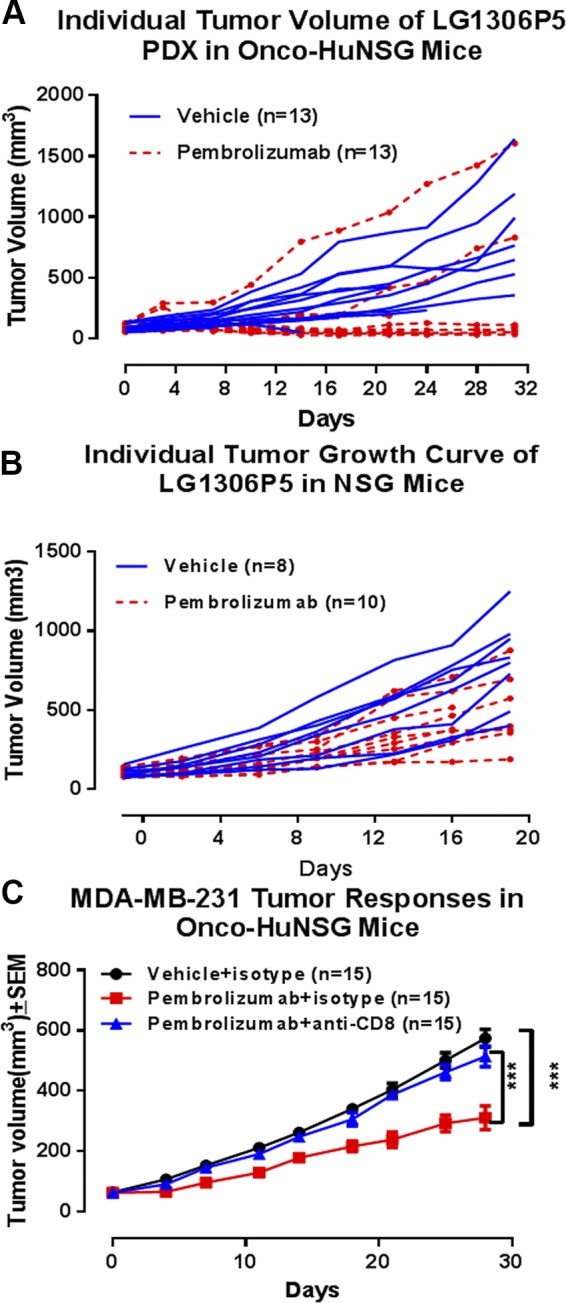
Efficacy of pembrolizumab is mediated by hCD8+ T cells in Onco-HuNSG mice. NSCLC PDX LG1306P5 was implanted in HuNSG mice (A) or NSG mice (B). A) Vehicle control saline or pembrolizumab (5 mg/kg) was injected intraperitoneally every 5 d for 30 d on d 0, 5, 10, 15, 20, and 25 in HuNSG mice. B) Vehicle control saline and pembrolizumab (5 mg/kg) was injected intraperitoneally every 5 d for 20 d on d 0, 5, 10, and 15 in NSG mice. C) MDA-MB-231 cells (5 × 106) were injected into the mammary fat pads in HuNSG mice. When the tumor reached 50–120 mm3, the mice were treated with pembrolizumab or vehicle on d 1, 6, 11, 16, 21, and 26. Isotype control antibody or anti-hCD8 antibody was injected intraperitoneally on d 0, 7, 14, 21, and 28. Tumor growth response curves were analyzed by 2-way ANOVA, followed by Dunnett’s posttests. ***P < 0.001.
DISCUSSION
Here, we show that the Onco-HuNSG model, using allogeneic but HLA partially matched CD34+ HPSC donors and tumors, might be used for cancer immunotherapy studies. Indeed, we demonstrated significant tumor growth delay following pembrolizumab (anti-PD-1) therapy in Onco-HuNSG mice. More CD8+ T cells were found infiltrating the center of the tumor post-anti-PD-1 treatment, whereas most CD8+ T cells in vehicle-treated control mice were scattered at the margin of the tumor. Our data further documented that the efficacy of pembrolizumab was mediated by hCD8+ T cells.
The immune responses in allogeneic Onco-HuNSG mice
The immune system functionality of human HPSC-engrafted NSG mice has been validated in studies, demonstrating the rejection of major histocompatibility complex (MHC) mismatched human islets allografts (35) and xenogeneic mouse skin grafts (36). The inability of Onco-HuNSG mice to reject partially HLA-matched tumors confirms the sensitivity of the humanized mouse immune system to regulation and provides a powerful, new model to identify immune-evasion mechanisms for cancers and to test immunotherapies to overcome tumor-mediated immune suppression.
The mechanisms for tumor rejection in Onco-HuNSG mice treated with anti-PD-1 remain to be elucidated. However, a large fraction of T cells developing in HPSC-engrafted mice are educated in the mouse thymus and are restricted to H2-MHC (37). Thus, we hypothesize that recognition of tumors in Onco-HuNSG mice is mediated by alloreactive or xenoreactive T cells, similar to the mechanisms involved in a “graft versus tumor” response. Clinical studies have suggested that immunotherapies also enhance graft versus tumor responses, and these findings support the clinical relevance of the Onco-HuNSG model (38). Ishikawa et al. (5) demonstrated that positive selection on human HLA+ HPSC-derived cells in the mouse thymus is possible, suggesting an allogeneic response to tumor recognition in the Onco-HuNSG model. Numerous studies have shown the existence of highly peptide-specific alloreactive T cells [reviewed in Felix and Allen (39)], indicating the generation of tumor peptide-specific T-cell activation in the allogeneic response. The abundant cytokines and T-cell costimulatory ligands generated by the allogeneic response might favor the development of T-cell responses to tumor peptides, overcoming T-cell anergy previously (40). The development of humanized mouse models to study HLA-restricted tumor antigen-specific T-cell responses is underway and will require the complete MHC matching between engrafting HPSCs and the tumor, as well as the use of HLA-expressing NSG mice for human T-cell development (41). Recent advancements in generating human HPSCs from induced pluripotent stem cells or embryonic stem cells will facilitate the generation of humanized Onco-HuNSG mice that are engrafted with autologous tumors and HPSCs in the future (42).
T-cell profile in Onco-HuNSG mice after pembrolizumab treatment
To evaluate the efficacy of the anti-PD-1 immune-checkpoint inhibitor pembrolizumab in the Onco-HuNSG model, we selected 3 tumor models with high PD-L1 surface-expression levels on tumors. Pembrolizumab exhibited a significant tumor growth inhibitory effect on all 3 tumor models tested in Onco-HuNSG mice (Fig. 2). Ashizawa et al. (43) has shown that anti-PD-1 treatment delayed the growth of 2 CDX tumors (i.e., head and neck squamous cell carcinoma SCC-3 and glioblastoma U87) in MHC class I and II double-knockout humanized peripheral blood mononuclear cell-NOD-scid/IL-2Rγ null mice. We demonstrated herein in Onco-HuNSG mice that pembrolizumab can inhibit tumor growth, not only in CDX but also in various PDX tumor models. Our results showed that the efficacy of pembrolizumab is dependent on the engraftment of an adaptive human immune system in Onco-HuNSG mice, specifically hCD8+ T cells, as the tumor inhibitory effect of pembrolizumab was absent in NSG mice that lacked human immune cells and in hCD8+ T-cell-depleted Onco-HuNSG mice (Fig. 6).
We found that pembrolizumab increased both CD4+ and CD8+ T-cell numbers in the blood of the 2 NSCLC (LG1306P5 and LG0978P5 responder) Onco-HuNSG models but decreased both CD4+ and CD8+ T-cell numbers in the blood of the TNBC (MDA-MB-231) Onco-HuNSG model (Figs. 3 and 4). Anti-PD-1 therapy has been demonstrated to decrease Treg cell populations (44, 45). It is possible that there are more Treg cells in the peripheral blood and tumor tissues of the MDA-MB-231 Onco-HuNSG model that are susceptible to pembrolizumab treatment. Therefore, anti-PD-1 therapy resulted in altered compositions of human T-cell populations in a tumor model-dependent manner.
We also found different distribution patterns of CD8+ T cells in pembrolizumab-treated tumors compared with the vehicle saline-treated group. In TNBC (MDA-MB-231) Onco-HuNSG tumors, successful pembrolizumab treatment resulted in more CD8+ T-cell infiltration into the center of the tumor, whereas most CD8+ T cells localized at the margin of the tumor in the vehicle saline group (Fig. 5C, D). The ratio of CD8+ T cells in the tumor center to invasive margin is positively correlated to overall survival rate in patients with colorectal cancer (46). Moreover, PD-L1 expression has been detected mainly at the invasive margin of the tumor, colocalizing with TILs in patients with cancer (47), possibly acting as a barrier for neighboring TILs, which could otherwise enter the center of the tumor. In this study, we did not detect proliferation or increased intratumoral infiltration of CD8+ T cells, as reported by others (34). The inconsistency between our data and the clinical data (48), in terms of intratumoral CD8+ T-cell infiltrates, could be a result of inefficient human lymphocyte trafficking to the tumor, as well as the dampened dendritic cell activation in the Onco-HuNSG mice (49, 50). This might be why we detected increased CD4+ and CD8+ T-cell populations in the blood of NSCLC (LG1306) Onco-HuNSG mice (Fig. 4A) but not at the tumor site (Fig. 4C). It is also possible that the release of the immune checkpoints by anti-PD-1 blockade, using pembrolizumab, can lead to more activated effector TILs on a per-cell basis.
HPSC donor variability in response to pembrolizumab
We observed variability from different CD34+ HPSC donors, which is in line with different response rates of patients to anti-PD-1 therapy in the clinics. The underlying mechanisms of donor variation in response to pembrolizumab treatment are unclear. In the NSCLC (LG0978P5) Onco-HuNSG model, we found higher CD4+ and CD8+ T-cell levels in the anti-PD-1 treatment group compared with vehicle control in responder donor mice peripheral blood (Fig. 3E), whereas no increased T-cell levels were observed in nonresponder donor mice (Fig. 3F). Furthermore, we did not find any correlation between HLA-matching status and response rate (Table 1). Immunotherapies that rely on T-cell activation against tumor cells are driven by recognition of neoantigens formed as a consequence of tumor-specific DNA mutations, where only a minority of the neoantigens was recognized by autologous T cells from the patients. In addition, even within healthy donor populations, only some donors can induce specific T-cell reactivity against the neoantigens from the tumor (51). Therefore, the donor variation exhibited in the Onco-HuNSG model may derive from the vast variety of the T-cell repertoire across the human population, which leads to antitumor responses in some but not all patients.
In summary, our Onco-HuNSG model provides a novel and promising platform for testing efficacy of immunotherapies, such as immune-checkpoint inhibitors in activation of T cells.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank the Flow Contract Site Laboratory (Bothell, WA, USA) for processing and staining samples for flow cytometry, and Pure Transplant Solutions (Oklahoma City, OK, USA) for HLA typing. This study was funded by The Jackson Laboratory. The authors declare no conflicts of interest.
Glossary
- CDX
cell line-derived xenograft
- HLA
human leukocyte antigen
- HPSC
hematopoietic progenitor and stem cell
- HuNSG
humanized nonobese diabetic (NOD).Cg-PrkdcscidIL2rgtm1Wjl/Sz
- MHC
major histocompatibility complex
- NOD
nonobese diabetic
- NSCLC
non–small cell lung cancer
- NSG
nonobese diabetic (NOD).Cg-PrkdcscidIL2rgtm1Wjl/Sz
- PD-1
programmed cell death protein 1
- PD-L1/2
programmed cell death protein 1/2 ligand
- PDX
patient-derived xenograft
- PE
phycoerythrin
- SBT
sequence-based typing
- scid
severe combined immunodeficiency
- TIL
tumor-infiltrating lymphocyte
- TNBC
triple-negative breast cancer
- Treg
regulatory T
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
M. Wang, K. Palucka, and J. G. Keck designed research. M. Wang, M. Cheng, D. Cai, and J. G. Keck developed methodology. M. Wang, L.-C. Yao, J. Martinek, A.-H. Ma, and W. Shi performed research. M. Wang, C.-X. Pan, and K. Palucka analyzed data. C.-X. Pan, R. W. De Vere White, S. Airhart, E. T. Liu, J. Banchereau, M. A. Brehm, and D. L. Greiner consulted and advised on the research. M. Wang, C.-X. Pan, R. W. De Vere White, S. Airhart, E. T. Liu, J. Banchereau, M. A. Brehm, D. L. Greiner, L. D. Shultz, K. Palucka, and J. G. Keck wrote and reviewed the manuscript.
REFERENCES
- 1.Hackam D. G., Redelmeier D. A. (2006) Translation of research evidence from animals to humans. JAMA 296, 1731–1732 https://doi.org/10.1001/jama.296.14.1731 [DOI] [PubMed] [Google Scholar]
- 2.Mestas J., Hughes C. C. (2004) Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 https://doi.org/10.4049/jimmunol.172.5.2731 [DOI] [PubMed] [Google Scholar]
- 3.Theocharides A. P., Rongvaux A., Fritsch K., Flavell R. A., Manz M. G. (2016) Humanized hemato-lymphoid system mice. Haematologica 101, 5–19 https://doi.org/10.3324/haematol.2014.115212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rongvaux A., Willinger T., Martinek J., Strowig T., Gearty S. V., Teichmann L. L., Saito Y., Marches F., Halene S., Palucka A. K., Manz M. G., Flavell R. A. (2014) Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 32, 364–372 https://doi.org/10.1038/nbt.2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishikawa F., Yasukawa M., Lyons B., Yoshida S., Miyamoto T., Yoshimoto G., Watanabe T., Akashi K., Shultz L. D., Harada M. (2005) Development of functional human blood and immune systems in NOD/SCID/IL2 receptor gamma chain(null) mice. Blood 106, 1565–1573 https://doi.org/10.1182/blood-2005-02-0516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka S., Saito Y., Kunisawa J., Kurashima Y., Wake T., Suzuki N., Shultz L. D., Kiyono H., Ishikawa F. (2012) Development of mature and functional human myeloid subsets in hematopoietic stem cell-engrafted NOD/SCID/IL2rγKO mice. J. Immunol. 188, 6145–6155 https://doi.org/10.4049/jimmunol.1103660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palucka A. K., Gatlin J., Blanck J. P., Melkus M. W., Clayton S., Ueno H., Kraus E. T., Cravens P., Bennett L., Padgett-Thomas A., Marches F., Islas-Ohlmayer M., Garcia J. V., Banchereau J. (2003) Human dendritic cell subsets in NOD/SCID mice engrafted with CD34+ hematopoietic progenitors. Blood 102, 3302–3310 https://doi.org/10.1182/blood-2003-02-0384 [DOI] [PubMed] [Google Scholar]
- 8.Strowig T., Gurer C., Ploss A., Liu Y. F., Arrey F., Sashihara J., Koo G., Rice C. M., Young J. W., Chadburn A., Cohen J. I., Münz C. (2009) Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J. Exp. Med. 206, 1423–1434 https://doi.org/10.1084/jem.20081720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shultz L. D., Brehm M. A., Garcia-Martinez J. V., Greiner D. L. (2012) Humanized mice for immune system investigation: progress, promise and challenges. Nat. Rev. Immunol. 12, 786–798 https://doi.org/10.1038/nri3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma P., Allison J. P. (2015) Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 https://doi.org/10.1016/j.cell.2015.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khalil D. N., Smith E. L., Brentjens R. J., Wolchok J. D. (2016) The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 13, 394 https://doi.org/10.1038/nrclinonc.2016.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gajewski T. F., Schreiber H., Fu Y. X. (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 https://doi.org/10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francisco L. M., Sage P. T., Sharpe A. H. (2010) The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242 https://doi.org/10.1111/j.1600-065X.2010.00923.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardoll D. M. (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 https://doi.org/10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullard A. (2013) New checkpoint inhibitors ride the immunotherapy tsunami. Nat. Rev. Drug Discov. 12, 489–492 https://doi.org/10.1038/nrd4066 [DOI] [PubMed] [Google Scholar]
- 16.Butte M. J., Keir M. E., Phamduy T. B., Sharpe A. H., Freeman G. J. (2007) Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 https://doi.org/10.1016/j.immuni.2007.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keir M. E., Butte M. J., Freeman G. J., Sharpe A. H. (2008) PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 https://doi.org/10.1146/annurev.immunol.26.021607.090331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmadzadeh M., Johnson L. A., Heemskerk B., Wunderlich J. R., Dudley M. E., White D. E., Rosenberg S. A. (2009) Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544 https://doi.org/10.1182/blood-2008-12-195792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freeman G. J., Long A. J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L. J., Malenkovich N., Okazaki T., Byrne M. C., Horton H. F., Fouser L., Carter L., Ling V., Bowman M. R., Carreno B. M., Collins M., Wood C. R., Honjo T. (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 https://doi.org/10.1084/jem.192.7.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latchman Y., Wood C. R., Chernova T., Chaudhary D., Borde M., Chernova I., Iwai Y., Long A. J., Brown J. A., Nunes R., Greenfield E. A., Bourque K., Boussiotis V. A., Carter L. L., Carreno B. M., Malenkovich N., Nishimura H., Okazaki T., Honjo T., Sharpe A. H., Freeman G. J. (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 https://doi.org/10.1038/85330 [DOI] [PubMed] [Google Scholar]
- 21.Dong H., Zhu G., Tamada K., Chen L. (1999) B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 5, 1365–1369 https://doi.org/10.1038/70932 [DOI] [PubMed] [Google Scholar]
- 22.Okazaki T., Honjo T. (2007) PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 19, 813–824 https://doi.org/10.1093/intimm/dxm057 [DOI] [PubMed] [Google Scholar]
- 23.Yokosuka T., Takamatsu M., Kobayashi-Imanishi W., Hashimoto-Tane A., Azuma M., Saito T. (2012) Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 209, 1201–1217 https://doi.org/10.1084/jem.20112741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou W., Chen L. (2008) Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 8, 467–477 https://doi.org/10.1038/nri2326 [DOI] [PubMed] [Google Scholar]
- 25.Garon E. B., Rizvi N. A., Hui R., Leighl N., Balmanoukian A. S., Eder J. P., Patnaik A., Aggarwal C., Gubens M., Horn L., Carcereny E., Ahn M. J., Felip E., Lee J. S., Hellmann M. D., Hamid O., Goldman J. W., Soria J. C., Dolled-Filhart M., Rutledge R. Z., Zhang J., Lunceford J. K., Rangwala R., Lubiniecki G. M., Roach C., Emancipator K., Gandhi L.; KEYNOTE-001 Investigators . (2015) Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 https://doi.org/10.1056/NEJMoa1501824 [DOI] [PubMed] [Google Scholar]
- 26.Robert C., Ribas A., Wolchok J. D., Hodi F. S., Hamid O., Kefford R., Weber J. S., Joshua A. M., Hwu W. J., Gangadhar T. C., Patnaik A., Dronca R., Zarour H., Joseph R. W., Boasberg P., Chmielowski B., Mateus C., Postow M. A., Gergich K., Elassaiss-Schaap J., Li X. N., Iannone R., Ebbinghaus S. W., Kang S. P., Daud A. (2014) Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 384, 1109–1117 https://doi.org/10.1016/S0140-6736(14)60958-2 [DOI] [PubMed] [Google Scholar]
- 27.Topalian S. L., Hodi F. S., Brahmer J. R., Gettinger S. N., Smith D. C., McDermott D. F., Powderly J. D., Carvajal R. D., Sosman J. A., Atkins M. B., Leming P. D., Spigel D. R., Antonia S. J., Horn L., Drake C. G., Pardoll D. M., Chen L., Sharfman W. H., Anders R. A., Taube J. M., McMiller T. L., Xu H., Korman A. J., Jure-Kunkel M., Agrawal S., McDonald D., Kollia G. D., Gupta A., Wigginton J. M., Sznol M. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 https://doi.org/10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamid O., Robert C., Daud A., Hodi F. S., Hwu W. J., Kefford R., Wolchok J. D., Hersey P., Joseph R. W., Weber J. S., Dronca R., Gangadhar T. C., Patnaik A., Zarour H., Joshua A. M., Gergich K., Elassaiss-Schaap J., Algazi A., Mateus C., Boasberg P., Tumeh P. C., Chmielowski B., Ebbinghaus S. W., Li X. N., Kang S. P., Ribas A. (2013) Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 369, 134–144 https://doi.org/10.1056/NEJMoa1305133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibney G. T., Weiner L. M., Atkins M. B. (2016) Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 17, e542–e551 https://doi.org/10.1016/S1470-2045(16)30406-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taube J. M., Klein A., Brahmer J. R., Xu H., Pan X., Kim J. H., Chen L., Pardoll D. M., Topalian S. L., Anders R. A. (2014) Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 20, 5064–5074 https://doi.org/10.1158/1078-0432.CCR-13-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shultz L. D., Lyons B. L., Burzenski L. M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S. D., King M., Mangada J., Greiner D. L., Handgretinger R. (2005) Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 174, 6477–6489 https://doi.org/10.4049/jimmunol.174.10.6477 [DOI] [PubMed] [Google Scholar]
- 32.Gerner M. Y., Kastenmuller W., Ifrim I., Kabat J., Germain R. N. (2012) Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 37, 364–376 https://doi.org/10.1016/j.immuni.2012.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fridman W. H., Pagès F., Sautès-Fridman C., Galon J. (2012) The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer 12, 298–306 https://doi.org/10.1038/nrc3245 [DOI] [PubMed] [Google Scholar]
- 34.Tumeh P. C., Harview C. L., Yearley J. H., Shintaku I. P., Taylor E. J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., West A. N., Carmona M., Kivork C., Seja E., Cherry G., Gutierrez A. J., Grogan T. R., Mateus C., Tomasic G., Glaspy J. A., Emerson R. O., Robins H., Pierce R. H., Elashoff D. A., Robert C., Ribas A. (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 https://doi.org/10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brehm M. A., Bortell R., Diiorio P., Leif J., Laning J., Cuthbert A., Yang C., Herlihy M., Burzenski L., Gott B., Foreman O., Powers A. C., Greiner D. L., Shultz L. D. (2010) Human immune system development and rejection of human islet allografts in spontaneously diabetic NOD-Rag1null IL2rgammanull Ins2Akita mice. Diabetes 59, 2265–2270 https://doi.org/10.2337/db10-0323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waldron-Lynch F., Henegariu O., Deng S., Preston-Hurlburt P., Tooley J., Flavell R., Herold K. C. (2012) Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients. Sci. Transl. Med. 4, 118ra12 https://doi.org/10.1126/scitranslmed.3003401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh N. C., Kenney L. L., Jangalwe S., Aryee K. E., Greiner D. L., Brehm M. A., Shultz L. D. (2017) Humanized mouse models of clinical disease. Annu. Rev. Pathol. 12, 187–215 https://doi.org/10.1146/annurev-pathol-052016-100332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merryman R. W., Kim H. T., Zinzani P. L., Carlo-Stella C., Ansell S. M., Perales M. A., Avigdor A., Halwani A. S., Houot R., Marchand T., Dhedin N., Lescaut W., Thiebaut-Bertrand A., François S., Stamatoullas-Bastard A., Rohrlich P. S., Labussière Wallet H., Castagna L., Santoro A., Bachanova V., Bresler S. C., Srivastava A., Kim H., Pesek E., Chammas M., Reynolds C., Ho V. T., Antin J. H., Ritz J., Soiffer R. J., Armand P. (2017) Safety and efficacy of allogeneic hematopoietic stem cell transplant after PD-1 blockade in relapsed/refractory lymphoma. Blood 129, 1380–1388 https://doi.org/10.1182/blood-2016-09-738385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felix N. J., Allen P. M. (2007) Specificity of T-cell alloreactivity. Nat. Rev. Immunol. 7, 942–953 https://doi.org/10.1038/nri2200 [DOI] [PubMed] [Google Scholar]
- 40.Zilberberg J., Feinman R., Korngold R. (2015) Strategies for the identification of T cell-recognized tumor antigens in hematological malignancies for improved graft-versus-tumor responses after allogeneic blood and marrow transplantation. Biol. Blood Marrow Transplant. 21, 1000–1007 https://doi.org/10.1016/j.bbmt.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shultz L. D., Saito Y., Najima Y., Tanaka S., Ochi T., Tomizawa M., Doi T., Sone A., Suzuki N., Fujiwara H., Yasukawa M., Ishikawa F. (2010) Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc. Natl. Acad. Sci. USA 107, 13022–13027 https://doi.org/10.1073/pnas.1000475107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J., Dykstra B., Spencer J. A., Kenney L. L., Greiner D. L., Shultz L. D., Brehm M. A., Lin C. P., Sackstein R., Rossi D. J. (2017) mRNA-mediated glycoengineering ameliorates deficient homing of human stem cell-derived hematopoietic progenitors. J. Clin. Invest. 127, 2433–2437 https://doi.org/10.1172/JCI92030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashizawa T., Iizuka A., Nonomura C., Kondou R., Maeda C., Miyata H., Sugino T., Mitsuya K., Hayashi N., Nakasu Y., Maruyama K., Yamaguchi K., Katano I.,, Ito M., Akiyama Y. (2016) Antitumor effect of programmed death-1 (PD-1) blockade in humanized the NOG-major histocompatibility complex (MHC) double knockout mouse. Clin Cancer Res. 23, 149–158 [DOI] [PubMed] [Google Scholar]
- 44.Rosenblatt J., Glotzbecker B., Mills H., Vasir B., Tzachanis D., Levine J. D., Joyce R. M., Wellenstein K., Keefe W., Schickler M., Rotem-Yehudar R., Kufe D., Avigan D. (2011) PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J. Immunother. 34, 409–418 https://doi.org/10.1097/CJI.0b013e31821ca6ce [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGee H. S., Yagita H., Shao Z., Agrawal D. K. (2010) Programmed death-1 antibody blocks therapeutic effects of T-regulatory cells in cockroach antigen-induced allergic asthma. Am. J. Respir. Cell Mol. Biol. 43, 432–442 https://doi.org/10.1165/rcmb.2009-0258OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galon J., Costes A., Sanchez-Cabo F., Kirilovsky A., Mlecnik B., Lagorce-Pagès C., Tosolini M., Camus M., Berger A., Wind P., Zinzindohoué F., Bruneval P., Cugnenc P. H., Trajanoski Z., Fridman W. H., Pagès F. (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964 https://doi.org/10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- 47.Taube J. M., Anders R. A., Young G. D., Xu H., Sharma R., McMiller T. L., Chen S., Klein A. P., Pardoll D. M., Topalian S. L., Chen L. (2012) Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 4, 127ra37 https://doi.org/10.1126/scitranslmed.3003689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ribas A., Shin D. S., Zaretsky J., Frederiksen J., Cornish A., Avramis E., Seja E., Kivork C., Siebert J., Kaplan-Lefko P., Wang X., Chmielowski B., Glaspy J. A., Tumeh P. C., Chodon T., Pe’er D., Comin-Anduix B. (2016) PD-1 blockade expands intratumoral memory T cells. Cancer Immunol. Res. 4, 194–203 https://doi.org/10.1158/2326-6066.CIR-15-0210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y., Mention J. J., Court N., Masse-Ranson G., Toubert A., Spits H., Legrand N., Corcuff E., Strick-Marchand H., Di Santo J. P. (2016) A novel Flt3-deficient HIS mouse model with selective enhancement of human DC development. Eur. J. Immunol. 46, 1291–1299 https://doi.org/10.1002/eji.201546132 [DOI] [PubMed] [Google Scholar]
- 50.Chen Q., Khoury M., Chen J. (2009) Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc. Natl. Acad. Sci. USA 106, 21783–21788 https://doi.org/10.1073/pnas.0912274106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strønen E., Toebes M., Kelderman S., van Buuren M. M., Yang W., van Rooij N., Donia M., Böschen M. L., Lund-Johansen F., Olweus J., Schumacher T. N. (2016) Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341 https://doi.org/10.1126/science.aaf2288 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.