

Abstract
The unified airway model has developed from indications that the upper and lower respiratory tracts share key elements of pathogenesis. These shared traits likely extend to similar niche characteristics that support bacterial communities, and as such, we suspect that similar microbes exist on upper and lower respiratory tract epithelium. Over the past decade and a half there have been significant improvements in microbiological identification and analysis due to the development of new molecular technologies, including next-generation sequencing. In this review, we provide an overview of the modern collection and sequencing methods involved in respiratory microbiota research, and outline the specific microbial communities that have been found to be associated with the healthy and diseased human respiratory tract. Demonstration of a remarkable similarity between the upper and lower respiratory tract in terms of microbiological presence adds further corroboration to the existence of a unified airway.
Keywords: Respiratory, Bacteria, Microbiota
1. Introduction
Rhinitis and asthma share key elements of pathogenesis and have long been noted to co-occur, suggesting that the upper and lower respiratory tracts are more than just physically connected [1,2]. Indeed, the unified airway model suggests that immunological responses in one section of the respiratory tract can be linked to responses in other areas [3–5]. Data from the Human Microbiome Project (HMP) presents further supportive evidence of this model. Bacterial communities in healthy lungs have been shown to be highly similar to those in the upper respiratory tract, and shifts in one anatomical location may be associated with changes in others [6,7]. This is perhaps not surprising due to the ecologically similar niches found throughout the respiratory tract, many of which are covered in a continuous mucosal layer, and bathed in mucus and saliva.
Research in the past few decades has shown that many of the roughly 1014 microbial cells that live in and on our bodies are necessary for our wellbeing [8]. This consortium of microbes, termed the microbiota, serve a number of functions including priming the immune system, digesting food, providing nutrients and vitamins, and protecting us from potential pathogens [9]. Bacteria are estimated to outnumber human cells 10 to 1, and contain more collective genetic content than that found in human cells. Indeed, every surface of the human body in contact with the outside world is coated in microbes, including the gastrointestinal tract from the mouth to the anus, the respiratory tract from the mouth to the lungs, the entirety of our skin, even our eyes [6,7,10,11]. Interestingly, out of the about 50 known bacterial phyla, humans generally only associate with members from ten of these phyla, including Actinobacteria, Bacteroidetes, Cyanobacteria, Firmicutes, Fusobacteria, Proteobacteria, Spirochaetes, Tenericutes, TM7, and Verrucomicrobia [12].
The methods used to study these bacterial communities has changed drastically in the past decade, due in large part to a vast increase in computing power coupled with the advent of new molecular technologies, often called “next-generation sequencing” (NGS), that generate thousands to millions of sequences per sample [13]. While standard culture-dependent work certainly laid the groundwork for this field of research, NGS has opened the door to a better understanding of the breadth and depth of the human microbiota. Understanding what grows attached to surfaces of the human body may give us not only a better view of what it means to be healthy, but also open avenues to a better understanding of chronic diseases that may be associated with an altered microbiota, and may aid in development of better treatments and perhaps even preventative measures.
The goals of this review are three-fold: 1) give a brief overview of how human microbiota research is conducted in general and specifically in the respiratory system; 2) review the microbial community found associated with the healthy and diseased human respiratory tract; and 3) discuss how this corroborates the unified airway model.
2. Human microbiota – modern sequencing methods
Study of the human microbiota starts with sample collection. Samples may be collected in many different ways, depending on the site being studied and the questions researchers are trying to answer. Swabs and brushes have both been used to physically remove bacteria from any surface the implement can reach, along with instruments that more literally scrap the epithelia. Likewise, liquids (saliva, sputum, vaginal secretions, and gastric juices) and solids (feces) can be used. Historically, and still today in many labs, these samples were used in culture based assays, where an attempt was made to grow bacteria on agar plates or in liquid media, and then identify the bacteria present in the original sample. However, today we know that many bacteria associated with humans are not easily cultured, and this older method of sampling missed the vast majority of bacteria associated with humans [14]. The inability to culture bacteria from some areas of the body, such as the stomach and lungs, led to the belief that these sites were sterile. This idea has been shown to be incorrect, though many of these bacteria remain elusive in terms of required culturing conditions [14].
The discovery of the 16S rRNA (16S) gene and the advent of polymerase chain reaction (PCR) birthed a new era in microbial ecology research. Pioneering work by Carl Woese demonstrated that distinct groups of bacteria could be identified based on the 16S gene [15]. The 16S gene, universally present in all bacteria, is about 1500 base pairs long and contains nine hypervariable regions flanked by highly conserved regions (Fig. 1). This gene structure is highly amenable for identifying bacteria; universal primers can be designed for the conserved regions, while the intervening regions can be used for sequencing and identification of bacteria. Out of this came a number of techniques to explore microbial communities using molecular techniques, such as clone libraries, denaturing gradient gel electrophoresis (DGGE), and terminal restriction fragment length polymorphism (tRFLP), amongst others [16,17]. The drawbacks of these techniques have made them less popular, and they have mostly been replaced by newer sequencing technologies.
Fig. 1.

Basic structure of the 16S rRNA gene found in all bacteria. The gene product of the 16S rRNA gene serves a structural role in the 30S small subunit of all bacterial ribosomes. The 16S gene is roughly 1500 base pairs long, and characterized by nine hypervariable regions, marked as V1-V9 in light gray, dispersed between highly conserved regions, denoted in dark gray. Hash marks are placed every 100 base pairs, and the diagram has been drawn to scale. Useful in identifying bacteria, primers can be designed for these highly conserved regions and the intervening sequences used for identification of bacteria. Commonly used primers are noted in the figure including arrows noting the direction they are used for amplification in PCR. Choice of primer pair, one forward and one reverse, is determined by a number of different options. Particularly important is production of an appropriate length amplicon for the chosen sequencing platform. Illumina platforms such as the HiSeq and MiSeq require shorter amplicons, such as those produced from using 519F and 806R. Primers are named based on their base pair position in the Escherichia coli 16S gene, with F for forward primers and R for reverse primers.
The advent of NGS techniques, so named because they further the ideas of Sanger sequencing, made possible the description and exploration of microbial communities like never before [18,19]. Where before a successful clone library may have yielded a few hundred clones from a handful of samples, we can now recover millions of sequences, easily multiplexed across numerous samples. Sequencing platforms such as the MiSeq and HiSeq from Illumina, and SMRT sequencing from Pacific Biosciences, in addition to others, make this possible. In short, for bacterial community analysis using the 16S gene, PCR products are produced targeting relatively short regions of the 16S gene using universal bacterial primers that also contain sequences specific for the sequencing platform being used. Primers are usually also designed with barcodes for each sample allowing each sample to be labeled with a unique 4-12 bp DNA fragment that, after sequencing, can be tied back to the original sample [20]. This also allows PCR products from numerous samples to be pooled, sequenced en masse, and the sequences for each sample to be separated later based on the assigned barcode using bioinformatics programs such as mothur or QIIME [21,22].
These current sequencing techniques are not without bias. Results from experiments may vary based on the sampling technique, DNA extraction protocol, polymerase used for PCR along with the primer choice and region of the 16S gene sequenced [23–25]. Likewise, multiple protocols and programs exist for processing sequences [21,22], checking sequence quality and removing noise [26,27], and detecting chimeras [28,29]. Sequence identification can also vary, as the array of databases for identification purposes each have their own strengths and weaknesses [30–32]. Even how sequences are aligned to these databases is important [33]. Each step in the overall protocol for how samples are handled introduces bias and studies done using different protocols are not always easily comparable to each other.
3. Sampling the respiratory system as a niche for bacteria
For the purposes of this review, we use the term ‘respiratory tract’ loosely to include all epithelial surfaces associated with respiration and the path air takes to reach the lungs, including surfaces from the anterior nares into the nasal cavity and sinuses, back to the nasopharynx, the soft tissue of the oral cavity, back to the oropharynx, down through the larynx, trachea, and finally, to the lungs (Fig. 2). Two types of epithelia dominate these surfaces. Ciliated pseudostratified columnar epithelium lines the sinuses, nasal cavity, nasopharynx, larynx, and trachea, while the oral cavity, oropharynx, and vocal folds are lined with stratified squamous epithelium. Mucin producing goblet cells are found only in ciliated pseudostratified columnar epithelium, while submucosal glands found throughout the respiratory tract are also known to produce mucus.
Fig. 2.
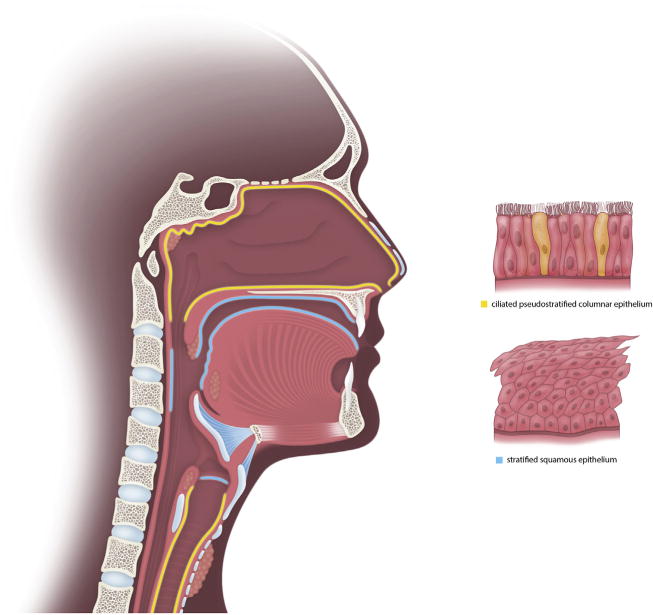
Illustration of the basic anatomy of the respiratory system with the corresponding epithelial types for each anatomical site. Ciliated pseudostratified columnar epithelium (yellow) is found in the sinuses, nasal cavity, nasopharynx, larynx, and trachea. Stratified squamous epithelium (blue) lines the oral cavity, oropharynx, and vocal cords.
Mucin, a heavily O-glycosylated protein, is found in both saliva and the continuous mucus layer that covers all epithelial surfaces of the respiratory tract. It likely serves as the primary source of nutrients for bacteria in the respiratory tract [34]. Multispecies bacterial communities are known to digest mucin, often in cooperation using overlapping patterns of enzyme activity [35]. Partial consumption by bacteria of mucins, which have no known antibacterial activity, suggests that part of their physiological function is to serve as nutrients for bacteria [34]. Saliva, despite containing antimicrobial compounds such as lysozyme, lactoferrin, lactoperoxidase, and IgA, also supports bacterial growth [36]. In addition to nutrients, mucus attachment to the epithelial surface provides an adhesion site for bacteria, decreasing the rate at which they are eliminated from the respiratory tract despite mucociliary beating that is generally accepted to move mucus out of the respiratory system.
Many surfaces in the respiratory tract are highly amenable to sampling for bacterial community analyses, including those in the oral cavity, nasopharynx, oropharynx, anterior nares, and nasal vestibule. Design of the Human Microbiome Project (HMP) was particularly cognizant of this, and chose to focus on seven easy to sample sites to describe the bacterial community of the oral cavity and respiratory system including the tongue dorsum, hard palate, buccal mucosa, keratinized gingiva, palatine tonsils, oropharynx, and saliva [37]. Conversely, sampling from other sites of the respiratory tract, such as the lungs, sinuses, and vocal folds are considered invasive. Large-scale studies, like the HMP, for these sites in the respiratory system remain rare due to these limitations.
Collection of samples in the respiratory tract can be taken in a number of ways. Swab and brush sampling can be done on any location that can be reached while minimizing damage to the epithelial surface. Additionally, fluids such as saliva and induced sputum have been used for many studies, as have washes from locations such as the oral cavity, sinus cavities, and lungs. However, cross contamination in sites such as the lungs can be problematic, where the bronchoscope required for sampling may contaminate the lower respiratory system with bacteria from the upper respiratory system. Charlson et al developed a two bronchoscope approach to address this concern, where samples taken from the glottis with the first bronchoscope were used to determine what, if any, contamination a bronchoscope might transport to the lungs, and then a second bronchoscope was used to take samples from the lungs [7]. While bacterial numbers were lower in the lungs, cross contamination did not appear to be a problem. Likewise, those sampling in the sinuses have noted awareness of preventing nasal community contamination when withdrawing the sampling device [38,39].
Additional questions remain about how well swabs and brushes remove bacteria that are well attached to epithelial surfaces. While swabbing to collect microbes is highly convenient, few studies have compared the resulting community from easily removed bacteria versus those more firmly attached. Research in the ileal pouch suggests that brush sampling is sufficiently similar to samples taken via biopsy [40].
4. Healthy human respiratory microbiota and the unified airway model
The unified airway model suggests that different areas of the respiratory tract share many similar characteristics [3]. These shared traits likely extend to similar niche characteristics that support bacterial communities in similar mucosal surfaces, and some have even suggested that the respiratory tract should be viewed as a single ecosystem [41]. Shared traits such as a continuous mucosal layer, exposure to inhaled air, and nutrient availability in the form of mucin, may support the growth of similar microbial communities, while the differences seen may be due to local characteristics such as increased presence of saliva, exposure to ingested substances, proximity to skin surfaces, or increased oxygen concentration. Despite the methodological problems of comparing studies done using different protocols, and ethical issues of invasively sampling healthy subjects, both discussed above, a broad view of what constitutes the healthy microbial community in the respiratory tract is emerging (Table 1).
Table 1.
Table of common microbiota found within the regions of the respiratory tract.
| Oral cavity | Nasopharynx | Oropharynx | Larynx | Lung |
|---|---|---|---|---|
| Prevotella | Corynebacterium | Prevotella | Prevotella | Prevotella |
| Streptococcus | Streptococcus | Streptococcus | Streptococcus | Streptococcus |
| Veillonella | Veillonella | Veillonella | Veillonella | Veillonella |
| Haemophilus | Propionibacterium | Haemophilus | Unclassified-Comamonoadaceae | Pseudomonas |
| Staphylococcus | Fusobacterium | Cloacibacterium | Fusobacterium | |
| Moraxella | Helicobacter |
In the largest study to date, roughly 300 healthy individuals were recruited for an in depth study by the Human Microbiome Project (HMP), a large multicenter study funded by the National Institute of Health [42]. Due to the stringent exclusion criteria [37], data from the HMP provides a large framework for what the healthy human microbiota constitutes at seven sites associated with the respiratory tract (described above). These sites could be characterized by two distinct, but similar, bacterial communities that differed more in the abundance of community members rather than what taxa were present in the community [6,43]. While there is little agreement on whether a “core” microbiota exists in any body site, Prevotella, Streptococcus, and Veillonella were consistently present in the HMP subjects across all sites tested in the oral cavity and oropharynx [6]. Segata et al surmised that the similarities in the bacterial communities from these seven body sites may be due to the buffering nature of saliva and high nutrient availability in the form of mucin [6]. Recent reanalysis of this data using oligotyping, a high resolution method that looks at very fine sequence differences, suggests that a core oral community may indeed exist [44,45]. Eren et al found 58 oligotypes in 95% of HMP oral samples sequenced using V3-V5, suggesting a highly similar community across these subjects [45]. Many of these oligotypes were identified as taxa found in the original data set including Neisseria, Streptococcus, and Veillonella.
Smaller studies corroborate both the original data from the HMP and the oligotype reanalysis. Charlson et al described highly similarly communities in the nasopharynx and oropharynx of 33 healthy nonsmokers [46]. Communities in the nasopharynx were distinguished by Corynebacterium, Propionibacterium, and Staphylococcus, taxa also associated with the skin [47], while the oropharynx was distinguished by Fusobacterium, Haemophilus, Neisseria,, Prevotella, and Veillonella [46]. Despite these differences, many shared taxa were also present including Lactococcus, Leuconostoc, Streptococcus, and Veillonella. A study by Jette et al, which analyzed false vocal fold tissue biopsies from 97 healthy individuals, identified that the most common bacterial communities in the larynx are unclassified genus of Comamonadaceae, Streptococcus, Cloacibacterium, Prevotella, Propionibacterium, Helicobater, and Veillonella [48]. Follow up work by Charlson et al expanded their sampling of the naso and oropharynx to the lungs, a notoriously difficult location to sample without cross contamination with bacteria from the upper respiratory tract as discussed above. Lung samples from six healthy patients were indistinguishable from upper respiratory sites such as the oropharynx, nasopharynx, and oral washes [7]. Most samples, regardless of sample location (oral versus lung), or technique (swab versus wash) were dominated by community members in the families Prevotellaceae, Streptococcaceae, and Veillonellaceae. Like their previous work, nasopharyngeal samples also had higher abundances of bacteria often associated with the skin, including members of the families Corynebacteriaceae, Propionobacteriacae, and Staphylococcaceae. Morris et al further confirmed the ubiquity of Streptococcus, Veillonella, and Prevotella in oral washes and bronchoalveolar lavage (BAL) in a larger cohort (n = 45) of healthy nonsmokers [49].
However, study of healthy patients remains problematic and limited. Parameters used by the HMP would exclude many patients used as healthy controls in other studies. While Jensen et al found the same genera as mentioned above present in their healthy subjects in a tonsillar crypt study, these patients were also undergoing surgery for removal of vocal fold polyps and a benign tumor in the throat, conditions that would exclude these patients as healthy in the HMP [37,50]. Likewise, similar community structures have been found in the middle meatus and maxillary sinus, but control populations in these studies would also not meet the criteria for healthy in the HMP due to pituitary tumor [51], obstructive sleep apnea [52], and fungal ball [53].
Despite these challenges, and the question of who is healthy enough to include as healthy subjects in these types of studies, Prevotella, Streptococcus, and Veillonella have been found as prominent community members in all respiratory sites except the anterior nares (Table 1). This is perhaps not surprising since the nares have different niche characteristics which is likely reflected by the similarity of their microbiota to skin [12]. It is likely that characteristics of these genera allows them to flourish in the respiratory tract while being rare in other locations such as the gastrointestinal tract [6].
5. Microbiota of the diseased respiratory system
While description of the microbial community of the healthy respiratory tract is indeed interesting, the comparison to communities found in diseased patients has begun to offer insights into factors affecting disease that were not even considered until about a decade ago. The first glimpses that microbial communities might affect human health came from studies comparing obese and lean twins, which showed that despite identical genetics, obesity could be correlated to changes in the microbial community of the gastrointestinal tract [61]. Likewise, alterations in the diversity of microbial communities have been identified within the respiratory tract of smokers compared to non-smokers [46,48], and disease processes such as cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD), and asthma.
Cystic fibrosis is perhaps one of the best studied diseases affecting the respiratory tract in terms of bacterial community composition and shifts that may be associated with health and disease. Traditionally, deaths from infection in this patient group have been associated with a handful of opportunistic pathogens, such as Pseudomonas aeruginosa and Staphylococcus aureus. However, numerous groups over the past few years have clearly demonstrated that cystic fibrosis lungs are microbially more complex than previously thought, much like the diverse array of bacteria that can be found in healthy lungs. Taxa well known to inhabit the healthy respiratory system also inhabit patients with CF, such as Gemella, Prevotella, Porphyromonas, Rothia, Stenotrophomonas, and Streptococcus [54]. Likewise, the diversity of bacteria found associated with the lungs of patients with CF is higher than previously suggested by cultureand cloning-based studies [54–56]. Additionally, recent work has shown that decreases in bacterial diversity in the lungs, specifically reductions in Gemella, Granulicatella, Prevotella, Streptococcus, and Veillonella, were highly correlated with poor lung function [57].
Nasal communities were also highly similar in healthy study participants and patients with COPD and asthma, regardless of health status [58]. Though this study used clone libraries with the acknowledgement of likely undersampling the community, asthmatic lungs were highly different from healthy lungs, though their oropharynx communities were not, while both the lung and oropharynx samples from COPD patients were markedly different from healthy. Increases in Haemophilus in both COPD and asthmatics coupled with a decrease in Prevotella seem to explain most of these differences. Further work in patients with moderate and severe COPD showed decreased community diversity compared to smokers, healthy never smokers, and two patients with mild COPD [59]. Despite this, Fusobacterium, Haemophilus, Porphyromonas, Prevotella, Pseudomonas, Streptococcus, and Veillonella were still common community members. Explanted lungs from COPD patients, while dominated by Pseudomonas, also showed microanatomic heterogeneity, much like every other body surface that has been sampled [59–61].
Similar shifts in community membership have been found in other respiratory maladies. Recurrent tonsillitis in adults was associated with increases in Fusobacterium necrophorum, Streptococcus intermedius, and Prevotella melaninogenica/histicola, while pathogens traditionally thought to be the etiological agent were not found, such as Staphylococcus aureus and Streptococcus pyogenes [50]. Likewise, rhinosinusitis was shown to be associated with a decrease in lactic acid bacteria coupled with an increase in Corynebacterium tuberculostearicum, a bacterium generally considered to be a member of the skin microbiota [52]. While sampling healthy vocal folds remains problematic, benign vocal fold lesions were found to have a significantly higher abundance of Streptococcus pseudopneumoniae compared to false vocal fold biopsies [48,62].
6. The unified airway and implications of the microbiota in respiratory diseases
The unified airway model suggests that disease in one part of the respiratory tract may be associated with disease in distal locations. One might hypothesize that taking, perhaps, an oropharynx swab could indicate shifts in the microbial communities in other regions of the respiratory tract and serve as a marker for disease. While healthy communities in the lungs are highly concordant with samples from the upper respiratory tract, studies matching disease in one location to samples in others remains scant.
Goddard et al found that while sputum samples matched lung samples in cystic fibrosis patients, throat swabs were discordant [56]. However, this study was only based on three patients, and as the HMP has demonstrated, larger numbers are needed to see the overall patterns of microbial communities. In a larger study, the exact opposite conclusion was reached, where the lungs of healthy smokers (n = 19) had highly similar communities compared to oral washes [49]. Large scale studies are needed to determine if this type of noninvasive sampling is indeed a sufficient representative sample for other locations in the respiratory tract. To date, most studies in the respiratory tract of diseased individuals rely on a single sample type or do not include a large enough patient population to determine this.
7. Future directions in respiratory microbial research
Large knowledge gaps remain in our understanding of the composition and function of the microbial community associated with the respiratory tract despite these large leaps in research in the last decade and a half. While the respiratory tract is composed of many smaller niches characterized by different cell types, nutrient and oxygen availability, and the presence of saliva and mucous, the similarity between communities in these locations is remarkable. The ubiquity of species in the genera Prevotella, Streptococcus, and Veillonella speaks to their ability to survive in respiratory niches despite variable conditions.
Research is still needed to tie together the complete picture of what a healthy microbial community can look like in the respiratory system. Large scale studies like the HMP are required for the respiratory tract before we can have a full grasp of the range of community composition that may constitute healthy. Work in the gastrointestinal tract has shown that no single community can be described as healthy, rather these healthy communities may have completely different composition while still serving the same function [12]. Likewise, additional research is needed to tease out whether perturbations in one respiratory niche are related to responses and changes in other areas as noted in the unified airway theory, and particularly if they are associated with disease. Current research has largely focused on localized responses to changes in community membership and structure. These changes could ripple out in systemic responses and changes in even further distal locations of the body, such as the gastrointestinal tract and skin, may be more interrelated than currently thought.
Acknowledgments
The authors thank our sources of funding, National Institutes of Health grants R01 DC009600 and T32 DC009401, and Hannah Sandvold for her contributions to figure illustration.
Financial disclosures
This work was supported by the National Institute of Health grants R01 DC009600 and T32 DC009401.
Footnotes
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- 1.Corren J. Allergic rhinitis and asthma: how important is the link? J allergy Clin Immunol. 1997;99(2):S781–S786. doi: 10.1016/s0091-6749(97)70127-1. [DOI] [PubMed] [Google Scholar]
- 2.Lipworth BJ, White PS. Allergic inflammation in the unified airway: start with the nose. Thorax. 2000;55(10):878–881. doi: 10.1136/thorax.55.10.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krouse JH. The unified airway–conceptual framework. Otolaryngologic Clin N Am. 2008;41(2):257–266. doi: 10.1016/j.otc.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Krouse JH. The unified airway, Facial plastic Surg. Clin N Am. 2012;20(1):55–60. doi: 10.1016/j.fsc.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Krouse JH, Veling MC, Ryan MW, Pillsbury HC, 3rd, Krouse HJ, Joe S, Heller AJ, Han JK, Fineman SM, Brown RW. Executive summary: asthma and the unified airway. Otolaryngology–Head and Neck Surgery: Official J Am Acad Otolaryngology-Head Neck Surg. 2007;136(5):699–706. doi: 10.1016/j.otohns.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 6.Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D, Huttenhower C, Izard J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012;13(6):R42. doi: 10.1186/gb-2012-13-6-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit care Med. 2011;184(8):957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A. 1998;95(12):6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA. Topographical and temporal diversity of the human skin microbiome. Sci (New York, NY) 2009;324(5931):1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong Q, Brulc JM, Iovieno A, Bates B, Garoutte A, Miller D, Revanna KV, Gao X, Antonopoulos DA, Slepak VZ, Shestopalov VI. Diversity of bacteria at healthy human conjunctiva. Investigative Ophthalmol Vis Sci. 2011;52(8):5408–5413. doi: 10.1167/iovs.10-6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Bella JM, Bao Y, Gloor GB, Burton JP, Reid G. High throughput sequencing methods and analysis for microbiome research. J Microbiol methods. 2013;95(3):401–414. doi: 10.1016/j.mimet.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Lagier JC, Hugon P, Khelaifia S, Fournier PE, La Scola B, Raoult D. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin Microbiol Rev. 2015;28(1):237–264. doi: 10.1128/CMR.00014-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A. 1977;74(11):5088–5090. doi: 10.1073/pnas.74.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muyzer G, Smalla K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Leeuwenhoek. 1998;73(1):127–141. doi: 10.1023/a:1000669317571. [DOI] [PubMed] [Google Scholar]
- 17.Liu WT, Marsh TL, Cheng H, Forney LJ. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol. 1997;63(11):4516–4522. doi: 10.1128/aem.63.11.4516-4522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci U S A. 2006;103(32):12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M, Fire AZ. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic acids Res. 2007;35(19):e130. doi: 10.1093/nar/gkm760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat methods. 2008;5(3):235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albertsen M, Karst SM, Ziegler AS, Kirkegaard RH, Nielsen PH. Back to the basicsThe influence of DNA extraction and primer choice on phylogenic analysis of activated sludge communities. PLoS One. 2015;10(7):e0132783. doi: 10.1371/journal.pone.0132783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn JH, Kim BY, Song J, Weon HY. Effects of PCR cycle number and DNA polymerase type on the 16S rRNA gene pyrosequencing analysis of bacterial communities. J Microbiol (Seoul, Korea) 2012;50(6):1071–1074. doi: 10.1007/s12275-012-2642-z. [DOI] [PubMed] [Google Scholar]
- 25.Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domain-specific 16S primers. J Microbiol methods. 2003;55(3):541–555. doi: 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Bakker MG, Tu ZJ, Bradeen JM, Kinkel LL. Implications of pyrosequencing error correction for biological data interpretation. PLoS One. 2012;7(8):e44357. doi: 10.1371/journal.pone.0044357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinforma. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinforma Oxf Engl. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methe B, DeSantis TZ, Petrosino JF, Knight R, Birren BW. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21(3):494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6(3):610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic acids Res. 2014;42:D633–D642. doi: 10.1093/nar/gkt1244. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glockner FO. The SILVA and “All-species living tree Project (LTP)” taxonomic frameworks. Nucleic acids Res. 2014;42:D643–D648. doi: 10.1093/nar/gkt1209. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schloss PD. The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput Biol. 2010;6(7):e1000844. doi: 10.1371/journal.pcbi.1000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derrien M, van Passel MW, van de Bovenkamp JH, Schipper RG, de Vos WM, Dekker J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut microbes. 2010;1(4):254–268. doi: 10.4161/gmic.1.4.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradshaw DJ, Homer KA, Marsh PD, Beighton D. Metabolic cooperation in oral microbial communities during growth on mucin. Microbiol Read Engl. 1994;140(Pt 12):3407–3412. doi: 10.1099/13500872-140-12-3407. [DOI] [PubMed] [Google Scholar]
- 36.Kolenbrander PE. Multispecies communities: interspecies interactions influence growth on saliva as sole nutritional source. Int J Oral Sci. 2011;3(2):49–54. doi: 10.4248/IJOS11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aagaard K, Petrosino J, Keitel W, Watson M, Katancik J, Garcia N, Patel S, Cutting M, Madden T, Hamilton H, Harris E, Gevers D, Simone G, McInnes P, Versalovic J. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J : Official Publ Fed Am Soc Exp Biol. 2013;27(3):1012–1022. doi: 10.1096/fj.12-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boase S, Foreman A, Cleland E, Tan L, Melton-Kreft R, Pant H, Hu FZ, Ehrlich GD, Wormald PJ. The microbiome of chronic rhinosinusitis: culture, molecular diagnostics and biofilm detection. BMC Infect Dis. 2013;13:210. doi: 10.1186/1471-2334-13-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan M, Pamp SJ, Fukuyama J, Hwang PH, Cho DY, Holmes S, Relman DA. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell Host Microbe. 2013;14(6):631–640. doi: 10.1016/j.chom.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huse SM, Young VB, Morrison HG, Antonopoulos DA, Kwon J, Dalal S, Arrieta R, Hubert NA, Shen L, Vineis JH, Koval JC, Sogin ML, Chang EB, Raffals LE. Comparison of brush and biopsy sampling methods of the ileal pouch for assessment of mucosa-associated microbiota of human subjects. Microbiome. 2014;2(1):5. doi: 10.1186/2049-2618-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, Beck JM, Curtis JL, Huffnagle GB. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio. 2015;6(2):e00037. doi: 10.1128/mBio.00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke-Reed P, Zakhari S, Read J, Watson B, Guyer M. The NIH human microbiome project. Genome Res. 2009;19(12):2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kraal L, Abubucker S, Kota K, Fischbach MA, Mitreva M. The prevalence of species and strains in the human microbiome: a resource for experimental efforts. PLoS One. 2014;9(5):e97279. doi: 10.1371/journal.pone.0097279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mark Welch JL, Utter DR, Rossetti BJ, Mark Welch DB, Eren AM, Borisy GG. Dynamics of tongue microbial communities with singlenucleotide resolution using oligotyping. Front Microbiol. 2014;5:568. doi: 10.3389/fmicb.2014.00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eren AM, Borisy GG, Huse SM, Mark Welch JL. Oligotyping analysis of the human oral microbiome. Proc Natl Acad Sci U S A. 2014;111(28):E2875–E2884. doi: 10.1073/pnas.1409644111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charlson ES, Chen J, Custers-Allen R, Bittinger K, Li H, Sinha R, Hwang J, Bushman FD, Collman RG. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One. 2010;5(12):e15216. doi: 10.1371/journal.pone.0015216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mathieu A, Delmont TO, Vogel TM, Robe P, Nalin R, Simonet P. Life Hum. surfaces skin metagenomics. PLoS One. 2013;8(6):e65288. doi: 10.1371/journal.pone.0065288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jette ME, Dill-McFarland KA, Hanshew AS, Suen G, Thibeault SL. The human laryngeal microbiome: effects of cigarette smoke and reflux. Sci Rep. 2016 Oct 24;6 doi: 10.1038/srep35882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L, Jablonski K, Kleerup E, Lynch SV, Sodergren E, Twigg H, Young VB, Bassis CM, Venkataraman A, Schmidt TM, Weinstock GM. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187(10):1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jensen A, Fago-Olsen H, Sorensen CH, Kilian M. Molecular mapping to species level of the tonsillar crypt microbiota associated with health and recurrent tonsillitis. PLoS One. 2013;8(2):e56418. doi: 10.1371/journal.pone.0056418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aurora R, Chatterjee D, Hentzleman J, Prasad G, Sindwani R, Sanford T. Contrasting the microbiomes from healthy volunteers and patients with chronic rhinosinusitis. JAMA Otolaryngology– Head Neck Surg. 2013;139(12):1328–1338. doi: 10.1001/jamaoto.2013.5465. [DOI] [PubMed] [Google Scholar]
- 52.Abreu NA, Nagalingam NA, Song Y, Roediger FC, Pletcher SD, Goldberg AN, Lynch SV. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci Transl Med. 2012;4(151):151ra124. doi: 10.1126/scitranslmed.3003783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feazel LM, Robertson CE, Ramakrishnan VR, Frank DN. Microbiome complexity and Staphylococcus aureus in chronic rhinosinusitis. Laryngoscope. 2012;122(2):467–472. doi: 10.1002/lary.22398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guss AM, Roeselers G, Newton IL, Young CR, Klepac-Ceraj V, Lory S, Cavanaugh CM. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J. 2011;5(1):20–29. doi: 10.1038/ismej.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Willner D, Haynes MR, Furlan M, Schmieder R, Lim YW, Rainey PB, Rohwer F, Conrad D. Spatial distribution of microbial communities in the cystic fibrosis lung. ISME J. 2012;6(2):471–474. doi: 10.1038/ismej.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goddard AF, Staudinger BJ, Dowd SE, Joshi-Datar A, Wolcott RD, Aitken ML, Fligner CL, Singh PK. Direct sampling of cystic fibrosis lungs indicates that DNA-based analyses of upper-airway specimens can misrepresent lung microbiota. Proc Natl Acad Sci U S A. 2012;109(34):13769–13774. doi: 10.1073/pnas.1107435109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flight WG, Smith A, Paisey C, Marchesi JR, Bull MJ, Norville PJ, Mutton KJ, Webb AK, Bright-Thomas RJ, Jones AM, Mahenthiralingam E. Rapid detection of emerging pathogens and loss of microbial diversity associated with severe lung disease in cystic fibrosis. J Clin Microbiol. 2015;53(7):2022–2029. doi: 10.1128/JCM.00432-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, Martinez FJ, Huffnagle GB. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6(2):e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9(4):244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Z, Geng J, Tang X, Fan H, Xu J, Wen X, Ma ZS, Shi P. Spatial heterogeneity and co-occurrence patterns of human mucosal-associated intestinal microbiota. ISME J. 2014;8(4):881–893. doi: 10.1038/ismej.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanshew AS, Jette ME, Thibeault SL. Characterization and comparison of bacterial communities in benign vocal fold lesions. Microbiome. 2014;2:43. doi: 10.1186/2049-2618-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]