

Isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) are tricarboxylic acid cycle enzymes that catalyze conversion of isocitrate to α-ketoglutarate, operating in the cytosol and mitochondria, respectively. Recently, Parsons et al.1 described that IDH1 is mutated in 12% of glioblastomas, followed by the Yan et al.'s2 report showing a high frequency (80%) of IDH1 mutations at the R132 residue and less frequent IDH2 mutations at the R172 residue in grade II–III gliomas and secondary glioblastomas. IDH1 and IDH2 were shown to be also mutated in 7.7–9.9% and 15.4% of acute myeloid leukemia (AML) cases, respectively, especially those having normal karyotypes. More recently, both gene mutations were found in other myeloid neoplasms, including myeloproliferative neoplasms and myelodysplastic syndrome (MDS), and were associated with a high rate of MDS–AML transformation and poor prognosis.3–5 In hematopoietic neoplasms, all reported IDH1 mutations involved the R132 residue, while IDH2 mutations involve either the R172 or the R140 residue.
These mutations are thought to result in loss-of-function of these enzymes and prevent α-ketoglutarate production from isocitrate in a dominant-negative manner, which in turn leads to activation of the HIF-1α pathway.6 On the other hand, Dang et al.7 and Ward et al. demonstrated that mutant IDH1 and IDH2 also showed gain-of-function that promotes production of 2-hydroxyglutarate (2HG) from α-ketoglutarate, which is thought to be oncogenic. However, the relative contribution of loss-of-function and gain-of-function to oncogenesis is still unclear.
Here, we report mutations of both IDH1 and IDH2 in a series of 171 cases with MDS or related myeloid neoplasms, where the complete status of genome-wide copy numbers and mutations of major MDS-related genes, including c-CBL, RUNX1, TP53, FLT3ITD, JAK2, NRAS and KRAS genes, were previously investigated.8 Our series included 28 cases with AML, 35 with chronic myelomonocytic leukemia (CMML), 3 with refractory anemia (RA), 69 with RA with excess of blasts (RAEB), 1 with RA with ringed sideroblasts (RARS), 25 with refractory cytopenia with multilineage dysplasia (RCMD), 4 with MDS-unclassified (MDS-U), and 6 with 5q-syndrome, according to the new World Health Organization classification. Mutations of IDH1 and IDH2 were examined with genomic DNA isolated from bone marrow by direct sequencing of the PCR-amplified exon 4 of both genes as previously described.2 This study was performed under the regulation of the ethics committee at the Faculty of Medicine, University of Tokyo.
In total, 9 IDH1 (5.2%) and 7 IDH2 (4.2%) mutations were identified (Table 1). IDH1/2 mutations were most frequently found in RAEB (14.4%), followed by RCMD (8%), transformed AML (7.1%) and CMML (5.7%). There were conflicting reports regarding the frequencies of IDH2 mutations in MDS. While similar frequencies of IDH1 and IDH2 mutations were observed by Kosmider et al.,5 Thol et al.3 reported no IHD2 mutations in their 193 MDS cases showing 3.6% mutation rate for IDH1. In our series, on the other hand, IDH2 mutations were found to have a frequency comparable to that of IDH1 mutations, although all IDH2 mutations were R140Q, with no R172 mutations that are common in brain tumors, and also have been reported in the previous literatures on AML, myeloproliferative neoplasm and MDS. All mutations were heterozygous and no sample had both mutations occurring simultaneously. All but one of the IDH1 mutations corresponded to amino-acid conversions previously reported in AML and glioblastoma, including four R132C, three R132 and one R132L mutations. The remaining one was a nonsense mutation (CGA>TGA) at the R100 residue (Figure 1). The R100X mutation was most likely to result in loss-of-enzymatic function of IDH1, because it truncates all the enzymatically active domains of the protein.
Table 1.
Mutational status of 16MDS or MDS-related disease with IDH mutations
| Patient number |
Sex/age | Type (WHO) | Cytogenetics | Results by 50k | IDH | cbl | RUNX1 | p53 | FLT3ITD | JAK2 | NRAS | KRAS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F/71 | RAEB1 | ND | Normal | IDH1 R132H | wt | wt | mut | wt | wt | wt | wt |
| 2 | M/75 | RAEB2 | 46,XY | +1q,der(1;7) | IDH1 R132H | wt | wt | wt | wt | wt | wt | wt |
| 3 | M/66 | RAEB1 | 46,XY | 9p UPD | IDH1 R132H | wt | wt | wt | wt | mut | wt | wt |
| 4 | M/65 | RAEB2 | ND | +8 | IDH1 R132C | wt | wt | wt | wt | wt | wt | wt |
| 5 | M/51 | RAEB2 | 46,XY | Normal | IDH1 R132C | wt | mut | wt | wt | wt | wt | wt |
| 6 | M/75 | RCMD | 46,XY,del(20)(q11) | add(3)(p26.3-24.3), del(20)(q11.21-13.13) | IDH1 R132C | wt | wt | wt | wt | wt | wt | wt |
| 7 | M/82 | RCMD | 46,XY,del(20)(q11) | del(20)(q11.21-13.2), 3qUPD | IDH1 R132L | wt | wt | wt | wt | wt | wt | wt |
| 8 | M/64 | RAEB2 | 46,XY | del(8)(q21.3) | IDH1 R132C | wt | wt | wt | wt | wt | wt | wt |
| 9 | M/— | AML with MLD | 46,XY[7] 49,XY,+4,+8,+10[12] | Normal | IDH1 R100X | wt | wt | wt | wt | wt | wt | wt |
| 10 | M/77 | RAEB2 | 46,XY | Normal | IDH2 R140Q | wt | wt | wt | wt | wt | wt | wt |
| 11 | M/80 | CMML2 | 47,XY,+8 | 11q UPD | IDH2 R140Q | mut | wt | wt | wt | wt | wt | wt |
| 12 | M/57 | AML with MLD | 46,XY | +8 | IDH2 R140Q | wt | wt | wt | wt | wt | wt | wt |
| 13 | M/63 | RAEB2 | 46,XY | Normal | IDH2 R140Q | wt | mut | wt | wt | wt | wt | wt |
| 14 | F/74 | RAEB1 | 45,XX,del(5)(q23q32), der(11;12)(q10;q10), del(20)(q11) | del(5)(q23.2-35.3), del(20)(q11.22-13.2) | IDH2 R140Q | wt | wt | mut | wt | wt | wt | wt |
| 15 | M/76 | CMML2 | 46,XY,+1, der(1;7) (q10;p10) | +1q,der(1;7) | IDH2 R140Q | wt | wt | wt | wt | wt | wt | wt |
| 16 | M/63 | RAEB2 | 46,XY | 7q UPD | IDH2 R140Q | wt | wt | wt | wt | wt | wt | wt |
Abbreviations: AML with MLD, acute myeloid leukemia with multilineage dysplasia; CMML, chronic myelomonocytic leukemia; IDH, isocitrate dehydrogenase; MDS, myelodysplastic syndrome; mut, mutation; RAEB, refractory anemia with excess blasts; RCMD, refractory cytopenia with multilineage dysplasia; wt, wild type.
Figure 1.
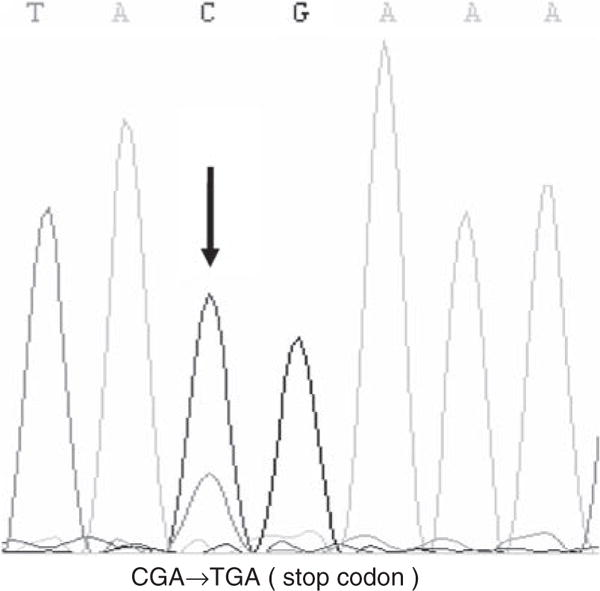
Sequence of IDH1 R100 mutation (case 9). The arrow indicates the mutated nucleotide.
IDH1/2 mutations were found in 8 out of 72 cases with normal karyotypes (cases 2, 3, 5, 8, 10, 12, 13 and 16), but 5 of them showed abnormalities in SNP array analysis, although none of them had poor-prognosis karyotypes such as complex chromosomal abnormalities. Pardanani et al.9 reported frequent IDH mutations in cases with simple del(5q) (4/16, or 25%), but in our series none of four such cases had the mutations. Coexisting mutations of other genes were found in 5 of the 16 cases with IDH mutations, including mutations in c-CBL in one case, RUNX1 in two cases, TP53 in another and JAK2 in the other case. RAS mutations were found in 13 cases, but none of them coexisted with IDH mutations. No statistical difference in the clinical outcomes was observed between cases with and without IDH mutations.
In the past reports, IDH1 mutations were confined to the R132 residue. It is predicted that the consequences of these mutations at this position will be the structural alteration of the protein to a closed, active conformation, which has a higher affinity to NADPH+, loss of three hydrogen bonds with isocitrate, and reorganization of the active sites via the positional shift of Y139 and K212′.7 These are thought to facilitate the synthesis of 2HG from α-ketoglutarate (gain of function) rather than conversion of isocitrate to α-ketoglutarate. However, the R100X mutation truncates most amino-acid residues that contact substrates, including R100 and R132, and, thus, is thought to abrogate all enzymatic activities without side effects, such as the synthesis of 2HG from α-ketoglutarate. This is the first example that represents pure loss-of-function of IDH1. Our findings will provide an interesting insight into the mechanism by which IDH1 mutations promote the development of human cancers.
Acknowledgments
This work was supported by a Grant-in-Aid from the Ministry of Health, Labor and Welfare of Japan (22140501) and from the Ministry of Education, Culture, Sports, Science and Technology, and NIH R01 CA026038-31.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thol F, Weissinger EM, Krauter J, Wagner K, Damm F, Wichmann M, et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica. 2010;95:1668–1674. doi: 10.3324/haematol.2010.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24:1094–1096. doi: 10.1038/leu.2010.52. [DOI] [PubMed] [Google Scholar]
- 6.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Gliomaderived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460:904–908. doi: 10.1038/nature08240. [DOI] [PubMed] [Google Scholar]
- 9.Pardanani A, Patnaik MM, Lasho TL, Mai M, Knudson RA, Finke C, et al. Recurrent IDH mutations in high-risk myelodysplastic syndrome or acute myeloid leukemia with isolated del(5q) Leukemia. 2010;24:1370–1372. doi: 10.1038/leu.2010.98. [DOI] [PubMed] [Google Scholar]