

Abstract
Objective
Mantle cell lymphoma (MCL) is a lymphoma characterized by aberrant activation of CCND1/Cyclin D1 followed by sequential genetic abnormalities. Genomic abnormalities in MCL have been extensively examined by classical cytogenetics and microarray-based comparative genomic hybridization techniques, pointing out a number of alterations in genomic regions that correlate with the neoplastic phenotype and survival. Recently, single nucleotide polymorphism genomic microarrays (SNP-chip) have been developed and used for analysis of cancer genomics. This technique allows the detection of genomic changes with higher resolution including loss of heterozygosity without changes of gene dosage, so-called acquired uniparental disomy, aUPD.
Materials and Methods
We have examined 33 samples of MCL (28 primary MCL and 5 cell lines) using the 250,000 SNP-chip from Affymetrix.
Results
Known alterations were confirmed by SNP arrays, including deletion of INK4A/ARF, duplication/amplification of MYC, deletion of ATM and deletion of TP53. We also identified a duplication/amplification that occurred at 13q involving oncogenic microRNA, miR17-92. We found other genomic abnormalities including duplication/amplification of cyclin D1, del(1p), dup(6p), dup(3q) and dup(22q). Our SNP-chip analysis detected these abnormalities at high resolution allowing us to narrow the size of the commonly deleted regions, including 1p and 6q. Our SNP-chip analysis detected a number of aUPD sites including whole chromosome 9 aUPD and 9p aUPD. We also found a MCL case with 19p, leading to homozygous deletion of TNFSF genes.
Conclusion
SNP-chip analysis detected in MCL very small genomic gains/losses, as well as aUPDs which could not be detected by more conventional methods.
Keywords: SNP-chip, uniparental disomy, BIM, TNF superfamily genes
Introduction
Mantle cell lymphoma (MCL) is one of the subtypes of B-cell non-Hodgkin lymphoma derived from the mantle cell [1–3]. This disease predominantly develops in elderly male patients [1–3]. The disease-free and progression-free survival of MCL is worse than many other types of lymphomas including diffuse large B-cell lymphoma. Although MCL usually responds to initial chemotherapy, the disease often relapses after a short duration of remission. Recently, an antibody (rituximub) targeting CD20 on the B-cell lymphoma cells has often been used with chemotherapy as initial therapy with some improvement in disease-free survival [4, 5].
This disease is characterized by the chromosomal translocation, t(11;14)(q13;q32), resulting in overexpression of the cyclin D1 gene on 11q23 driven by the enhancer region of immunoglobulin heavy chain (IgH) gene on 14q32 [1–3]. Of interest, transgenic mice mimicking this chanage only develop lymphoid hyperplasia, suggesting that overexpression of cyclin D1 in lymphoid cells is not sufficient to cause MCL [6, 7]. Additional oncogenic abnormalities must co-operate with cyclin D1 in development of MCL [6, 7].
Previous data including karyotyping and comparative genomic hybridization showed that MCL has a number of additional genomic abnormalities including deletion of ATM, p53 and p16INK4/p14ARF [8 - 11, 12]. Further, expression microarray and protein microarray data revealed dysregulation of apoptosis-associated genes, including Pim1 and MDM2 in this disease [13–16].
However, genomic abnormalities in this disease have not been fully explored. Recently, high resolution single nucleotide polymorphism genomic microarray (SNP-chip) has been developed and employed in analysis of the cancer genome [17–18]. This novel technique allows us to detect very small gain and/or loss of genomic materials as well as acquired uniparental disomy (aUPD, loss of heterozygosity with neutral gene dosage) [20, 21]. We named this new technology molecular allelokaryotyping since SNP-chip is able to detect genomic abnormalities at the molecular level and identify each parental allelic dosage [17, 18]. To explore genomic abnormalities in MCL, we employed this new technique and analyzed 33 MCL samples including 28 primary MCL and 5 MCL cell lines.
Materials and Methods
Samples and DNA extraction
We collected MCL cells from 28 newly diagnosed patients with MCL. All cases fulfilled World Health Organization diagnostic criteria for MCL, including overexpression of cyclin D1, presence of t(11;14)(q13; q32) as determined by fluorescence in situ hybridization (FISH) and/or chromosomal analysis [22]. For cases #6505, #22073, #11527, #19358 and #22601, MCL cells were collected from peripheral blood since a large number of the lymphoma cells were detected in the peripheral blood. Except for these three cases, all MCL cells were collected from LNs. This study was approved by the institutional review board of the University of Navarra and Cedars-Sinai Medical Center. Patients gave their informed consent, per Declaration of Helsinki. Five MCL cell lines were also studied: HBL-2, SP-49, REC-1, NCEB-1, and Jeko1. DNA was extracted using Qiagen DNA extraction kit (Qiagen, Valencea, CA) according to the manufacturer’s protocol.
SNP-chip analysis
DNA from the samples was analyzed on Affymetrix® GeneChip® Human mapping 250K Nsp arrays (Affymetrix Japan, Tokyo, Japan) according to the manufacturer’s protocol. Microarray data were analyzed for determination of both total and allelic-specific copy numbers using the CNAG program as previously described with minor modifications, where the status of copy numbers as well as UPD at each SNP was inferred using the algorithms based on Hidden Markov Models [20, 21].
Allele specific gene dosage using non-matched reference samples was measured as follows: SNP-typing on the SNP-chip uses two distinct sets of SNP-specific probes named “A-” and “B-” SNPs at all SNP sites. At each SNP-site, perfectly matched probes (PMAs or PMBs) and mismathced probes (MMAs or MMBs) are placed on the chip. For the allele-specific gene dosage measurement, the sums of perfectly matched probes (PMAs or PMBs) for the i th SNP site in the tumor and reference samples (ref1, ref2, ref3, …, refN),
and
are compared at each SNP site, according to the concordance of the SNP calls in the tumor sample (Otumi), and the SNP calls in a given reference sample (OrefIi),
and the total CN ratio was calculated as follows:
To obtain adjusted values (RrefIAB, i, RrefIA, i and RrefIB,i), the calculated signals were compensated as previously reported [20]. Then, these values were averaged over the references that have a concordant genotype for each SNP in a given set of references (K), and RKAB, i RKA, i and RKB, i were obtained. To minimize noise in allele-specific gene dosage analysis, we selected the optimized references using stepwise reference selection as previously reported [20].
Results
Five common well-known abnormalities detected by SNP-chip in MCL
Representative results of SNP-chip analysis in 33 MCL including homozygous and hemizygous deletions, duplications, amplifications and aUPDs are shown in Figures 1a–f. A overview of all of the genomic gains and/or losses detected by SNP-chip analysis is shown in Figure 1g.
Figure 1. Results of SNP-chip analysis.
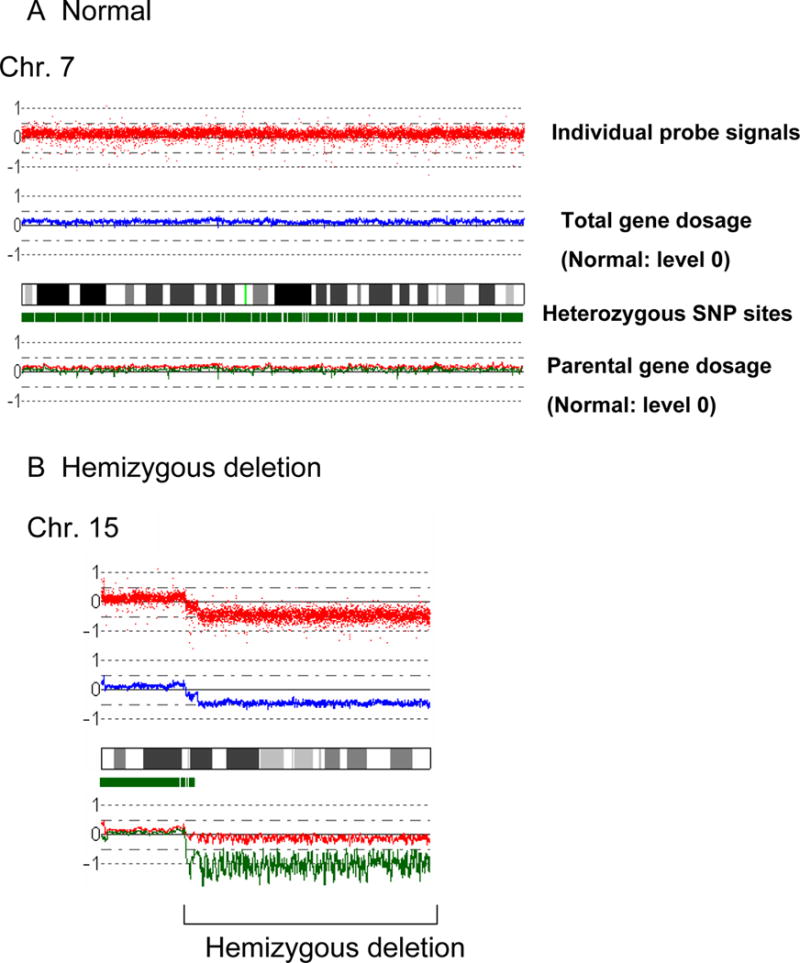

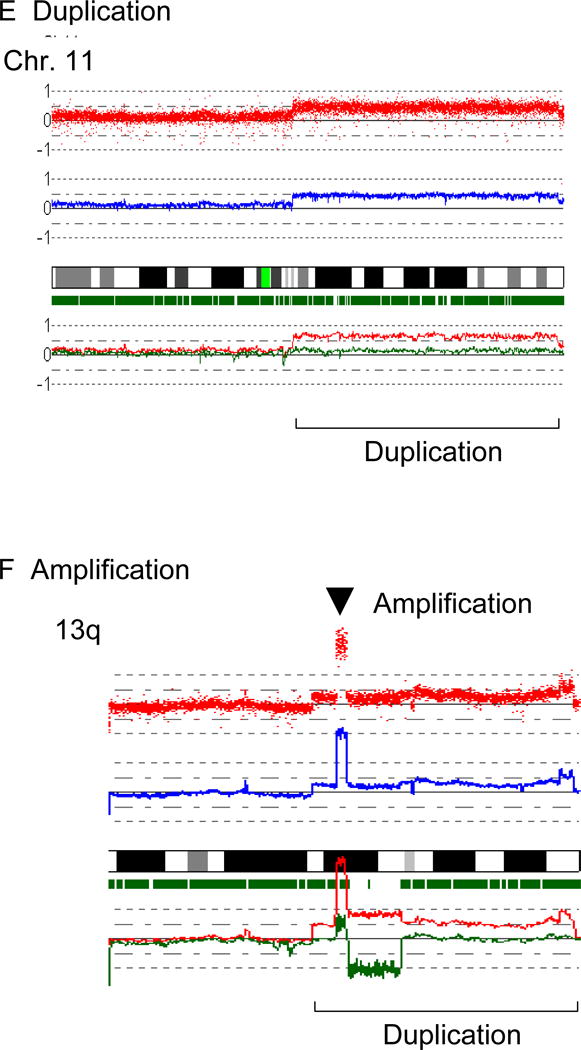
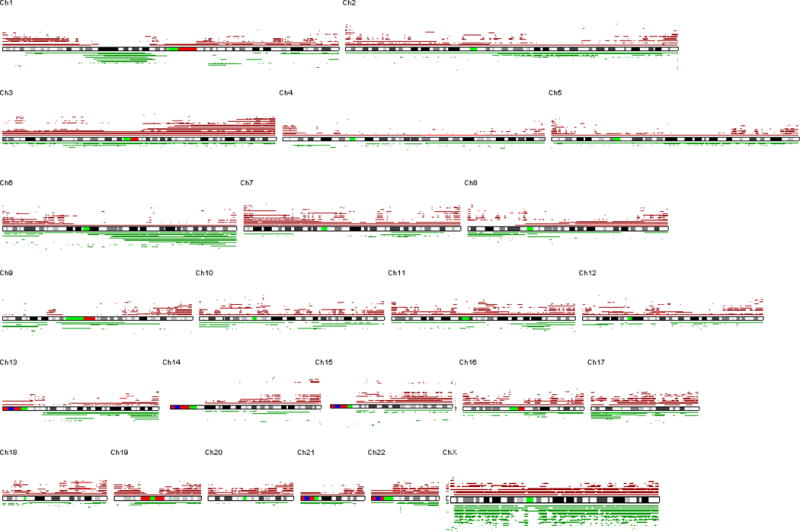
Representative results and the overview of genomic status of 33 MCL identified by SNP-chip are shown.
A: Normal chromosome. Top panel shows individual SNP probe signals; intensity of the signals are almost the same over the entire chromosome. Second panel indicates gene dosage level; gene dosage is normal over the chromosome. Third panel shows heterozygosity SNP sites by green rectangles. Heterozygous SNP site are frequently detected over the chromosome. Fourth panel indicates gene dosage of each parental allele (green or red line); level of each allele is identical over the chromosome.
B: Hemizygous deletion. Region of hemizygous deletion is delineated by brackets.
C: Homozygous deletion. Arrow indicates a site of homozygous deletion; each parental allele (red or green line) has a deletion at this site.
D: Acquired uniparental disomy (aUPD). aUPD is a site which has loss of heterozygosity with normal gene dosage; one of the parental alleles is missing (green line) and the other allele is duplicated (red line). aUPD region is indicated by brackets.
E: Duplication. Duplicated region is indicated by a parenthesis.
F: Amplification. Arrowhead indicates the sites of high copy number amplification. This case also has duplication at 13q as indicated by the brackets.
G: SNP-chip data of 33 cases of MCL. Deleted regions are indicated by green lines under each chromosome panel; and duplications/amplifications are indicated by brown lines above each chromosome panel.
We have found four common genomic abnormalities in MCL including deletion of 9p, duplication/amplification of 8q, deletion/aUPD of 11q and deletion/aUPD of 17p (Fig. 2 and Table 1). A commonly deleted gene at 9p13 was the INK4A/ARF locus (Fig. 2A); deletion of these genes was found in 4/5 cell lines and 6/28 primary MCL samples. SNP-chip was able to identify a homozygous region as small as 200 kb in the REC1 cell line (one allele had deletion of 9p21.3-pter; the other allele had deletion of nucleotide 21,884,299 – 22,093,813: http://genome.ucsc.edu/). A commonly duplicated/amplified region in 8q was 8q24 involving the c-MYC gene (Fig. 2B), occurring in 2/5 cell lines and 3/28 primary MCL samples. Although deletion/aUPD of 11q involved relatively large regions, ATM (11q22) is one of the genes always involved in this abnormal region (Fig. 2C), and deletion/aUPD of this gene was detected in 3/5 cell lines (2 deletions and 1 aUPD) and 4/28 primary MCL samples (3 deletions and 1aUPD). Deletion/aUPD of 17p encompassed a large region including TP53 (17p13) (Fig. 2D). Deletion/aUPD of 17p13 was detected in 3/5 cell lines (all deletions) and 9/28 primary MCL (6 deletions and 3 aUPDs). We found duplication/amplification of 13q in 4/5 cell lines, and the commonly amplified/duplicated region involved miR17-92 (Fig. 2E).
Figure 2. Common genomic alterations in MCL.
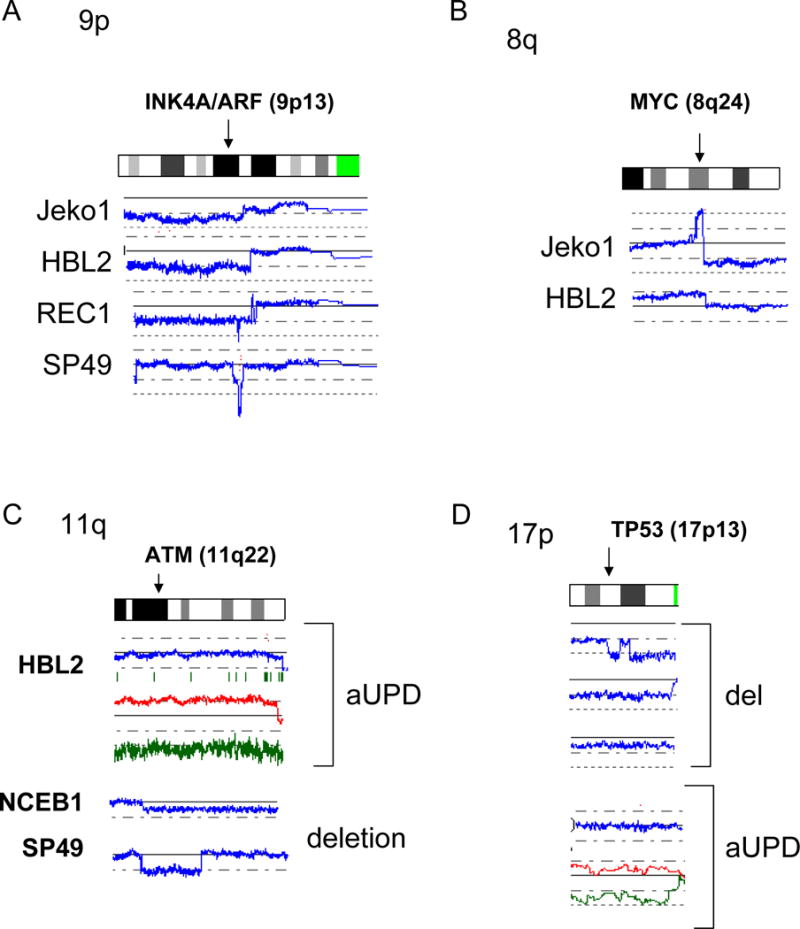
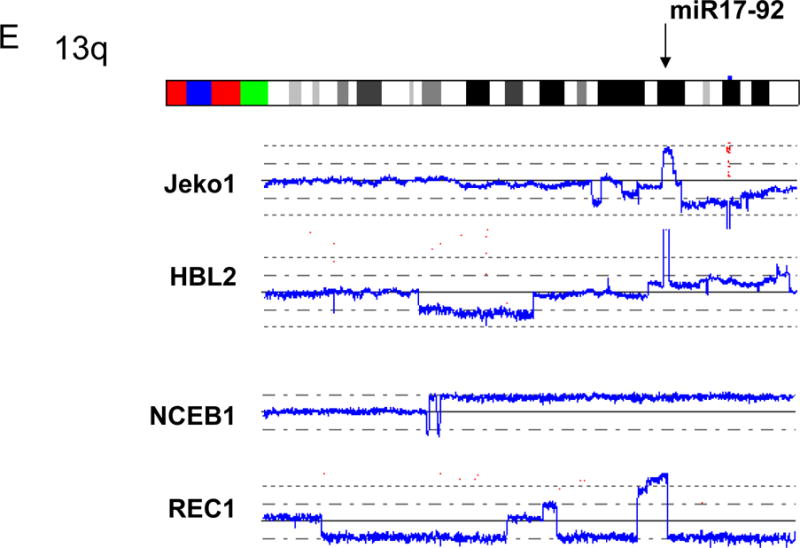
Representative SNP-chip results of common genomic alterations found in MCL are shown. Blue lines indicate gene dosage of each case. A: Deletion of INK4A/ARF (9p13). B: Amplification and duplication of c-MYC (8q24). C: aUPD and deletion of 11q involving ATM. Gene dosages are indicated by blue lines. NCEB1 and SP49 have deletion of 11q22. HBL2 shows normal gene dosage (blue line), but allele-specific gene dosage levels (green and red lines) indicate that one of the parental alleles (green line) is deleted and the other parental allele (red line) is duplicated, suggesting acquired uniparental disomy (aUPD). D: Deletion and aUPD of 17p involving TP53. Top three cases show deletion of 17p13. The bottom case demonstrates normal gene dosage at 17p13 (blue line), but allele-specific gene dosage levels (green and red lines) indicate that one of the parental alleles (green line) is deleted and the other parental allele (red line) is duplicated, suggesting acquired uniparental disomy (aUPD). E: Amplification and duplication of 13q involving miR17-92. Mantle cell lines: Jeko1, HBL2, REC1, SP49, NCEB1. Refer to Supplement Figure 1 and its legend for detailed interpretation of SNP-chip data.
Table 1.
Common genomic alterations found in MCL by SNP-chip
| Chromosome locus | Type of alteration | Frequency | Candidate gene |
|---|---|---|---|
| 1p | Deletion/aUPD | 9/33 | |
| 1q | Deletion | 7/33 | |
| 3q | Duplication | 13/33 | PIK3CA, BCL6 |
| 6q | Deletion | 13/33 | |
| 8q | Duplication/amplification | 2/33 | c-MYC |
| 9p | Deletion | 4/33 | INK4A/ARF |
| 9q | aUPD | 6/33 | |
| 11q | Deletion/aUPD | 8/33 | ATM |
| 11q | Duplication/amplification | 4/33 | CCND1 |
| 17p | Deletion | 6/33 | TP53 |
| 18q | Duplication/amplification | 4/33 | MALT1, BCL2 |
aUPD: acquired uniparental disomy
Recurrent deleted regions with unidentified target genes
In addition to the five well-known abnormal regions in MCL described above, we have found a number of abnormal regions (Fig. 1g and Table 1). Deletions involving 1p, 1q, 2q, 6q, 7q, 8p, 10p, 13q, and X chromosome were common in MCL (Fig. 1g and Table 1). Deletion of 1p was found in 3/5 cell lines and 6/28 primary MCL, and the commonly deleted region was 1p22.1 - 21.2 (nt. 93,123,689 – nt. 103,840,794) involving 34 genes (Fig. 3A). Deletions involving 1q were detected in 2/5 cell lines and 5/28 primary MCL (Fig. 3B). However, we could not identify a commonly deleted region in this abnormality. This region may contain multiple target genes deleted in MCL.
Figure 3. Recurrent deletions in MCL.
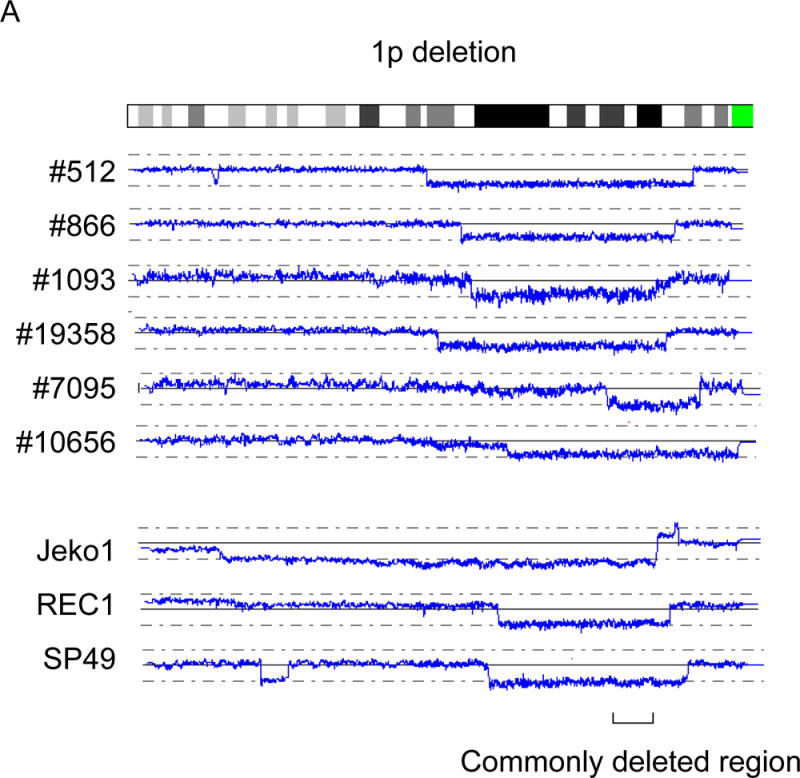
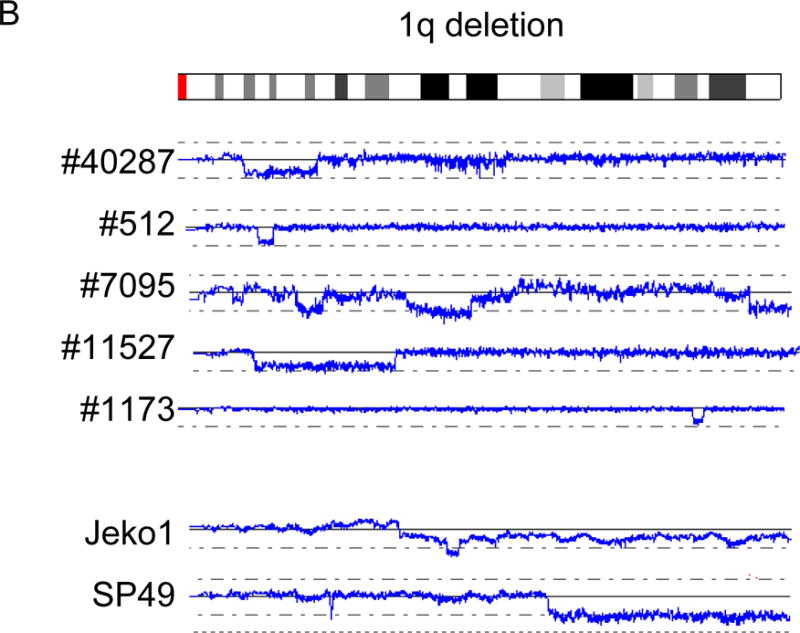
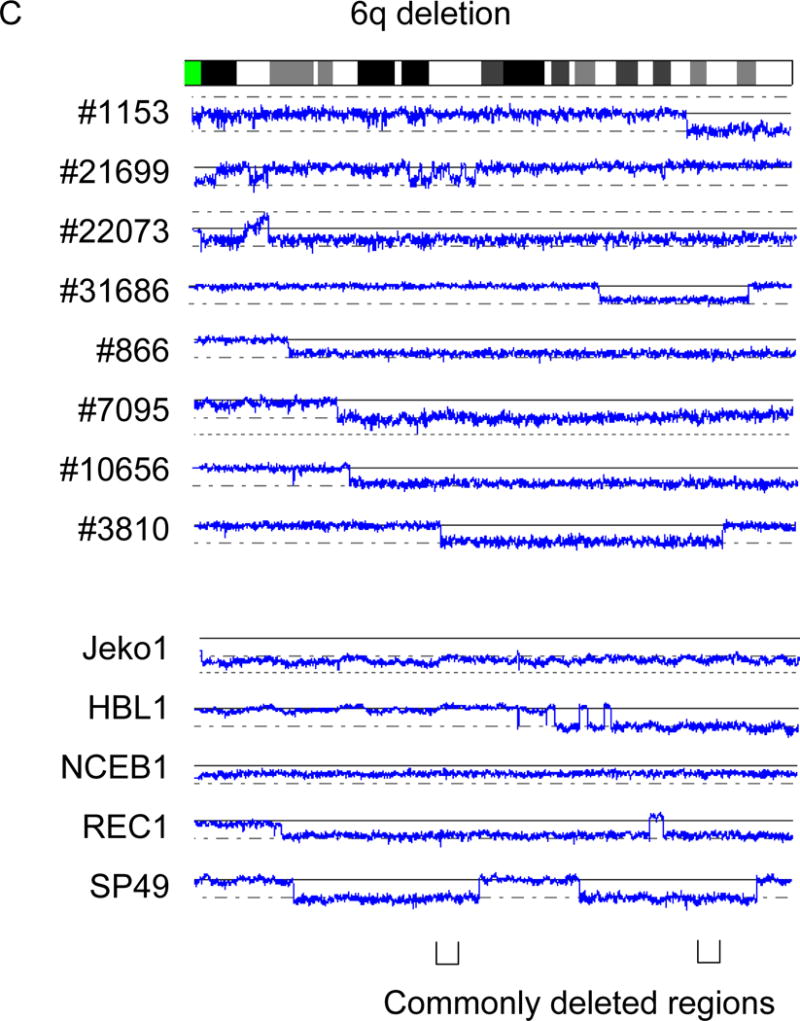
Recurrent deletions of 1p, 1q and 6q are shown. A: Deletion of 1p. B: Deletion of 1q. C: Deletion of 6q. Commonly deleted regions are indicated by parenthesis. Cell line names and sample numbers are indicated. Blue lines indicate gene dosage levels of each case.
We found deletions of 6q in 5/5 cell lines and 8/28 primary MCL samples. The data of these 13 cases allowed us to identify 2 commonly deleted regions, 6q21 (nt. 108,581,628 – nt. 113,358,672) which involves 31 genes including FYN, TRAF2IP2 and WISP3 (CCN6); and 6q25.1-25.3 (nt. 151,908,208 – nt. 157,547,535) which encompasses 17 genes including FBX05 (Fig. 3C).
Recurrent duplicated/amplified regions
We found duplication of 3q (8/28 primary MCL), trisomy 3 (4/28 primary MCL) and tetrasomy 3 (1/28 primary MCL), leading to gain of genomic material in 3q in 45 % of the primary MCL (Fig. 4a). This region was relatively large, and involved PIK3CA (3q26) and BCL6 (3q27).
Figure 4. Recurrent duplication/amplification in MCL.
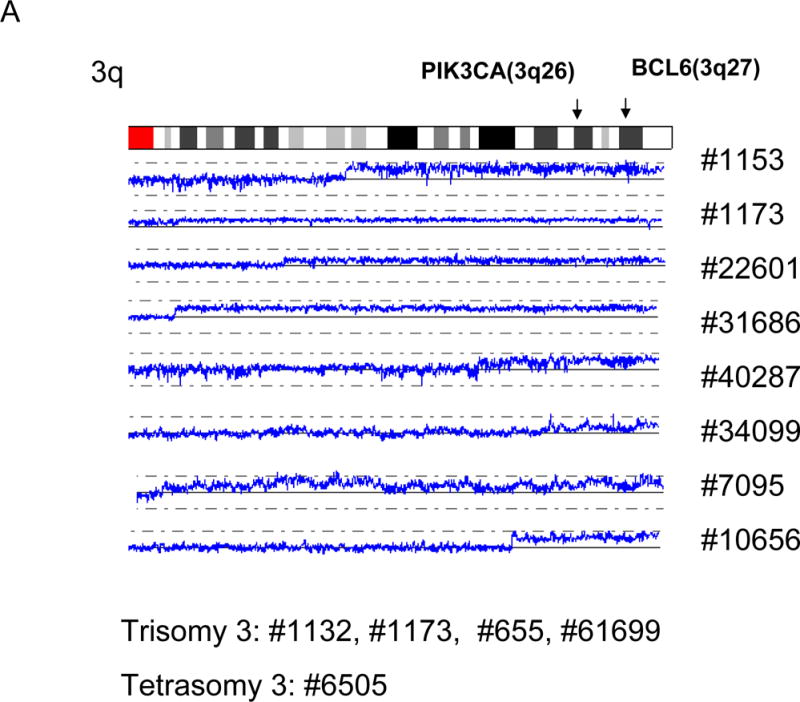
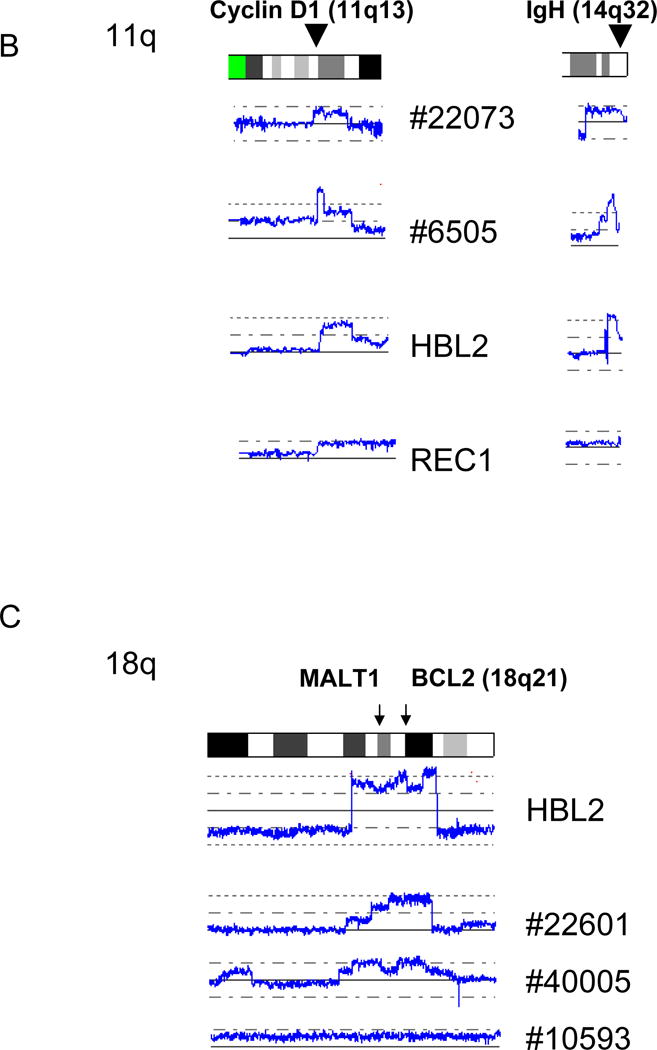
A: Duplications of 3q are shown. Positions of PIK3CA (3q26) and BCL6 (3q27) are indicated by arrows. Sample numbers with trisomy 3 and tetrasomy 3 are also shown. B: Amplifications/duplications of cyclin D1 and IgH. The positions of cyclin D1 and IgH are indicated by arrowheads. C: Amplifications/duplications of 18q. Positions of MALT1 and BCL2 are indicated by arrows. Sample numbers and cell line names are shown.
We also found duplication/amplification of 11q in 2/28 primary MCL and 2/5 MCL cell lines. This commonly amplified/duplicated region involved cyclin D1. All of the cases with duplication/amplification of 11q also had duplication/amplification of the IgH gene (Fig. 4B), suggesting that rearranged IgH-CCND1 region was duplicated/amplified.
We identified duplication/amplification of 18q (1 cell line and 3 primary MCL samples), and this region is relatively large. This DNA segment contains MALT1 (18q21) and BCL2 (18q21) which are frequently overexpressed in low grade lymphomas and associated with anti-apoptosis.
Besides the above mentioned common regions of duplications/amplifications, other less frequent duplications and amplifications were also detected in a number of chromosomal loci (Fig. 1).
aUPD in MCL
We found a number of novel aUPD regions in MCL (Fig. 5) including 1p, 9p, 9q, 11q, 17p, and 19p. As described above, aUPD of 17p was detected in 3 primary MCL, and aUPD of 11q was found in 2 MCL (1 cell line and 1 primary tumor). aUPD involving chromosome 9 was the most common aUPD in our study; whole chromosome 9 aUPD was found in 3 MCL, and aUPD of the whole short arm of chromosome 9 was found in 1 MCL (Figure 5). Two cases had aUPD of whole long arm of chromosome 9. None of the cases with aUPD involving whole chromosome 9 or 9p had homozygous deletion of INK4A/ARF (9p21.3) (data not shown).
Figure 5. Acquired uniparental disomy in mantle cell lymphoma.
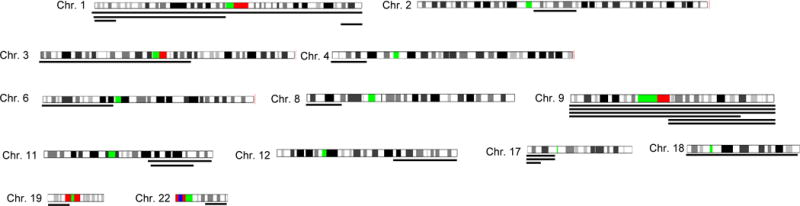
Regions of acquired uniparental disomy (aUPD) are shown by lines under each chromosome. Each line under a chromosome reflects a MCL sample. Only the chromosomes having aUPD are shown.
In primary MCL #21699, we found a homozygous deletion of 19p13.1 caused by aUPD. This homozygously deleted region contained 8 genes including three TNF superfamily genes (TNFSF9, TNFSF7 and TNFSF14) (Figure 6).
Figure 6. Homozygous deletion of 19p13.3 by acquired uniparental disomy in MCL.
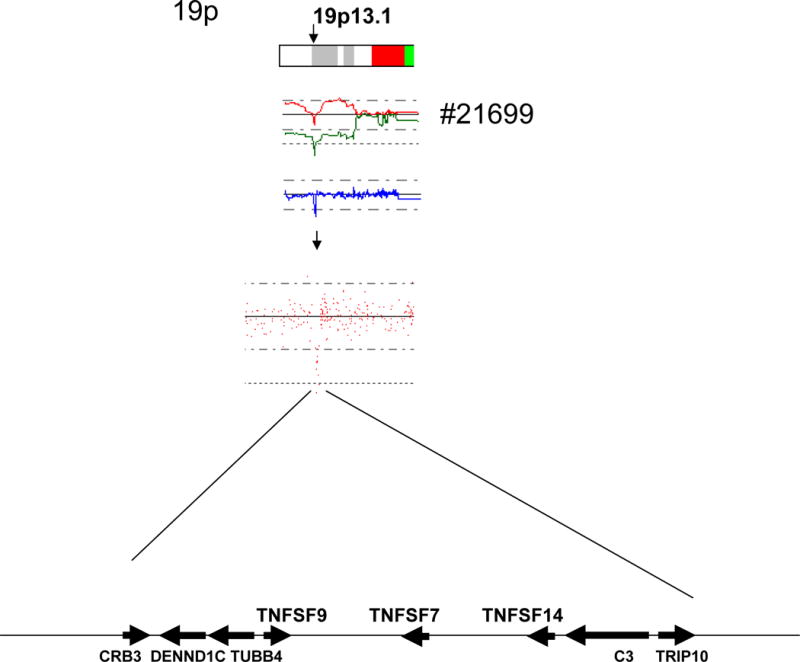
Top panel: aUPD was found on 19p leading to homozygous deletion of 19p13.1 in MCL case #21699. Middle panel: SNP-chip probe signals on 19p. Bottom panel: Eight genes involved in homozygous deletion caused by aUPD. TNF superfamily genes (TNFSF 7, 9, 14) are homozygously deleted in this case. Arrows indicate the directions and approximate relative sizes of the genes located in this region.
Discussion
Most MCL samples have genomic abnormalities in addition to t(11;14) [8, 9], and a transgenic murine model carrying overexpression of cyclin D1 suggests that additional genetic events are needed for development of MCL [6, 7]. Nevertheless, genomic alterations of MCL have not been fully examined as yet. In this study, we employed high resolution SNP-chip analysis on 33 MCL samples to explore genomic abnormalities in this disease. This technique can detect alterations as small as 200 kb.
Previously, we have validated the reliability of SNP-chip analysis by fluorescence in situ hybridization and genomic quantitative PCR, which showed that this method has low background, and the data are reliable [17, 18]. In this study, we have found well-known genomic abnormalities including deletion of INK4A/ARF [23, 24], validating that the SNP-chip analysis can sensitively detect genomic abnormalities in MCL.
Copy number variants (CNV) are polymorphic difference observed in normal individuals [25–27]. This change may be misinterpreted as a deletion and/or a duplication/amplification. Copy number variants (CNV) have been reported by a number of investigators; and the sites and sizes of CNV have been collected in a database (http://genome.ucsc.edu/, http://projects.tcag.ca/variation/). We compared all altered genomic sites that we found in this study with CNV sites in the databases and excluded CNVs from our results. Therefore, all genomic altered sites in this study are most likely pathogenic and acquired during the development of MCL. However, rare CNV sites which have not been reported, may be included in our results. Accumulation of further data of CNV sites will further exclude the false-positive sites of deletions/duplications.
We found gain of the 3q region in 45 % of primary MCL. This 3q region is relatively large and contains a number of genes, including PIK3CA and BCL6. PIK3CA is one of the important molecules in signal-transduction for cellular proliferation [28]. Slight increase of protein levels of PIK3CA by gain of genomic material in this region may cause stimulation of this signal-transduction pathway in MCL [29]. Another interesting duplicated/amplified site found in MCL is 18q involving MALT1 and BCL2. Overexpression of these genes by duplication/amplification may co-operate with cyclin D1 in development of MCL. We also found duplication/amplification of cyclin D1; and these same samples had duplication/amplification of IgH. Therefore, these MCL probably have increased expression of cyclin D1, similar to duplication of der(22)t(9;22) carrying the BCR-ABL fusion gene in chronic myelogenous leukemia (CML) transforming to blastic crisis [30]. A duplication of IgH-cyclin D1 may also play a role in transforming MCL to an even more aggressive form of the disease [31].
We also found a number of aUPD sites in MCL. These aUPDs may contain an activation mutation of an oncogene; the wild-type allele becomes deleted and the mutated (activated) allele is duplicated as is often observed with JAK2 mutations [20, 32]. Also, these aUPDs are detected in the regions of tumor suppressor genes. Duplication of a chromosome which has a point mutation or segmental deletion involving a tumor suppressor gene, leads to homozygous deletions of the tumor suppressor gene. This is often observed with homozygous deletion of INK4A/ARF (9p21.3) by aUPD in a variety of cancers [17]. Interestingly, although we found four cases with aUPD involving whole chromosome 9 or 9p, none of these four MCL samples had homozygous deletion of INK4A/ARF. In our previous study analyzing 399 samples of pediatric acute lymphoblastic leukemia by SNP-chip, 9p aUPD usually was associated with homozygous deletion of INK4A/ARF [17]. However, cases with whole chromosome 9 aUPD rarely had homozygous deletion of this gene [19]. In MCL, an unidentified altered tumor suppressor gene or oncogene may be duplicated by aUPD involving chromosome 9.
In development of cancer, a number of functional cellular pathways are dysregulated including those involved in proliferation, apoptosis and differentiation. Dysregulation of multiple pathways by multiple genomic alterations may be needed for development of MCL. Cyclin D1 plays a key role in the cell cycle, and over-expression of this molecule inappropriately drives the cell cycle. Dysregulation of cyclin D1 is necessary but not sufficient to cause MCL. Additionally, deletion of genes associated with detection of DNA damage including p53 and ATM, have also frequently been detected in MCL. These latter abnormalities might accelerate accumulation of genomic alterations transforming MCL cells to a more aggressive disease.
In this study, we found a homozygous deletion of 19p13.3 caused by aUPD, and this region contains seven genes; three of these include members of the TNF superfamily genes. Likely, one of the genes in this region behaves as a tumor suppressor gene, and is homozygously deleted by aUPD. Accumulation of additional MCL SNP-chip data should lead to identification of the tumor suppressor gene in this region.
In this study, we found candidate genomic events which could contribute to development of MCL in concert with overexpression of cyclin D1. These genes include tumor suppressor genes whose function is impaired by deletions and/or mutations, including INK4A/ARF, TP53, ATM1, and TNF superfamily genes on 19p13.3. We also identified commonly deleted regions on chromosome 1p, 1q and 6q, where unidentified tumor suppressor genes may be localized. The other collaborating abnormalities are overexpression of oncognes which may occur by their duplication or amplification. Examples from our study include c-MYC, miR17-92, PIK3CA, BCL6, MALT1 and BCL2.
Accumulation of additional SNP-chip data will complement our study, leading to identification of novel target signal transduction pathways that can become targets for treatment of this frequently fatal disease.
Acknowledgments
We thank NIH grants as well as Sheryl Weissberg Lymphoma Research Foundation for supporting this study. NK is supported by the fellowship from The Tower Cancer Research Foundation. HPK holds the Mark Goodson Chair in Oncology Research at Cedars Sinai, and is a member of the Jonsson Cancer Center and the Molecular Biology Institute of UCLA. This work was also supported by grant-in-aid from Department of Health, Welfare and Labor and from MEXT of the Japanese government.
We also thank Dr. M.A.Piris and the Spanish National Tumor Bank (CNIO, Madrid) for providing selected MCL samples.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
No financial interest/relationships with financial interest relating to the topic of this article have been declared.
References
- 1.Fernàndez V, Hartmann E, Ott G, et al. Pathogenesis of mantle-cell lymphoma: all oncogenic roads lead to dysregulation of cell cycle and DNA damage response pathways. J Clin Oncol. 2005;23:6364–9. doi: 10.1200/JCO.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 2.Bertoni F, Zucca E, Cotter FE. Molecular basis of mantle cell lymphoma. Br J Haematol. 2004;124:130–40. doi: 10.1046/j.1365-2141.2003.04761.x. [DOI] [PubMed] [Google Scholar]
- 3.Weisenburger DD, Armitage JO. Mantle cell lymphoma–an entity comes of age. Blood. 1996;87:4483–94. [PubMed] [Google Scholar]
- 4.Meusers P, Hense J, Brittinger G. Mantle cell lymphoma: diagnostic criteria, clinical aspects and therapeutic problems. Leukemia. 1997;11(Suppl 2):S60–4. [PubMed] [Google Scholar]
- 5.Witzig TE. Current treatment approaches for mantle-cell lymphoma. J Clin Oncol. 2005;23:6409–14. doi: 10.1200/JCO.2005.55.017. [DOI] [PubMed] [Google Scholar]
- 6.Bodrug SE, Warner BJ, Bath ML, et al. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J. 1994;13:2124–30. doi: 10.1002/j.1460-2075.1994.tb06488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lovec H, Grzeschiczek A, Kowalski MB, et al. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J. 1994;13:3487–95. doi: 10.1002/j.1460-2075.1994.tb06655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuneo A, Bigoni R, Rigolin GM, et al. Cytogenetic profile of lymphoma of follicle mantle lineage: correlation with clinicobiologic features. Blood. 1999;93:1372–80. [PubMed] [Google Scholar]
- 9.Beà S, Ribas M, Hernández JM, et al. Increased number of chromosomal imbalances and high-level DNA amplifications in mantle cell lymphoma are associated with blastoid variants. Blood. 1999;93:4365–74. [PubMed] [Google Scholar]
- 10.Schraders M, Pfundt R, Straatman HM, et al. Novel chromosomal imbalances in mantle cell lymphoma detected by genome-wide array-based comparative genomic hybridization. Blood. 2005;105:1686–93. doi: 10.1182/blood-2004-07-2730. [DOI] [PubMed] [Google Scholar]
- 11.Rubio-Moscardo F, Climent J, Siebert R, et al. Mantle-cell lymphoma genotypes identified with CGH to BAC microarrays define a leukemic subgroup of disease and predict patient outcome. Blood. 2005;105:4445–54. doi: 10.1182/blood-2004-10-3907. [DOI] [PubMed] [Google Scholar]
- 12.Kohlhammer H, Schwaenen C, Wessendorf S, et al. Genomic DNA-chip hybridization in t(11;14)-positive mantle cell lymphomas shows a high frequency of aberrations and allows a refined characterization of consensus regions. Blood. 2004;104:795–801. doi: 10.1182/blood-2003-12-4175. [DOI] [PubMed] [Google Scholar]
- 13.Hofmann WK, de Vos S, Tsukasaki K, et al. Altered apoptosis pathways in mantle cell lymphoma detected by oligonucleotide microarray. Blood. 2001;98:787–94. doi: 10.1182/blood.v98.3.787. [DOI] [PubMed] [Google Scholar]
- 14.Rizzatti EG, Falcão RP, Panepucci RA, et al. Gene expression profiling of mantle cell lymphoma cells reveals aberrant expression of genes from the PI3K-AKT, WNT and TGFbeta signalling pathways. Br J Haematol. 2005;130:516–26. doi: 10.1111/j.1365-2141.2005.05630.x. [DOI] [PubMed] [Google Scholar]
- 15.Ghobrial IM, McCormick DJ, Kaufmann SH, et al. Proteomic analysis of mantle-cell lymphoma by protein microarray. Blood. 2005;105:3722–30. doi: 10.1182/blood-2004-10-3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Vos S, Krug U, Hofmann WK, et al. Cell cycle alterations in the blastoid variant of mantle cell lymphoma (MCL-BV) as detected by gene expression profiling of mantle cell lymphoma (MCL) and MCL-BV. Diagn Mol Pathol. 2003;12:35–43. doi: 10.1097/00019606-200303000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Kawamata N, Ogawa S, Zimmermann M, et al. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood. 2008;111:776–84. doi: 10.1182/blood-2007-05-088310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann S, Ogawa S, Raynaud SD, et al. Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia. Cancer. 2008;112:1296–305. doi: 10.1002/cncr.23270. [DOI] [PubMed] [Google Scholar]
- 19.Walsh CS, Ogawa S, Karahashi H, et al. ERCC5 is a novel biomarker of ovarian cancer prognosis. J Clin Oncol. 2008;26:2952–8. doi: 10.1200/JCO.2007.13.5806. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto G, Nannya Y, Kato M, et al. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet. 2007;81:114–26. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nannya Y, Sanada M, Nakazaki K, et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–9. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 22.Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835–49. doi: 10.1200/JCO.1999.17.12.3835. [DOI] [PubMed] [Google Scholar]
- 23.Pinyol M, Hernandez L, Cazorla M, et al. Deletions and loss of expression of p16INK4a and p21Waf1 genes are associated with aggressive variants of mantle cell lymphomas. Blood. 1997;89:272–80. [PubMed] [Google Scholar]
- 24.Hutter G, Scheubner M, Zimmermann Y, et al. Differential effect of epigenetic alterations and genomic deletions of CDK inhibitors [p16(INK4a), p15(INK4b), p14(ARF)] in mantle cell lymphoma. Genes Chromosomes Cancer. 2006;45:203–10. doi: 10.1002/gcc.20277. [DOI] [PubMed] [Google Scholar]
- 25.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–8. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 26.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakobsson M, Scholz SW, Scheet P, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 28.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 29.Kawamata N, Chen J, Koeffler HP. Suberoylanilide hydroxamic acid (SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells. Blood. 2007;110:2667–73. doi: 10.1182/blood-2005-11-026344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitelman F. The cytogenetic scenario of chronic myeloid leukemia. Leuk Lymphoma. 1993;11(Suppl 1):11–5. doi: 10.3109/10428199309047856. [DOI] [PubMed] [Google Scholar]
- 31.Gruszka-Westwood AM, Atkinson S, et al. Unusual case of leukemic mantle cell lymphoma with amplified CCND1/IGH fusion gene. Genes Chromosomes Cancer. 2002;33:206–12. doi: 10.1002/gcc.1216. [DOI] [PubMed] [Google Scholar]
- 32.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]