

Abstract
Levonorgestrel (LNG) is a synthetic progestin that is available in oral contraceptive tablets, a subdermal implant, and an intrauterine system for contraception. LNG pharmacokinetics are a pivotal determinant of contraceptive efficacy and essential in assessing drug-drug interactions influencing LNG exposure following different routes of LNG administration. A highly sensitive LC-MS/MS method was developed and validated to quantify levonorgestrel in human plasma. Liquid-liquid extraction was utilized with a sample volume of 500 μL to extract levonorgestrel from plasma. Chromatographic separation of LNG was achieved with a Fortis™ C18 (3 μm: 100mm × 2.1mm) reverse phase analytical column. The mobile phases consisted of de-ionized water plus 0.1% NH4OH (100:0.1%, v/v) (A), and methanol plus 0.1% NH4OH (100:0.1%, v/v) (B) delivered as a gradient at a flow rate of 400 μL/min. Detection of LNG and internal standard (D-(-)-norgestrel-d7) was achieved using positive polarity mode monitoring at 313.2-245.2 amu and 320.1-251.2 amu, respectively. The assay was linear over the calibration range of 49.6 to 1500 pg/mL. This method was used to quantify plasma LNG released by subdermal implant in support of a drug interaction study among women with HIV receiving efavirenz- or nevirapine-based antiretroviral therapy.
Keywords: LC-MS/MS, liquid-liquid extraction, levonorgestrel, subdermal implant, plasma
1. Introduction
Levonorgestrel (LNG) is a synthetic progestin and is the biologically active levorotatory enantiomer of norgestrel. As a progestin, its contraceptive effect stems from inhibition of ovulation, as well as thickening cervical mucus and altering endometrial implantation [1]. LNG is used in various formulations of hormonal contraception, including combined oral contraceptives co-formulated with ethinyl estradiol, alone as emergency contraceptive tablets, LNG-releasing intrauterine systems, and subdermal implants [2]. After systemic distribution, LNG is metabolized by cytochrome P450 (CYP) 3A4 and further biotransformed by glucuronidation and sulfation, then eliminated primarily in the urine as metabolites (40% to 68%) [3]. Due to differences in administered dose, peak plasma LNG concentrations (Cmax) following non-oral administration via subdermal implants or intrauterine systems are lower as compared to oral administration. For example, the subdermal implant yields peak LNG concentrations that are substantially lower than peak concentrations following the oral tablet (Cmax 723 ± 344 pg/mL versus 19,100 ± 9700 pg/mL, respectively) [4, 5]. The subdermal implant is approved for use up to 5 years after insertion, at which time LNG concentrations have been reported to be 279 ± 123 pg/mL [1].
Detection of plasma LNG released by subdermal implantation requires an analytical assay that is highly sensitive and specific for LNG. To date, radioimmunoassays (RIA) have provided the most sensitive method for quantification of LNG released from subdermal implants [6], with an assay lower limit of quantification (LLOQ) of 50 pg/mL [7]. While highly sensitive, RIA lacks specificity, as the assays have cross reactivity with other steroids and metabolites, as well as with non-water soluble metabolites that remain following extraction [8, 9].
While other reports describe LC-MS/MS methods for quantifying LNG, most are validated to detect plasma LNG after oral administration with LLOQs ranging from 100 to 265 pg/mL, which is inadequate for accurately detecting LNG from a subdermal implant [10–13].
Herein, we describe a highly sensitive, highly specific, non-automated, rapid assay using liquid-liquid plasma LNG extraction with LC-MS/MS detection, developed and validated for quantification of LNG from subdermal implant in women living with human immunodeficiency virus (HIV) infection enrolled in a clinical pharmacokinetic (PK) study [14].
2. Materials and Methods
2.1 Chemicals
LNG (MW: 312.45, 98% pure) was purchased from Sigma, UK. The internal standard D-(−)-norgestrel-d7 (MW: 319.49, 98% pure) was purchased from Toronto Research Chemicals (North York, Ontario, Canada). The chemical structures of LNG and IS are shown in Figure 1. Acetonitrile (LC/MS grade), methanol (HPLC grade), and tert-Butyl methyl ether (reagent grade >98% pure) were purchased from Sigma, UK. Ammonia in aqueous solution (25% AnalaR NORMAPUR®) was purchased from VWR International (Lutterworth, UK). HPLC grade de-ionized water was produced from an Elga Option S water purifier (Elga LabWater, High Wycombe, UK) and was further purified to 18.2 MΩ with a Purelab Ultra system (Elga LabWater, High Wycombe, UK). Blank human plasma (K2EDTA) was obtained from the National blood service (Liverpool, UK).
Figure 1.
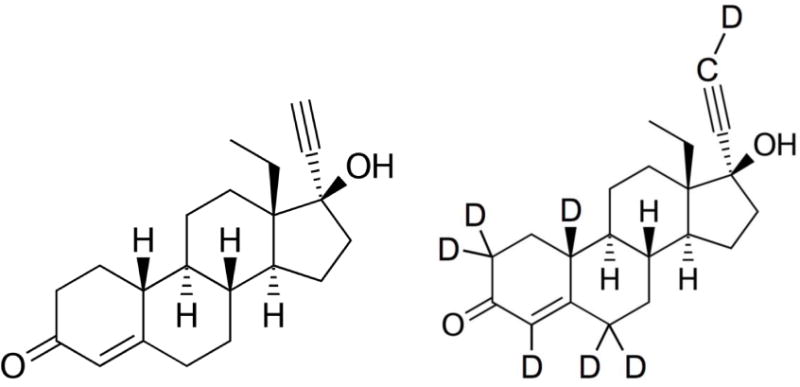
Chemical structures of levonorgestrel and D-(−)-norgestrel-d7 (IS)
2.2 Instruments
An Accela HPLC autosampler and pump were interfaced with a Thermo Quantum Access triple quadrupole mass spectrometer operating in the electrospray ionization (ESI), positive polarity mode (Thermo Scientific, Hemel Hempstead, UK). Chromatographic separation of LNG was achieved with a Fortis™ C18 (3 μm: 100mm × 2.1mm) reverse phase analytical column (Fortis Technologies, Cheshire, UK) at an oven temperature of 40°C and interfaced with a 2 μm Quest pre-column (Thermo Scientific, Hemel Hempstead, UK). The mobile phases consisted of de-ionized water plus 0.1% NH4OH (100:0.1%, v/v) (Mobile Phase A), and methanol plus 0.1% NH4OH (100:0.1%, v/v) (Mobile Phase B) delivered as a gradient at a flow rate of 400 μL/min. Needle wash solvent consisted of acetonitrile and de-ionized water plus formic acid (80:20:0.1%, v/v/v). All reagent solutions were stored at room temperature. TSQ Tune software was used for tuning and optimizing LNG. Both compound dependent parameters (S-lens, collision energy) and instrument parameters (electrospray voltage, capillary temperature, vaporizer temperature, sheath gas and auxiliary gas pressures) were optimized. Data acquisition and processing was performed using LC Quan™ software (Version 2.7, Thermo Scientific, Hemel Hempstead, UK).
2.3 Calibration, IS, and QC Solutions
Two weighed 1 mg/mL stock solutions of LNG (calibration primary stock, and QC primary stock) were prepared independently by separately dissolving 2 mg aliquots of LNG, weighed on an AnD GR-202 balance (Scientific Laboratory Supplies, East Yorkshire, UK), in appropriate volumes of methanol. The primary LNG stocks (1 mg/mL) were used for preparation of working calibration standards and quality controls (QCs). From the calibration standard primary stock (1 mg/mL), an intermediate solution (1 μg/mL) was prepared by dilution with an appropriate volume of methanol. Three calibration standard spiking solutions were prepared by diluting the 1 μg/mL intermediate solution in methanol to 75 ng/mL, which was further diluted in methanol to 37.5 ng/mL and 6.2 ng/mL. Blank human plasma was spiked with each solution to yield working calibration standard concentrations of 1500, 750, and 124, pg/mL, respectively. On the day of analysis, the working calibration standard solutions were diluted with drug-free plasma to yield final LNG plasma concentrations of 49.6, 120, 250, 500, 750, 1010, 1280, and 1500 pg/mL. QCs were prepared from LNG QC primary stock. These consisted of High QC (1200 pg/mL; 80% of the highest calibration standard), Medium QC (420 pg/mL), Low QC (134 pg/mL; 3× the lowest calibration standard) and LLOQ (49.6 pg/mL; equivalent to the lowest calibration standard). They were prepared with blank human plasma spiked with appropriate volumes of the QC primary stock. All standards and QC were prepared using calibrated air-displacement pipets. All stock solutions were stored between 2 to 8°C in tightly sealed glass tubes. Working calibration standards and QCs in plasma were stored at −40°C.
The primary stock of 1 mg/mL internal standard (IS) solution was prepared by dissolving the calculated quantity of D-(−)-norgestrel-d7 in methanol. A 10 μg/mL intermediate-stock was prepared by diluting the primary stock in the appropriate volume of methanol. The intermediate solution was stored at 4°C in a 10 mL tightly sealed glass tube for up to 6 months. The working IS solution (5 ng/mL) was prepared by dilution of the intermediate in the appropriate volume of methanol. Working IS was prepared fresh for each analytical run.
2.4 Sample Preparation
Calibrators, blanks, QCs and unknowns (500 μL) were added to labeled glass tubes and working IS solution (50 μL) was added to each extraction tube using a calibrated positive displacement pipet and vortexed. To normalize the volume of the blank tubes, 50 μL of methanol was added to each blank. Liquid-liquid extraction was performed by adding tert-Butyl methyl ether (4 mL) to all tubes using a repeater pipette, followed by prompt capping and tumbling for 30 minutes at room temperature. All tubes were centrifuged in a Heraeus Multifuge 3SR Plus (Scientific Laboratory Supplies, East Yorkshire, UK) at a relative centrifugal force (RCF) of 850 for 5 minutes at room temperature. After centrifugation, all tubes were placed in a cryogenic bath containing solid CO2 and methanol until the aqueous layer was frozen (~45 seconds). The organic layer containing the analytes of interest was poured into appropriately labeled 12 mm × 75 mm glass tubes. All samples were evaporated to dryness under a stream of nitrogen at 40°C for 25 minutes. The residue was reconstituted with 100 μL reconstitution solvent (methanol:water:formic acid, 50:50:0.1 v/v/v) and transferred to appropriately labeled autosampler vials (Chromacol, Herts, UK). The samples were centrifuged for 10 minutes at 1700 RCF at room temperature. The vials were loaded onto the autosampler trays and 25 μL was injected into the LC-MS/MS system for analysis.
2.5 LC-MS/MS Conditions
A chromatographic step-gradient at a flow rate of 400 μl/min was used. Initial conditions consisted of 80% mobile phase A, increasing in organic content to 80% mobile phase B in 0.8 min, maintained over 3.7 min and equilibrated back to the initial conditions over a total run time of 6 minutes. The column temperature was 40°C. The mass spectrometer used heated ESI with positive polarity. The electrospray voltage was 5000 V, and the capillary temperature and vaporizer temperature were set at 300°C and 350°C, respectively. The sheath and auxiliary gas pressures were set at 50 and 20 (arbitrary units). Argon, used as the collision gas, was delivered at a pressure of 20 bar. Selective reaction monitoring (SRM) scan was used for quantification by monitoring the precursor- product ion transitions (Table 1).
Table 1.
Mass transitions for LNG and D-(−)-norgestrel-d7
| Analyte | Parent ion (m/z) | Product ion (m/z) | Tube Lens (V) | Collision Energy (V) |
|---|---|---|---|---|
| LNG | 313.2 | 245.2 | 98 | 23 |
| D-(−)-norgestrel-d7 | 320.1 | 251.2 | 92 | 23 |
Abbreviations: LNG, Levonorgestrel
2.6 Method Validation
The assay was validated in accordance with the United States Food and Drug Administration’s (US FDA) guidance on Bioanalytical Method Validation [15].
2.6.1 Specificity and Selectivity
Selectivity was assessed by screening six different lots of blank human plasma to test for any interference at the specific retention times of LNG and IS. The peak area responses of interference (%) at the LNG or IS retention times were acceptable if <20% of the mean LLOQ (49.6 pg/mL) area response, and <5% of the mean IS area response, respectively (n=6). In addition, potential cross talk between the LNG and IS m/z transition windows was evaluated by injecting samples spiked with LNG in the absence of IS (and vice versa) and monitoring the level of interference in each transition window.
2.6.2 Accuracy and Precision
Five separate batches consisting of the calibration curve and six LQC, MQC, and HQC were analyzed to examine inter and intra-assay accuracy and precision. Three separate batches consisting of six LLOQs were analyzed. Assay precision was measured over each QC concentration by the percent coefficient of variation [CV% = (standard deviation/mean)*100]. Assay accuracy (% bias) was defined as the absolute value of the ratio of the calculated mean values of QC concentrations to the respective nominal values. Acceptance criteria for accuracy and precision were 15% for LQC, MQC, and HQC and 20% for LLOQ (49.6 pg/mL).
2.6.3 Stability
Stability of LNG in plasma was assessed at low (134 pg/mL), medium (420 pg/mL) and high (1200 pg/mL) QC concentrations (n=6 per concentration) for all conditions tested. QC samples were analyzed on a freshly prepared calibration curve. Multiple conditions were used to determine stability of LNG in plasma before and after processing. These included stability of LNG in plasma during freeze-thaw cycles, bench-top stability, long-term stability over 39 days at −40°C, processed sample stability, and stability after heat inactivation at 58°C for 40 minutes. Reinjection reproducibility was also assessed. LNG instability was considered if detected concentrations deviated from the nominal value by at least 15%.
2.6.4 Dilution Integrity
Dilution integrity of LNG in plasma was also determined. Aliquots of blank plasma were spiked at a concentration of 2600 pg/mL. Dilutions of 1:2 and 1:4 were prepared with blank plasma (n=6 per dilution), resulting in concentrations within the assay calibration range. Each sample concentration was back calculated with the appropriate dilution factor against a fresh calibration curve. Accuracy was determined if the percent deviation from the nominal value was <15% the nominal concentration. At least 67% (n=8) of the total diluted samples needed to meet these acceptance criteria.
2.6.5 Carry-over
Analyte carry-over (%) was determined by injecting calibrators at the LLOQ and the upper limit of quantification (ULOQ) (1500 pg/mL) immediately followed by injection of a blank plasma sample. This sequence was run 6 times, and the peak areas of the LLOQ, ULOQ and blank sample were measured. The carry-over was considered to be negligible if the LNG peak area of the blank sample was <20% of the LLOQ (49.6 pg/mL) peak area, and <5% of the IS peak area.
2.6.6 Matrix Effects and Recovery
Matrix effects were tested using a modification of the Matuszewski et al. method [16]. Six different plasma lots were spiked to LQC (134 pg/mL), MQC (420 pg/mL), and HQC (1200 pg/mL) concentrations. Three QC sets were prepared by spiking LNG and IS in blank plasma samples before extraction andafter extraction, and in mobile phase. To assess recovery (RE), LNG peak areas from pre-extracted QCs were compared to post- extracted QC samples. To assess process efficiency (PE), LNG peak areas from pre- extracted QCs were compared to mobile phase spiked at an equivalent LNG concentration. Matrix effects (ME) were assessed by comparing QCs spiked post- extraction to mobile phase spiked at an equivalent LNG concentration. The IS- normalized recovery (analysis RE) was calculated by comparing the LNG:IS peak area ratios of the pre-extracted (C2) and post-extracted (B2) QCs.
2.7 Clinical Application
Plasma aliquots obtained from HIV-infected women using LNG-releasing subdermal implant (150 mg LNG) alone or in combination with oral efavirenz- or nevirapine-based antiretroviral therapy, prior and then at weeks 1, 4, 12, 24, 36, and 48 after implant placement were analyzed using the method described herein [14]. All plasma samples originating from participants of known HIV positive status were heat inactivated in a water bath (at 58°C for 40 minutes) prior to sample pre-treatment and analysis.
3. Results
The chromatographic conditions were optimized using different compositions of mobile phase and buffers. Both acidic and basic conditions were tested during early development, with a basic mobile phase and positive ionization mode providing a greater degree of sensitivity as compared with acidic conditions. An atmospheric pressure chemical ionization (APCI) source was assessed versus ESI during development, and it was determined that ESI gave the greater signal to noise ratio. Monitored m/z values for precursor and product ions are listed in Table 1. Calibration standards were analyzed by back calculating the concentration of LNG to IS ratio using a weighted 1/(concentration)2 linear regression. This value was compared to the nominal value of each calibration standard, and %CV and percent deviation from the nominal concentration were determined to assess assay precision and accuracy, respectively. Linearity was demonstrated over the analytical range (49.6 to 1500 pg/mL) by the coefficient of determination (R2), with all calibration standard curves achieving R2 of ≥ 0.991.
3.1 Specificity and Selectivity
Figure 2 depicts a blank sample chromatogram and a typical chromatogram obtained when the LLOQ calibrator (49.6 pg/mL) was analyzed. After screening six different blank plasma lots, the amount of background interference at the analyte retention times was minimal; <20% of the signal response at the LLOQ for LNG (for five of the six samples) and <5% of the IS response.
Figure 2.
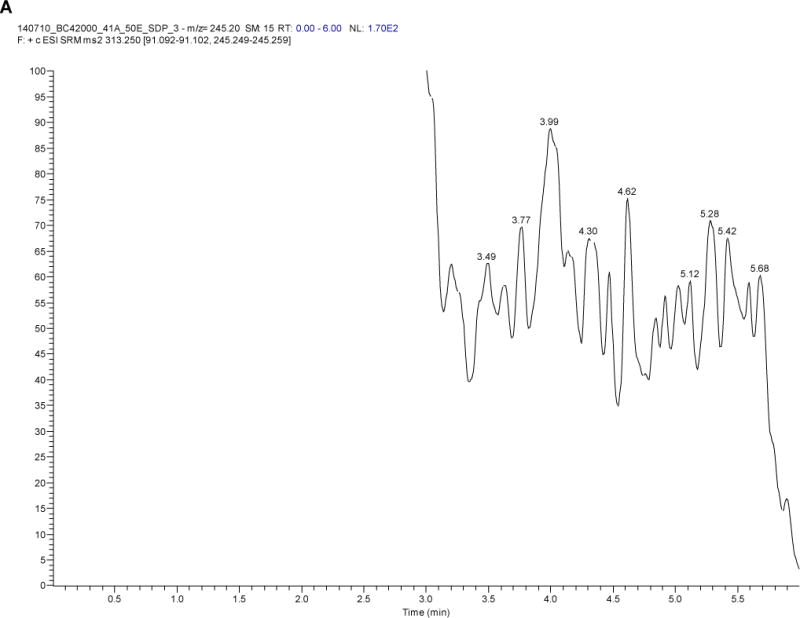
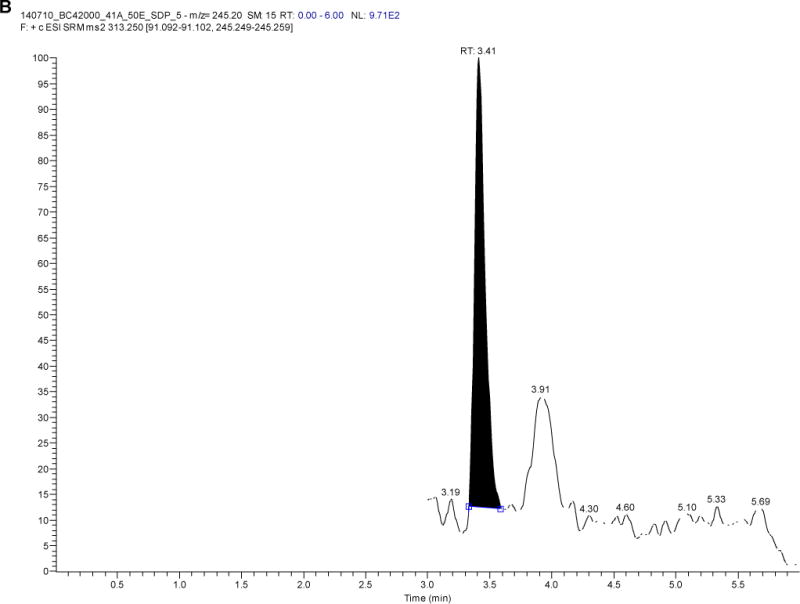
Levonorgestrel chromatograms of A) a blank sample and B) a sample at the lower limit of quantification (LLOQ; 49.6 pg/mL)
3.2 Accuracy and Precision
Inter- and intra-day accuracy and precision were assessed using day-to-day (n=30, LLOQ n=18) and within-batch (n=12, LLOQ n=6) LNG measurements from all QC concentrations and LLOQs. Five independent validation batches were run over four different days for all QC levels, and three additional batches including the LLOQs were run over three different days. Inter- and intra-day accuracy and precision were within ±15% for all QC levels and ±20% for LLOQs, as shown in Table 2.
Table 2.
Precision (%CV) and accuracy (%bias) data for LNG (inter- and intra-assay statistics)
| Interday | Mean (pg/mL) | S.D. (pg/mL) | CV (%) | Bias (%) | n |
|---|---|---|---|---|---|
| LLOQ (49.6 pg/mL) | 49.91 | 2.84 | 5.68 | −0.63 | 18 |
| LQC (134 pg/mL) | 123.70 | 9.42 | 7.62 | −7.69 | 30 |
| MQC (420 pg/mL) | 414.43 | 16.44 | 3.97 | −1.33 | 30 |
| HQC (1200 pg/mL) | 1263.42 | 65.13 | 5.15 | 5.28 | 30 |
|
| |||||
| Intraday | Mean (pg/mL) | S.D. (pg/mL) | CV (%) | Bias (%) | n |
|
| |||||
| LLOQ (49.6 pg/mL) | 50.57 | 2.83 | 5.59 | −1.95 | 6 |
| LQC (134 pg/mL) | 124.45 | 7.59 | 6.10 | −7.12 | 12 |
| MQC (420 pg/mL) | 421.17 | 15.23 | 3.62 | 0.28 | 12 |
| HQC (1200 pg/mL) | 1309.60 | 50.75 | 3.88 | 9.13 | 12 |
Abbreviations: LNG, levonorgestrel; LLOQ, lower limit of quantification; LQC, low quality control; MQC, medium quality control; HQC, high quality control.
Each analytical run consisted of the standard curve, blanks and 6 quality control samples at 3 specified concentrations (LQC, MQC, HQC); assays were run on 5 separate occasions. The LLOQ samples were run on 3 separate occasions.
3.3 Stability
Stability of LNG extracted from plasma samples was determined under multiple conditions and analyzed using a freshly prepared calibration curve (Table 3). LNG was stable during 3 freeze-thaw cycles, with the greatest deviation from the nominal concentration of −4.1% (LQC 134 pg/mL). Bench-top stability was demonstrated over 5 hours with the largest deviation from the nominal concentration of −13.4% (LQC 134 pg/mL). Processed plasma samples demonstrated stability for at least 24 hours at 2- 8°C, with the greatest deviation from the nominal concentration of −10.1% (LQC 134 pg/mL). Stability of LNG following heat inactivation was demonstrated with the greatest deviation from the nominal concentration of −4.12% (LQC 134 pg/mL). The impact of heat inactivation was determined due to laboratory procedures for safe handling of plasma specimens acquired from participants with HIV infection. Long-term LNG stability was demonstrated, with the greatest deviation from nominal concentration of 7.2% (HQC 1200 pg/mL). Reinjection reproducibility was demonstrated over 72 hours at 10°C, with the greatest deviation from nominal concentration of 10.0% (HQC 1200 pg/mL). These stability data indicate that no significant LNG decomposition occurred under the conditions tested. With the exception of heat inactivation stability data, which was specifically assessed for purposes of handling bio-hazardous clinical specimens for analysis with this method, all stability data fell within similar ranges described previously [9, 10, 12, 13].
Table 3.
Short and long-term stability data of levonorgestrel under different conditions
| Condition | QC | Nominal concentration (pg/mL) | Mean (pg/mL) | S.D. (pg/mL) | CV (%) | Bias (%) |
|---|---|---|---|---|---|---|
| Heat inactivation | LQC | 134 | 128.48 | 4.57 | 3.56 | −4.12 |
| MQC | 420 | 406.28 | 19.43 | 4.78 | −3.27 | |
| HQC | 1200 | 1248.05 | 66.86 | 5.36 | 4.00 | |
|
| ||||||
| Bench-top | LQC | 134 | 116.01 | 7.74 | 6.67 | −13.42 |
| MQC | 420 | 415.89 | 14.67 | 3.53 | −0.98 | |
| HQC | 1200 | 1303.72 | 18.15 | 1.39 | 8.64 | |
|
| ||||||
| Processed | LQC | 134 | 120.45 | 10.01 | 8.31 | −10.11 |
| MQC | 420 | 415.66 | 26.82 | 6.45 | −1.03 | |
| HQC | 1200 | 1278.22 | 31.39 | 2.46 | 6.52 | |
|
| ||||||
| Freeze-thaw | LQC | 134 | 126.52 | 6.38 | 5.05 | −5.59 |
| MQC | 420 | 412.92 | 8.88 | 2.15 | −1.69 | |
| HQC | 1200 | 1255.96 | 21.92 | 1.75 | 4.66 | |
|
| ||||||
| Long-Term | LQC | 134 | 127.58 | 9.37 | 7.35 | −4.79 |
| MQC | 420 | 416.19 | 9.53 | 2.29 | −0.91 | |
| HQC | 1200 | 1286.23 | 29.03 | 2.26 | 7.19 | |
|
| ||||||
| Re-injection | LQC | 134 | 140.47 | 6.00 | 4.27 | 4.83 |
| MQC | 420 | 453.90 | 8.67 | 1.91 | 8.07 | |
| HQC | 1200 | 1320.81 | 54.06 | 4.09 | 10.07 | |
Abbreviations: LNG, levonorgestrel; LQC, low quality control; MQC, medium quality control; HQC, high quality control.
3.4 Dilution Integrity
Dilution integrity was demonstrated for both 1:2 and 1:4 plasma dilutions of LNG, with deviation from the nominal concentration and precision of −5.9% and 3.9%, respectively, for both dilution factors.
All six diluted samples at both 1:2 and 1:4 dilutions demonstrated individual accuracy within 15% of the nominal value.
3.5 Carry-over
LNG carry-over was determined to be negligible after injecting the ULOQ (1500 pg/mL) calibration standard immediately followed by injection of a blank plasma sample. The LNG peak areas of the blank samples were <14.4% of the corresponding LLOQ (49.6 pg/mL) area response, and <5% of the IS response across all six batches.
3.6 Matrix Effect and Recovery
Plasma matrix effect testing found no significant variation in LNG peak area among six separate plasma lots, suggesting that endogenous plasma components did not affect analyte recovery (Table 4). The mean overall recovery (RE) for LNG and IS from plasma was 85.0% and 82.6%, respectively, with precision of 4.72% and 3.29%, respectively.
Table 4.
Recovery (%) and matrix effect (%) data for LNG (n=6)
| %Recovery (RE) | %Matrix Effect (ME) | %Process Efficiency (PE) | %Analysis RE | ||
|---|---|---|---|---|---|
| LQC | Mean | 88.94 | 97.85 | 87.04 | 93.62 |
| (134 pg/mL) | % CV | 8.33 | 3.25 | 4.22 | 3.54 |
|
| |||||
| MQC | Mean | 89.95 | 92.39 | 83.10 | 87.04 |
| (420 pg/mL) | % CV | 3.29 | 3.23 | 5.64 | 6.87 |
|
| |||||
| HQC | Mean | 80.93 | 97.70 | 79.07 | 94.78 |
| (1200 pg/mL) | % CV | 6.10 | 0.49 | 3.18 | 2.14 |
|
| |||||
| Overall | Mean | 85.03 | 98.78 | 83.07 | 91.81 |
| % CV | 4.71 | 1.75 | 4.80 | 4.55 | |
|
| |||||
| LNG-d7 (IS) | Mean | 83.94 | 92.35 | 82.98 | – |
| % CV | 3.29 | 3.23 | 4.36 | – | |
Abbreviations: LNG, levonorgestrel; LQC, low quality control; MQC, medium quality control; HQC, high quality control.
3.7 Clinical Application
Plasma samples from 60 women living with HIV and using LNG-releasing subdermal implant (2 rods, 75 mg LNG/rod) alone or with efavirenz- or nevirapine-based antiretroviral therapy (ART) over one year were analyzed for LNG using this method and have been previously published [14]. Figure 3 depicts the chromatogram of LNG and concentrations detected ranged from 89.3 to 3298 pg/mL (n=414 samples, median concentration 579.9 pg/mL).
Figure 3.
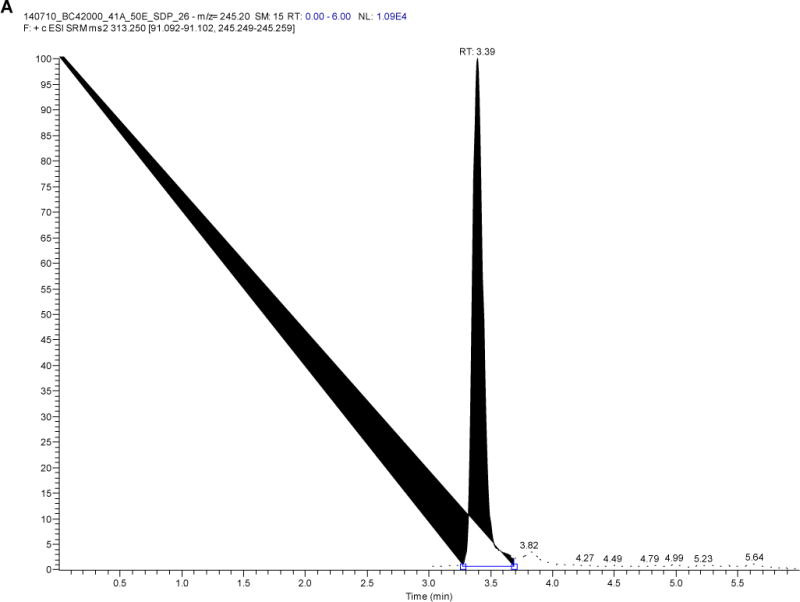
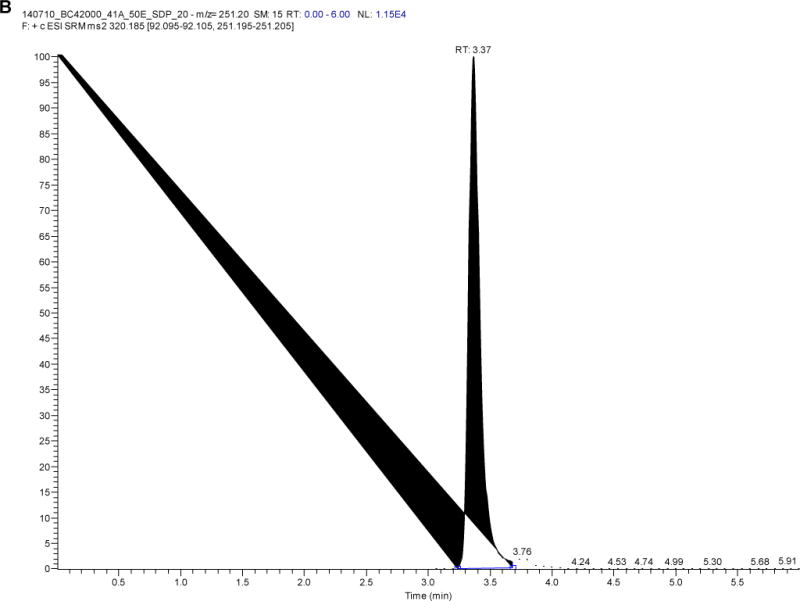
Chromatogram of A) levonorgestrel and B) D-(−)-norgestrel-d7 from a study participant with a levonorgestrel plasma concentration of 821 pg/mL)
4. Discussion
LNG is an exogenous progestin included in many formulations of contraception that, depending on the dose and route of administration used, has highly variable concentrations in plasma. For example, LNG 1.5 mg administered orally as a single dose for emergency contraception results in peak concentrations twenty-six times higher than LNG-releasing subdermal implants used as long acting reversible contraception [4]. Clinically significant drug-drug interactions between progestin-releasing contraceptives and efavirenz have been demonstrated [14, 17–19]. Furthermore, LNG concentrations may be approximately 50% lower when combined with other CYP inducers, including anticonvulsants [20], and significant decreases are expected with agents like rifamycins [21]. In fact, the European and British guidelines for emergency contraception have recently been updated to recommend a two-fold higher dose of LNG (3 mg once) for women receiving CYP3A4 inducers [22, 23], highlighting the importance of understanding LNG pharmacokinetics in relationship to contraceptive effectiveness. In addition, LNG exposure may be increased by strong CYP inhibitors such as azole antifungals or HIV protease inhibitors [1], although the clinical significance of inhibition of LNG metabolism is unclear.
A simple, rapid and highly sensitive analytical assay is necessary for determining plasma LNG concentrations in the context of drug-drug interactions. High sensitivity is particularly critical, as CYP3A enzyme inducers like efavirenz further decrease systemic LNG. While RIA provided an early method for sensitive LNG quantification, radioactive tracers complicate its use, and it is both labor intensive and lacks high throughput. To this end, LC-MS/MS has emerged as a preferable method for plasma LNG quantification. Comparison between LNG extraction techniques found that solid-phase extraction (SPE) produces more signal interferences and has decreased specificity compared to liquid-liquid extraction [11, 24]. Liquid-liquid extraction of plasma LNG has been described [25–27], however these methods are less sensitive, with LLOQs greater than or equal to 100 pg/mL. Furthermore, specialized semi-automated processes are required by one liquid-liquid extraction assay [25], which may potentially limit the application of this method in other laboratories. A method using an online SPE LC- MS/MS system following liquid-liquid extraction of LNG from plasma has been described, with a quantifiable linear range from 50 to 1500 pg/mL [24]. However online SPE LC-MS/MS requires additional costly equipment and maintenance that may not be available in all laboratory settings. Furthermore, the risk of carryover may complicate online SPE [28].
Steroidal drugs are poorly ionizable compounds and hence detection on a mass spectrometer poses analytical challenges. For example, LNG possesses only a single - OH functional group that can be ionized. Another challenge in measuring steroidal compounds is the potential for interference from endogenous components. With this in mind, of the mobile phases that were tested during early development, we opted for basic mobile phase conditions as this achieved increased separation of LNG from endogenous interferences (background interference) and improved the sensitivity of the method.
Using the method described herein, plasma extracts were prepared using a rapid and highly sensitive liquid-liquid extraction with good recovery. Similar to Liceo-Perez et al. [25], we developed a LC-MS/MS method using a liquid-liquid extraction technique; however, we did not utilize semi-automated instruments during sample processing and the analyte extraction procedure, which permits our method to be utilized in laboratories without automated resources. Furthermore, we achieved similar sensitivity as Moser et al., with a lower limit of detection of 49.6 pg/mL [24]. Vieria et al. used a similar LC- MS/MS method to determine concentrations of the subdermal implant progestin, etonogestrel that was linear over a range of 49.6 to 1500 pg/mL [17]. One limitation of our current method is the large plasma sample volume required for analysis (500 μL). Plasma sample volumes of 200 μL have been utilized [26, 27], but this in turn corresponded with substantially higher assay LLOQs (625 pg/mL and 5000 pg/mL). Moser et al. similarly used a plasma sample volume of 500 μL [24].
5. Conclusion
We have developed and validated a rapid and highly sensitive and specific LC-MS/MS method to quantify plasma LNG concentrations. Given the demonstrated high sensitivity of this assay, this approach is ideal for application in PK studies involving low systemic circulating LNG during placement of LNG-releasing subdermal implants, including detection of LNG in the context of drug-drug interactions.
Highlights.
Development of sensitive LC-MS/MS method for quantification of LNG in human plasma.
Application of method for determination of LNG released by subdermal impant.
Method used in pharmacokinetic study in women living with HIV receiving ART.
Acknowledgments
Funding
The authors acknowledge support from grants from the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development [grant numbers 1R01HD085887 and 5R21HD074462 (Scarsi)] and National Institute of Allergy and Infectious Diseases [grant number UM1AI06701 (Fletcher)]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
MS has received research grants from Janssen Pharmaceutica NV and ViiV. SHK and DJB have received research grants and travel bursaries from Gilead, ViiV, Merck, Bristol Myers Squibb, Boehringer Ingelheim and Janssen Pharmaceuticals. DJB has board membership and consultancy with ViiV, Merck, and Janssen Pharmaceuticals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All other authors: None to declare.
References
- 1.Jadelle (levonorgestrel implants) [package insert] Auckland, New Zealand: Bayer New Zealand Limited Company; Aug, 2010. [Google Scholar]
- 2.Scarsi KK, Darin KM, Chappell CA, Nitz SM, Lamorde M. Drug–Drug Interactions, Effectiveness, and Safety of Hormonal Contraceptives in Women Living with HIV. Drug Safety. 2016;39:1053–1072. doi: 10.1007/s40264-016-0452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanczyk FZ, Roy S. Metabolism of levonorgestrel, norethindrone, and structurally related contraceptive steroids. Contraception. 1990;42:67–96. doi: 10.1016/0010-7824(90)90093-b. [DOI] [PubMed] [Google Scholar]
- 4.Plan B One-Step (levonorgestrel) [package insert] Pomona, NY: Durmed Pharmaceuticals, Inc.; 2009. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/021998lbl.pdf. Accessed 12 Sept 2017. [Google Scholar]
- 5.Sivin I, Lahteenmaki P, Mishell DR, Jr, Alvarez F, Diaz S, Ranta S, Grozinger C, Lacarra M, Brache V, Pavez M, Nash H, Stern J. First week drug concentrations in women with levonorgestrel rod or Norplant capsule implants. Contraception. 1997;56:317–321. doi: 10.1016/s0010-7824(97)00153-4. [DOI] [PubMed] [Google Scholar]
- 6.Weiner E, Johansson ED. Plasma levels of d-norgestrel, estradiol and progesterone during treatment with silastic implants containing d-norgestrel. Contraception. 1976;14:81–92. doi: 10.1016/s0010-7824(76)80011-x. [DOI] [PubMed] [Google Scholar]
- 7.Sivin I, Wan L, Ranta S, Alvarez F, Brache V, Mishell DR, Jr, Darney P, Biswas A, Diaz S, Kiriwat O, Anant MP, Klaisle C, Pavez M, Schechter J. Levonorgestrel concentrations during 7 years of continuous use of Jadelle contraceptive implants. Contraception. 2001;64:43–49. doi: 10.1016/s0010-7824(01)00226-8. [DOI] [PubMed] [Google Scholar]
- 8.Cherala G, Edelman A, Dorflinger L, Stanczyk FZ. The elusive minimum threshold concentration of levonorgestrel for contraceptive efficacy. Contraception. 2016;94:104–108. doi: 10.1016/j.contraception.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Moser C, Zoderer D, Luef G, Rauchenzauner M, Wildt L, Griesmacher A, Seger C. Simultaneous online SPE-LC-MS/MS quantification of six widely used synthetic progestins in human plasma. Analytical and bioanalytical chemistry. 2012;403:961–972. doi: 10.1007/s00216-011-5612-0. [DOI] [PubMed] [Google Scholar]
- 10.Theron HB, Coetzee C, Sutherland FC, Wiesner JL, Swart KJ. Selective and sensitive liquid chromatography-tandem mass spectrometry method for the determination of levonorgestrel in human plasma. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2004;813:331–336. doi: 10.1016/j.jchromb.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 11.Wang R, Tian Y, Zhang L, Zhang Z. Simultaneous determination of levonorgestrel and two endogenous sex hormones in human plasma based on LC-MS/MS. Bioanalysis. 2016;8:1133–1144. doi: 10.4155/bio-2015-0036. [DOI] [PubMed] [Google Scholar]
- 12.Zhao LZ, Zhong GP, Bi HC, Ding L, Deng Y, Guan S, Chen X, Huang ZY, Huang M. Determination of levonorgestrel in human plasma by liquid chromatography- tandem mass spectrometry method: application to a bioequivalence study of two formulations in healthy volunteers. Biomedical chromatography: BMC. 2008;22:519–526. doi: 10.1002/bmc.963. [DOI] [PubMed] [Google Scholar]
- 13.Kumar V, Ashish S, Amol P, SMN M, R V, Ravisekhar K, Pratima A, Ashawat MS. A Rapid and Sensitive UPLC–ESI-MS/MS Method for Determination of Levonorgestrel by Chemical Derivatization in Human Plasma and its Application to Pharmacokinetic Study. J Anal Bioanal Techniques. doi: 10.4172/2155-9872.s6-003. [DOI] [Google Scholar]
- 14.Scarsi KK, Darin KM, Nakalema S, Back DJ, Byakika-Kibwika P, Else LJ, Penchala SD, Buzibye A, Cohn SE, Merry C, Lamorde M. Unintended Pregnancies Observed With Combined Use of the Levonorgestrel Contraceptive Implant and Efavirenz-based Antiretroviral Therapy: A Three-Arm Pharmacokinetic Evaluation Over 48 Weeks. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2016;62:675–682. doi: 10.1093/cid/civ1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.United States Food and Drug Administration. Bioanalytical Method Validation. 2013 Sep; Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM368107.pdf. Accessed 18 June 2017.
- 16.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Matrix effect in quantitative LC/MS/MS analyses of biological fluids: a method for determination of finasteride in human plasma at picogram per milliliter concentrations. Analytical chemistry. 1998;70:882–889. doi: 10.1021/ac971078+. [DOI] [PubMed] [Google Scholar]
- 17.Vieira CS, Bahamondes MV, de Souza RM, Brito MB, Rocha Prandini TR, Amaral E, Bahamondes L, Duarte G, Quintana SM, Scaranari C, Ferriani RA. Effect of antiretroviral therapy including lopinavir/ritonavir or efavirenz on etonogestrel-releasing implant pharmacokinetics in HIV-positive women. Journal of acquired immune deficiency syndromes. 1999;66:378–385. doi: 10.1097/QAI.0000000000000189. 2014. [DOI] [PubMed] [Google Scholar]
- 18.Carten ML, Kiser JJ, Kwara A, Mawhinney S, Cu-Uvin S. Pharmacokinetic interactions between the hormonal emergency contraception, levonorgestrel (Plan B), and Efavirenz. Infectious diseases in obstetrics and gynecology. 2012;2012:137192. doi: 10.1155/2012/137192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chappell CA, Lamorde M, Nakalema S, Chen BA, Mackline H, Riddler SA, Cohn SE, Darin KM, Achilles SL, Scarsi KK. Efavirenz decreases etonogestrel exposure: a pharmacokinetic evaluation of implantable contraception with antiretroviral therapy. AIDS (London, England) 2017;31:1965–1972. doi: 10.1097/QAD.0000000000001591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crawford P, Chadwick DJ, Martin C, Tjia J, Back DJ, Orme M. The interaction of phenytoin and carbamazepine with combined oral contraceptive steroids. British Journal of Clinical Pharmacology. 1990;30:892–896. doi: 10.1111/j.1365-2125.1990.tb05457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faculty of Sexual & Reproductive Healthcare Clinical Effectiveness Unit. Drug Interactions with Hormonal Contraception. 2017 Available at https://www.fsrh.org/standards-and-guidance/documents/ceu-clinical-guidance-drug-interactions-with-hormonal/drug-interactions-final-15feb.pdf. Accessed 9 Oct 2017.
- 22.European Consortium for Emergency Contraception. Emergency contraception: A guideline for service provision in Europe. 2016 Available at http://www.ec-ec.org/custom-content/uploads/2016/05/ECEC-Guidelines-2nd-edition-May2016.pdf. Accessed 9 Oct 2016.
- 23.Faculty of Sexual & Reproductive Healthcare Clinical Effectiveness Unit. Emergency Contraception. 2017 Available at http://www.ec-ec.org/custom-content/uploads/2013/07/fsrh-guideline-emergency-contraception-may-2017-1.pdf. Accessed 9 Oct 2017.
- 24.Moser C, Gschliesser A, Mattle V, Wildt L, Griesmacher A, Seger C. An ultra- sensitive online SPE-LC-MS/MS method for the quantification of levonorgestrel released from intrauterine devices. Analytical and bioanalytical chemistry. 2011;400:2655–2662. doi: 10.1007/s00216-011-4790-0. [DOI] [PubMed] [Google Scholar]
- 25.Licea-Perez H, Wang S, Bowen CL, Yang E. A semi-automated 96-well plate method for the simultaneous determination of oral contraceptives concentrations in human plasma using ultra performance liquid chromatography coupled with tandem mass spectrometry. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2007;852:69–76. doi: 10.1016/j.jchromb.2006.12.052. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q, Wu Z, Wang Y, Luo G, Wu E, Gao X, Li G. Determination of levonorgestrel in human serum by liquid chromatographic-electrospray tandem mass spectrometry. Analytical Letters. 2001;34:103–112. [Google Scholar]
- 27.Liu F, Xu Y, Liu A, Xu F, Hu W, Guo Q. LC–Tandem-MS Validation for the Quantitative Analysis of Levonorgestrel in Human Plasma. Chromatographia. 2008;68:707–712. [Google Scholar]
- 28.Mullett WM. Determination of drugs in biological fluids by direct injection of samples for liquid-chromatographic analysis. Journal of biochemical and biophysical methods. 2007;70:263–273. doi: 10.1016/j.jbbm.2006.10.001. [DOI] [PubMed] [Google Scholar]