

Abstract
Tissue transglutaminase (tTG) is a multifunctional protein with a wide range of enzymatic and non-enzymatic functions. We have recently demonstrated that tTG expression is upregulated in the pressure-overloaded myocardium and exerts fibrogenic actions promoting diastolic dysfunction, while preventing chamber dilation. Our current investigation dissects the in vivo and in vitro roles of the enzymatic effects of tTG on fibrotic remodeling in pressure-overloaded myocardium. Using a mouse model of transverse aortic constriction, we demonstrated perivascular and interstitial tTG activation in the remodeling pressure-overloaded heart. tTG inhibition through administration of the selective small molecule tTG inhibitor ERW1041E attenuated left ventricular diastolic dysfunction and reduced cardiomyocyte hypertrophy and interstitial fibrosis in the pressure-overloaded heart, without affecting chamber dimensions and ejection fraction. In vivo, tTG inhibition markedly reduced myocardial collagen mRNA and protein levels and attenuated transcription of fibrosis-associated genes. In contrast, addition of exogenous recombinant tTG to fibroblast-populated collagen pads had no significant effects on collagen transcription, and instead increased synthesis of matrix metalloproteinase (MMP)3 and tissue inhibitor of metalloproteinases (TIMP)1 through transamidase-independent actions. However, enzymatic effects of matrix-bound tTG increased the thickness of pericellular collagen in fibroblast-populated pads. tTG exerts distinct enzymatic and non-enzymatic functions in the remodeling pressure-overloaded heart. The enzymatic effects of tTG are fibrogenic and promote diastolic dysfunction, but do not directly modulate the pro-fibrotic transcriptional program of fibroblasts. Targeting transamidase-dependent actions of tTG may be a promising therapeutic strategy in patients with heart failure and fibrosis-associated diastolic dysfunction.
Keywords: tissue transglutaminase, cardiac fibrosis, extracellular matrix, fibroblast
1. INTRODUCTION
Tissue transglutaminase (tTG, transglutaminase 2), the most ubiquitous and best-studied member of the transglutaminase family, is a multifunctional protein with a wide range of enzymatic and non-enzymatic functions [1],[2]. As a transglutaminase, tTG is capable of mediating Ca2+-dependent isopeptide bond formation, in which free amines (from protein-bound lysine residues, or small molecules such as serotonin) are covalently linked to glutamine residues of an acceptor protein [3]. Transamidase-dependent actions of tTG have been implicated in the function, stability and immunogenicity of its substrate proteins, such as collagen and fibronectin [4]. In addition to its enzymatic functions, tTG has been suggested to exert a wide range of non-enzymatic effects. Following tissue injury, tTG is secreted to the extracellular matrix (ECM), where it binds with high affinity to fibronectin and heparin sulfate proteoglycans (HSPGs) [5]. Matrix-bound tTG may serve as a bridging protein that activates syndecan-4 signaling – in a transamidase-independent manner [6], or may function as a co-receptor for integrin-mediated cell adhesion [7],[8],[9]. The relative contributions of the enzymatic and non-enzymatic functions of tTG to cellular responses in vivo remain unknown.
Cardiac pressure overload activates a hypertrophic and fibrogenic program in the myocardium leading to the development of heart failure [10],[11]. Using a mouse model of transverse aortic constriction (TAC), we have recently demonstrated that tTG is the only member of the transglutaminase family that is upregulated in the pressure-overloaded myocardium [12]. Using mice with global loss of tTG, we found that tTG promotes diastolic dysfunction, while protecting the pressure-overloaded heart from dilation and systolic dysfunction [12]. In addition to its effects on collagen crosslinking, tTG also restrained fibroblast proliferation and attenuated activation of matrix metalloproteinase (MMP)2 [12]. Whether the in vivo functions of tTG in the remodeling myocardium are mediated through enzymatic effects, or through transamidase-independent actions remains unknown. In this study, we dissected the role of enzymatic tTG actions in the remodeling myocardium, by treating mice undergoing pressure-overload protocols with ERW1041E, a specific, irreversible pharmacological inhibitor of enzymatically-active tTG[13]. Our findings provide the first direct evidence of tTG activation in the pressure-overloaded myocardium and demonstrate that enzymatic actions of tTG are involved in activation of a fibrogenic program and in the pathogenesis of diastolic dysfunction in the pressure-overloaded heart. Moreover, our in vitro experiments demonstrate that stimulation with matrix-bound, recombinant tTG does not affect collagen transcription in cardiac fibroblasts, but induces MMP3 and TIMP1 synthesis through non-enzymatic actions. The fibrogenic enzymatic in vivo actions of tTG may reflect effects on collagen processing.
2. METHODS
2.1. Animal Protocols
Animal experiments were conducted in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals and were approved by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee. Male and female, 3–4 month-old wildtype (WT) mice on a C57BL/6J background (purchased from Jackson labs) underwent TAC protocols to induce pressure overload, as previously described [14],[10],[15]. Mice were anesthetized with inhaled isoflurane. Aortic banding was achieved by creating a constriction between the right innominate and left carotid arteries. The degree of pressure overload was assessed by measuring right-to-left carotid artery flow velocity ratio after constricting the transverse aorta. Mice with a very low (<4) or a very high (>15) carotid flow ratio were excluded from further analysis. In order to investigate the role of enzymatic tTG-mediated actions, age- and gender-matched male and female wild type C57BL/6J mice treated with either a small molecule tTG inhibitor or vehicle control underwent TAC protocols (n= 25–26 per group) for 28 days. As a control, a sham operation without aortic constriction was performed on gender and age-matched mice treated with either a small molecule tTG inhibitor (n=12) or vehicle control (n=12). ERW1041E ((S)-Quinolin-3-ylmethyl 2-((((S)-3-Bromo-4, 5-dihydroisoxazol-5-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate) is a specific, irreversible small molecule inhibitor of tTG that pharmacologically blocks activated tTG in vivo. This compound was synthesized as described previously [16]. The efficacy and duration of blockade of tTG activity have been already determined and published [16]. ERW1041E solution was formulated as follows: 10 mg/mL ERW1041E was reconstituted in 90% ultrapure water, 10% DMSO (biotechnology, endotoxin free grade), 2% Tween-80 detergent, and 2.5% (m/v) 2-hydroxypropyl beta-cyclodextrin and then it was filter sterilized. This solution was freshly prepared before every injection to ensure there were no solubility issues or degradation. Control animals were injected with the vehicle without ERW1041E. Both the inhibitor and vehicle-treated group animals from TAC and sham groups underwent echocardiography at baseline and after 28 days of TAC or sham surgery. Based on the estimated t1/2 of tTG inhibition by ERW1041E (12±1 hr.) [16], a twice daily dose of 25 mg/kg was administered to the mice intraperitoneally for 28 days following TAC, with the first IP injection administered on the day of TAC. At the end of the experiment, mice were euthanized, the hearts were harvested and the basal 2/3 were fixed in Z-fix (Anatech Ltd, Fisher Scientific) and processed for paraffin histology. The apical 1/3 was snap frozen and used for protein extraction.
2.2. Echocardiography and strain rate imaging
Short axis M-mode echocardiography was performed prior to instrumentation and before the end of each experiment, after 28 days of TAC or sham surgery, using the Vevo 2100 system (VisualSonics, Toronto ON), as previously described [17]. The following parameters were assessed as indicators of function and remodeling: left ventricular end-diastolic volume (LVEDV), ejection fraction, and left ventricular mass (LV mass). Speckle tracking echocardiography was performed at baseline and after 7–28 days of pressure overload in order to study the effects of the tTG inhibitor on radial strain rates using parasternal long axis views. Analysis was performed using the VevoStrain package (VisualSonics).
2.3. In vivo transglutaminase assay and quantitation of transglutaminase activity
5-biotinamidopentylamine (5BP) is a synthetic substrate of tTG that can be used to probe tTG activity in vivo [18]. 5-BP·HCl was synthesized as previously described [16] and was used to localize the in vivo transglutaminase activity of tTG in the pressure-overloaded heart and to assess the effectiveness of the tTG inhibitor. Age- and gender-matched male and female wild type C57BL/6J mice, treated with either a tTG inhibitor ERW1041E or vehicle control, underwent 14 days of TAC (n=6 per group, 3 males and 3 females). Solutions were formulated just prior to the experiment and used the same day for each mouse experiment. 5BP was dissolved in PBS (20 mg/mL) and was then filtered through a disposable 0.2 μm sterile polyvinylidenefluoride (PVDF) membrane filter prior to injection. Ninety minutes prior to sacrifice, mice were injected with 5BP (100 mg/kg i.p.). To ensure that 5BP incorporation depended on the tTG activity, ERW1041E (25 mg/kg i.p) or vehicle control was contemporaneously administered. The 5BP injection was repeated at 60 and 30 mins before the mice were sacrificed. Subsequently, the heart was dissected into two segments. The basal two-third portion of the heart was directly embedded in optimal cutting temperature (OCT) (Tissue-Plus O.C.T compound, Fisher Scientific), frozen in −80°C, and used for assessment of enzymatic activity. The apical one-third segment was used for RNA extraction.
Frozen mouse tissue cryomolds were transferred to a cryostat equilibrated to −20 °C. Tissue blocks were removed from the cryomolds, cut into 10 μm sections, and transferred to Superfrost Plus slides (ThermoFisher Scientific). Tissue sections were stored at −80 °C and used for assessment of enzymatic activity using a peroxidase-based method. Sections were fixed in 4% (w/v) paraformaldehyde for 15 min at room temperature and thoroughly washed with PBS. Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide for 15 mins followed by thorough washing with PBS. The 5BP signal was amplified using the Vectastain Elite ABC Peroxidase Kit (PK-6100, Vector Labs). DAB (3, 3′-diaminobenzidine) was used to detect peroxidase (SK-4100). The slides were then rinsed in distilled water and counterstained either with Hematoxylin or Eosin. Coverslips were then mounted using Cytoseal XYL (Richard Allen Scientific) mounting medium. Images were then obtained at 200X on Zeiss Axio Imager M2.
For quantitation of transglutaminase activity, stained slides from both the ERW1041E-treated and vehicle-treated control groups were analyzed by light microscopy. 8 fields (200X) from each mouse (n=5/group) were scored individually for intensity and extent of staining. For intensity of staining, the following scale was used: no staining: 0, low intensity of staining: 1, moderate intensity of staining: 2, strong intensity of staining: 3. For extent of staining: no staining: 0, staining in <25% of the section:1, staining in 25–75% of the section: 2, staining in >75% of the section: 3. Then the average of the intensity and extent of staining was used as a measure of tTG activity in each field.
2.4. Assessment of cardiomyocyte size
Cardiomyocytes were outlined using wheat germ agglutinin (WGA) histochemistry, as previously described, using an Alexa Fluor® 594 Conjugate (Life Technologies) (dilution 1:100) [19]. Cardiomyocyte size was quantified using AxioVision LE 4.8 software (Zeiss). A total of 80 cells from ten fields from three non-adjacent stained sections at 3 different levels were used for analysis (n=13–14 hearts/group). Cardiomyocyte area was expressed in μm2.
2.5. Cardiac fibroblast isolation, culture, and RNA extraction
Cardiac fibroblasts were isolated from C57BL/6J animals using enzymatic digestion as previously described [20] and were cultured in DMEM/F12 (GIBCO Invitrogen Corporation, Carlsbad, CA) with 10% Fetal Calf Serum (FCS). Cells were serum-starved at passage 2 for 16h and subsequently used in collagen pad assays. Total RNA was isolated from the collagen pads using GeneJET RNA Purification Kit (ThermoFisher Scientific) and the RNA obtained was used for quantitative PCR.
2.6. Stimulation of cardiac fibroblasts populating collagen pads with recombinant and inactive tTG
Cardiac fibroblasts isolated from both male and female adult wild-type C57BL/6J mice and tTG global knockout mice in a C57BL/6J background (from our colony) [21], [12] were cultured to passage 2 and serum-starved overnight (16 h). Collagen matrix was prepared on ice by diluting a stock solution of rat collagen I (3.0 mg/mL, GIBCO Invitrogen Corporation, Carlsbad, CA) with 2X MEM and distilled water to a final concentration of 1 mg/ml collagen. Cell suspensions in 2X MEM were mixed with collagen solution to achieve the final concentration of 3×105 cells/ml. Subsequently, 500 μl of this suspension was aliquoted to a 24-well culture plate (BD Falcon, San Jose, CA) and allowed to polymerize at 37°C for 30 min. Following polymerization, pads were released from wells, transferred to 6-well culture plate (BD Falcon, San Jose, CA) and cultured in 0% FCS DMEM/F12 for 24 h. After incubation, pads were used either for RNA extraction, or for histological processing, as previously described [22]. For experiments examining the effects of tTG on fibroblast phenotype, enzymatically active recombinant mouse tTG (T040, Zedira GmbH), or the C277S active site null mutant of human recombinant tTG (T018, Zedira GmbH) was incorporated into collagen pads at a concentration of 50 μg/ml (pads used for RNA analysis: n=12–16/group for WT cells, n=4–6/group for tTG KO cells, pads used for histological analysis: n=10–13 pads/group for WT cells, n=6–7 pads for tTG KO cells).
In additional experiments examining effects of tTG on fibroblast phenotype, three site-specific inhibitors of tTG were used: Boc-DON (B003, Zedira GmbH), which is a potent cell impermeable irreversible site-specific inhibitor [23], Z-DON (616467, Calbiochem), a potent cell-permeable peptide-based irreversible site-specific inhibitor [23], and TAMRA-DON (R002, Zedira GmbH), a fluorescent blocker of tTG [24]. These inhibitors were added at a final concentration of 100 μm to 0% FCS DMEM/F12 media in which collagen pads prepared as described above were cultured for 24 h, following which the pads were used for RNA extraction (5–9 pads/group).
2.7. Quantitative real-time PCR
Isolated total RNA from mouse hearts or collagen pads was reverse transcribed to cDNA using the iScript™ cDNA synthesis kit (Bio-Rad) following the manufacturer’s guidelines. Quantitative PCR was performed using the SsoFast™ EvaGreen® Supermix (Bio-Rad) method on the CFX384™ Real-Time PCR Detection System (Bio-Rad). Primers were synthesized by Integrated DNA Technologies. The following sets of primers were used in the study: TIMP1 forward GCCTGAACACTGTCTACTT reverse TTGCTGCTGTCTGATAGTT; TIMP2 forward CTTCTGTCTCTTAATGTC reverse CTGTCTATATTGTTAGTGAT; MMP2 forward TCCGCTGCATCCAGACTT, reverse GGTCCTGGCAATCCCTTTGTATA; MMP3 forward ATTTGGGTTTCTCTACTT, reverse GAAGAACTATAAGCATCAG; MMP8 forward TTAGGATGAGCCATAAGT, reverse TTGCTTGGTCTCTTCTAT; MMP9 forward TCTTACATTGGAGAACAC, reverse GAAGGAAGAACCAACATT; collagen I forward GATACTTGAAGAATATGAAC, reverse AATGCTGAATCTAATGAA; collagen III forward TACTCATTCACCAGCATA, reverse GTATAGTCTTCAGGTCTCA; GAPDH forward AACGACCCCTTCATTGACCT, reverse CACCAGTAGACTCCACGACA.
2.8. Protein extraction and western blotting
The apical 1/3 of the hearts undergoing 28-day TAC protocols were used for protein extraction. Protein was isolated using T-PER tissue protein extraction reagent (78510, ThermoFisher), 10% protease inhibitor cocktail (Roche), 10% phosphatase inhibitor cocktail (Roche). Protein concentrations were determined and proteins (25 μg) in denaturing conditions were subjected to SDS-PAGE (4%-20%) and then electrotransferred onto polyvinylidene difluoride membranes (Bio-Rad). The blots were then blocked with 5% BSA dissolved in Tris-buffered saline containing 0.1% Tween 20 (TTBS) either overnight or 2 h at room temperature, washed three times with TTBS for 5 min each and incubated overnight at 4°C with specific primary antibodies anti-tTG (ab109200, Abcam, 1:2000 dilution), anti-Collagen III (ab7778 Abcam; 1:1000 dilution), anti-MMP2 (ab37150, Abcam 1:1000 dilution), in TTBS containing 3% BSA. On the next day, membranes were washed (3 times for 5 min each) with TTBS and incubated for 1h at room temperature with horseradish peroxidase-conjugated secondary anti-rabbit IgG antibody (1:5000 dilution) in TTBS containing 2% nonfat dry milk. Protein bands were visualized by enhanced chemiluminescence using Super Signal West Pico or Femto reagents (Pierce). Signal intensity was measured with a Bio-Rad Imaging Station. Membranes were then stripped and reprobed with GAPDH antibody to verify equal loading and densitometric analysis was performed using Image J software.
In order to study the time course of tTG protein expression in pressure-overloaded hearts, we used myocardial protein samples from C57BL/6J mice undergoing 3–28 day TAC protocols and from corresponding sham-operated controls (n=4–7/group) [12]. The specificity of the tTG antibody was validated using protein samples from tTG KO mice and from cardiac tTG KO fibroblasts.
2.9. Sirius red staining and polarized light microscopy for assessment of collagen content
Polarized light microscopy was used to assess collagen deposition, as previously described [25], [26]. Briefly, paraffin sections (5 μm thick) were stained using sirius red. Circularly polarized images were obtained using Axio Imager M2 for polarized light microscopy (Zeiss). When the collagen fibers are stained with sirius red and viewed with polarized light, depending on the thickness of the collagen fibers, the hues range from green to yellow to orange to red. The respective proportions of different hues were assessed using ImageJ software. The 8-bit hue images contain 256 colors, and we distinguished different colors based on visible hues. We used the following hue definitions; red 2–9 and 230–256, orange 10–38, yellow 39–51, green 52–128. The number of pixels within each hue range was expressed as a percentage of the total number of collagen pixels, which in turn was expressed as a percentage of the total number of pixels in the image. At least 15 different fields from three non-adjacent stained sections per mouse at 3 different levels were obtained and analyzed per heart sample (n=13–14 hearts/group).
In a separate experiment, enzymatic actions of tTG on collagen processing and structure were studied using collagen pads which incorporated rtTG and itTG, as described above. Polarized light microscopy was used to visualize the pericellular “halo” of orange/red thick fibers around fibroblasts embedded in the collagen pad. This halo represents new synthesis and/or processing of collagen by the cells. Using AxioVision LE4.8 software (Zeiss), the pericellular collagen area was traced and quantitatively assessed. 15 cells per WT (n=10–13 pads/condition) or tTG KO pad (n=6–7 pads/condition) were used for quantitation.
2.10. Statistical analysis
Comparisons between more than 2 groups were performed using one-way ANOVA followed by t-test corrected for multiple comparisons (Sidak’s test). For non-Gaussian distributions, non-parametric ANOVA was used (Kruskal-Wallis). Comparisons between 2 groups were performed using unpaired t-test, or the Mann-Whitney U test (for nonparametric distributions). Mortality was compared using the log rank test. Data were expressed as mean±SEM. Statistical significance was set at p=0.05.
3. RESULTS
3.1. Upregulation of tTG in the pressure-overloaded myocardium
We have previously demonstrated that cardiac pressure overload is associated with myocardial tTG mRNA upregulation [12]. In the current study, we used western blotting experiments to assess expression of tTG protein in the pressure-overloaded heart in WT C57Bl6J mice. Myocardial tTG protein levels were significantly increased after 3 days of TAC in comparison to corresponding shams (Supplemental figure 1A, C). Samples from tTG KO sham hearts and from tTG KO fibroblasts served as a negative control, exhibiting absence of tTG (Supplemental figure 1A). After 28 days of TAC, there was a trend towards increased tTG levels (Supplemental figure 1B, D).
3.2. Activation of tTG in the pressure-overloaded myocardium is attenuated by infusion of the tTG inhibitor ERW1041E
Next, we used 5-BP, an established tTG activity probe[18], to localize tTG activity in the pressure overloaded myocardium and to assess the effectiveness of the tTG inhibitor. Negligible tTG activity was found in sham hearts (Figure 1A, D, G, J). After 14 days of pressure overload, tTG activity was localized in the perivascular areas (Figure 1B, E), in cardiomyocytes (Figure 1H) and in the cardiac interstitium (Figure 1K). Treatment with the tTG inhibitor ERW1041E markedly attenuated tTG activity in the pressure overloaded heart (Figure 1C, F, I, L). Quantitative analysis suggested intense activation of tTG in the pressure-overloaded myocardium that was markedly reduced after treatment with the tTG inhibitor ERW1041E (***p<0.001, Figure 1M).
Figure 1. tTG activation in the pressure-overloaded myocardium is markedly attenuated following daily injection of the specific small molecule inhibitor ERW1041E (ERW).
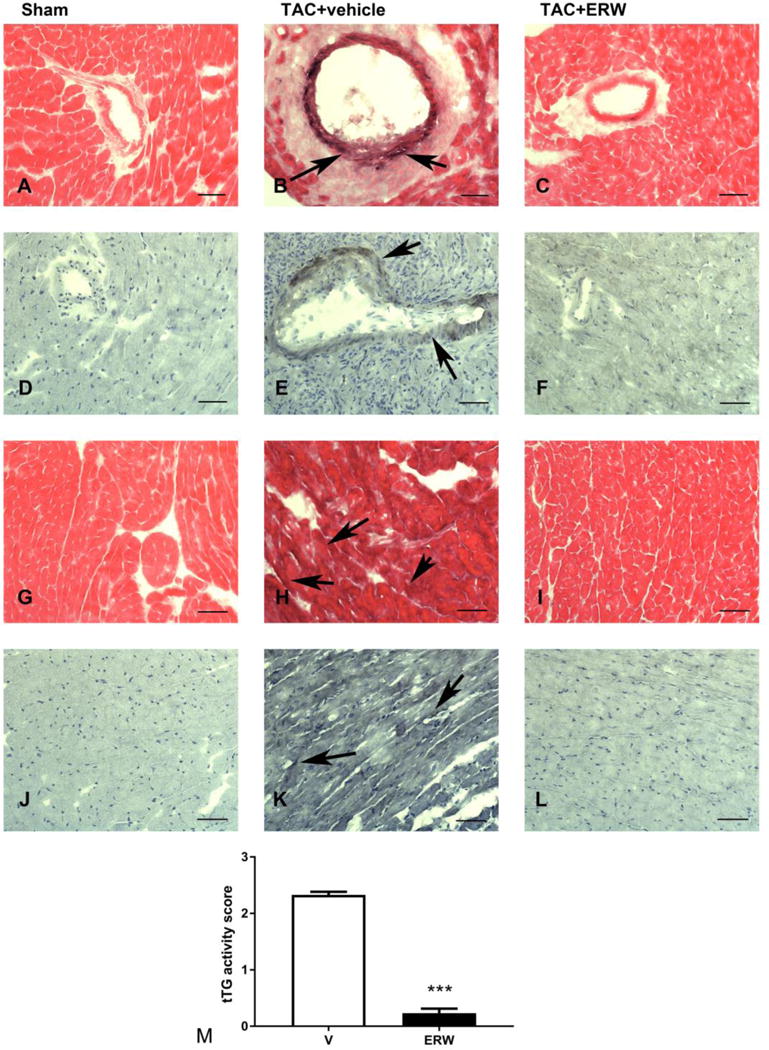
5-biotinamidopentylamine (5BP), a synthetic substrate of tTG, was used as a probe for tTG activity in tissues. When administered systemically, 5BP is covalently attached to extracellular matrix proteins in tissues in which tTG is enzymatically active. 5BP injections were performed in mice undergoing TAC protocols and sham controls. 5BP was visualized in frozen sections using a biotin-avidin-peroxidase method and was developed with DAB+nickel (black). Representative images from sham (A, D, G, J), TAC (B, E, H, K) and ERW-treated TAC animals (C, F, I, L) are shown. Sham hearts showed negligible tTG activity (A, D, G, J). After 14 days of TAC, intense staining, reflecting tTG activation was noted in areas of perivascular fibrosis (B, E, arrows), in the interstitium (H, K, arrows) and in cardiomyocytes. Treatment with ERW markedly attenuated tTG activity (C, F, I. L). M: Semiquantitive analysis showed marked reduction in tTG activity in ERW-treated animals after 14 days of TAC (***p<0.001, n=5/group). Scalebar=50μm.
3.3. Treatment with tTG inhibitor attenuates development of left ventricular hypertrophy without affecting chamber dimensions and ejection fraction following cardiac pressure overload
TAC induced a marked increase in the R:L carotid flow ratio in comparison to sham-operated mice (Supplemental figure 2A). In pressure-overloaded mice, the carotid flow ratio was comparable between vehicle- and inhibitor-treated animals (ERW), suggesting comparable pressure loads between groups (Supplemental figure 2A). Two vehicle-treated mice had very high R:L carotid flow ratios (>15), indicating an extremely high pressure load, and were excluded from further analysis. Mortality following TAC was not affected by tTG inhibition (Supplemental figure 2B). The survival rate at 28 days was 84% for vehicle-treated mice and 73.1% for inhibitor-treated animals (p=NS, n=25–26/group). Vehicle-treated controls and inhibitor-treated animals had no significant differences in body weight (BW) at baseline and after 28 days of TAC (Figure 2A). Pressure overload markedly increased the heart weight to body weight ratio (HW:BW) in both vehicle- and ERW-treated animals. ERW1041E-treated animals exhibited a trend towards reduced HW:BW after 28 days of TAC (Figure 2B). In contrast, the lung weight:body weight ratio (LW:BW) was comparable between vehicle and ERW-treated animals (Figure 2C). Echocardiographic analysis showed that tTG inhibition did not significantly affect LVEDV (Figure 2D) following pressure overload. Ejection fraction was significantly increased in sham animals, likely reflecting a hyperdynamic circulation due to the daily injections. Cardiac pressure overload was associated with a significantly reduced ejection fraction in both vehicle and ERW-treated groups, in comparison to shams (Figure 2E, G). However, tTG inhibition did not significantly affect ejection fraction in sham or pressure-overloaded animals. Cardiac pressure overload was associated with a significantly increased echocardiographically-derived LV mass in vehicle-treated, but not in ERW-treated animals (Figure 2F, H), suggesting that tTG inhibition attenuated cardiac hypertrophy. In contrast, ERW administration had no significant effects on LV mass in sham animals.
Figure 2. Effects of tTG inhibition on cardiac geometry and function following pressure overload.
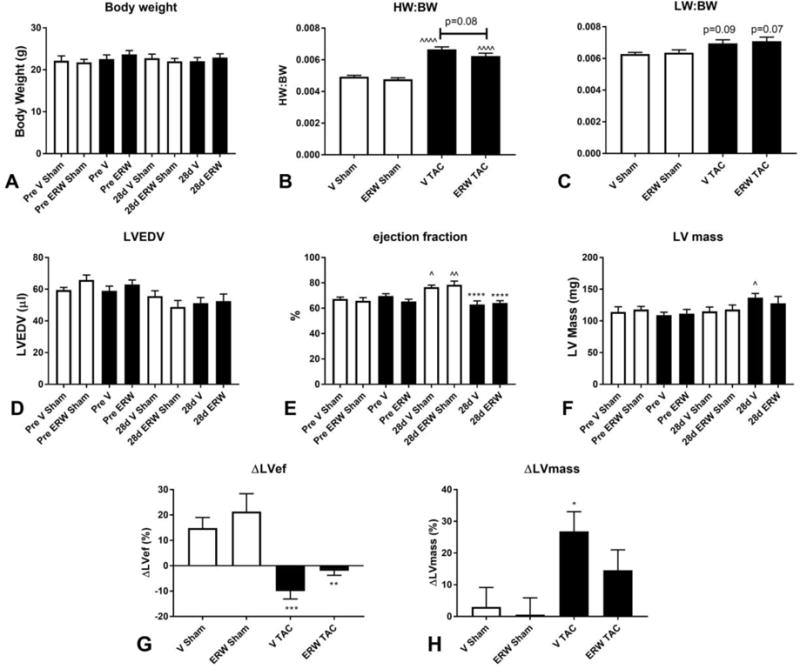
A. tTG inhibition had no significant effects on body weight (BW) in sham or pressure-overloaded mice. B. Cardiac pressure overload significantly increased the heart weight to body weight ratio (HW:BW) after 28 days of TAC (^^^^p<0.0001 vs. sham, n=12–19/group). There was a trend towards a lower HW:BW in animals treated with tTG inhibitor (p=0.08, n=19/group). C: The lung weight to body weight ratio (LW:BW) was comparable between groups. D: Echocardiographic analysis showed that treatment with the tTG inhibitor did not affect LVEDV after 28 days of TAC. E: Ejection fraction was increased in sham animals receiving vehicle or ERW, likely reflecting a hyperdynamic circulation due to the stress of daily injection protocols (^p<0.05, ^^p<0.01 vs. corresponding baseline). Ejection fraction was significantly lower in both vehicle and ERW-treated groups after 28 days of TAC (****p<0.0001 vs. corresponding sham); however, treatment with the inhibitor did not affect ejection fraction. F: TAC significantly increased LV mass in vehicle-treated controls (^p<0.05), but not in inhibitor-treated animals. G. Treatment with the inhibitor did not affect the reduction in ejection fraction (ΔLVef) following pressure overload (**p<0.01, ***p<0.001 vs. corresponding sham). H: Vehicle-treated mice exhibited a 25% increase in LV mass (ΔLVmass) in comparison to baseline levels (*p<0.05 vs. sham). In contrast, the increase in LV mass in ERW-treated mice did not reach statistical significance (p=NS vs. sham), suggesting that tTG inhibition attenuates cardiac hypertrophy (n=12–19/group).
3.4. Gender-specific effects of tTG inhibition in cardiac remodeling
Next, we examined whether tTG inhibition exerts distinct effects on cardiac function and remodeling in male and female mice. Male and female pressure-overloaded mice receiving vehicle or ERW had comparable R:L carotid flow ratio (Supplemental figure 2C–D). Treatment with the tTG inhibitor did not significantly affect BW in either male or female animals (Supplemental figure 3A–B). ERW-treated male mice had a trend towards decreased HW:BW ratio than vehicle-treated controls; in contrast, in female mice the reduction in HW:BW in inhibitor-treated animals did not reach statistical significance (Supplemental figure 3C–D). Gender-specific analysis of the echocardiographic data showed no significant effects of the inhibitor on LVEDV or ejection fraction (Supplemental figure 3E–H). When treated with vehicle, both male and female mice undergoing TAC exhibited a significant increase in LV mass in comparison to baseline values. In contrast, no significant increase in LV mass was noted in male or female inhibitor-treated animals undergoing TAC. After 28 days of pressure overload, ERW-treated female mice had significantly lower LV mass than corresponding vehicle-treated controls (Supplemental figure 3I–J).
3.5. tTG inhibition attenuates diastolic dysfunction in the pressure overloaded myocardium
Speckle tracking echocardiography demonstrated that male mice treated with tTG inhibitor had preserved systolic strain rate after 28 days of pressure overload, suggesting improved systolic function (Figure 3A–C). In contrast, no significant effects were noted in female mice (Figure 3B). In order to assess the effects of tTG inhibition on diastolic dysfunction, we measured diastolic strain rate. When compared with gender-matched vehicle-treated animals, mice treated with tTG inhibitor exhibited higher diastolic strain rate, suggesting preserved diastolic function (Figure 3D–F).
Figure 3. tTG inhibition attenuates diastolic dysfunction in the pressure-overloaded myocardium.
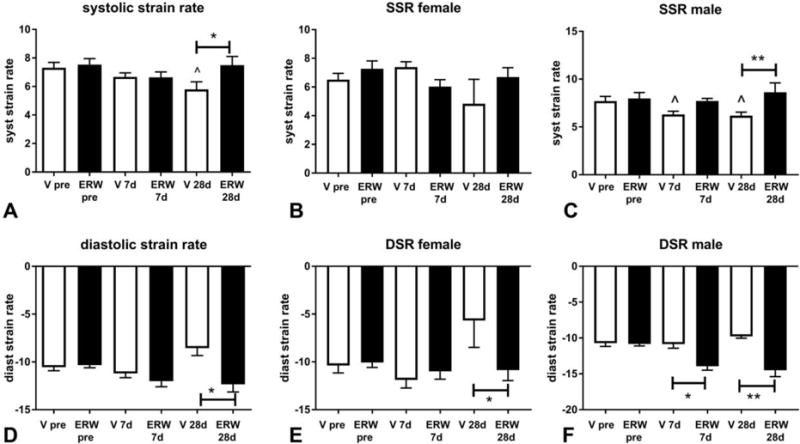
A: Speckle tracking echocardiography demonstrated that in comparison to vehicle-treated pressure overloaded animals (V), mice treated with the tTG inhibitor (ERW) had higher systolic strain rate (SSR) after 28 days of TAC. The effects of the inhibitor were noted in male (C), but not in female animals (B). D: tTG inhibition preserved diastolic strain rate (DSR) after 28 days of TAC, suggesting attenuated diastolic dysfunction. E–F: The protective effects of the inhibitor were noted in both female and male animals (*p<0.05, **p<0.01, n=14–15/group; male: n=5–9/group; female: n=5–10/group).
3.6. tTG inhibition attenuates cardiomyocyte hypertrophy and reduces collagen deposition in the pressure-overloaded heart
WGA lectin histochemistry was used to assess cardiomyocyte size in the pressure-overloaded myocardium (Figure 4A–B). Treatment with the tTG inhibitor significantly reduced cardiomyocyte size after 28 days of TAC (Figure 4C). Gender-specific analysis showed a trend towards larger cardiomyocyte size in male animals, when compared with female mice (male: 371.3μm2±9.5 n=8, vs. female 324μm2±24.3, n=5, p=0.057). Although treatment with the inhibitor had more impressive effects in male mice (reduction in cardiomyocyte size: 31% in males vs. 20% in females), tTG inhibition significantly attenuated cardiomyocyte hypertrophy in both male and female animals (Supplemental figure 4A–B). Sirius red staining was used to label collagen fibers in the remodeling myocardium (Figure 4D–E). Quantitative analysis demonstrated that tTG inhibition attenuated collagen deposition in the pressure-overloaded myocardium after 28 days of TAC (Figure 4F). Gender-specific analysis demonstrated that although the extent of fibrosis following TAC was comparable between male and female animals (p=NS), the tTG inhibitor significantly attenuated collagen deposition only in male mice (*p<0.05). Female mice had a trend towards reduced collagen content, when treated with the tTG inhibitor (p=0.10, Supplemental figure 4C–D).
Figure 4. tTG inhibition attenuates cardiomyocyte hypertrophy and reduced fibrosis in the pressure-overloaded myocardium.
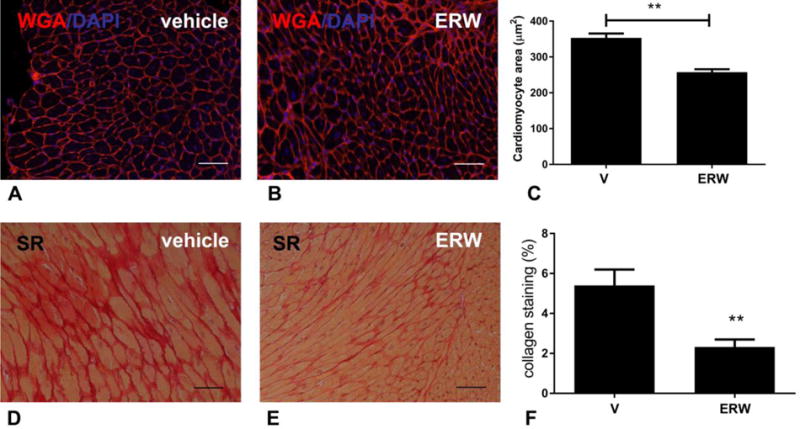
A–C. Sections stained for WGA lectin were used to assess cardiomyocyte size. 80 cells from 10 fields from 3 non-adjacent sections from 3 different levels were used for analysis. When compared to vehicle-treated controls (A), inhibitor-treated animals (B) had a significant reduction in cardiomyocyte size (C) after 28 days of TAC. D–F: Sirius red (SR) staining was used to label collagen. In comparison to vehicle-treated animals (D), inhibitor-treated animals (E) had significantly reduced collagen content (F). (**p<0.01 vs. vehicle (V)-treated, n=13–14/group). Scalebar=50μm.
3.7. tTG inhibition reduces collagen thickness in the pressure-overloaded myocardium
Sirius red staining followed by polarized light microscopy was used to investigate the effects of tTG inhibition on collagen structure. When visualized under polarized microscopy, thicker cross-linked fibers show orange or red birefringence, whereas thinner fibers appear green (Figure 5A–H). tTG inhibition significantly reduced deposition of both thin and thick collagen fibers in the pressure overloaded myocardium after 28 days of TAC (Figure 5I).
Figure 5. tTG inhibition reduces thickness of collagen fibers in the pressure-overloaded myocardium.
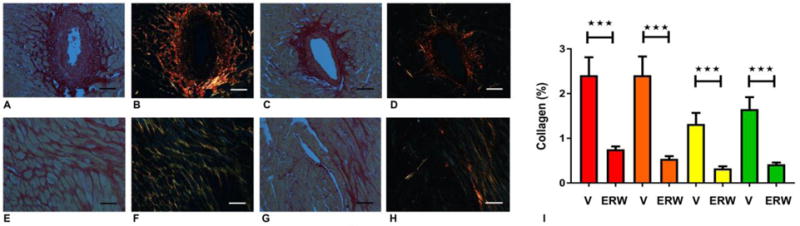
Polarized light microscopy in sirius red-stained sections was used to assess the morphological characteristics of collagen fibers. Panels A–D show areas of perivascular fibrosis, whereas panels E–H show interstitial fibrosis (A, C, E, G: light microscopy; B, D, F, H: polarized microscopy). After 28 days of TAC, vehicle treated animals showed extensive perivascular (A, B) and interstitial (E, F) fibrosis with many fibers exhibiting red birefringence, suggestive of increased thickness. Treatment with the tTG inhibitor attenuated deposition of both thicker (red) and thinner (yellow, green) fibers in perivascular (C, D) and interstitial (G, H) areas. I: Quantitative analysis showed that tTG inhibition (ERW) markedly reduced deposition of fibers with red, orange, yellow and green birefringence (***p<0.001 vs. vehicle-treated (V), n=13–14). Scalebar=50μm.
3.8. tTG inhibition reduces collagen and TIMP transcription in the pressure-overloaded myocardium
The global attenuation of both thin and thick collagen fibers by tTG inhibition suggested that the in vivo enzymatic actions of tTG may extend beyond its known effects on collagen cross-linking. Accordingly, we investigated the in vivo effects of tTG inhibition on transcription of genes associated with matrix synthesis and metabolism. Mice treated with ERW1041E exhibited attenuated myocardial levels of type I and type III collagen mRNA after 14 days of TAC (Figure 6A–B). Moreover, tTG inhibition modestly, but significantly, reduced MMP2 mRNA expression (Figure 6C), but had no statistically significant effects on MMP3 (Figure 6D), MMP8 (Figure 6E) and MMP9 (Figure 6F) synthesis. Animals treated with tTG inhibitor had markedly reduced expression of TIMP1 and TIMP2 mRNA (Figure 6G–H). Western blotting studies showed that inhibitor-treated animals had a significant reduction in collagen III (Figure 6I–J) and MMP2 protein levels (Figure 6K–L), when compared with vehicle-treated mice. These findings suggest that in vivo, tTG inhibition reduces ECM protein synthesis and attenuates expression of genes associated with matrix metabolism.
Figure 6. tTG inhibition attenuates expression of collagens and genes associated with matrix metabolism in the pressure-overloaded myocardium.
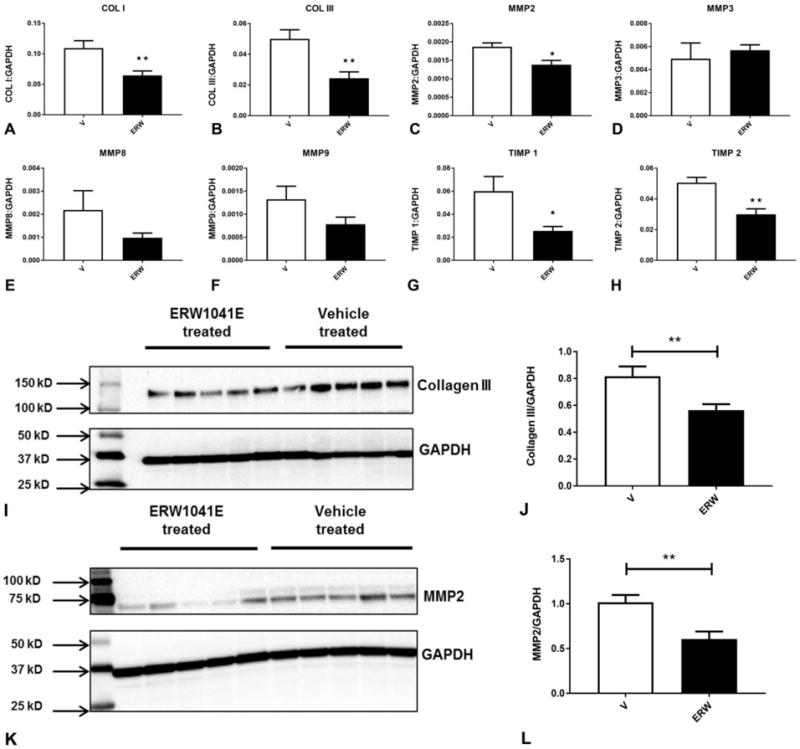
qPCR showed that inhibitor treatment reduced collagen I (A) and collagen III mRNA (B) levels. Treatment with the inhibitor also reduced MMP2 levels (C), but had no statistically significant effects on MMP3, MMP8 and MMP9 expression. TIMP1 and TIMP2 levels were markedly reduced in inhibitor-treated animals (*p<0.05, **p<0.01, n=6/group). Western blotting showed that treatment with the tTG inhibitor markedly reduced myocardial collagen III (I–J) and MMP2 (K–L) protein expression levels following 28 days of pressure overload. (**p<0.01 vs. vehicle-treated, n=8–15/group).
3.9. The in vitro effects of exogenous tTG on expression of genes associated with matrix synthesis and metabolism are predominantly non-enzymatic
Our in vivo findings suggested that tTG activity is localized in the interstitial and perivascular space following pressure overload, and that enzymatic actions of tTG mediate synthesis of fibrosis-associated genes and collagen deposition. Accordingly, we used an in vitro model of cardiac fibroblasts populating collagen pads to examine whether extracellular tTG activates a fibrogenic program in fibroblasts through enzymatic actions. In order to dissect enzymatic and non-enzymatic actions of tTG on fibroblast gene expression, we used 2 independent strategies: a) we compared the effects of recombinant tTG (rtTG) and inactive tTG (bearing a C277S mutation that renders the active site catalytically inactive, itTG) and b) we examined the effects of 3 different tTG inhibitors on tTG-mediated modulation of fibroblast genes. Neither rtTG nor itTG affected collagen I or III expression in cardiac fibroblasts populating collagen pads (Figure 7A–B). Both rtTG and itTG markedly induced MMP3 expression, suggesting that this effect was mediated through non-enzymatic actions (Figure 7C). In contrast, MMP8 mRNA synthesis was not affected by stimulation with rtTG or itTG (Figure 7D). rtTG induced a 4.0 fold increase in TIMP1 mRNA levels through actions that were independent of its enzymatic activity (Figure 7E). Because stimulated fibroblasts are known to produce and secrete tTG, we examined the effects of rtTG and itTG on gene expression by tTG KO cells. rtTG and itTG induced MMP3 and TIMP1 mRNA synthesis in both WT and tTG KO cells, suggesting that these actions are independent of endogenous cell-derived tTG, and reflect non-enzymatic effects of matrix-bound tTG on the fibroblasts (Figure 7F–J). Moreover, incubation with 3 different tTG inhibitors (Z-DON, Boc-DON and TAMRA-DON) did not affect rtTG-induced gene expression (Figure 7K–O), further supporting the notion that the effects of rtTG are non-enzymatic. The absence of enzymatic actions of tTG on fibroblast gene expression in vitro suggest that the in vivo observations indicating profound effects of the tTG inhibitor on transcription of fibrosis-associated genes (Figure 6) may result from actions on other cell types (such as macrophages, cardiomyocytes, mast cells, or vascular cells), or may reflect enzymatic effects of tTG on collagen processing.
Figure 7. Transamidase-independent actions of tTG modulate fibroblast gene expression.
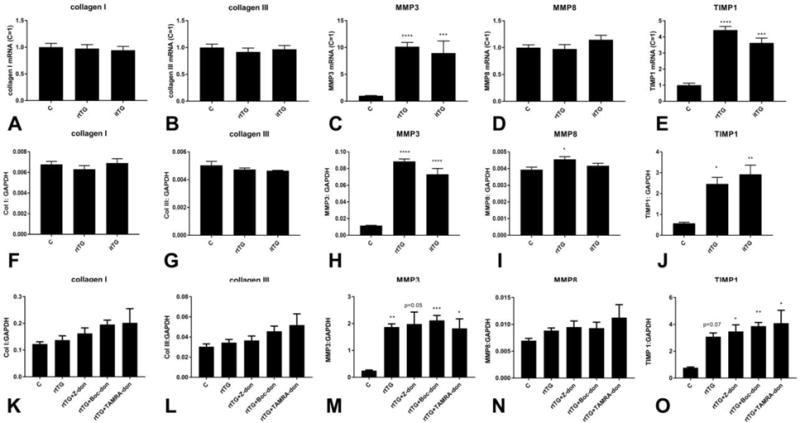
A–J: qPCR was used to assess the effects of recombinant tTG (rtTG) and inactive mutant tTG (itTG) on gene expression in WT or tTG KO cardiac fibroblasts populating collagen pads. In WT cells, rtTG and itTG had no effects on collagen I (A) and III (B) transcription. C: Both rtTG and itTG markedly induced synthesis of MMP3, suggesting a non-enzymatic effect. D: rtTG and itTG had no significant effects on MMP8 expression. E: Both itTG and rtTG upregulated TIMP1 expression by cardiac fibroblasts, suggesting non-enzymatic actions (***p<0.001, ****p<0.0001 vs. control, n=12–16/group). F–J: In order to examine whether these effects are independent of endogenous tTG, we used tTG KO cells. rtTG and itTG did not significantly affect collagen I (F), and collagen III (G) mRNA levels in tTG KO cells, but (much like in WT cells) markedly induced MMP3 (H), and TIMP1 (J) synthesis. rtTG had a modest, but significant effect on MMP8 expression by tTG KO cells (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 vs. control, n=4–6/group). K–O: We examined the effects of 3 different tTG inhibitors (Z-DON, Boc-DON and TAMRA-DON) on rtTG-mediated fibroblast gene expression. The inhibitors did not affect collagen I (K), collagen III (L) and MMP8 (N) mRNA expression. Moreover, rtTG-mediated induction of MMP3 (M) and TIMP1 (O) was not affected by enzymatic inhibition (*p<0.05, **p<0.01, ***p<0.001 vs. control, n=5–9/group).
3.10. Recombinant tTG increases pericellular collagen deposition in collagen pads through enzymatic actions
In order to examine whether enzymatic actions of tTG in vivo reflect effects on collagen processing, we studied the effects of rtTG and itTG on collagen structure in fibroblast-populated collagen pads. When visualized under polarized microscopy, fibroblasts in collagen pads show a pericellular “halo” of orange/red thick fibers, reflecting new synthesis and/or processing of collagen by the cells (Figure 8A–F). In contrast to itTG, rtTG markedly increased the thickness of the pericellular collagen in both WT and tTG KO cells (Figure 8A–F). Quantitative analysis showed that the area of pericellular collagen was markedly increased by rtTG stimulation, but was not affected by itTG, suggesting that matrix-bound tTG increases deposition of collagen fibers through enzymatic actions (Figure 8G).
Figure 8. The effects of tTG on pericellular collagen deposition in fibroblast-populated collagen pads are dependent on transamidase activity.
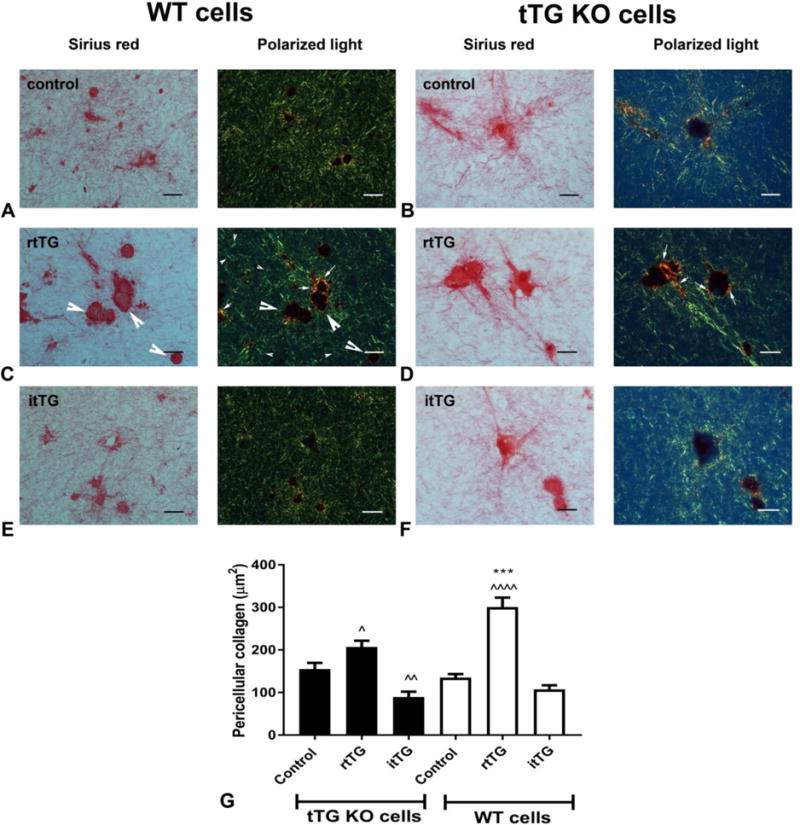
Collagen pads populated with WT (A, C, E) or tTG KO cardiac fibroblasts (B, D, F) were stimulated with rtTG (C, D) or itTG (E, F) for 24h. Paraffin-embedded sections of the pads were stained with sirius red and visualized under polarized light (A–F). Intracellular procollagen localized in cardiac fibroblasts is dark red under light microscopy, but is not visualized under polarized light (C – large arrowheads). Polarized light microscopy identifies the green fibers of the collagen pad (C – small arrowheads), and also labels the pericellular network of thicker orange/red collagen fibers (C-small arrows), that reflects processing of collagen fibers by the cells. Images are representative of 5 independent experiments. G: Quantitative analysis of the pericellular collagen area showed that rtTG (C), but not itTG (E) increased the area of pericellular collagen in both WT (C, E) and tTG KO (D, F) cells (^p<0.05, ^^p<0.01, ^^^p<0.001 vs. corresponding control; ***p<0.001 vs corresponding tTG KO, n=90–150 cells from 6–10 different pads from each condition). The findings demonstrate that matrix-bound extracellular tTG promotes collagen deposition and organization through transamidase-independent actions. Scalebar=20μm.
4. DISCUSSION
We report for the first time that the enzymatic functions of tTG mediate fibrosis and diastolic dysfunction in the pressure-overloaded myocardium. Using a mouse model of cardiac pressure overload induced through TAC, and in vitro experiments investigating the effects of active and inactive tTG on isolated cardiac fibroblasts, we found that: a) tTG activity is localized in the interstitial and perivascular areas of the remodeling myocardium, b) inhibition of the enzymatic actions of tTG with ERW1041E, a specific small molecule inhibitor, attenuates diastolic dysfunction following pressure overload, c) the protective effects of tTG inhibition are associated with decreased cardiomyocyte hypertrophy, attenuated interstitial fibrosis, and reduced thickness of collagen fibers, d) although enzymatic inhibition of tTG in vivo attenuates expression of collagen and other fibrosis-associated genes, the in vitro effects of tTG on cardiac fibroblast gene expression are transamidase-independent, e) enzymatic actions of matrix-bound tTG promote collagen deposition in fibroblasts populating collagen pads. Our observations highlight the complexity of the in vivo effects of tTG, and the distinct effects of enzymatic and non-enzymatic actions.
4.1. Activation of tTG in the remodeling myocardium
Our findings demonstrate that tTG is upregulated and activated in the remodeling pressure-overloaded myocardium (Fig 1, Supplemental figure 1). Although tTG is constitutively expressed in isolated cardiomyocytes [27] and in adult mammalian hearts (Supplemental figure 1) [28], minimal tTG activity was noted in normal mouse myocardium (Figure 1A). In mammalian tissues, tTG activity is tightly regulated to prevent spontaneous or excessive activation and subsequent intracellular cross-linking. Low Ca2+ concentrations prevent intracellular transamidation activity. Moreover, intracellular GTP/GDP act as allosteric inhibitors of tTG, inducing a closed conformation, reducing accessibility of the catalytic domain, and preventing inappropriate intracellular tTG activation [29],[30],[31]. In the pressure-overloaded myocardium, tTG activity is present in the interstitial and perivascular space (Figure 1B, E, H, K), but the mechanisms of secretion remain unknown. In the extracellular space, high levels of Ca2+ may contribute to activation of tTG-mediated crosslinking activity. Induction of inflammatory stimuli, such as Toll-like receptor signaling cascades and pro-inflammatory cytokines, and upregulation of the antioxidant protein thioredoxin [32] may also promote extracellular tTG activation [33],[13],[34].
4.2. Enzymatic and non-enzymatic functions of tTG in the pressure-overloaded myocardium
Perturbations in transglutaminase-mediated ECM cross-linking have been previously associated with the development of cardiomyopathy in senescent mice lacking the matricellular protein thrombospondin (TSP)-2 [35]. Recently, we showed that tTG KO mice exhibit increased mortality following pressure overload, associated with chamber dilation and increased MMP2 activity, but are protected from diastolic dysfunction, and have attenuated collagen crosslinking [12]. The effects of tTG may be due to enzymatic or non-enzymatic functions. In order to dissect the role of tTG-dependent transamidation in the pressure-overloaded myocardium, we treated mice undergoing TAC protocols with the specific tTG small molecule inhibitor ERW1041E. Effective tTG inhibition was documented in vivo (Figure 1). In contrast to the increased mortality and dilation associated with global loss of tTG, enzymatic inhibition exerted protective actions, attenuating cardiomyocyte hypertrophy, interstitial fibrosis and diastolic dysfunction (Figures 3–5). Our findings suggest that transamidase-dependent tTG actions are involved in fibrotic and hypertrophic remodeling of the pressure-overloaded heart (Figure 9), but do not contribute to protection from chamber dilation and death.
Figure 9. Schematic cartoon illustrating the enzymatic and non-enzymatic actions of tTG in the pressure-overloaded myocardium.
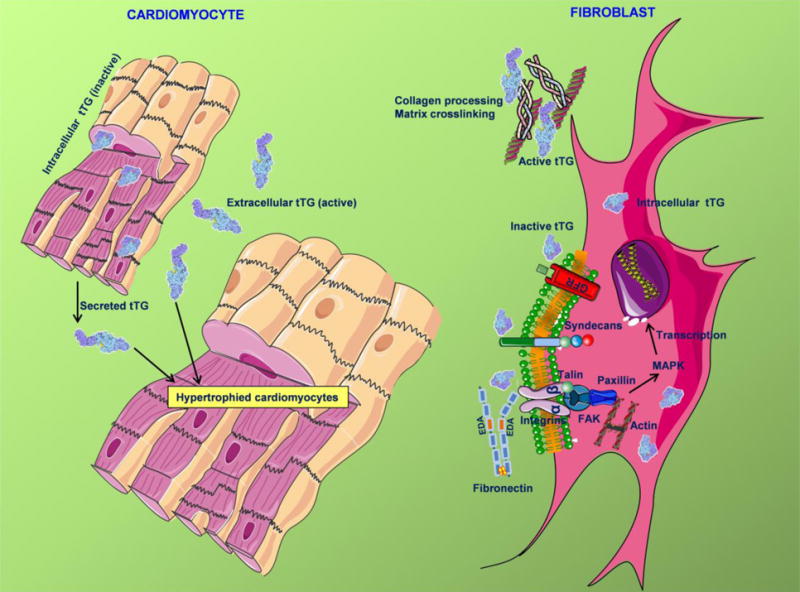
tTG is upregulated, secreted and activated in the pressure-overloaded myocardium and exerts a wide range of enzymatic and non-enzymatic actions in cardiomyocytes and fibroblasts. In vivo, the enzymatic effects of tTG promote cardiomyocyte hypertrophy, fibrosis and diastolic dysfunction. In vitro, extracellular matrix-bound tTG induces synthesis of fibrosis-associated genes (such as MMP3 and TIMP1) through a transamidase-independent mechanism. These effects may involve binding of inactive tTG to fibronectin and facilitation of integrin or syndecan-mediated signaling pathways, or stimulation of growth factor receptor (GFR)-dependent cascades. Matrix-bound tTG also acts through enzymatic actions that may involve extracellular matrix protein crosslinking, or effects on collagen processing. Additional symbols: MAPK, mitogen-activated protein kinase; FAK, focal adhesion kinase.
4.3. The cellular basis for the fibrogenic actions of tTG
Attenuated collagen deposition in mice treated with the tTG inhibitor is associated with marked reductions in myocardial collagen mRNA and protein levels (Figure 6). In order to examine whether attenuated collagen synthesis reflects direct effects of tTG on fibroblast-derived collagen expression, we performed in vitro experiments investigating the effects of tTG on isolated cardiac fibroblasts. Surprisingly, tTG did not affect collagen expression by cardiac fibroblasts (Figure 7A–B), suggesting that the observed in vivo effects of tTG inhibition may reflect indirect actions on other cell types. tTG is known to modulate the phenotype of macrophages, cardiomyocytes, vascular cells and mast cells. In vitro and in vivo studies have suggested that tTG is implicated in macrophage-dependent efferocytosis, and may regulate transition of macrophages to an anti-inflammatory phenotype upon ingestion of apoptotic cells [36],[37],[38]. Moreover, recent investigations in human and mouse tissues have demonstrated that tTG is a conserved marker for M2 macrophages [39]. Whether tTG plays a role in acquisition of a fibrogenic phenotype by macrophages remains unknown. Moreover, tTG has been implicated in mast cell-mediated macrophage activation [40]. Considering the important role of mast cells as a source of cytokines and growth factors in the injured and remodeling myocardium [41], and their involvement as cellular effectors of cardiac fibrosis [42], tTG may exert fibrogenic actions by orchestrating interactions between inflammatory cells in the remodeling heart. In cardiomyocytes, tTG has been implicated as a mediator of hypertrophic and pro-survival responses [27],[43]. Endothelial tTG has been suggested to regulate inflammatory activation, and may also promote angiogenic responses [44] by regulating Vascular Endothelial Growth Factor (VEGF) signaling [45], [46], [47]. Whether the pro-fibrotic enzymatic effects of tTG are due to activation of a fibrogenic program in cardiomyocytes and vascular cells has not been investigated.
4.4. Transamidase-independent effects of tTG modulate transcription of genes associated with matrix remodeling in cardiac fibroblasts, whereas enzymatic actions may regulate collagen processing
Our in vitro experiments demonstrated that recombinant tTG induces expression of genes associated with matrix metabolism in isolated cardiac fibroblasts, inducing a 10-fold increase in MMP3 and a 4-fold increase in TIMP1 expression (Figure 7). We used 2 independent strategies to examine whether the effects of tTG on fibroblast gene expression are mediated through enzymatic actions. First, recombinant tTG and enzymatically inactive mutant tTG (itTG) had similar effects on MMP3 and TIMP1 synthesis (Figure 7). Second, incubation with 3 different tTG inhibitors did not affect tTG-mediated gene expression. Both approaches suggested that the effects of tTG on fibroblast gene expression are non-enzymatic. In contrast, stimulation with recombinant active, but not with inactive tTG increased pericellular collagen deposition in fibroblast-populated pads (Figure 8), suggesting that enzymatic effects of tTG may be implicated in pericellular collagen processing. Experiments using tTG KO cells demonstrated that both the enzymatic and transamidase-independent effects of extracellular, matrix-bound tTG are independent of endogenous tTG expression (Figure 7–8).
Obviously, our study has significant limitations. First, the in vitro model of fibroblast-populated pads does not recapitulate the complex in vivo environment, in which tTG may exert a wide range of actions on all cell types involved in cardiac remodeling. Second, collagen deposition in fibrotic tissues is a complex process. Large amounts of pro-collagen undergo intracellular degradation in fibroblasts before secretion and deposition in the ECM. Thus, mRNA levels do not necessarily correlate with collagen deposition. Moreover, net accumulation of collagen is dependent on post-translational modifications, the presence of other cross-linking enzymes, and on the balance between proteases and anti-proteases in the interstitial space [48], [49]. Our previously reported effects of global tTG loss on lysyl-oxidase levels in the pressure-overloaded myocardium [12] highlight the complex in vivo interactions between proteins involved in ECM regulation.
4.5. The enzymatic effects of tTG: a therapeutic opportunity for patients with heart failure with preserved ejection fraction (HFpEF)?
HFpEF currently accounts for the majority of heart failure cases and its prevalence continues to rise [50]. Interstitial fibrosis is commonly found in patients with HFpEF and is associated with increased left ventricular stiffness [51],[52],[53]. Inhibition of enzymes that crosslink the ECM, such as the members of the lysyl oxidase family, reduces cardiac fibrosis, attenuating diastolic dysfunction in experimental models of fibrotic cardiac remodeling that recapitulate aspects of HFpEF [54],[55].[56]. Our findings suggest that although non-enzymatic actions of tTG transduce protective signals that preserve myocardial geometry in the remodeling heart [12],[57] the enzymatic effects of tTG contribute to fibrosis and diastolic dysfunction (Figure 9). Considering the availability of effective and selective tTG inhibitors [58],[59], targeting tTG-dependent transamidation may be a promising therapeutic strategy for subsets of HFpEF patients with a prominent fibrotic phenotype.
4.6. Conclusions
tTG is a ubiquitously expressed, multifunctional protein with an important role in regulating inflammation, repair and tissue remodeling. tTG is upregulated and activated in the remodeling pressure-overloaded myocardium and exerts fibrogenic actions dependent on transamidase activity, and protective non-enzymatic actions that regulate matrix metabolism and MMP activity [12]. The significant effects of pharmacologic inhibition of the enzymatic actions of tTG on diastolic dysfunction in mice may suggest a promising new therapeutic approach for patients with HFpEF. Clinical studies are needed in order to assess myocardial tTG expression and activity in various subpopulations of heart failure patients, and to identify subjects with excessive deposition of crosslinked ECM. Moreover, considering the broad functional repertoire of tTG, its wide distribution in myocardial cells and in the interstitial ECM, and the diversity of tTG interactions with ECM proteins, growth factors, integrins and syndecans, there is a need to dissect the in vivo mechanisms of tTG actions, focusing on cell biological targets, molecular interactors and the relative significance of enzymatic and non-enzymatic effects.
Supplementary Material
HIGHLIGHTS.
Tissue transglutaminase (tTG) is activated in the pressure-overloaded myocardium.
tTG inhibition attenuates diastolic dysfunction, and reduces hypertrophy and fibrosis.
In vivo, inhibition of the enzymatic actions of tTG markedly reduces collagen synthesis.
In vitro, recombinant tTG modulates MMP and TIMP mRNA through non-enzymatic actions.
Enzymatic actions of tTG increase pericellular collagen thickness and may be involved in collagen processing.
Acknowledgments
FUNDING
This work was supported by grants from the National Institutes of Health (R01 HL76246 and R01 HL85440), and by the Department of Defense (PR151134 and PR151029).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS
None
References
- 1.Kanchan K, Fuxreiter M, Fesus L. Physiological, pathological, and structural implications of non-enzymatic protein-protein interactions of the multifunctional human transglutaminase 2. Cell Mol Life Sci. 2015;72:3009–35. doi: 10.1007/s00018-015-1909-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nurminskaya MV, Belkin AM. Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol. 2012;294:1–97. doi: 10.1016/B978-0-12-394305-7.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckert RL, Kaartinen MT, Nurminskaya M, Belkin AM, Colak G, Johnson GV, et al. Transglutaminase regulation of cell function. Physiol Rev. 2014;94:383–417. doi: 10.1152/physrev.00019.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–56. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Griffin M. TG2, a novel extracellular protein with multiple functions. Amino Acids. 2012;42:939–49. doi: 10.1007/s00726-011-1008-x. [DOI] [PubMed] [Google Scholar]
- 6.Telci D, Wang Z, Li X, Verderio EA, Humphries MJ, Baccarini M, et al. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired cell adhesion through syndecan-4 and beta1 integrgn co-signaling. J Biol Chem. 2008;283:20937–47. doi: 10.1074/jbc.M801763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zemskov EA, Loukinova E, Mikhailenko I, Coleman RA, Strickland DK, Belkin AM. Regulation of platelet-derived growth factor receptor function by integrin-associated cell surface transglutaminase. J Biol Chem. 2009;284:16693–703. doi: 10.1074/jbc.M109.010769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verderio EA, Telci D, Okoye A, Melino G, Griffin M. A novel RGD-independent cel adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J Biol Chem. 2003;278:42604–14. doi: 10.1074/jbc.M303303200. [DOI] [PubMed] [Google Scholar]
- 9.Akimov SS, Belkin AM. Opposing roles of Ras/Raf oncogenes and the MEK1/ERK signaling module in regulation of expression and adhesive function of surface transglutaminase. J Biol Chem. 2003;278:35609–19. doi: 10.1074/jbc.M303488200. [DOI] [PubMed] [Google Scholar]
- 10.Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol. 2009;131:471–81. doi: 10.1007/s00418-008-0541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shinde AV, Dobaczewski M, de Haan JJ, Saxena A, Lee KK, Xia Y, et al. Tissue transglutaminase induction in the pressure-overloaded myocardium regulates matrix remodelling. Cardiovasc Res. 2017;113:892–905. doi: 10.1093/cvr/cvx053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dafik L, Albertelli M, Stamnaes J, Sollid LM, Khosla C. Activation and inhibition of transglutaminase 2 in mice. PLoS One. 2012;7:e30642. doi: 10.1371/journal.pone.0030642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia Y, Dobaczewski M, Gonzalez-Quesada C, Chen W, Biernacka A, Li N, et al. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension. 2011;58:902–11. doi: 10.1161/HYPERTENSIONAHA.111.175323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frunza O, Russo I, Saxena A, Shinde AV, Humeres C, Hanif W, et al. Myocardial galectin-3 expression is associated with remodeling of the pressure-overloaded heart and may delay the hypertrophic response without affecting survival, dysfunction, and cardiac fibrosis. Am J Pathol. 2016;186:1114–27. doi: 10.1016/j.ajpath.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiRaimondo TR, Klock C, Warburton R, Herrera Z, Penumatsa K, Toksoz D, et al. Elevated transglutaminase 2 activity is associated with hypoxia-induced experimental pulmonary hypertension in mice. ACS Chem Biol. 2014;9:266–75. doi: 10.1021/cb4006408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, Saxena A, Li N, Sun J, Gupta A, Lee DW, et al. Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts, Arterioscler. Thromb Vasc Biol. 2012;32:2598–608. doi: 10.1161/ATVBAHA.112.300310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plugis NM, Palanski BA, Weng CH, Albertelli M, Khosla C. Thioredoxin-1 selectively activates Transglutaminase 2 in the extracellular matrix of the small intestine: IMPLICATIONS FOR CELIAC DISEASE. J Biol Chem. 2017;292:2000–2008. doi: 10.1074/jbc.M116.767988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christia P, Bujak M, Gonzalez-Quesada C, Chen W, Dobaczewski M, Reddy A, et al. Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J Histochem Cytochem. 2013;61:555–70. doi: 10.1369/0022155413493912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–28. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol. 2001;21:148–55. doi: 10.1128/MCB.21.1.148-155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shinde AV, Humeres C, Frangogiannis NG. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta. 2017;1863:298–309. doi: 10.1016/j.bbadis.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McConoughey SJ, Basso M, Niatsetskaya ZV, Sleiman SF, Smirnova NA, Langley BC, et al. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med. 2010;2:349–70. doi: 10.1002/emmm.201000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basso M, Berlin J, Xia L, Sleiman SF, Ko B, Haskew-Layton R, et al. Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci. 2012;32:6561–9. doi: 10.1523/JNEUROSCI.3353-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res Cardiol. 1994;89:397–410. doi: 10.1007/BF00788278. [DOI] [PubMed] [Google Scholar]
- 26.Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, et al. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation (2017) 2017 doi: 10.1161/CIRCULATIONAHA.117.029622. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Wei XL, Meng LL, Chi MG, Yan JQ, Ma XY, et al. Involvement of tissue transglutaminase in endothelin 1-induced hypertrophy in cultured neonatal rat cardiomyocytes. Hypertension. 2009;54:839–44. doi: 10.1161/HYPERTENSIONAHA.109.130161. [DOI] [PubMed] [Google Scholar]
- 28.Oh YJ, Pau VC, Steppan J, Sikka G, Bead VR, Nyhan D, et al. Role of tissue transglutaminase in age-associated ventricular stiffness. Amino Acids. 2017;49:695–704. doi: 10.1007/s00726-016-2295-z. [DOI] [PubMed] [Google Scholar]
- 29.Achyuthan KE, Greenberg CS. Identification of a guanosine triphosphate-binding site on guinea pig liver transglutaminase. Role of GTP and calcium ions in modulating activity. J Biol Chem. 1987;262:1901–6. [PubMed] [Google Scholar]
- 30.Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010;285:25402–9. doi: 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Begg GE, Carrington L, Stokes PH, Matthews JM, Wouters MA, Husain A, et al. Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc Natl Acad Sci U S A. 2006;103:19683–8. doi: 10.1073/pnas.0609283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest. 2003;112:1395–406. doi: 10.1172/JCI17700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diraimondo TR, Klock C, Khosla C. Interferon-gamma activates transglutaminase 2 via a phosphatidylinositol-3-kinase-dependent pathway: implications for celiac sprue therapy. J Pharmacol Exp Ther. 2012;341:104–14. doi: 10.1124/jpet.111.187385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin X, Stamnaes J, Klock C, DiRaimondo TR, Sollid LM, Khosla C. Activation of extracellular transglutaminase 2 by thioredoxin. J Biol Chem. 2011;286:37866–73. doi: 10.1074/jbc.M111.287490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swinnen M, Vanhoutte D, Almen GC Van, Hamdani N, Schellings MW, D’Hooge J, et al. Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation. 2009;120:1585–97. doi: 10.1161/CIRCULATIONAHA.109.863266. [DOI] [PubMed] [Google Scholar]
- 36.Szondy Z, Sarang Z, Molnar P, Nemeth T, Piacentini M, Mastroberardino PG, et al. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc Natl Acad Sci U S A. 2003;100:7812–7. doi: 10.1073/pnas.0832466100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falasca L, Iadevaia V, Ciccosanti F, Melino G, Serafino A, Piacentini M. Transglutaminase type II is a key element in the regulation of the anti-inflammatory response elicited by apoptotic cell engulfment. J Immunol. 2005;174:7330–40. doi: 10.4049/jimmunol.174.11.7330. [DOI] [PubMed] [Google Scholar]
- 38.Chrobok NL, Sestito C, Wilhelmus MM, Drukarch B, van Dam AM. Is monocyte- and macrophage-derived tissue transglutaminase involved in inflammatory processes? Amino Acids. 2017;49:441–452. doi: 10.1007/s00726-016-2334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood. 2013;121:e57–69. doi: 10.1182/blood-2012-06-436212. [DOI] [PubMed] [Google Scholar]
- 40.Eom S, Kim Y, Kim M, Park D, Lee H, Lee YS, et al. Transglutaminase II/microRNA-218/-181a loop regulates positive feedback relationship between allergic inflammation and tumor metastasis. J Biol Chem. 2014;289:29483–505. doi: 10.1074/jbc.M114.603480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 42.Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension. 2009;53:1041–7. doi: 10.1161/HYPERTENSIONAHA.108.123158. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Z, Vezza R, Plappert T, McNamara P, Lawson JA, Austin S, et al. COX-2-dependent cardiac failure in Gh/tTG transgenic mice. Circ Res. 2003;92:1153–61. doi: 10.1161/01.RES.0000071749.22027.45. [DOI] [PubMed] [Google Scholar]
- 44.Bijli KM, Kanter BG, Minhajuddin M, Leonard A, Xu L, Fazal F, et al. Regulation of endothelial cell inflammation and lung polymorphonuclear lymphocyte infiltration by transglutaminase 2. Shock. 2014;42:562–9. doi: 10.1097/SHK.0000000000000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13:1787–95. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 46.Beckouche N, Bignon M, Lelarge V, Mathivet T, Pichol-Thievend C, Berndt S, et al. The interaction of heparan sulfate proteoglycans with endothelial transglutaminase-2 limits VEGF165-induced angiogenesis. Sci Signal. 2015;8:ra70. doi: 10.1126/scisignal.aaa0963. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Perez M, Caja S, Melino G, Johnson TS, Lindfors K, et al. A novel extracellular role for tissue transglutaminase in matrix-bound VEGF-mediated angiogenesis. Cell Death Dis. 2013;4:e808. doi: 10.1038/cddis.2013.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horn MA, Trafford AW. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J Mol Cell Cardiol. 2016;93:175–85. doi: 10.1016/j.yjmcc.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El Hajj EC, El Hajj MC, Ninh VK, Gardner JD. Cardioprotective effects of lysyl oxidase inhibition against volume overload-induced extracellular matrix remodeling. Exp Biol Med. 2016;241:539–49. doi: 10.1177/1535370215616511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, et al. Phenotype-specific treatment of heart failure with preserved ejection fraction. A multiorgan roadmap, Circulation. 2016;134:73–90. doi: 10.1161/CIRCULATIONAHA.116.021884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rommel KP, von Roeder M, Latuscynski K, Oberueck C, Blazek S, Fengler K, et al. Extracellular volume fraction for characterization of patients with heart Failure and preserved ejection fraction. J Am Coll Cardiol. 2016;67:1815–25. doi: 10.1016/j.jacc.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 52.Schelbert EB, Sabbah HN, Butler J, Gheorghiade M. Employing extracellular volume cardiovascular magnetic resonance measures of myocardial fibrosis to foster novel therapeutics. Circ Cardiovasc Imaging. 2017;10 doi: 10.1161/CIRCIMAGING.116.005619. [DOI] [PubMed] [Google Scholar]
- 53.Jia G, Jia Y, Sowers JR. Role of mineralocorticoid receptor activation in cardiac diastolic dysfunction. Biochim Biophys Acta. 2017;1863:2012–2018. doi: 10.1016/j.bbadis.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun. 2016;7:13710. doi: 10.1038/ncomms13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez-Martinez E, Rodriguez C, Galan M, Miana M, Jurado-Lopez R, Bartolome MV, et al. The lysyl oxidase inhibitor (beta-aminopropionitrile) reduces leptin profibrotic effects and ameliorates cardiovascular remodeling in diet-induced obesity in rats. J Mol Cell Cardiol. 2016;92:96–104. doi: 10.1016/j.yjmcc.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 56.Lopez B, Gonzalez A, Hermida N, Valencia F, de Teresa E, Diez J. Role of lysyl oxidase in myocardial fibrosis: from basic science to clinical aspects. Am J Physiol Heart Circ Physiol. 2010;299:H1–9. doi: 10.1152/ajpheart.00335.2010. [DOI] [PubMed] [Google Scholar]
- 57.Shinde AV, Frangogiannis NG. Tissue transglutaminase in the pathogenesis of heart failure. Cell Death Differ. 2017 Dec 13; doi: 10.1038/s41418-017-0028-9. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, et al. Chemistry and biology of dihydroisoxazole derivatives: selective inhibitors of human transglutaminase 2. Chem Biol. 2005;12:469–75. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 59.Badarau E, Wang Z, Rathbone DL, Costanzi A, Thibault T, Murdoch CE, et al. Development of potent and selective tissue transglutaminase inhibitors: their effect on TG2 function and application in pathological conditions. Chem Biol. 2015;22:1347–61. doi: 10.1016/j.chembiol.2015.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.