

Abstract
Background & Aims
The innate immune system responds not only to bacterial signals, but also to non-infectious danger-associated molecular patterns that activate the NLRP3 inflammasome complex after tissue injury. Immune functions vary over the course of the day, but it is not clear whether these changes affect the activity of the NLRP3 inflammasome. We investigated whether the core clock component nuclear receptor subfamily 1 group D member 1 (NR1D1, also called Rev-erbα) regulates expression, activity of the NLRP3 inflammasome, and its signaling pathway.
Methods
We collected naïve peritoneal macrophages and plasma, at multiple times of day, from Nr1d1−/− mice and their Nr1d1+/+ littermates (controls) and analyzed expression of NLR family pyrin domain containing 3 (NLRP3), interleukin 1 beta (IL1B, in plasma), and IL18 (in plasma). We also collected bone marrow-derived primary macrophages from these mice. Levels of NR1D1 were knocked down with small hairpin RNAs in human primary macrophages. Bone marrow-derived primary macrophages from mice and human primary macrophages were incubated with lipopolysaccharide (LPS) to induce expression of NLRP3, IL1B and IL18; cells were incubated with LPS and ATP to activate the NLRP3 complex. We analyzed caspase 1 activity and cytokine secretion. NR1D1 was activated in primary mouse and human macrophages by incubation with SR9009; some of the cells were also incubated with an NLRP3 inhibitor or inhibitors of caspase 1. Nr1d1−/− mice and control mice were given intraperitoneal injections of LPS to induce peritoneal inflammation; plasma samples were isolated and levels of cytokines were measured. Nr1d1−/− mice, control mice, and control mice given injections of SR9009 were given LPS and galactosamine to induce fulminant hepatitis and MCC950 to specifically inhibit NLRP3; plasma was collected to measure cytokines and a marker of liver failure (ALAT); liver tissues were collected and analyzed by quantitative PCR, immunohistochemistry, and flow cytometry.
Results
In peritoneal macrophages, expression of NLRP3 and activation of its complex varied with time of day (circadian rhythm)—this regulation required NR1D1. Primary macrophages from Nr1d1−/− mice and human macrophages with knockdown of NR1D1 had altered expression patterns of NLRP3, compared to macrophages that expressed NR1D1, and altered patterns of IL1B and 1L18 production. Mice with disruption of Nr1d1 developed more-severe acute peritoneal inflammation and fulminant hepatitis than control mice. Incubation of macrophage with the NR1D1 activator SR9009 reduced expression of NLRP3 and secretion of cytokines. Mice given SR9009 developed less-severe liver failure and had longer survival times than mice given saline (control).
Conclusions
In studies of Nr1d1−/− mice and human macrophages with pharmacologic activation of NR1D1, we found NR1D1 to regulate the timing of NLRP3 expression and production of inflammatory cytokines by macrophages. Activation of NR1D1 reduced the severity of peritoneal inflammation and fulminant hepatitis in mice.
Keywords: immune regulation, biological clock, acute liver failure, rev-erb-alpha
Graphical abstract
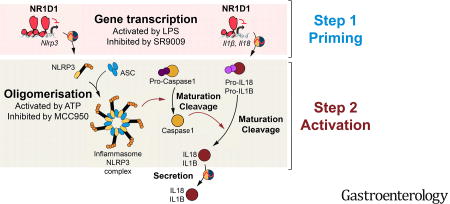
Introduction
Fulminant hepatitis (FH) is a life-threatening pathological condition characterized by a fast evolving hepatic dysfunction associated with encephalopathy and coagulopathy1. FH is triggered by numerous etiological factors such as viral infection, absorption of toxic compounds, metabolic and genetic diseases1. However, the overdose of drugs such as acetaminophen is still the main cause of FH. Acetaminophen accumulation leads to P450-mediated overproduction of toxic metabolites provoking oxidative stress, mitochondrial membrane potential loss and hepatocellular death. This entrains the secretion of Danger-Associated Molecular Pattern (DAMPs) and activation of the innate immune system, leading to secretion of a large number of pro-inflammatory mediators1. So far, no specific treatment is available apart from liver transplantation, pleading for an urgent need to identify pharmacological targets and to uncover the underlying mechanisms.
Organisms rely on a well-conserved molecular clockwork in order to anticipate changes imposed by the rotation of the Earth and gate physiological processes to the most appropriate time window2. Immune functions vary according to the time-of-day, and clock disruption leads to inflammatory diseases3–5. Indeed, mounting evidences argue for the existence of a circadian rhythmicity in several immune functions including trafficking and abundance of blood leukocytes, and their recruitment to tissues3, 6–9, although the underlying mechanisms are not elucidated yet. The importance of appropriate timing of the immune response is supported by the more severe response to endotoxin, i.e. increased macrophage cytokine production and increased mortality, upon clock dysfunction during chronic jetlag10.
The biological clock is a complex network of transcription factors and interlocked transcriptional feedback loops that orchestrate circadian rhythms of various physiological processes2, 3. Clock dysfunction is associated with the development of metabolic and cardio-vascular disorders as well as cancers, and, as described above, immune defects, underscoring its central role in behaviour and physiology. Among the core clock components, the ligand-activated nuclear receptor and transcriptional repressor NR1D1 participates in the circadian control of hepatic glucose, lipid and bile acid metabolism11–16, muscle mitochondrial activity17 and body temperature18. Lymphoid organs and immune cells such as macrophages harbour an intrinsic clockwork that drives circadian transcription of genes involved, for instance, in interleukin (IL) secretion and the Toll-like Receptor (TLR)-4 inflammatory pathway, leading to marked circadian variations in the response to endotoxin or bacterial challenge19–21. Interestingly, NR1D1 has previously been linked to the immune system as it represses TLR4 and Cx3cr1 expression in macrophages22, 23. In addition, LPS-treated peritoneal macrophages from Nr1d1−/− mice display increased Ccl2 (Mcp1) expression, whereas pharmacological activation of NR1D1 represses the expression and release of IL-620.
The innate immune system recognizes and responds not only to bacterial components but also to non-infectious, structurally diverse DAMPs, such as crystals or extracellular ATP24–26, mounting an immune response to clear these factors27. The NLRP3 inflammasome complex is central to this response and is activated by a two-step mechanism. Nlrp3 gene expression is first primed by pro-inflammatory stimuli such as LPS or Neutrophil Extracellular Traps (NET)24, 26. DAMPs then act as secondary signals to trigger several pathways activating the NLRP3 inflammasome including ATP-activated potassium efflux, translocation to mitochondria, reactive oxygen species production and release of mitochondrial DNA, cardiolipin and crystal-activated lysosomal cathepsins24, 25, 28–31. Upon activation by infection or cellular stresses, the NLRP3 inflammasome complex elicits the maturation of the pro-inflammatory cytokines IL1B and IL1832. Importantly, deregulation of NLRP3 participates in the development of IL18 and IL1B-driven auto-inflammatory diseases including atherosclerosis, gout and rheumatoid arthritis where accumulation of crystals of cholesterol, uric acid or hydroxyapatite is perceived as an endogenous sterile stress24, 33–36, calling for therapeutic strategies to modulate inflammasome signaling. In addition, NLRP3 expression is increased in viral and drug-induced fulminant hepatitis, although its actual role in these pathological conditions is still underinvestigated37, 38. Although the precise regulation of the NLRP3 inflammasome complex is of utmost importance in innate inflammation, whether it is controlled by the biological clock has not yet been demonstrated. Here, we show that susceptibility to LPS/Galactosamine-induced fulminant hepatitis depends on the time of challenge in mice. Interestingly, we demonstrate that expression and activation of the NLRP3 inflammasome pathway oscillate in a daily manner under the direct control of NR1D1 in primary mouse and human macrophages. Macrophages from Nr1d1−/− mice or human macrophages in which Nr1d1 has been silenced mature and secrete higher amount of IL1B and IL18 in a NLRP3-dependent manner in response to DAMPs. The patho-physiological relevance of these observations has been tested in vivo. We demonstrate that the inflammasome is activated during peritonitis and acute hepatic liver failure. Furthermore, NR1D1 deletion results in a higher NLRP3 inflammasome pathway activation, leading to exacerbated peritonitis and fulminant hepatitis, whereas pharmacological NR1D1 activation attenuates leucocyte infiltration, inflammasome activation, and liver injury thereby delaying death and improving the rate of survival from fulminant hepatitis. Collectively, these data identify NR1D1 as a pharmacological target for NLRP3-dependent inflammatory disorders and in particular in the context of acute fulminant hepatitis.
EXPERIMENTAL PROCEDURES
See the supplementary Materials and Methods section
Results
Diurnal variations in the rate and duration of survival after fulminant hepatitis induction
Fulminant hepatitis is an acute liver injury resulting from massive hepatocyte apoptosis, hemorrhagic necrosis and inflammation. Low doses of LPS in combination with the specific hepatotoxic agent D-galactosamine (GalN) promote specific liver injury in mice and induce the production of inflammatory cytokines (TNF-α, IL1B and IL-6)39 thus recapitulating the clinical picture of acute liver injury in humans39. For these reasons, the LPS/GalN-induced hepatic injury is a widely used mouse model to understand fulminant hepatitis and its pharmacological treatment39–41. Because the clock machinery controls the circadian behavior of the immune system, we hypothesized that the outcome of fulminant hepatitis may depend on the time of hepatitis induction. Mice were thus challenged with LPS/Galactosamine every 4 hours and their survival duration and rate was monitored for 48 hours. Strikingly, we demonstrate that death occurs after only 5 hours for all mice challenged with LPS/GalN at ZT16 while the survival duration of mice injected at ZT20 or ZT0 increases up to 8.5 hours with a survival rate reaching 40% (Figure 1A–B). Thus, mice display dramatic daily differences in their susceptibility to LPS/GalN-induced fulminant hepatitis, indicating a role of the circadian clock therein. We then searched for the oscillating pathways involved in these inflammatory processes39–42.
Figure 1. NR1D1 controls daily variations in Nlrp3 expression and activation, and IL1B and IL18 secretion.

(A) Survival time and (B) Survival rate after FH induction every for 4 hours. Data are means ± SEM (n=4). ***p<0.001 as determined by Log-rank Mantel Cox (A) and Log-rank (B) test. (C) Nlrp3, (D) Il18, (E) Il1β and (H) Nr1d1 mRNA levels in CD11b+ PECs from unstimulated Nr1d1+/+ or Nr1d1−/− mice obtained by peritoneal lavage around the clock (ZT4→ZT4). (F) Secreted IL18 and (G) IL1B in peritoneal lavages from unstimulated Nr1d1+/+ or Nr1d1−/− mice around the clock (ZT4→ZT4). Data are means ± SEM (n=4–7). *p<0.05, **p<0.01, ***p<0.001 as determined by two-way ANOVA and Bonferroni post-hoc test to compare Nr1d1+/+ and Nr1d1−/−. °p<0.05, °°p<0.01, °°°p<0.001 as determined by one-way ANOVA test to compare Nr1d1+/+. $p<0.05, $$p<0.01, $$$p<0.001 as determined by one-way ANOVA test to compare Nr1d1−/−. Cosinor analysis is given in Supplementary Figure 1. ZT0: lights on, ZT12: lights off.
The NLRP3 inflammasome pathway displays circadian rhythmicity
Because the NLRP3 inflammasome pathway is the central sensor of DAMPs released from necrotic tissues and may therefore play an essential role in fulminant hepatitis pathophysiology, we next determined whether Nlrp3, Il1β and Il18 gene expression as well as cytokine levels vary during the course of the day in macrophages isolated every 4 hours from the peritoneum of non-stimulated mice. Analysis of mRNA levels from CD11b+ peritoneal exudate cells (PECs) reveals that Nlrp3, Il18, Il1β gene expression display strong daily variations with a minimum expression peak (nadir) at ZT4-8 and a maximum expression level (zenith) at ZT16-20 (Figure 1C–E and Supplementary Figure 1). Accordingly, IL1B and IL18 secretion also shows cyclic variations in vivo (Figure 1F–G and Supplementary Figure 1). Accordingly, similar oscillations were observed in synchronized mouse bone marrow-derived macrophages (BMDM) (Supplementary Figure 2A–E and Supplementary Figure 1) and human monocyte-derived macrophages (hMDM) (Supplementary Figure 2F–J and Supplementary Figure 1).
NR1D1 governs circadian rhythmicity of the NLRP3 inflammasome pathway
Interestingly, Nlrp3 gene expression is maximal at the time Nr1d1 reaches a nadir (ZT20) (Figure 1C vs H, Supplementary Figure 1A vs D and Supplementary Figure 2F vs I). Thus, we wondered whether the rhythmicity observed in NLRP3 inflammasome expression and IL1B and IL18 expression and secretion is under the direct control of NR1D1. Analysis of Nr1d1−/− PECs and synchronized BMDM revealed an altered rhythmicity of Nlrp3 expression compared to the one in Nr1d1+/+ PECs and synchronized BMDM (Figure 1C and Supplementary Figure 2D), demonstrating that NR1D1 is essential to drive circadian Nlrp3 gene expression. Accordingly, oscillations in Il1β and Il18 mRNA levels were blunted or phase-shifted in Nr1d1−/− PECs and synchronized BMDM compared to Nr1d1+/+ cells (Figure 1D–E and Supplementary Figure 2E). Bmal1 (Arntl) was measured as a control for NR1D1 activity (Supplementary Figure 2C, H). The secretion of IL1B and IL18, which is rhythmic in Nr1d1+/+ mice (Figure 1F–G), was found phase-delayed in Nr1d1−/− mice. Collectively, our data demonstrate that NR1D1 drives circadian Nlrp3 expression and ensuing rhythmic IL1B and IL18 secretion.
NR1D1 directly regulates Nlrp3 gene expression and activation in macrophages
NLRP3 expression is enhanced by exposure to pathogens and is activated by specific, structurally diverse molecular patterns associated to danger such as, for instance, extracellular ATP released by injured cells, the presence of microbial toxins including nigericin or accumulated crystals (eg cholesterol, alum)24, 25. We thus investigated whether NR1D1 directly controls NLRP3 expression and activation in human MDMs and mouse primary bone marrow-derived macrophages (BMDMs) after LPS priming. Nlrp3 mRNA levels were significantly higher in LPS-primed mouse and human macrophages in absence of Nr1d1 (Figure 2A–B). Interestingly, analysis of ChIPseq and 5’GROseq data from RAW264.7 cells23 indicated that NR1D1 occupies two sites located at −0.2 and −1.2 kb from the transcription start site (TSS), and inhibits de novo transcription from the −1.2 kb site (Supplementary Figure 3A). ChIP experiments showed that NR1D1 occupies these two sites also in wild-type BMDMs as well as two additional response elements at −4.4 and −4.2kb identified by in silico analysis (Figure 2C). Consistent with an active repression of these sites by NR1D1, we observed an increase of H3K27me3, a histone mark associated with a transcriptional repression43, in wild-type compared to Nr1d1-deficient BMDMs (Figure 2D). Interestingly, the lack of a functional DNA binding domain in NR1D1 mimics the effect of Nr1d1 deficiency on Nlrp3 mRNA levels in LPS-treated BMDM (Figure 2E), thus emphasizing the importance of the direct binding of NR1D1 to DNA in this regulatory mechanism. Interestingly, 1 hour-LPS treatment strongly decreased NR1D1 binding to these sites, likely allowing a de-repression of the NLRP3 pathway in response to inflammatory stimuli, when a full response is needed to restore homeostasis (Figure 2C). Accordingly, short exposure to LPS increased Nlrp3, Il1β and Il18 mRNA expression but did not alter neither Nr1d1 nor Nr1d2 mRNA levels (Supplementary Figure 3B–F), thus suggesting that LPS priming triggers NR1D1 removal from its response elements without affecting its expression. However, prolonged exposure to LPS decreases Nr1d1 and Nr1d2 gene expression (Supplementary Figure 4A–B and Supplementary Figure 4E–F) but does not change their phase in synchronised BMDMs (data not shown). It is thus likely that both phenomena concur in vivo. In addition, Nlrp3 protein levels were higher in BMDMs from Nr1d1−/− mice (Figure 2F and Supplementary Figure 3G–H). Upon activation by DAMPs, the NLRP3 protein associates with the Apoptosis-associated Speck-like protein containing a Caspase recruitment domain protein (ASC) to form ASC specks44, a hallmark of cellular activation of the NLRP3 inflammasome. Nr1d1-deficiency led to a marked elevation of the number of cells displaying ASC specks upon ATP or nigericin activation (Figure 2G–H), indicating that NR1D1 regulates NLRP3 inflammasome complex formation. These findings implicate NR1D1 as a negative regulator of NLRP3 expression and activation in vitro. Furthermore, NR1D2 knock-down did not affect Nlrp3 mRNA levels in BMDM (Figure 2I and Supplementary Figure 4A–B), but enhanced NLRP3 as well as IL1β and IL18 expression in LPS-primed human MDMs (Supplementary Figure 4C–F) suggesting that the Nr1d2 isotype is not involved in this process in mouse macrophages, but displays redundant, and not synergistic, activity with NR1D1 in human macrophages. Finally, NR1D1 deletion did not affect other inflammasomes such as AIM2 (Supplementary Figure 5A), or AIM2-mediated IL1B and IL18 maturation and secretion in LPS-primed BMDM challenged with the AIM2 activator poly(dA:dT) (Supplementary Figure 5B–C).
Figure 2. NR1D1 directly regulates the NLRP3 inflammasome in macrophages.

Nlrp3 mRNA levels in control (CTRL) and LPS-primed (LPS) (A) BMDMs from Nr1d1+/+ and Nr1d1−/− mice and in (B) MDMs transfected with siRNA against Nr1d1 (siNr1d1) or a scrambled siRNA (siCTRL) (n=3). Data are represented as means ± SD. ***p<0.001 as determined by two-way ANOVA followed by a Bonferroni post-hoc test. (C) ChIP analysis of NR1D1 occupancy to the Nlrp3 promoter in control (Vehicle) or LPS-primed (for 1 hour) BMDMs. The top diagram represents putative Rev-erb Response Elements (RevREs) unveiled by MatInspector promoter analysis. Down, ChIP analysis from one representative experiment. (D) ChIP analysis of H3K27me3 at the Nlrp3 promoter in Nr1d1+/+ and Nr1d1−/− BMDMs. (E) Nlrp3 mRNA levels in BMDMs from Nr1d1Floxed/Floxed;LysM+/+ (Nr1d1Floxed/Floxed) or Nr1d1Floxed/Floxed; LysMCre/+ (Nr1d1DBDmut) mice mice. (F) NLRP3 protein expression in control (CTRL) and LPS-primed ATP-activated (LPS ATP) BMDMs from Nr1d1+/+ or Nr1d1−/− mice (n=3). Top, the western blot and down, its quantification. ***p<0.001 as determined by two-way ANOVA followed by a Bonferroni post-hoc test. (G) ASC specks formation and (H) its quantification in LPS-primed BMDMs from Nr1d1+/+ and Nr1d1−/− mice treated with ATP, Nigericin or vehicle (CTRL). Results are presented as mean percentage of cells displaying ASC specks ± SD. *p<0.05, ***p<0.001 as determined by two-way ANOVA followed by a Bonferroni post-hoc test. (I) Nlrp3 mRNA levels in BMDMs from Nr1d1+/+ and Nr1d1−/− mice transfected with siRNA against Nr1d2 (siNr1d2) or a scrambled siRNA (siCtrl) (n=3). Data are represented as means ± SD. ***p<0.001 as determined by two-way ANOVA followed by a Bonferroni post-hoc test.
NR1D1 controls NLRP3-dependent maturation and secretion of IL1B and IL18 in a Caspase-1-dependent manner
The NLRP3 inflammasome complex regulates IL18 and IL1B processing and secretion at the post-transcriptional level through the oligomerization and auto-cleavage of an inactive p45 pro-Caspase-1 precursor into active p20 and p10 Caspase-1 subunits responsible for the subsequent maturation of IL1B and IL1824, 25, 44. Nr1d1 deficiency did not affect casp1 gene expression (Supplementary figure 6A) nor p45 protein amount (Figure 3A–B, Supplementary Figure 6B–C), but enhanced Caspase-1 maturation in LPS-primed BMDMs that were either unstimulated or ATP-stimulated to activate the NLRP3 inflammasome (Figure 3A and Supplementary Figure 6D). Consistently, NR1D1 knockdown in human MDMs (Supplementary Figure 7A) and NR1D1 deletion in BMDMs (Figure 3C and supplementary Figure 7B) increased IL1B secretion after LPS priming and activation with ATP. Similarly, IL18 secretion in culture medium from activated macrophages was increased (Figure 3D and supplementary Figure 7C). In addition, NR1D1 activation by its natural ligand hemin16 decreased LPS-primed ATP-stimulated IL1B and IL18 secretion (Figure 3E–F and Supplementary Figure 8A) in BMDMs and human MDMs. Although IL1B and IL18 may be processed by other caspases such as Caspase-8 or Caspase-1144, 45, the increase in IL1B and IL18 secretion in Nr1d1−/− BMDMs is Caspase-1 and NLRP3-dependent, since it was abolished by Caspase-1 inhibition (Figure 3C–D and Supplementary Figure 9A–B) and Nlrp3 silencing (Figure 3G–H). Accordingly, addition of MCC950, a specific NLRP3 inhibitor46, blunted the effect of NR1D1 activation by hemin on IL1B and IL18 secretion (Figure 3E–F). Altogether, these results demonstrate that NR1D1 regulates macrophage IL1B and IL18 maturation and secretion in a NLRP3-dependent manner in vitro.
Figure 3. NR1D1 regulates IL18 and IL1B maturation and secretion in a NLRP3 inflammasome-dependent manner.

Caspase-1 protein (sub-unit p10, p20, p35, p45) expression in supernatant (A) or lysate (B) from LPS-primed (LPS) and ATP-activated (LPS/ATP) or not (/) BMDMs from Nr1d1+/+ (Rα+/+) and Nr1d1−/− (Rα−/−) mice (representative of 3 independent experiments). (C) IL1B and (D) IL18 secreted by LPS-primed and ATP-activated BMDMs isolated from Nr1d1+/+ or Nr1d1−/− mice that were treated with low dose of caspase inhibitor z-VAD-fmk (z-VAD-fmk) or not (Vehicle) (n=3). (E) IL1B secreted by LPS-primed and ATP activated (ATP) or not (CTRL) BMDMs treated with (hemin) or its vehicle (vehicle) and with NLRP3 inhibitor MCC950 or not (Saline) (n=3). (F) IL18 secreted by LPS-primed and ATP-activated (ATP) or not (CTRL) MDMs treated with (hemin) or its vehicle (vehicle) and with NLRP3 inhibitor MCC950 or not (Saline) (n=3). (G) IL1B and (H) IL18 secreted by LPS-primed and ATP-activated BMDMs isolated from Nr1d1+/+ or Nr1d1−/− mice in which NLRP3 was silenced (siRNA NLRP3) or not (siRNA CTRL) (n=3). The top western blot (G) shows NLRP3 protein silencing. Data are represented as means ± SD. ***p<0.001 as determined by two-way ANOVA followed by a Bonferroni post-hoc test.
NR1D1 also regulates Il1β and Il18 at the gene expression level
In addition to its role on NLRP3-dependent IL18 and IL1B maturation, we also assessed whether NR1D1 regulates Il18 and Il1β expression at the transcriptional level in primary macrophages. Nr1d1-deficiency in BMDMs or siRNA-mediated NR1D1 knockdown in human MDMs led to higher IL1B and IL18 mRNA and protein levels after LPS priming (Figure 4A–D and Supplementary Figure 9C). ChIPseq and 5’GRO-seq data analysis from NR1D1-overexpressing RAW264.7 macrophages23 indicate that NR1D1 binds to the Il1β promoter and inhibits its de novo transcription (Supplementary Figure 10). ChIP-qPCR analysis demonstrated that endogenous NR1D1 binds to the Il1β promoter also in BMDMs, thus showing that NR1D1 directly regulates Il1β expression (Figure 4E). In addition, impairment of NR1D1 direct DNA binding enhances Il1β gene expression (Figure 4F), which, together with the ChIP experiment results, suggests that NR1D1 directly inhibits Il1β gene transcription. Finally, NR1D2 knockdown did not affect Il1β gene expression in mouse macrophages (Figure 4G), but enhanced Il1β gene expression in human MDM (Supplementary Figure 4D) as well as IL1B and IL18 secretion (Supplementary Figure 4G–H). However, no synergistic effect with Nr1d1-deficiency was observed, highlighting the redundant role of NR1D2 in the regulation of this pathway.
Figure 4. NR1D1 regulates IL18 and IL1B expression.

(A–D) Il18 (A,B) and Il1β (C,D) mRNA levels in LPS-primed BMDMs isolated from Nr1d1+/+ or Nr1d1−/− mice (A,C) and in LPS-primed human MDMs transfected with siRNA against Nr1d1 (siNr1d1) or a scrambled siRNA (siCTRL) (B,D) (n=3). Data are represented as means ± SD. **p<0.01, ***p<0.001 as determined by two-way ANOVA followed by a Bonferroni post-hoc test. (E) ChIP analysis of NR1D1 occupancy at the Il1β and Arntl promoter (as control) in control (Vehicle) or LPS-primed (for 1 hour) BMDMs (n=3). The top diagram represents putative Rev-erb Response Elements (RevREs) unveiled by MatInspector promoter analysis. Data are represented as means ± SD. **p<0.01, ***p<0.001 as determined by an unpaired t-test. Il1β mRNA levels in LPS-primed (F) BMDMs isolated from Nr1d1Floxed/Floxed;LysM+/+ (Nr1d1Floxed/Floxed) or Nr1d1Floxed/Floxed; LysMCre/+ (Nr1d1DBDmut) mice mice and (G) BMDMs from Nr1d1+/+ and Nr1d1−/− mice transfected with siRNA against Nr1d2 (siNr1d2) or a scrambled siRNA (siCtrl) (n=3). Data are represented as means ± SD. ***p<0.001 as determined by two-way ANOVA followed by a Bonferroni post-hoc test.
Pharmacological activation of NR1D1 inhibits the NLRP3 inflammasome pathway
Since Nr1d1-deficiency enhances the NLRP3 inflammasome pathway, we hypothesized that ligand-mediated activation of NR1D1 inhibits NLRP3 expression and subsequent maturation of IL1B. Indeed, pharmacological activation of NR1D1 with SR900915 down-regulated Nlrp3 gene expression in LPS-primed BMDMs (Figure 5A) and lowered NLRP3 protein amount (Figure 5B, Supplementary Figure 8B). Similar effects were observed in SR9009- and hemin-stimulated human MDMs (Figure 5C and Supplementary Figure 8C). In addition, SR9009- and hemin-activation of NR1D1 decreased Il1β and Il18 mRNA levels (Figure 5D–F, Supplementary Figure 8D–E), as well as IL1B and IL18 protein amount, maturation and secretion (Figures 3E–F, 5G–J) in BMDMs and human MDMs. Altogether, these results emphasize the pharmacological potential of NR1D1 to control the inflammasome and pro-inflammatory pathways in innate immune cells.
Figure 5. NR1D1 pharmacological activation inhibits the NLRP3 inflammasome pathway in primary macrophages.

(A) Nlrp3 mRNA levels in LPS-primed (LPS) or not (NT) BMDMs treated with SR9009 (n=3). (B) NLRP3 protein levels in LPS-primed (LPS) or not (/) BMDMs treated with SR9009 or DMSO (representative of 3 independent experiments). (C) NLRP3 and (E) IL1β mRNA levels in LPS-primed human MDMs pre-treated with SR9009 or not (Vehicle) (n=3). (D) Il1β and (F) Il18 mRNA levels in LPS-primed BMDMs pre-treated or not with SR9009 (n=3). (G) pro- and matured IL1B protein expression in LPS-primed BMDM or not (NT) and treated with SR9009 or not (Vhc). (H) IL1B secretion in LPS-primed and ATP-activated BMDMs pre-treated with SR9009 or not (Vehicle) (n=3) (I) IL1B and (J) IL18 secretion in LPS-primed and ATP-activated human MDMs pre-treated with SR9009 or not (Vehicle) (n=3). Data are represented as means ± SD. °°°p<0.001 in LPS vs unstimulated vehicle condition (A); *p<0.05, **p<0.01, ***p<0.001 as determined by one-way ANOVA (A) or two-way ANOVA (C–F, H–J) followed by a Bonferroni post-hoc test.
NR1D1 attenuates NLRP3-mediated inflammation in acute peritonitis
We next determined whether NR1D1 regulates the NLRP3 inflammasome in vivo in an acute model of sterile peritoneal inflammation. Mice were pre-treated or not with the NLRP3 inhibitor MCC950, and challenged by intraperitoneal administration of LPS and alum to specifically activate the NLRP3 signaling pathway47. IL1B in the peritoneum and IL1B and IL18 plasma levels were higher in Nr1d1−/− compared to Nr1d1+/+ mice, especially upon LPS-priming and alum activation of NLRP3 (Figure 6A–C). Remarkably, this effect was totally abolished in presence of MCC950 (Figure 6A–C).
Figure 6. NR1D1 modulates the development of acute innate inflammation through regulation of the NLRP3 inflammasome in a peritonitis model.

(A) Secreted IL1B from peritoneal lavage of Nr1d1+/+ or Nr1d1−/− mice treated with LPS, LPS + Alum, LPS + Alum + MCC950 or not (Ctrl) at ZT8 (n=6–12). (B) Plasma IL1B and (C) IL18 from Nr1d1+/+ or Nr1d1−/− mice treated as in (A) (n=6–12). (D) Secreted IL1B and (E) IL18 from peritoneal macrophages isolated around the clock from LPS-stimulated mice treated with SR10067 (Ligand) or vehicle (n=10). (F) Secreted IL1B from peritoneal lavage at ZT8 and ZT20 of Nr1d1+/+ or Nr1d1−/− mice treated with LPS + Alum (n=6–12). Data are represented as means ± SEM. *p<0.05, **p<0.01, ***p<0.001 as determined by two-way ANOVA and Bonferroni post-hoc test. (G) NR1D1 protein expression in CD11b+ PECs at ZT8 and ZT20 of Nr1d1+/+ (Rα+/+) or Nr1d1−/− (Rα−/−) mice treated with LPS + Alum (n=6–12).
In response to infection or injection of LPS, macrophage cytokine secretion displays a circadian pattern with a higher response toward the end of the light phase compared to ZT08, 20. Accordingly, IL1B and IL18 secretion displayed daily variations in peritoneal exudates isolated from mice intraperitoneally challenged with LPS every 6 hours (Figure 6D–E). Pharmacological activation of NR1D1 with SR10067 decreased IL1B and IL18 secretion in LPS-injected mice around the clock and reduced the time-of-day variations in LPS-stimulated cytokine secretion (Figure 6D–E) indicating that NR1D1 activation normalizes the daily fluctuations of the NLRP3 inflammasome pathway in conditions of an overstimulated inflammatory response. In addition, the higher IL1B secretion observed in LPS/Alum challenged Nr1d1−/− mice sacrificed at ZT8 was completely blunted when the peritonitis was induced 12-hour apart in mice that were sacrificed at ZT20 (Figure 6F), ie. when NR1D1 is nearly absent (Figure 6G), consistent with the concept that NR1D1 regulates the NLRP3 inflammasome pathway in a circadian manner. Altogether these data indicate that NR1D1 inhibits peritonitis through its action on the NLRP3 inflammasome in vivo.
NR1D1 prevents NLRP3-mediated fulminant hepatitis
Interestingly, the NLRP3 inflammasome is enhanced in MHV-3-induced viral fulminant hepatitis37, and in LPS/GalN-induced acute liver failure38. In addition, low doses of LPS in combination with GalN increase NLRP3 protein and Caspase1 activity38, 42. Therefore, we tested whether NR1D1 may ameliorate acute liver failure using this experimental setting39.
Strikingly, liver damage was strongly increased in LPS/GalN-injected Nr1d1−/− mice compared to littermate controls, while NLRP3 inhibition by MCC950 treatment protected Nr1d1−/− mice from liver failure (Figure 7A–B). NR1D1 deletion induced hepatic F4/80 and Ccl2 gene expression suggesting enhanced macrophage infiltration (Figure 7C and Supplementary Figure 11A). Finally, the induction of caspase 1 activity (Figure 7D), liver Nlrp3 and Il1β gene expression and IL1B secretion (Figure 7E–F, Supplementary Figure 11B) by LPS/GalN treatment, was further enhanced upon Nr1d1-deficiency. TNFα is enhanced in fulminant hepatitis and plays an important role in the regulation of innate inflammatory responses. We thus verified whether the TNFα pathway alters the Nr1d1-mediated regulation of the NLRP3 inflammasome pathway. As expected, hepatic TNFα gene expression was increased in LPS/GalN-challenged mice. However, it was not affected by NR1D1 deletion (Supplementary Figure 11C). In addition, although TNFα treatment enhances Il1β and Nlrp3 gene expression in BMDM, Nr1d1-deficiency did not affect the effect of TNFα on this pathway (Supplementary Figure 11D–E). We next assessed whether NR1D1 activation can inhibit fulminant hepatitis. As expected, control mice died 6.5 hours after LPS/GalN injection at ZT2 and ~90% were dead by 9 hours. Strikingly, SR9009-treated mice lived longer compared to vehicle-treated mice and the majority of mice survived from fulminant hepatitis (~70% survival rate of SR9009 pre-treated mice) (Figure 7G). The increased survival rate upon SR9009 treatment was associated with decreased LPS/GalN-induced hepatic hemorrhage (Supplementary Figure 11F). SR9009 pre-treatment strongly decreased the induction of ALAT, a marker of hepatic dysfunction, by LPS/GalN (Figure 7H) and improved liver appearance (Supplementary Figure 11G). In addition, SR9009 prevented the increase of hepatic F4/80 (Figure 7I) and Ccl2 (Supplementary Figure 11I) gene expression, Caspase 1 activity (Figure 7J), hepatic Nlrp3 and Il1β gene expression (Figure 7K–L) and IL1B blood levels (Supplementary figure 11H) after LPS/GalN injection. It is noteworthy that co-treatment with MCC950 and SR9009 resulted in similar, but not additive, effects on ALAT activity and inflammasome pathway gene expression compared to treatment with MCC950 or SR9009 alone, indicating that NLRP3 mediates the effect of NR1D1 on these parameters (Figure 7 and Supplementary Figure 11). In addition, SR9009 treatment did not alter LPS/GalN-induced Tnfα gene expression in liver (Supplementary Figure 11J). Finally, SR9009, but not MCC950, decreased the total number of hepatic leucocytes recruited upon LPS/GalN injection (Supplementary Figure 12A). More precisely, we performed FACS analysis, which demonstrates that SR9009 impaired the recruitment of infiltrating monocytes and macrophages (Supplementary Figure 12B–E) as well as the hepatic recruitment of neutrophils (Supplementary Figure 12F–G). As expected, SR9009, but not MCC950, inhibited Nlpr3, Il1β and Il18 gene expression in hepatic F4/80+ cells isolated from LPS-GalN challenged mice (Supplementary Figure 12H–J). Our results indicate that NR1D1 activation improves mice survival during LPS/GalN-induced fulminant hepatitis by decreasing CCL2-mediated hepatic infiltration of innate immune cells and by inhibiting the activation of the NLRP3 inflammasome pathway.
Figure 7. NR1D1 prevents the development of fulminant hepatitis through regulation of the NLRP3 inflammasome.

(A–F) Nr1d1+/+ or Nr1d1−/− mice were pre-treated with MCC950 or Saline and then intraperitoneally injected with LPS plus GalN or PBS as control (Ctrl). (A) Representative liver appearance, (B) serum ALAT activity, (C) liver F4/80 mRNA levels, (D) Caspase-1 activity in tissue sections (FMI: Fluorescence Mean Intensity) and (E) Nlrp3 and (F) Il1β mRNA levels in liver from each group as described. Data are represented as means ± SEM (n=5–11). *p<0.05, **p<0.01, ***p<0.001 as determined by two-way ANOVA and Bonferroni post-hoc test. (G–L) C57/Bl6 mice were pre-treated with SR9009 or vehicle and/or MCC950 and then challenged with LPS/GalN or PBS as control (Ctrl) as indicated. (G) Kaplan-Meier survival curve of mice challenged with LPS/GalN at ZT2 (n=15 per group). ***p<0.0001 as determined by log rank test. (H) serum ALAT activity, (I) liver F4/80 mRNA levels, (J) Caspase 1 activity in tissue sections (FMI: Fluorescence Mean Intensity), and (K) Nlrp3 and Il1β mRNA levels in liver from each group as described. Data are represented as means ± SEM (n=5–6) **p<0.01, ***p<0.001 as determined by two-way ANOVA and Bonferroni post-hoc test.
Discussion
The nuclear receptors NR1D1 and NR1D2 are now considered as core components of the clock machinery. Indeed, Nr1d1-deficiency leads to a circadian period shortening by half-an-hour. Although this triggers a subtle activity phase shift compared to wild-type mice after one subjective day, this results in for a dramatic shift after a prolonged period12, 48. Interestingly, whereas NR1D2 deletion alone does not trigger any effect on the circadian activity in mice, the circadian activity of Nr1d1−/− Nr1d2−/− mice is abolished, emphasizing the redundant activity of both nuclear receptors in this context12. Because their expression itself displays daily variations, NR1D1 and NR1D2 drive oscillatory expression of their target genes involved, for instance, in hepatic lipid synthesis and storage11, 12, bile acid synthesis13 and thermogenesis18. In addition to its role in the control of metabolism, Nr1d1’s circadian pattern has also an important impact on the daily behavior of the immune system by controlling the circadian variations in cytokine expression such as MCP-1/CCL2 and IL-6 in mouse macrophages20. We provide here new insights on the regulatory role of NR1D1 in circadian immunity in human and mouse primary macrophages.
Our results identify NR1D1 as a novel regulator of the NLRP3 inflammasome, driving circadian rhythmicity in its expression, activation, and ensuing IL1B and IL18 maturation and secretion (Supplementary Figure 13). We demonstrate that NR1D1 deletion or silencing in primary mouse and human MDMs results in exacerbated NLRP3-dependent IL1B and IL18 production. Our results further show that NR1D1 attenuates NLRP3-driven inflammation in vivo in models of acute peritonitis and fulminant hepatitis and demonstrate that pharmacological NR1D1 activation reduces NLRP3 expression and IL1B and IL18 production by macrophages, presenting a promising strategy to treat NLRP3-dependent inflammatory diseases.
Compared to other inflammasomes, NLRP3 is activated by a plethora of signals of different biological origin such as bacterial, fungal and viral pathogens, pore-forming toxins, crystals, aggregates (β-amyloid) and DAMPs (ATP)24. As such, NLRP3 is a key platform centralizing the detection of different signals to activate pro-inflammatory cytokine maturation and secretion. Because of this central role, a better understanding of the mechanisms that govern NLRP3 inflammasome activation is an important step toward the development of novel therapeutic strategies to combat over-activation of the immune system. Circadian regulation of immunity has been proposed as a mechanism to prepare the organism for a higher risk of tissue damage and infection during the active phase and as a process to anticipate the regeneration of the immune system during the resting phase3. Our data demonstrate for the first time that the molecular clock controls the NLRP3 inflammasome and subsequent cytokine responses in activated macrophages, hence supporting the idea that the circadian clock not only regulates oscillations in blood leucocyte trafficking7, but is also involved in the very first step of the immune response such as pro-inflammatory signal sensing.
The response to pro-inflammatory stimuli such as LPS depends on the time of pro-inflammatory challenge8, 20. In line, our data demonstrate that FH severity depends on the induction time (Figure 1). Accordingly, treatment of mice at ZT8 leads to a more pronounced secretion of IL1B and IL18 than at ZT20 (Figure 6), which furthermore is inversely correlated with NR1D1 expression (Figure 1). Since the enhanced immune response at ZT10-ZT12 is also responsible for a higher risk of septic shock and death, a well-balanced circadian immunity appears to be essential to preserve immune homeostasis3. In this context, it is important that Nr1d1 activity can be modulated by small synthetic molecules49. Here, we show that pharmacological activation of NR1D1 has the potential to restrain immune responses at specific times of the day (Figure 6).
IL1B secretion has been shown to oscillate diurnally with a peak during the onset of the active phase50. Our data obtained on mouse peritoneum corroborate this finding (Figure 1G), and we additionally show that secretion of IL18, the other IL-1 family member matured by the NLRP3 inflammasome pathway, also displays daily fluctuations in macrophages isolated from the peritoneum (Figure 1F). According to our data obtained from synchronized isolated human and mouse macrophages, we suggest that these oscillations are mainly due to the internal clock machinery of macrophages themselves (Figure 1 and Supplementary Figures 1 and 2). It is noteworthy that despite the fact that Il1β and Il18 gene expression was not in phase at the transcriptional level (Figure 1D–E), IL1B and IL18 secretion oscillates with the same peaks and nadirs (Figure 1F–G), suggesting that the mere regulation of cytokine gene transcription is not sufficient to account for the daily fluctuations of cytokine levels and that an additional step, namely post-translational modifications and maturation, is required to modulate this circadian pattern. In our study, Nlrp3 expression oscillates around the clock under the control of NR1D1, supporting the idea that NLRP3 circadian rhythmicity may account for the daily pattern of IL1B and IL18 secretion.
Although the priming of Nlrp3 expression is known as a key step of the inflammatory response51, 52, the transcriptional control of Nlrp3 is poorly understood. Here, we demonstrate that Nr1d1-deficiency strongly alters Nlrp3, Il1β and Il18 expression in unstimulated, but also in LPS-challenged peritoneal macrophages and in injured livers from LPS/GalN-treated mice (Figure 1, Figure 7). In addition, NR1D1 controls NLRP3 complex assembly, Caspase-1 maturation of pro-IL1B and pro-IL18 and their secretion in response to NLRP3 priming and activation in both human and murine primary macrophages. Thus, NR1D1 acts through a two-pronged mechanism, acting at the transcriptional level to repress Nlrp3, Il1β and Il18 gene expression, and by modulating IL1B and IL18 protein maturation and secretion through the activation of the inflammasome complex and Caspase 1.
Aberrant activation of the NLRP3 inflammasome is implicated in the pathogenesis of numerous diseases including type 2 diabetes, atherosclerosis, gout, rheumatoid arthritis and kidney diseases24, 34, 36, 45, some of which have also been linked to disruption of the clock3. Our study points to NR1D1 as a plausible link between a dysregulated clock and NLRP3-driven pathologies. Indeed, NR1D1 deletion led to an exacerbated response to acute inflammation such as sterile peritonitis (Figure 6A–C) and drug-induced FH (Figure 7) for which no therapy other than liver transplantation is effective at improving patient survival. Current treatments for NLRP3-dependent diseases include IL-1-targeting strategies53 and glyburide54. We now show that Rev-erb activation protects against acute liver failure and improves the survival rate in a mouse model of FH (Figure 7, Supplementary Figure 11), identifying NR1D1 as a novel therapeutic target for the treatment of FH. It is noteworthy that, compared to MCC950 which inhibits NLRP3, NR1D1 targets not only the NLRP3 inflammasome, but also Ccl2 expression (Supplementary Figure 12), macrophage infiltration (Figure 7, Supplementary Figure 12) and likely LPS signalling through TLR4 regulation22, and therefore may lead to a stronger efficiency compared to the mere inhibition of the NLRP3 pathway and IL-1 maturation by MCC950. In conclusion, NR1D1 represents an interesting target to control the DAMP-mediated NLRP3-dependent inflammatory process in a circadian manner.
Supplementary Material
Acknowledgments
We thank Sami Lasri and Emmanuelle Vallez for their technical assistance. We also thank John Walker for overseeing the chemical synthesis of SR9009 and SR10067. We thank the BICeL Facility for access to systems and technical advice. BS is recipient of an ERC Advanced Grant (European Research Council Advanced grant 694717).
Fundings
This work was supported by research grants from Région Nord Pas-de-Calais (to BP), Fondation pour la Recherche Médicale FRM (FDT20170739031) (to MZ), Fédération Francophone de Recherche sur le Diabète FFRD, sponsored by Fédération Française des Diabétiques (AFD), AstraZeneca, Eli Lilly, Merck Sharp & Dohme (MSD), Novo Nordisk & Sanofi, (to HD), Fondation de France (to HD), Région Nord-Pas-de-Calais (CPER) starting grant (to HD), the European Commission (FP7-HEALTH) consortium Eurhythdia (grant number 278397) (to BS and HD), the ‘European Genomic Institute for Diabetes’ (EGID, ANR-10-LABX-46) (to HD and BS), an unrestricted ITMO/Astra Zeneca grant (to BS), and from the American Heart Association (to SS; 14PRE1826000) and the National Institutes of Health (to TPB; MH093429).
Abbreviations used in this paper
- ALAT
alanine aminotransferase
- ASC
Apoptosis-associated Speck-like protein containing a Caspase recruitment domain protein
- ATP
adenosine triphosphate
- BMDMs
bone marrow-derived macrophages
- CASP1
caspase-1
- DAMPs
danger-associated molecular patterns
- D-GalN
D-galactosamine
- ELISA
enzyme-linked immunosorbent assay
- FH
Fulminant Hepatitis
- IL
interleukin
- LPS
lipopolysaccharide
- MDMs
monocyte-derived macrophages
- NET
Neutrophil Extracellular Traps
- NLRP3
NACHT, LRR, and pyrin domain–containing protein 3
- PBS
phosphate-buffered saline
- PECs
Peritoneum Exudate Cells
- siRNA
small interfering RNA
- ZT
zeitgeber
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
BP, MZ and HD conceived the project, designed and performed experiments, analysed the results and wrote the manuscript; LF and JB performed key experiments, analysed the results and read the manuscript; SS, CB and TPB designed and performed key experiments, analysed the results and read the manuscript; SD, JV, AML, QT, DD, CD, AB, YS, SL performed experiments and read the manuscript; JH performed cosinor analysis; JE analysed ChIPseq and 5’GROseq databases and read the manuscript; SD was in charge of the mouse colonies and read the manuscript; BS discussed the experiments and read the manuscript.
Conflict of interest statements
The authors disclose no conflicts
Supplemental information includes Supplemental Experimental Procedures, one table and thirteen figures.
References
- 1.Cardoso FS, Marcelino P, Bagulho L, et al. Acute liver failure: An up-to-date approach. J Crit Care. 2017;39:25–30. doi: 10.1016/j.jcrc.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Bass J. Circadian topology of metabolism. Nature. 2012;491:348–356. doi: 10.1038/nature11704. [DOI] [PubMed] [Google Scholar]
- 3.Curtis AM, Bellet MM, Sassone-Corsi P, et al. Circadian clock proteins and immunity. Immunity. 2014;40:178–186. doi: 10.1016/j.immuni.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Early JO, Curtis AM. Immunometabolism: Is it under the eye of the clock? Semin Immunol. 2016 doi: 10.1016/j.smim.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Man K, Loudon A, Chawla A. Immunity around the clock. Science. 2016;354:999–1003. doi: 10.1126/science.aah4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibbs J, Ince L, Matthews L, et al. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat.Med. 2014;20:919–926. doi: 10.1038/nm.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendez-Ferrer S, Lucas D, Battista M, et al. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen KD, Fentress SJ, Qiu Y, et al. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science. 2013;341:1483–8. doi: 10.1126/science.1240636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheiermann C, Kunisaki Y, Lucas D, et al. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity. 2012;37:290–301. doi: 10.1016/j.immuni.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castanon-Cervantes O, Wu M, Ehlen JC, et al. Dysregulation of inflammatory responses by chronic circadian disruption. J.Immunol. 2010;185:5796–5805. doi: 10.4049/jimmunol.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugge A, Feng D, Everett LJ, et al. Rev-erbalpha and Rev-erbbeta coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012;26:657–667. doi: 10.1101/gad.186858.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho H, Zhao X, Hatori M, et al. Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature. 2012;485:123–127. doi: 10.1038/nature11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duez H, van der Veen JN, Duhem C, et al. Regulation of bile acid synthesis by the nuclear receptor Rev-erbalpha. Gastroenterology. 2008;135:689–698. doi: 10.1053/j.gastro.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 14.Raspe E, Duez H, Mansen A, et al. Identification of Rev-erbalpha as a physiological repressor of apoC-III gene transcription. J.Lipid Res. 2002;43:2172–2179. doi: 10.1194/jlr.m200386-jlr200. [DOI] [PubMed] [Google Scholar]
- 15.Solt LA, Wang Y, Banerjee S, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62–68. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin L, Wu N, Curtin JC, et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–1789. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 17.Woldt E, Sebti Y, Solt LA, et al. Rev-erb-alpha modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat.Med. 2013;19:1039–1046. doi: 10.1038/nm.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerhart-Hines Z, Feng D, Emmett MJ, et al. The nuclear receptor Rev-erbalpha controls circadian thermogenic plasticity. Nature. 2013;503:410–413. doi: 10.1038/nature12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellet MM, Deriu E, Liu JZ, et al. Circadian clock regulates the host response to Salmonella. Proc.Natl.Acad.Sci.U.S.A. 2013;110:9897–9902. doi: 10.1073/pnas.1120636110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibbs JE, Blaikley J, Beesley S, et al. The nuclear receptor REV-ERBalpha mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc.Natl.Acad.Sci.U.S.A. 2012;109:582–587. doi: 10.1073/pnas.1106750109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller M, Mazuch J, Abraham U, et al. A circadian clock in macrophages controls inflammatory immune responses. Proc.Natl.Acad.Sci.U.S.A. 2009;106:21407–21412. doi: 10.1073/pnas.0906361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontaine C, Rigamonti E, Pourcet B, et al. The nuclear receptor Rev-erbalpha is a liver × receptor (LXR) target gene driving a negative feedback loop on select LXR-induced pathways in human macrophages. Mol.Endocrinol. 2008;22:1797–1811. doi: 10.1210/me.2007-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lam MT, Cho H, Lesch HP, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 26.Warnatsch A, Ioannou M, Wang Q, et al. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–20. doi: 10.1126/science.aaa8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naik E, Dixit VM. Modulation of inflammasome activity for the treatment of auto-inflammatory disorders. J.Clin.Immunol. 2010;30:485–490. doi: 10.1007/s10875-010-9383-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munoz-Planillo R, Kuffa P, Martinez-Colon G, et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian N, Natarajan K, Clatworthy MR, et al. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–61. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 32.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat.Rev.Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masters SL, Latz E, O'Neill LA. The inflammasome in atherosclerosis and type 2 diabetes. Sci.Transl.Med. 2011;3:81ps17. doi: 10.1126/scitranslmed.3001902. [DOI] [PubMed] [Google Scholar]
- 35.Sheedy FJ, Grebe A, Rayner KJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat.Immunol. 2013;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vande Walle L, Van Opdenbosch N, Jacques P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512:69–73. doi: 10.1038/nature13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo S, Yang C, Diao B, et al. The NLRP3 Inflammasome and IL-1beta Accelerate Immunologically Mediated Pathology in Experimental Viral Fulminant Hepatitis. PLoS Pathog. 2015;11:e1005155. doi: 10.1371/journal.ppat.1005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seo MJ, Hong JM, Kim SJ, et al. Genipin protects d-galactosamine and lipopolysaccharide-induced hepatic injury through suppression of the necroptosis-mediated inflammasome signaling. Eur J Pharmacol. 2017;812:128–137. doi: 10.1016/j.ejphar.2017.07.024. [DOI] [PubMed] [Google Scholar]
- 39.Furuya S, Kono H, Hara M, et al. Interleukin 17A plays a role in lipopolysaccharide/D-galactosamine-induced fulminant hepatic injury in mice. J Surg Res. 2015;199:487–93. doi: 10.1016/j.jss.2015.05.060. [DOI] [PubMed] [Google Scholar]
- 40.Nakama T, Hirono S, Moriuchi A, et al. Etoposide prevents apoptosis in mouse liver with D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure resulting in reduction of lethality. Hepatology. 2001;33:1441–50. doi: 10.1053/jhep.2001.24561. [DOI] [PubMed] [Google Scholar]
- 41.Shirozu K, Hirai S, Tanaka T, et al. Farnesyltransferase inhibitor, tipifarnib, prevents galactosamine/lipopolysaccharide-induced acute liver failure. Shock. 2014;42:570–7. doi: 10.1097/SHK.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16:1467–81. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broderick L, De Nardo D, Franklin BS, et al. The inflammasomes and autoinflammatory syndromes. Annu.Rev.Pathol. 2015;10:395–424. doi: 10.1146/annurev-pathol-012414-040431. [DOI] [PubMed] [Google Scholar]
- 45.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat.Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat.Med. 2015;21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–23. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Preitner N, Damiola F, Lopez-Molina L, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–60. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 49.Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev Drug Discov. 2014;13:197–216. doi: 10.1038/nrd4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sherman H, Frumin I, Gutman R, et al. Long-term restricted feeding alters circadian expression and reduces the level of inflammatory and disease markers. J Cell Mol Med. 2011;15:2745–59. doi: 10.1111/j.1582-4934.2010.01160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–91. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol. 2009;183:792–6. doi: 10.4049/jimmunol.0900173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat.Rev.Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J.Cell Biol. 2009;187:61–70. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.