

Abstract
Purpose
To create a model of an ischemic retina with temporary ischemia and reperfusion (IR) and to examine the possible antiapoptotic and neurodegenerative effects of a vascular endothelial growth factor (VEGF) antagonist.
Methods
Three groups were formed. Rats were subjected to continued ischemia for 45 min, and then reperfusion was allowed for 2 days. For the first group, ischemia was induced, but an anti-VEGF agent was not administered. For the second group, 2 days before ischemia, 0.005 ml (0.125 mg) of bevacizumab was administered intravitreally, and then the ischemic model was created. The last group’s intraocular pressure was not increased as in the control group, and only a cannula was introduced into the anterior chamber through the cornea. Six animals from each group were subjected to histomorphometry, and four were subjected to immunohistochemical and histopathologic examinations. For a histomorphometric examination, the number of cells in the retinal ganglion cell (RGC) layer was counted using the optical dissector method. For immunohistochemistry, the vascular endothelial growth factor receptor-2 (VEGFR-2) levels and apoptosis were examined in the retinal and choroidal tissue.
Results
It was observed that in an IR injury, bevacizumab reduces the death and apoptosis of cells in the RGC layer. It was also identified that although bevacizumab is a large molecule, the agent affects the choroid and reduces the amount of VEGFR-2 in this tissue.
Conclusions
IR may be used as a model of ischemic retinopathy that includes VEGF-dependent vascular permeability and neurodegeneration. Although VEGF is a neurotrophic molecule, in IR injury, treatment with bevacizumab, which is an anti-VEGF agent, decreases apoptosis, showing that excess function of this molecule can be hazardous.
Introduction
Macular edema and retinal neovascularization are associated with visual loss in several diseases, such as ischemic retinopathies (including diabetic retinopathy and retinal vascular occlusive diseases). Although vascular components are the most obvious pathology of these diseases, neurodegeneration and some degree of inflammation also contribute to the pathogenesis [1,2]. The pathology in diabetic retinopathy is related not only to vascular problems but also to microglial activation, increased expression of inflammatory cytokines, adherence of leukocytes to retinal microvasculature, and death of neurons [3-6]. There is a strong connection between vascular, inflammatory, and neuronal components in the pathogenesis of these retinopathies. It has been hypothesized that by causing leukocytosis, subsequent microvascular occlusion, increased vascular endothelial growth factor (VEGF), and inflammation contribute to the development of vascular abnormalities resulting in retinal ischemia, which becomes the initiating event in neurodegeneration and neovascularization [5-9].
VEGF, a neuroprotective molecule, might also be secreted as an adaptive response to inflammation, in turn creating vascular permeability and angiogenesis. VEGF is one of the main causes of neovascularization and increased vessel permeability associated with many retinopathies. VEGF antagonism can diminish edema and neovascularization in ischemic and inflammatory retinopathies. This has been shown in clinical trials and case studies [10,11]. However, there is a concern that the inhibition of VEGF, a neuroprotective molecule, might cause neurodegeneration [12]. In one clinical trial, the constant blocking of VEGF caused the death of retinal ganglion cells (RGCs) [13]. Nonetheless, in other trials the opposite was shown, and animal studies suggest this risk is minimal [14-16].
Bevacizumab (BEV) is a human monoclonal recombinant antibody that attaches specifically to VEGF and binds all isoforms of VEGF-A. BEV is the first anti-VEGF molecule used in clinical settings. BEV prevents the VEGF molecule from binding to its receptor on endothelial cells. BEV has two antigen-binding sites and weighs 140 kDa. It is frequently used in ophthalmology with off-label status for the treatment of retinal diseases. Repeated clinical trials have shown that 0.125 mg of BEV is effective with minimal side effects, and subsequent injections can be 4 or 5 weeks apart due to the antibody’s long half-life [17].
The model of ischemia and reperfusion (IR) by increasing intraocular pressure includes temporary ischemia followed by reperfusion, which causes inflammation and degeneration in the retina. This procedure is a model of the neuronal damage in transient retinal vessel occlusion. In most IR studies, ischemia lasting from 45 to 120 min is employed, and the retinal function and histological changes are observed after 7 to 10 days of reperfusion; an electroretinogram (ERG) shows reduced a- and b-waves, indicating a decrease in neuronal function, and the reduced thickness of the retinal layers, including the ganglion cell layer (GCL), inner nuclear layer (INL), and inner plexiform layer (INP), are shown by histological analysis after IR [18-21]. Terminal deoxynucleotidyl transferase-mediated dUTP Nick-end labeling (TUNEL) of the retina after ischemia shows neuronal apoptosis in all retinal layers. One study also mentioned that IR might be a model for diabetic retinopathy. The study suggested the hypothesis that diabetic retinopathy damage occurring to the vasculature, in turn, damages the retina is not entirely correct; the neuronal retina is damaged first. This, in turn, damages the vasculature and augments the pathology. The results were coherent with the study hypothesis, showing there was a loss in the neuronal retina but no apparent damage to the vasculature 2 days after ischemia. The number of degenerated vascular cells was observed only after 7 and 8 days [22].
In this study, we investigated the effects of an intravitreal injection of BEV on the retina in a model of IR. The retinal cells were counted, and the apoptotic index was calculated in the GCL. The VEGFR-2 level was analyzed qualitatively in the RGC layer and in the choroid. The results demonstrated that although decreased VEGFR-2 and subsequent action of VEGF due to BEV had no adverse effect on the retina, VEGFR-2 reduced apoptosis and ganglion cell death compared to the other group that did not receive BEV. This study demonstrates that although VEGF is a neurotrophic factor when it is secreted in high amounts in response to ischemia, VEGF might become harmful. Thus, reducing it with an antagonist in ischemic situations helps to limit the extent of the pathology.
Methods
Animal model
In total, 18 Wistar albino rats weighing 200–300 g were used. The rats were obtained from the Yeditepe University Experimental Animal Laboratory, Istanbul, Turkey, were housed under a constant light-dark cycle, were fed ad libitum with normal rat chow, and were given free access to water. Animals were treated in accordance with the guidelines of the Yeditepe University Laboratory Animals Ethical Committee (Permission No: 184) and in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Visual Research.
VEGF antibody BEV
A pharmaceutical-grade formulation of humanized VEGF antibody BEV (25 mg/ml solution: Avastin; Genentech Inc., South San Francisco, CA) was used. The drug was injected intravitreally (0.005 ml/eye) with a 30-gauge needle 48 h before IR or the sham treatment.
Ischemia-reperfusion
Deep anesthesia was induced in the rats with an intraperitoneal injection of ketamine (100 mg/kg) and chlorpromazine (25 mg/kg). Ischemia was applied to the eye by increasing the intraocular pressure and thus, cutting off the blood supply from the retinal artery. Increased pressure was achieved through the introduction of sterile saline through a 30-gauge needle that was inserted into the anterior chamber of the eye through the cornea. The sterile saline reservoir was raised to a 100-cm height to achieve a 74 mmHg pressure in the anterior chamber, and the reservoir was maintained at that height for 45 min. The measurement of intraocular pressure was conducted using a TonoPen XL tonometer (Mentor, Inc., Norwell, MA) calibrated according to the manufacturer’s instructions. The retina was monitored for blanching, indicating loss of blood flow. After 45 min, the saline reservoir was lowered, allowing for natural eye reperfusion for 48 h. Reperfusion was confirmed by observing the retina returning to its original color. Sham-treated eyes were treated by briefly inserting a 30-gauge needle into the anterior chamber of the eye through the cornea.
The rats were divided into three groups of six animals each: Group 1 was the control group with the sham treatment and was euthanized after 48 h. Group 2 was the IR group but did not receive BEV and was euthanized 48 h after IR. Group 3 received 0.125 mg (0.005 ml) of BEV (BEV) 48 h before IR and was euthanized 48 h after IR.
Euthanasia and cardiac perfusion tissue processing
Euthanasia was achieved by thoracotomy and intracardiac perfusion of 2.5% glutaraldehyde in a cacodylate buffer solution (0.1 M cacodylic acid sodium salt, pH 7.4) under deep anesthesia with an intramuscular injection of ketamine hydrochloride (100 mg/kg) and chlorpromazine (25 mg/kg). Right eyes of all rats were enucleated after euthanasia and were used for histological evaluation. The eye was fixed with immersion in 10% neutral formaldehyde in 0.1 M PBS (140 mM NaCl, 10 mM Phosphate Buffer, 3 mM KCl, pH 7.4) at 4 °C for about 48 h. Dehydration was performed by treatment with a graded ethanol series (50%, 70%, 80%, 96%, and 100% ethanol) followed by treatment with 125 xylene, and the eye from each animal was embedded in paraffin wax. A paraffin-embedded 20-μm-thick section was cut from each block in six animals.
Stereological analysis
The nuclei numbers of cells in the RGC layer per eye were estimated using the optical fractionator. This technique is composed of an optical dissector with the fractionator sampling design [23].
Equipment, delineation of the region of interest and cell counting
The total nuclei number of cells in the RGC layer was counted using the Stereo Investigator version 7.5 (MicroBrightField, Colchester, VT) and observed on a computer screen connected to a Leica DM 4000 microscope (Leica, Weltzlar, Germany). The x and y planes had a stage that moved a motorized connected joystick. To move the z-axis, the focus button on the microscope was used manually. This axis distance was measured using the Heidenhain electronic microcator (Heidenhain, Traunreut, Germany). A general examination of the sections was observed using a 4X objective lens, but the counting of the nuclei was performed using a 63X Plan Apo objective (NA = 1.40).
Sampling
The total nuclei number of cells in the RGC layer was counted using an unbiased estimation with systematic random sampling of 1 to 10. All of the retinal tissue was included in the counting. In focusing continually to a depth of 16 µm in the tissue section, a 2-μm zone at the uppermost part of the section was counted at every step as the upper guard zone. The mean thickness was a thickness-sampling fraction, which was 16 μm/t.
Estimate of total cell numbers
The total nuclei number (N) of cells in the RGC layer was estimated, as described by West et al. [23,24]. The equation in this study was **n = ΣQ- × 1/ hsf × 1 / asf × 1 / ssf [1]. ΣQ- is the total number of cells in the RGC layer counted in all optically sampled fields of the retina (ssf is the section sampling fraction [1/10], asf is the area sampling fraction [900/40,000], and hsf is the height sampling fraction [defined by dissector height (16 μm)] separated by the estimated mean section thickness).
Eye histology
Paraffin-embedded eye blocks from six animals were inspected for histological procedures. A paraffin-embedded 20-μm-thick section was cut from each block. We used poly-L-lysine-coated slides for these sections.
The eye sections were deparaffinized in xylene and rehydrated with descending amounts in an ethanol series (100%, 96%, 80%, 70%, and 50%). Then, they were brought to the distillated water. Sections were immersed in filtered Harris hematoxylin (St Louis, MO) for 1 min. Then, the sections were rinsed with tap water until the water was clear. They were immersed in an eosin stain for 5 min, and the eye slides were washed in tap water until the water was clear. Next, the sections were dehydrated by ascending into ethanol solutions (50%, 70%, 80%, 95% × 2, and 100% × 2). After dehydration, the slides were cleared with xylene. The sections were mounted on a labeled glass slide with Permount. (Milan, Italy) The sections were analyzed with stereological rules.
Immunohistochemical processing
Paraffin-embedded eye blocks in six animals for each group were inspected for two immunohistochemistry procedures. In total, 36 serial 20-μm-thick sections of paraffin-embedded blocks were inspected. First, to detect apoptotic cells, 18 sections were chosen for the in situ cell death detection kit, POD (Roche Diagnostics; Mannheim, Germany) according to the instructions supplied by the manufacturer. The other 18 sections were used for the VEGF Receptor 2 (Cell Signaling; Danvers, MA).
In situ cell death detection
The staining procedure was performed according to the manufacturer’s instructions. Twenty-micrometer-thick eye sections were deparaffinized in xylene and rehydrated in ethanol. The sections were washed with PBS (0.1 mM, pH 7.2) and were incubated with detergent (Triton X-100 [Acros Organics; Geel, Belgium] was employed at 0.1% [v/v] in 0.1% [w/v] sodium citrate [Merck; Darmstadt, Germany] for 4 min on ice, and then, the slides were washed twice in PBS at room temperature [RT]). Next, the sections were kept in the citrate buffer (retrieval solution) in a 750 W microwave (Bosch HMT 812C domestic oven, Stuttgart, Germany, operating at a frequency of 2.45 GHz with five power-level settings) at 45 s to 1 min, and the eye slides were then quickly immersed in PBS at RT (rapid cooling). The slides were placed in a humidified container and incubated with a Ready to Use (R.T.U.) VECTASTAIN Universal Kit (Vector Labs; Burlingame, CA, Cat No: PK 7800) in double-distillated water for 30 min at RT. Then, a terminal deoxynucleotidyl transferase-mediated dUTP Nick-end labeling (TUNEL) kit Label 1 (Stuttgart, Germany) was applied to the sections and incubated for 60 min in a humidified container and then in a 37 °C incubator. After incubation, the slides were washed twice with PBS for 5 min each time. This time, the sections were treated with a POD converter (horseradish peroxidase) for 30 min in a humidified container and then in the 37 °C incubator. The slides were washed using the same procedure. They were stained with 0.05% 3,3′-diaminobenzidine tetrahydrochloride chromogen concentrate and diaminobenzidine substrate buffer (Thermo Scientific, Waltham,MA). Next, they were counterstained with Mayer’s hematoxylin (Darmstadt, Germany).
In situ cell death detection and cell counting
The staining procedure was performed according to the manufacturer’s instructions. Twenty micrometer-thick eye sections were deparaffinized in xylene and rehydrated in ethanol. The sections were washed with PBS (0.1 mM, pH 7.2) and were incubated with detergent (Triton X- 100 [Acros Organics; Geel, Belgium] was employed at 0.1% [v/v] in 0.1% [w/v] sodium citrate [Merck; Darmstadt, Germany] for 4 min on ice, and then, the slides were washed twice in PBS atroom temperature [RT]). Next, the sections were kept in the citrate buffer (retrieval solution) in a750 W microwave (Bosch HMT 812C domestic oven operating at a frequency of 2.45 GHz with five power-level settings) at 45 s to 1 min, and the eye slides were 182 then quickly immersed in PBS at RT (rapid cooling). The slides were placed in a humidified container and incubated with a Ready to Use (R.T.U.) VECTASTAIN Universal Kit (Vector Labs; Burlingame, CA, Cat No: PK 7800) in double-distillated water for 30 min at RT. Then, a terminal deoxynucleotidyl transferase-mediated dUTP Nick-end labeling (TUNEL) kit Label 1 was applied to the sections and incubated for 60 min in a humidified container and then in a 37 °C incubator. After incubation, the slides were washed twice with PBS for 5 min each time. This time, the sections were treated with a POD converter (horseradish peroxidase) for 30 min in a humidified container and then in the 37 °C incubator. The slides were washed using the same procedure. They were stained with 0.05% 3.3′-diaminobenzidine tetrahydrochloride chromogen concentrate and diaminobenzidine substrate buffer (Thermo Scientific, Waltham, MA). Next, they were counterstained with Mayer’s hematoxylin. The apoptotic, normal cells were marked using three different markers in each sampling frame.
Immunohistochemical labeling was scored considering the intensity and distribution of specific staining. Intensity of staining was reported based on H-score method, using 0 for negative staining, 1+ for weak staining, 2+ for moderate staining, and 3+ for strong staining. From these semiquantitative estimates of immunostaining, the H-score was derived according to a modification of a previously reported method [23]. The following formula produces an H-score in the range of 0–300, where 300 equals 100% of NF-κB positive cells stained strongly (i.e., 3+): H-score = (% of cells stained at intensity category 1 × 1) + (% of cells stained at intensity category 2 × 2) + (% of cells stained at intensity category 3 × 3) [25].
Statistical analysis
All data are expressed as means ± standard error of the mean (SEM). Parametric test assumptions were available for the total cell number in the RGC layer and the apoptotic index of the RGC layer of the eye. They were analyzed with one-way ANOVA, and then multiple comparisons between the pairs of groups were performed according to Tukey’s test. The sections were stained for VEGFR-2 and scored accordingly, and then they were analyzed with a non-parametric Mann–Whitney U test. The SPSS version 18.0 system for PC (SPSS, Chicago, IL) was used, and a p value of less than 0.05 was considered statistically significant.
Results
Stereological analysis for ganglion cell count
The neurodegenerative effects of IR and the neuroprotective effects of BEV were examined by inducing 45 min of ischemic insult and following reperfusion for 48 h. There were 203.712±27.7490 cells in the RGC layer in the control group and 131.133±16.0250 cells in the IR group, showing a 36% decrease in living cells as a strong sign of neurodegeneration (p<0.0001). When we compared the cell numbers in the BEV group (173.289±5.33600) to the IR group (131.133±16.0250), we clearly saw there was a higher number of cells in the BEV group, and the difference was statistically significant (p<0.01). This is a strong clue that BEV has a neuroprotective effect in the event of ischemia (Figure 1, Figure 2, and Table 1).
Figure 1.

Stereological analysis of the rat retina demonstrates surviving cells in the ganglion cell layer. The control group has the highest number of living cells as expected since they did not receive any manipulation. The important data in this graph is that the bevacizumab (BEV) group has more living cells than the ischemia and reperfusion (IR) group. This is an important finding that shows bevacizumab is neuroprotective in the event of ischemia and reperfusion. *p<0.01, **p<0.001 and ***p<0.0001. Error bars are median ± standard error of the mean (SEM), where n=6.
Figure 2.

Comparison of histological cross sections of the control, IR, and BEV groups. White arrows indicate the cells at the retinal ganglion cell layer. Staining: Hematoxylin and eosin. Scale bar: 50 µm. C = control; IR = ischemia and reperfusion; BEV = bevacizumab.
Table 1. Total cell count in RGCL and optical dissector parameters.
| Groups name | Control n=6 | IR n=6 | Bevacizumab n=6 |
|---|---|---|---|
| Mean Total Retinal Ggl Cell Count ± SEM |
203.712±27.749 |
131.133±16.025 |
173.289±5.336 |
| Dissector Particle Number |
446 |
319 |
393 |
| Section Thickness (µm) |
20,5 |
19,5 |
19,7 |
| Number of Sampled Sections |
25,2 |
23 |
24 |
| CE |
0,05 |
0,05 |
0,05 |
| CV | 0,09 | 0,14 | 0,09 |
Mean total ganglion cell count ±SEM, mean dissector number, section thickness, number of sampled sections, coefficient of error (CE), and coefficient of variation (CV) of stereological analysis between subjected to ischemia/reperfusion and treatment groups in the rodents. The stastical difference between all the groups is significant ; between C and IR p<0.0001, C and BEV p<0.01 and IR and BEV p<0.01. The neurodegenerative effects of IR and the neuroprotective effects of BEV were examined by inducing 45 min of an ischemic insult and following a reperfusion of 48 h. There were 203.712±27.749 cells in the RGCL in the control group and 131.133±16.025in the IR group, showing a 36% decrease in living cells as a strong sign of neurodegeneration (p<0.0001). When we compared the cell numbers in the BEV group 173.289±5.336to the IR group 131.133±16.025we clearly saw there was a higher number of cells in the BEV group, and the difference was statistically significant (p<0.01). This is a strong clue that BEV has a neuroprotective effect in the event of ischemia.
Immunohistochemical analysis for apoptosis
TUNEL-positive stained cells were counted only in the RGC layer. Apoptotic and normal, and total cell numbers were calculated separately for each group. The apoptotic index was calculated by dividing the apoptotic cell number by the total cell number. The median values for the control, IR, and BEV groups were 0.018±0.004, 0.647±0.0203, and 0.243±0.0203, respectively. The control group had the lowest apoptotic index compared to the other groups. The statistical difference between the control and IR groups (p<0.001) and the control and BEV groups (p<0.01) was statistically significant. When the BEV and IR groups were compared, the BEV group had a lower apoptotic index, and the difference was statistically significant (p<0.01). This result was another strong sign showing the neuroprotective effect of BEV in the event of ischemia (Figure 3, Figure 4, and Table 2).
Figure 3.
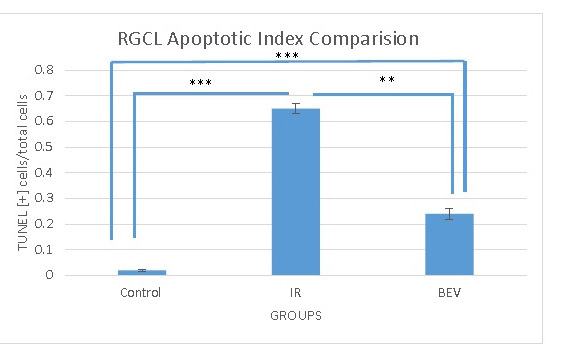
The apoptotic indices of the three groups. As expected, the lowest apoptosis is in the control group since they did not receive any insult. The important finding in this graph is the significantly lower apoptosis in the bevacizumab (BEV) group compared with the ischemia-reperfusion (IR) group. **p<0.01 and ***p<0.001. Error bars are median ± standard error of the mean (SEM), where n=6.
Figure 4.

Immunohistochemical evaluation of TUNEL-stained tissue samples: control, IR, and BEV. The black arrows point out the apoptotic ganglion cells in the retina layers. Scale bar: 50 µm. C = control; IR = ischemia and reperfusion; BEV = bevacizumab.
Table 2. Apoptotıc ındex of the experımental groups.
| Group name | Control (n=6) | BEV (n=6) | IR (n=6) |
|---|---|---|---|
| Mean apoptotic index % (TUNEL [+] cells/total cells) ±SEM | 0.018±0.004 | 0.243±0.203 | 0.647±0.212 |
The comparison of apoptotic indices of control, bev and IR groups. Among the groups that were manipulated IR group has higher apoptotic index compared to the BEV group. This is an indication showing the probable neuroprotective effect of bevacizumab. The statistical difference between all groups is significant; between C and IR p<0.001, C and BEV p<0.01 and IR and BEV p<0.01.
RGC layer VEGFR-2 analysis
The VEGFR-2 concentration scores in the RGC layer for the IR, BEV, and control groups were 53.3±2.97, 29.6±2.09, and 46.8±3.73, respectively. The differences between the groups were statistically significant. Given that VEGF is a neurotrophic factor for the retina, we wanted to determine whether the VEGFR-2 levels correlated with apoptosis and live cell numbers. However, these findings showed a negative correlation because as the VEGFR-2 levels increased, the number of living cells decreased, thus, increasing apoptosis. This result suggests that BEV by way of VEGF and VEGFR-2 antagonism might have an antiapoptotic effect (Figure 5 and Figure 6).
Figure 5.

VEGFR-2 staining intensity of the RGC layer. There are two important points in this graph. The first one is the high VEGF response to the ischemia and reperfusion and its significant suppression by bevacizumab (** p<0.01 and *** p<0.001). Data expressed as median ± standard error of the mean (SEM; n=6). BEV = bevacizumab; IR = ischemia-reperfusion.
Figure 6.

Immunohistochemical evaluation of VEGFR-2-stained tissue samples: C, IR, and BEV. VEGFR-2 staining intensity increased in the retina and choroid layers of the ischemia and reperfusion (IR) group, but this result was decreased in the bevacizumab (BEV) group. Stars indicate the choroid layer. C = control. Scale bar: 100 µm.
Choroid-level VEGFR-2 analysis
The VEGFR-2 concentration scores for the IR, BEV, and control groups were 47±2.28, 30.7±3.12, and 53.40±4.50, respectively. The differences between the groups were statistically significant. The results are the exact same as the results in the RGC layer; the only difference is that it is less suppressed in the choroid compared to the retina, which might indicate that BEV, because of its large size, penetrates the choroid less (Figure 6 and Figure 7).
Figure 7.

VEGFR-2 intensity of the choroid layer of the eye. Data expressed as median ± standard error of the mean (SEM; n=6). The response is the same as in the ganglion cell layer (GCL) as VEGFR-2 is suppressed by bevacizumab (BEV) statistically significantly (** p<0.01 and *** p<0.001). IR = ischemia and reperfusion.
Discussion
In this present study, we used pressure-induced retinal IR injury to model the pathology in an ischemic retinal disease. This is a useful model of the damage and responses that occur on a smaller scale during focal ischemic events caused by vessel dropout or vessel occlusion during diseases, such as diabetic retinopathy [22]. Although the vascular components are the most obvious pathology in ischemic diseases, various degrees of inflammation and neurodegeneration precede the vascular changes [1,2]. In this IR model, by increasing the ocular pressure, the blood supply to the retina is blocked because the pressure exceeds the systolic pressure of the retinal artery. Although not tested independently in this experimental model, it is reported in the literature that this model also causes neurodegeneration through the direct effect of increased intraocular pressure [26]. This neurodegeneration and subsequent inflammation cause microvascular occlusion, production of vasoactive compounds, such as VEGF, followed by vascular permeability increase and cell dropout. These events can initiate neovascularization [8,9]. Alternatively, adaptive responses to neurodegeneration and inflammation might cause the secretion of vasoactive compounds, causing neovascularization [10].
The retina responds to an ischemic insult with neurodegeneration and increased secretion of VEGF. VEGF contributes to vascular angiogenesis and permeability associated with many ischemic retinopathies [27]. In many clinical and case studies, it has been shown that VEGF antagonists reduce edema and neovascularization in ischemic and inflammatory retinopathies, supporting the previous statement [11,27]. VEGF is also a neurotrophic molecule, which is why it is likely that inhibition of this molecule might cause neurodegeneration [12,13,28]. In some animal studies, it has been observed that VEGF inhibition causes neurodegeneration [13]. Especially when used in high doses, VEGF antagonism creates a high degree of neurodegeneration, but in small doses, the toxic effects do not cause any important pathology [14,16,29]. In the present study, we used 0.005 ml (0.125 mg) of BEV intravitreally, which reduced cell death and apoptosis after the IR insult.
We also measured the VEGFR-2 amounts in the retina and the choroid to assess their responses to ischemic insult, as well as whether this affects apoptosis and cell death. VEGFR-2 levels increased mostly in the IR group. This finding is in accordance with other clinical studies in the literature, which show an increase in VEGF and VEGFR-2 expression [30,31]. In a clinical study of rats, after 90 min of ischemia, VEGF levels were found to be 2 times the normal level after 7 days [32]. In another study conducted with rats, VEGF mRNA and protein expression was increased after cerebral ischemia [33-35].
There is not much information in the literature about the role of VEGFR in IR. It has been shown that in some cerebral IR models, VEGF inhibition limits vascular permeability. In one study, the inhibition of VEGF receptor kinase reduced edema after cerebral ischemia [11]. In another study with an opposite result, Nishijima et al. mentioned that after optical sheath occlusion, VEGF receptor agonist infusion minimized neuronal death [13]. In a similar study, increasing the expression of VEGF through an adenovirus also reduced neuronal death [36]. Abocouwer et al. showed that BEV administered intravitreally in a retinal IR model did not change apoptosis but diminished vascular permeability [37]. In the present study, we found that BEV decreased the apoptosis of cells in the RGC layer. It has been extensively shown that BEV reduces vascular permeability. This reduction prohibits the excessive amount of oxygen during the reperfusion phase, which creates free oxygen radicals, thus reducing damage. In addition, when vascular permeability is limited, leukocyte infiltration is reduced, thus decreasing the inflammation that damages the tissue. It is also known that VEGF has different effects on different tissues. After cerebral ischemia, the VEGF expressed by microglial cells might have a neurotrophic effect [38], but the VEGF expressed after retinal ischemia might have a neurodegenerative effect [39].
Tsuchihashi et al. studied the effects of an anti-VEGF serum in an IR model of a rat liver [30]. The results showed that the group that received the anti-VEGF serum had less apoptosis, fewer proinflammatory cells, and more proteins that were antiapoptotic. The same study also showed that the anti-VEGF serum inhibits leukocytes migrating to the damage zone. The conclusion was that the anti-VEGF serum, being antiapoptotic, anti-inflammatory, and a leukocyte migration inhibitor, is neuroprotective in IR injuries. However, Tsuri et al. showed that in an IR injury of the liver, VEGF serum had cytoprotective effects [39]. Similar findings were observed in an animal model of myocardial ischemia. In light of these findings, it has been hypothesized that systematically given VEGF saturates leukocyte receptors and limits their responses to locally secreted VEGF, thus limiting the damage.
Another important finding of the present study regards tissue penetration by BE . Mordenti et al. showed that molecules larger than 70 kDa do not cross the internal limiting membrane (ILM) to the subretinal space [40]. In light of this study, we should assume that BEV, which is larger than 70 kDa, will be unable to go to the subretinal space when given intravitreally. However, in the medical literature it has been shown that BEV can penetrate the ILM and go into the subretinal space. The reasons behind this finding might be the increased permeability of the ILM with old age, the high dosage used in ILM permeability studies, and the increased permeability in the macula compared to the peripheral retina. In the present study, we found that BEV reduced the amount of VEGFR-2 in the retina and the choroid, indirectly showing that VEGFR-2 penetrates both tissues. We also observed that the reduction in the level of VEGFR-2 in the choroid was lower than the level in the retina. In our opinion, this suggests the amount of BEV that reached the choroid was smaller than the amount that reached retina. The one reason behind this finding is the retina being a semipermeable barrier for BEV [41].
Anti-VEGF therapies are commonly used to treat retinopathies [42]. Clinical studies of diabetic macular edema BEV and ranibizumab increased vision more when compared with laser therapies [43]. Similar results were obtained in studies of central or branch vein occlusion [44-48]. Although in some animal studies repeated VEGF injections caused the death of cells in the RGC layer [13], other studies have been unable to demonstrate this adverse effect [15,16]. In human trials, no significant adverse effects were observed [14].
The present study demonstrated that VEGF function contributes to the pathology in retinal IR and thus, suggests the IR model can be a useful tool in the study of VEGF-induced retinopathies. The finding that the inhibition of VEGF activity by BEV decreases apoptosis after retinal IR further supports the conclusion that this treatment is not neurodegenerative; however, it might be neuroprotective. These data are highly important regarding the concerns that treatments, including blocking VEGF and VEGFR-2 expression, might cause neuronal degeneration by blocking neurotrophic effects. The present study results also suggest that starting anti-VEGF therapies earlier, such as before any significant vascular pathology, might be beneficial to reducing the neuronal damage in ischemic diseases, such as diabetic retinopathy, and the IR model might prove useful for preclinical testing of treatments for such diseases.
FUNDING
This research was conducted using departmental support, no external funding was used.
References
- 1.Stem MS, Gardner TW. Neurodegeneration in the pathogenesis of diabetic retinopathy: molecular mechanisms and therapeutic implications. Curr Med Chem. 2013;20:3241–50. doi: 10.2174/09298673113209990027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen QD, Tatlipinar S, Shah SM, Haller JA, Quinlan E, Sung J, Zimmer-Galler I, Do DV, Campochiaro PA. Vascular endothelial growth factor is a critical stimulus for diabetic macular edema. Am J Ophthalmol. 2006;142:961–9. doi: 10.1016/j.ajo.2006.06.068. [DOI] [PubMed] [Google Scholar]
- 3.Zeng HY, Green WR, Tso MO. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. 2008;126:227–32. doi: 10.1001/archophthalmol.2007.65. [DOI] [PubMed] [Google Scholar]
- 4.Adamis AP, Berman AJ. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol. 2008;30:65–84. doi: 10.1007/s00281-008-0111-x. [DOI] [PubMed] [Google Scholar]
- 5.Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, Kester M, Kimball SR, Krady JK, LaNoue KF, Norbury CC, Quinn PG, Sandirasegarane L, Simpson IA, JDRF Diabetic Retinopathy Center Group Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55:2401–11. doi: 10.2337/db05-1635. [DOI] [PubMed] [Google Scholar]
- 7.Chibber R, Ben-Mahmud BM, Chibber S, Kohner EM. Leukocytes in diabetic retinopathy. Curr Diabetes Rev. 2007;3:3–14. doi: 10.2174/157339907779802139. [DOI] [PubMed] [Google Scholar]
- 8.Yeung L, Lima VC, Garcia P, Landa G, Rosen RB. Correlation between spectral domain optical coherence tomography findings and fluorescein angiography patterns in diabetic macular edema. Ophthalmology. 2009;116:1158–67. doi: 10.1016/j.ophtha.2008.12.063. [DOI] [PubMed] [Google Scholar]
- 9.Kaur C, Foulds WS, Ling EA. Blood-retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res. 2008;27:622–47. doi: 10.1016/j.preteyeres.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Jardeleza MS, Miller JW. Review of anti-VEGF therapy in proliferative diabetic retinopathy. Semin Ophthalmol. 2009;24:87–92. doi: 10.1080/08820530902800330. [DOI] [PubMed] [Google Scholar]
- 11.Lott MN, Schiffman JC, Davis JL. Bevacizumab in inflammatory eye disease. Am J Ophthalmol. 2009;4:4. doi: 10.1016/j.ajo.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saint-Geniez M, Maharaj AS, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, Darland DC, Young MJ, D’Amore PA. Endogenous VEGF is required for visual function: evidence for a survival role on muller cells and photoreceptors. PLoS One. 2008;3:e3554. doi: 10.1371/journal.pone.0003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, Shima DT. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziemssen F, Lüke M, Messias A, Beutel J, Tatar O, Zrenner E, Bartz-Schmidt KU, Tuebingen Bevacizumab Study Group Safety monitoring in bevacizumab (Avastin) treatment: retinal function assessed by psychophysical (visual fields, colour vision) and electrophysiological (ERG/EOG) tests in two subgroups of patients. Int Ophthalmol. 2008;28:101–9. doi: 10.1007/s10792-007-9122-1. [DOI] [PubMed] [Google Scholar]
- 15.Cheng CK, Peng PH, Tien LT, Cai YJ, Chen CF, Lee YJ. Bevacizumab is not toxic to retinal ganglion cells after repeated intravitreal injection. Retina. 2009;29:306–12. doi: 10.1097/IAE.0b013e3181909404. [DOI] [PubMed] [Google Scholar]
- 16.Ueno S, Pease ME, Wersinger DM, Masuda T, Vinores SA, Licht T, Zack DJ, Quigley H, Keshet E, Campochiaro PA. Prolonged blockade of VEGF family members does not cause identifiable damage to retinal neurons or vessels. J Cell Physiol. 2008;217:13–22. doi: 10.1002/jcp.21445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manzano Rp. Peyman Ga, Khan P, Kivilcim M. Testing Intravitreal Toxicity of Bevacizumab (Avastin). Retina. 2006;26:257–61. doi: 10.1097/00006982-200603000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Jehle T, Wingert K, Dimitriu C, Meschede W, Lasseck J, Bach M, Lagrèze WA. Quantification of ischemic damage in the rat retina: a comparative study using evoked potentials, electroretinography, and histology. Invest Ophthalmol Vis Sci. 2008;49:1056–64. doi: 10.1167/iovs.07-1050. [DOI] [PubMed] [Google Scholar]
- 20.Özden S, Müftüoğlu S, Tatlıpınar S, Kaymaz F, Yıldırım C, Yaylalı V, Ozbay D. Protective effects of antithrombin III on retinal ischemia/reperfusion injury in rats: a histopathologic study. Eur J Ophthalmol. 2005;15:367–73. [PubMed] [Google Scholar]
- 21.Konno T, Sato A, Uchibori T, Nagai A, Kogi K, Nakahata N. Adenosine A2A receptor mediated protective effect of 2-(6-cyano- 1-hexyn-1-yl)adenosine on retinal ischaemia/reperfusion damage in rats. Br J Ophthalmol. 2006;90:900–5. doi: 10.1136/bjo.2006.091496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007;48:361–7. doi: 10.1167/iovs.06-0510. [DOI] [PubMed] [Google Scholar]
- 23.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–97. doi: 10.1002/ar.1092310411. PMID: 1793176. [DOI] [PubMed] [Google Scholar]
- 24.West MJ, Ostergaard K, Andreassen OA, Finsen B. Estimation of the number of somatostatin neurons in the striatum: an in situ hybridization study using the optical fractionator method. J Comp Neurol. 1996;370:11–22. doi: 10.1002/(SICI)1096-9861(19960617)370:1<11::AID-CNE2>3.0.CO;2-O. PMID: 8797153. [DOI] [PubMed] [Google Scholar]
- 25.Abueid L, Uslu Ü, Cumbul A, Velioğlu Öğünç A, Ercan F, Alican İ. Inhibition of 5-lipoxygenase by zileuton in a rat model of myocardial infarction. Anatol J Cardiol. 2016;17:269–75. doi: 10.14744/AnatolJCardiol.2016.7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison JC, Johnson EC, Cepurna W, Jia L. Understanding mechanisms of pressure-induced optic nerve damage. Prog Retin Eye Res. 2005;24:217–40. doi: 10.1016/j.preteyeres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Özbay D, Özden S, Müftüoğlu S, Kaymaz F, Yaylalı V, Yıldırım C, Tatlipinar S. Protective effects of ischemic preconditioning on retinal ischemia-perfusion injury in rats. Can J Ophthalmol. 2004;39:727–32. [PubMed] [Google Scholar]
- 28.Philipp W, Speicher L, Humpel C. Expression of vascular endothelial growth factor and its receptors in inflamed and vascularized human corneas. Invest Ophthalmol Vis Sci. 2000;41:2514–22. [PubMed] [Google Scholar]
- 29.Ruiz de Almodovar C, Lambrechts D, Mazzone M, Carmeliet P. Role and therapeutic potential of vegf in the nervous system. Physiol Rev. 2009;89:607–48. doi: 10.1152/physrev.00031.2008. [DOI] [PubMed] [Google Scholar]
- 30.Tsuchihashi S, Ke B, Kaldas F, Flynn E. Busuttil Rw, Briscoe Dm, Kupiec-Weglinski JW. Vascular endothelial growth factor antagonist modulates leukocyte trafficking and protects mouse livers against ischemia/reperfusion injury. Am J Pathol. 2006;168:695–705. doi: 10.2353/ajpath.2006.050759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozaki H, Ay Yu, Della N, Ozaki K, Luna JD, Yamada H, Hackett SF, Okamoto N, Zack DJ, Semenza GL, Campochiaro PA. Hypoxia inducible factor-1alpha is increased in ischemic retina: Temporal and spatial correlation with vegf expression. Invest Ophthalmol Vis Sci. 1999;40:182–9. [PubMed] [Google Scholar]
- 32.Adamis AP, Miller JW, Bernal MT, D’Amico DJ, Folkman J, Yeo TK, Yeo KT. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol. 1994;118:445–50. doi: 10.1016/s0002-9394(14)75794-0. [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto K, Khosrof S, Bursell SE, Moromizato Y, Aiello LP, Ogura Y, Adamis AP. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (Icam-L). Am J Pathol. 2000;156:1733–9. doi: 10.1016/S0002-9440(10)65044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamoreaux WJ. Fitzgerald Me, Reiner A, Hasty Ka, Charles St. Vascular endothelial growth factor increases release of gelatinase A and decreases release of tissue inhibitor of metalloproteinases by microvascular endothelial cells Invitro. Microvasc Res. 1998;55:29–42. doi: 10.1006/mvre.1997.2056. [DOI] [PubMed] [Google Scholar]
- 35.Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and schwann cell proliferation in the peripheral nervous system. J Neurosci 1999; 19: 5731–40. 61. [DOI] [PMC free article] [PubMed]
- 36.Shen F, Su H, Fan Y, Chen Y, Zhu Y, Liu W, Young WL, Yang GY. Adeno-associated viral-vector-mediated hypoxia-inducible vascular endothelial growth factor gene expression attenuates ischemic brain injury after focal cerebral ischemia in mice. Stroke. 2006;37:2601–6. doi: 10.1161/01.STR.0000240407.14765.e8. [DOI] [PubMed] [Google Scholar]
- 37.Abcouwer Sf, Lin Cm, Wolpert Eb, Shanmugam S. Schaefer Ew, Freeman Wm, Barber AJ, Antonetti DA. Effects of ischemic preconditioning and bevacizumab on apoptosis and vascular permeability following retinal ischemia-reperfusion injury. Invest Ophthalmol Vis Sci. 2010;51:5920–33. doi: 10.1167/iovs.10-5264. [DOI] [PubMed] [Google Scholar]
- 38.Plate Kh, Beck H, Danner S. Allegrini Pr, Wiessner C. Cell type specific upregulation of vascular endothelial growth factor in a mca-occlusion model of cerebral infarct. J Neuropathol Exp Neurol. 1999;58:654–66. doi: 10.1097/00005072-199906000-00010. [DOI] [PubMed] [Google Scholar]
- 39.Ogata N, Yamanaka R, Yamamoto C, Miyashiro M, Kimoto T, Takahashi K, Maruyama K, Uyama M. Expression of vascular endothelial growth factor and its receptor, kdr, following retinal ischemia-reperfusion injury in the rat. Curr Eye Res. 1998;17:1087–96. doi: 10.1076/ceyr.17.11.1087.5234. [DOI] [PubMed] [Google Scholar]
- 40.Tsurui Y, Sho M, Kuzumoto Y, Hamada K, Akashi S, Kashizuka H, Ikeda N, Nomi T, Mizuno T, Kanehiro H, Nakajima Y. Dual role of vascular endothelial growth factor in hepatic ischemia-reperfusion injury. Transplantation. 2005;79:1110–5. doi: 10.1097/01.tp.0000161627.84481.5e. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen QD, Shah SM, Tatlıpınar S, Do D, Van Anden E, Campochiaro PA. Bevacizumab suppresses choroidal neovascularization due to pathologic myopia. Br J Ophthalmol. 2005;•••:891368–70. [PMC free article] [PubMed] [Google Scholar]
- 42.Ta C, Pj R. Anti-vascular endothelial growth factor therapy for neovascular ocular diseases other than age-related macular degeneration. Curr Opin Ophthalmol. 2009;20:166–74. doi: 10.1097/ICU.0b013e328329d173. [DOI] [PubMed] [Google Scholar]
- 43.Soheilian M, Ramezani A, Obudi A, Bijanzadeh B, Salehipour M, Yaseri M, Ahmadieh H, Dehghan MH, Azarmina M, Moradian S, Peyman GA. Randomized trial of intravitreal bevacizumab alone or combined with triamcinolone versus macular photocoagulation in diabetic macular edema. Ophthalmology. 2009;116:1142–50. doi: 10.1016/j.ophtha.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen QD, Shah SM, Heier JS, Do DV, Lim J, Boyer D, Abraham P, Campochiaro PA, READ-2 Study Group Primary end point (six months) results of the ranibizumab for edema of the macula in diabetes (Read-2) study. Ophthalmology. 2009;21:21. doi: 10.1016/j.ophtha.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 45.Rabena Md, Pieramici Dj, Castellarin Aa. Nasir Ma, Avery Rl. Intravitreal bevacizumab (Avastin) in the treatment of macular edema secondary to branch retinal vein occlusion. Retina. 2007;27:419–25. doi: 10.1097/IAE.0b013e318030e77e. [DOI] [PubMed] [Google Scholar]
- 46.Iturralde D, Spaide RF, Meyerle CB, Klancnik JM, Yannuzzi LA, Fisher YL, Sorenson J, Slakter JS, Freund KB, Cooney M, Fine HF. Intravitreal bevacizumab (Avastin) treatment of macular edema in central retinal vein occlusion: A short-term study. Retina. 2006;26:279–84. doi: 10.1097/00006982-200603000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Stahl A, Agostini H, Hansen Ll, Feltgen N. Bevacizumab in retinal vein occlusion-results of a prospective case series. Graefes Arch Clin Exp Ophthalmol. 2007;245:1429–36. doi: 10.1007/s00417-007-0569-6. [DOI] [PubMed] [Google Scholar]
- 48.Costa Ra, Jorge R, Calucci D. Melo La Jr, Cardillo Ja, Scott Iu. Intravitreal bevacizumab (Avastin) for central and hemicentral retinal vein occlusions: Ibevo study. Retina. 2007;27:141–9. doi: 10.1097/IAE.0b013e31802eff83. [DOI] [PubMed] [Google Scholar]
- 49.Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008;27:331–71. doi: 10.1016/j.preteyeres.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mordenti J, Cuthbertson RA, Ferrara N, Thomsen K, Berleau L, Licko V, Allen PC, Valverde CR, Meng YG, Fei DT, Fourre KM, Ryan AM. Comparisons of the intraocular tissue distribution, pharmacokinetics, and safety of 125 L–Labeled full-Iength and Fab antibodies in Rhesus monkeys following intravitreal administration. Toxicol Pathol. 1999;27:536–44. doi: 10.1177/019262339902700507. [DOI] [PubMed] [Google Scholar]