

Abstract
A decline in cognitive ability is a typical feature of the normal aging process, and of neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s diseases. Although their etiologies differ, all of these disorders involve local activation of innate immune pathways and associated inflammatory cytokines. However, clinical trials of anti-inflammatory agents in neurodegenerative disorders have been disappointing, and it is therefore necessary to better understand the complex roles of the inflammatory process in neurological dysfunction. The dietary phytochemical curcumin can exert anti-inflammatory, antioxidant and neuroprotective actions. Here we provide evidence that curcumin ameliorates cognitive deficits associated with activation of the innate immune response by mechanisms requiring functional tumor necrosis factor α receptor 2 (TNFR2) signaling. In vivo, the ability of curcumin to counteract hippocampus-dependent spatial memory deficits, to stimulate neuroprotective mechanisms such as upregulation of BDNF, to decrease glutaminase levels, and to modulate N-methyl-D-as-partate receptor levels was absent in mice lacking functional TNFRs. Curcumin treatment protected cultured neurons against glutamate-induced excitotoxicity by a mechanism requiring TNFR2 activation. Our results suggest the possibility that therapeutic approaches against cognitive decline designed to selectively enhance TNFR2 signaling are likely to be more beneficial than the use of anti-inflammatory drugs per se.
Keywords: Curcumin, Tumor necrosis factor α, Cognition, Inflammation, Lipopolysaccharide
Introduction
The activation of innate immune cells in the central nervous system (CNS), principally microglia, and subsequently the generation of cytokines [e.g. tumor necrosis factor α (TNF-α), interleukin (IL)-1β, and IL-6] and reactive oxygen species are major components of the neuroinflammatory response to injury, infection and neurodegenerative disease [1]. Increasing evidence suggests that local inflammatory processes contribute to neuronal dysfunction and associated neurological symptoms in neurodegenerative disorders, stroke, traumatic brain injury and HIV dementia [2–4]. However, the assumption that pro-inflammatory molecules exert only detrimental effects on CNS function has been challenged by evidence that cytokines play roles in cognition and synaptic plasticity [5]. For example, it was recently reported that IL-1β is essential for physiological regulation of memory [6]. Genetic ablation or blockage of the IL-1β receptor impairs long-term potentiation and hippocampus-dependent memory [7, 8], while lL-1β administration improves avoidance memory and contextual fear conditioning [7]. Mice deficient in IL-6 display impaired hippocampus-dependent and -independent memory [9]. TNF-α, tumor necrosis factor α receptor 1 (TNFR1) and TNFR2 knockout mice all exhibit learning and memory retention impairment, with a more pronounced phenotype in mice devoid of TNF-α signaling [5, 10]. These findings indicate that the role played by glia and neuroinflammatory molecules is far more complex than was previously appreciated.
Although neuroinflammation is associated with neuropathologies and cognitive decline, clinical trials using anti-inflammatory drugs have not been encouraging. For example, although epidemiological data suggested that nonsteroidal anti-inflammatory drugs may reduce the risk for Alzheimer’s disease (AD) [11–13], large randomized, placebo-controlled clinical trials using both steroidal and nonsteroidal drugs yielded negative results [14] and in a few cases accelerated progression of the disease [15, 16].
The therapeutic use of natural compounds in Western medicine has increased as a result of controlled studies substantiating beneficial effects of some such natural products in experimental disease models [17, 18]. Curcumin, the principal polyphenol of the spice turmeric (Curcuma longa), exhibits anti-inflammatory, antioxidant, antitumor, metal-chelating, neuroprotective and antiamyloidogenic properties [19]. Because of its pleiotropic actions [20] and potential ability to preserve cognitive performance in the elderly [21], we assessed the effect of curcumin on cognitive impairment elicited by activation of toll-like receptor 4 (TLR4), a prominent component of the innate immune system, by bacterial lipopolysaccharide (LPS). TLR4 plays important roles in the response of CNS cells to infection and tissue injury [22], and is also implicated in local inflammatory processes in neurodegenerative diseases [23]. Here we show that curcumin alleviates LPS-induced cognitive impairment by a mechanism requiring TNF-α signaling via TNFR2.
Animals and Methods
Animals and Treatments
Adult 12- to 14-week-old male TNFR1 and TNFR2 double knockout (DKO) and control background (C57BL/6J) (WT) mice were kept under a 12-hour light/12-hour dark cycle and allowed free access to food and water. All treatments were administered between 8: 00 and 11: 00 AM. Each animal received 50 mg/kg of curcumin (Sigma-Aldrich, St. Louis, Mo., USA) intraperitoneally for 4 days followed by a single 250 μg/kg LPS (O111:B4) (Sigma-Aldrich) dose 2 h after the last curcumin administration. The LPS and curcumin doses used in this study have been reported to induce memory impairment and neuroprotection, respectively, in murine experimental models [24, 25]. Based on the experimental end point, mice were euthanized either 4 h or 7 days after LPS administration. This research was performed according to guidelines in the NIH Guide for the Care and Use of Laboratory Animals.
Morris Water Maze
The test was performed as previously described [26]. Mice were trained in a circular tank containing water (22 ± 1°C) rendered opaque by the addition of nontoxic paint (Palmer Paints Products Inc., Mich., USA). Spatial visual clues were provided in the form of differently shaped objects on the walls of each quadrant. An escape platform (10 × 10 cm) was submerged approximately 1 cm below the surface of the water, 10 cm from the edge of the tank at a position designated as quadrant 3 (target quadrant). The swimming path length was monitored with a Videomex tracking system, and data were collected using Videomex Water Maze Software (Columbus Instruments, Ohio, USA). The training period consisted of four 60-second trials per day, with each mouse released into the water at a different quadrant for each trial. Learning retention (spatial memory) was measured in two probe trials at 4 and 24 h after the last training session. For both probe trials, the platform was removed and mice were allowed to swim freely in the tank for 60 s.
Open Field Test
Mice were placed in the center of an open field apparatus (40 × 40 cm), and ambulation scores (the number of squares crossed) and the number of rearings and peepings were measured during a 15-min period over 3 days after LPS treatment.
Rotarod
Motor skills were tested using the ENV-577M Rotarod system (Med Associates, Georgia, Vt., USA). The time spent on the rod and the number of falls from the rotating rod was measured for each animal during 300-second trials (the speed of rod rotation was increased progressively from 3 to 30 rpm during the 300-second period).
Fear Conditioning
Mice were habituated to the testing room for 5 consecutive days. During the training session, mice were placed in a contextual conditioning chamber (model MED-VFC-NIR-M; Med Associates) and allowed to explore for 2 min. The mice were subjected to 3 sessions of a 30-second neutral discrete stimulus (conditioned stimulus, audio tone 6 kHz, 70 dB), followed by 2 s of a motivationally significant stimulus (unconditioned stimulus; foot shock, 0.5 mA). Each session of conditioned and unconditioned stimulus pairings was separated by 30 s. On the following day during the contextual fear session, mice were returned in the same conditioning cage for 5 min without any stimulus. In the cued session, mice were placed in the chamber in a different context and allowed to explore for 5 min before being subjected to 5 audio tones over a 5-min period. The percentage of time freezing was recorded in both sessions and used as an index of either contextual or cued memory.
Primary Cultures and Experimental Treatments
Primary hippocampal and cortical neuronal cells were prepared as described [27]. For all in vitro experiments cells were pretreated for 24 h with vehicle dimethyl sulfoxide or 10 μM curcumin and then exposed to 200 μM glutamate. Recombinant TNFR2 (1 μg/ml) (A.G. Scientific, San Diego, Calif., USA) and 1 μg/ml TNFR2 neutralizing antibody (Santa Cruz Biotechnology, Calif., USA) or control IgG were added at 24 h and 30 min before glutamate challenge, respectively. Cell survival was assessed 24 h after addition of glutamate using a CellTiter 96 AQueous Assay System (Promega, Madison, Wisc., USA) and lactate dehydrogenase activity assay (Roche, Nutley, N.J., USA).
Protein Extraction and Immunoblot Analysis
Tissue, whole-cell extracts and nuclear fractions were prepared as described [28]. The primary antibodies used in this study were: Actin, NR1 and RelA (Sigma-Aldrich); EAAT2 and pSer897-NR1 (Cell Signaling, Danvers, Mass., USA); hnRNP C1/C2, TNFR1 and TNFR2 (Santa Cruz Biotechnology); EAAT3 (Abbiotec, San Diego, Calif., USA); GFAP (AbCam, Cambridge, Mass., USA); GluR1 (Upstate, Lake Placid, N.Y., USA); pSer845GluR1 (Millipore, Billerica, Mass., USA), and NOS (BD Transduction, Franklin Lakes, N.J., USA).
RNA Extraction and Real-Time PCR
RNA was isolated using Trizol (Invitrogen) and purified with an RNA Micro Kit (Qiagen, Valencia, Calif., USA). Real-time PCR analysis was performed with a PTC 200 Pelthier Thermo Cycler and Chromo 4 Fluorescent Detector (BioRad, Hercules, Calif., USA), and Sybr® Green PCR Master Mix according to the manufacturer’s instructions (Applied Biosystems, Foster City, Calif., USA). Each reaction included 3 μl of diluted (1: 4) cDNA and was performed in duplicate. The comparative Ct method was used to determine the normalized changes of the target gene relative to a calibrator reference. The genes analyzed in this study were: mBDNF (I:NM_007540, II:NM_001048139, III:NM_001048141, IV:NM_001048142), mGLS1 (NM_001081081), mHPRT (NM_013556), and mTrkB (NM_001025074).
Statistical Analysis
Data are expressed as mean and SEM. Statistical comparisons were performed by analysis of variance followed by post hoc Newman-Keuls test, using a Prism software package (Graphpad Software, San Diego, Calif., USA).
Results
Curcumin Ameliorates LPS-Induced Memory Impairment
The experimental design with regard to timing of administration of curcumin and LPS is shown in figure 1a. Mice pretreated with vehicle or curcumin were administered one dose of vehicle or LPS to activate immunocompetent brain cells [24]. The effect of curcumin and LPS administration on learning and memory was evaluated using the water maze test. No differences were observed among any of the experimental groups in terms of latency to find the platform, swimming distance or swimming speed during training (online suppl. fig. 1a–c; for all online suppl. material, see www.karger.com/doi/10.1159/000336074), nor in the probe trial performed 4 h after the last training section (fig. 1b). Thus all animals were able to learn the location of the platform, and showed comparable short-term memory retention. However, LPS-treated mice displayed impaired long-term memory retention, as indicated by a significantly reduced time spent in the correct quadrant compared to control mice when tested in the probe trial 24 h after training (fig. 1c). These findings suggest that curcumin pretreatment ameliorates a memory consolidation deficit induced by LPS. Although the comparable performances during the learning and the first probe trials argued against any interference due to LPS-induced motor function impairment, we further evaluated the motor skills of the mice using a rotarod apparatus. LPS-treated mice displayed motor problems during the first 2 days after treatment, with a reduced time spent on the rotating rod (fig. 1d) and an increased number of falls (fig. 1e). Their performance became indistinguishable from the other groups by 72 h after LPS administration (fig. 1d). Notably, curcumin pretreatment prevented the LPS-induced deficits in rotarod performance (fig. 1d, e), as well as the anxiety-like behaviors exhibited by the LPS-treated mice in the open field test (fig. 1f, g). Similarly to what was observed in the Morris water maze, all animals equally learned to associate a tone with an electric shock in the fear conditioning paradigm (online suppl. fig. 1d), and performed identically during the amygdala-dependent cued test (online suppl. table 1). However, in the hippocampus-dependent contextual fear paradigm the LPS-treated mice froze less than the control group, an effect again ameliorated by curcumin pretreatment (fig. 1h). Collectively, our results suggest that LPS causes cognitive impairment, specifically a deficit in long-term memory retention, which can be ameliorated by a short-term curcumin pretreatment.
Fig. 1.
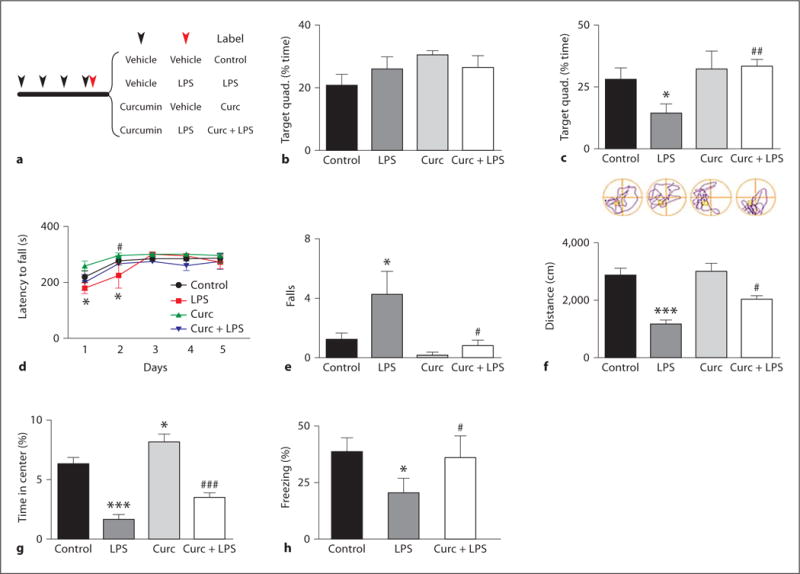
Curcumin ameliorates LPS-induced memory impairment. a Experimental design. Mice were treated with either vehicle or curcumin for 4 consecutive days (black arrow), followed by a single injection of vehicle or LPS (red arrow). Mice were trained in the Morris water maze for 5 consecutive days. The time spent in the target quadrant is expressed as percentage of the total time for the probe trial performed 4 h (b) and 24 h (c) after training (n = 10). Beginning 24 h after LPS injection, mice were tested daily for motor performance on a rotarod, the latency to fall (d) and the total number of falls during the testing period (e) are shown (n = 10). Results of analysis of behavior in the open field test showing the total distance traveled (f) and time spent in the center of the open field (g) (n = 10). h Percentage of time spent freezing during the contextual phase of the fear conditioning paradigm (n = 4–5). * p < 0.05 versus control; *** p < 0.001 versus control; # p < 0.05 versus LPS; ## p < 0.01 versus LPS; ### p < 0.001 versus LPS. For colors, see online version.
Curcumin Reduces Peripheral and CNS Inflammation
Long-term curcumin administration has been shown to exert anti-inflammatory [29] and neuroprotective effects in several experimental models [30]. Because we opted for a short-term pretreatment, we examined the extent of inflammation by measuring pro-inflammatory markers in the serum and hippocampus at early (4 h) and late (7 days) time points after drug administration. LPS caused a significant increase of TNF-α and IL-1β levels which was prevented by curcumin pretreatment administered either systemically (fig. 2a, b) or in the brain (fig. 2c, d). In the hippocampus, the protein levels of nitric oxide synthase (NOS), glial fibrillary acidic protein (GFAP), and the nuclear translocation of RelA (NF-κB activation) were elevated in response to LPS, and curcumin prevented the latter inflammatory cell responses (fig. 2f). One week after LPS exposure, the levels of TNF-α, IL-1β and NOS returned to basal levels (online suppl. table 2), while markers of NF-κB and glial cell activation remained significantly elevated only in the LPS-treated group (fig. 2e).
Fig. 2.
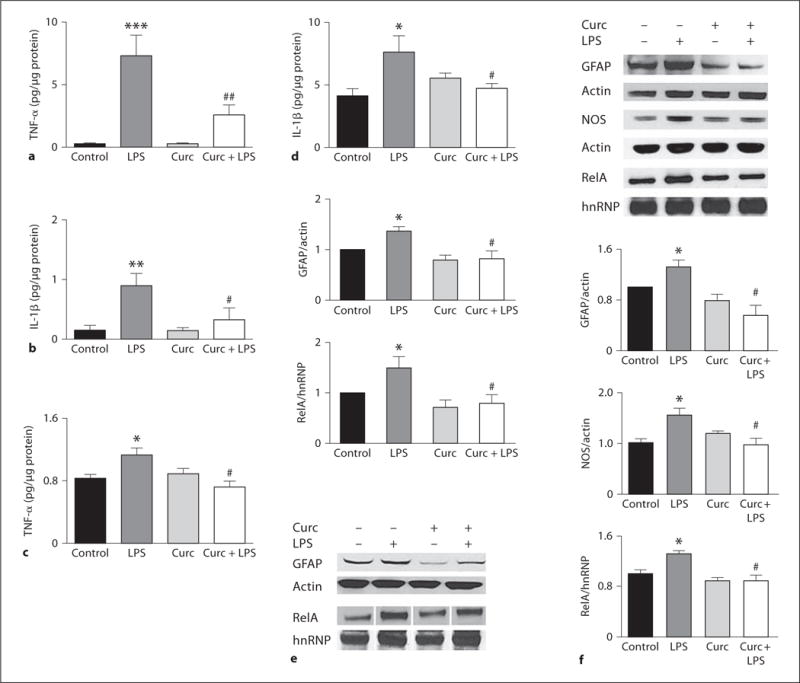
Effects of LPS and curcumin on pro-inflammatory markers. Levels of TNF-α and IL-1β were measured 4 h after treatment in serum (a, b) and in hippocampal extracts (c, d) by ELISA (n = 10). Representative immunoblots and densitometric analysis of multiple blots showing relative levels of GFAP, NOS, and nuclear RelA in hippocampi of mice killed either 4 h (f) or 7 days (e) after treatment. Levels of β-actin and hnRNP were used as internal loading controls for whole-cell extracts and purity of the nuclear fraction, respectively (n = 10). * p < 0.05 versus control; ** p < 0.01 versus control; *** p < 0.001 versus control; # p < 0.05 versus LPS; ## p < 0.01 versus LPS.
Curcumin Requires TNF-α Signaling to Prevent the Adverse Effects of LPS on Cognitive Function
Cytokines can influence cognition, and TNF-α is considered a key cytokine in the pro-inflammatory process [31]. To investigate the role played by TNF-α signaling in the LPS-induced cognitive impairment and curcumin protection, we performed behavioral testing using a TNFR1 and TNFR2 DKO mouse model which is completely devoid of TNF-α signaling [32]. The effects of LPS on rotarod performance in DKO mice were comparable to those observed in WT mice with decreased latency time to fall (fig. 3a) and increased number of falls (data not shown) during the first 2 days after treatment, and full recovery by the third day. However, in DKO mice, curcumin failed to prevent LPS-induced sickness (fig. 3a). As recently reported [33], in the open field, DKO mice were overall less anxious than WT mice, spending more time in the center of the field (WT, C = 6.26 ± 0.61%; DKO, C = 15.8 ± 1.56%). LPS treatment diminished the distance traveled regardless of the type of pretreatment (fig. 3b). During the nonspatial visible platform variant of the water maze, DKO mice spent most of the time floating in the pool, and swam toward the visible platform only when prompted. This lack of motivation agrees with the open field observation of an anxiolytic effect of TNFR deficiency, yet prevented us from testing hippocampus-dependent memory in the water maze. During the learning and cued portions of the fear conditioning test, no differences were observed between groups (online suppl. table 1), while in the contextual phase LPS caused a significant decrease in freezing which was not modified by curcumin pretreatment (fig. 3c).
Fig. 3.
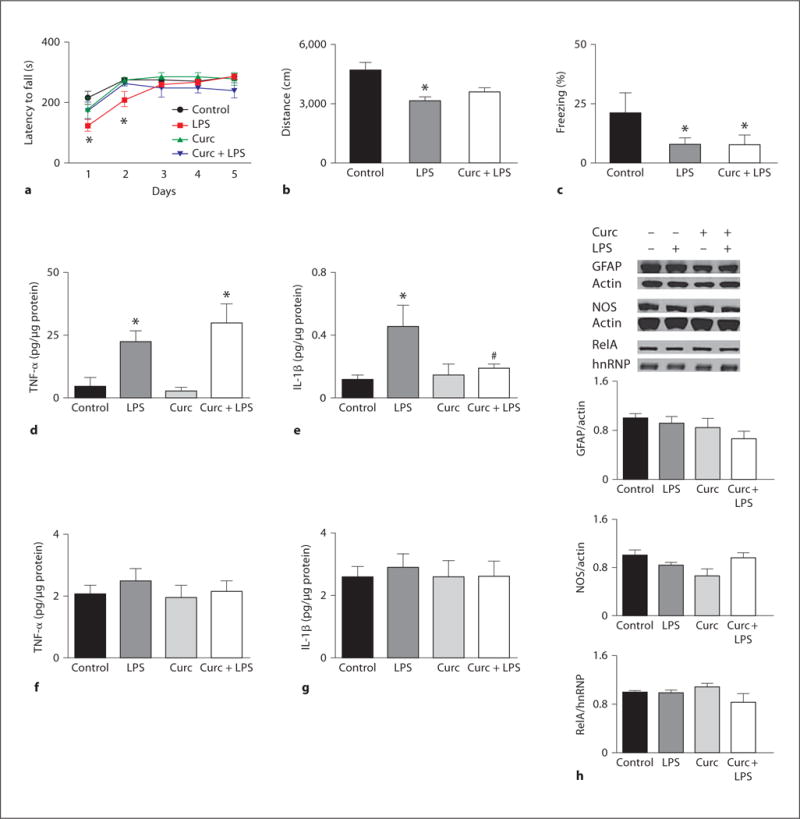
Amelioration of LPS-induced behavioral impairment by curcumin requires TNF receptors. DKO mice were tested on various behavioral test following treatment with curcumin and/or LPS. a Latency to fall off the rotarod over a 5-day testing period. b Distance traveled in the open field 48 h after LPS injection. c Percentage of time spent freezing in the contextual phase of the fear conditioning test (n = 4–6). Levels of TNF-α and IL-1β were measured 4 h after treatments in serum (d, e) and hippocampal extracts (f, g) (n = 4–11). h Representative immunoblots and densitometric analysis of multiple blots showing relative levels of GFAP, NOS and nuclear RelA in the hippocampus of DKO animals treated as indicated (n = 4–6). * p < 0.05 versus control; # p < 0.05 versus LPS.
TNF-α Signaling Is Required for CNS LPS-Induced Inflammatory Responses but Is Dispensable for Peripheral Inflammatory Responses
The results of behavioral testing indicated that LPS-induced cognitive impairment does not require TNF-α signaling, while curcumin’s counteractive actions do. To better understand the latter findings in the context of cellular inflammatory signaling, we evaluated peripheral and CNS inflammatory responses to LPS in DKO mice. As previously reported [34], we found that compared to WT mice, DKO mice have higher basal serum levels of TNF-α (WT = 0.28 ± 0.07; DKO = 4.34 ± 3.55 pg/μg proteins). The serum levels of TNF-α and IL-1β were both increased by LPS administration with curcumin only preventing IL-1β upregulation (fig. 3d, e). These data demonstrate that in DKO mice the peripheral inflammatory response to LPS is not impaired, that curcumin anti-inflammatory properties are overall unchanged, and that the decreased TNF-α levels found in the curcumin + LPS group among WT mice are likely not the result of reduced TNF-α production. In DKO mice, hippocampal levels of TNF-α and IL-1β (fig. 3f, g), as well as GFAP, NOS and nuclear RelA (fig. 3h) were not significantly affected by LPS or curcumin, indicating that contrary to the systemic response, CNS glia activation and cytokine production in response to LPS depends upon TNF-α signaling. Taken together with the behavioral data, our findings suggest that: (1) pro-inflammatory cytokines may not be responsible for the LPS-induced cognitive impairment; (2) the ability of curcumin to prevent the action of LPS on behavior does not depend upon its anti-inflammatory properties, and (3) curcumin’s mechanism of action requires intact TNFR signaling.
Curcumin Modifies Glutamate Signaling
Glutamate signaling is essential for hippocampus-dependent spatial working memory [35]. However, high extracellular levels of glutamate and overactivation of glutamate receptors may result in sustained elevation of intracellular calcium levels, neuronal dysfunction and cell death [36]. Increased glutamate levels have been shown following LPS administration [37]. We tested how LPS and/or curcumin impacted glutamate signaling by measuring levels of glutaminase, glutamate transporters and glutamate receptors in hippocampal tissue samples from WT and DKO mice. Glutaminase (GLS1) is a mitochondrial enzyme essential for the production of glutamate present in neurons and glia [38]. GLS1 deregulation and increased glutamate production have been demonstrated in vitro for TNFα- and LPS-activated microglia cells [39–41]. We found an LPS-induced upregulation of hippocampal GLS1 transcript levels in both mouse strains, which was prevented by curcumin only in WT mice (fig. 4a, b).
Fig. 4.
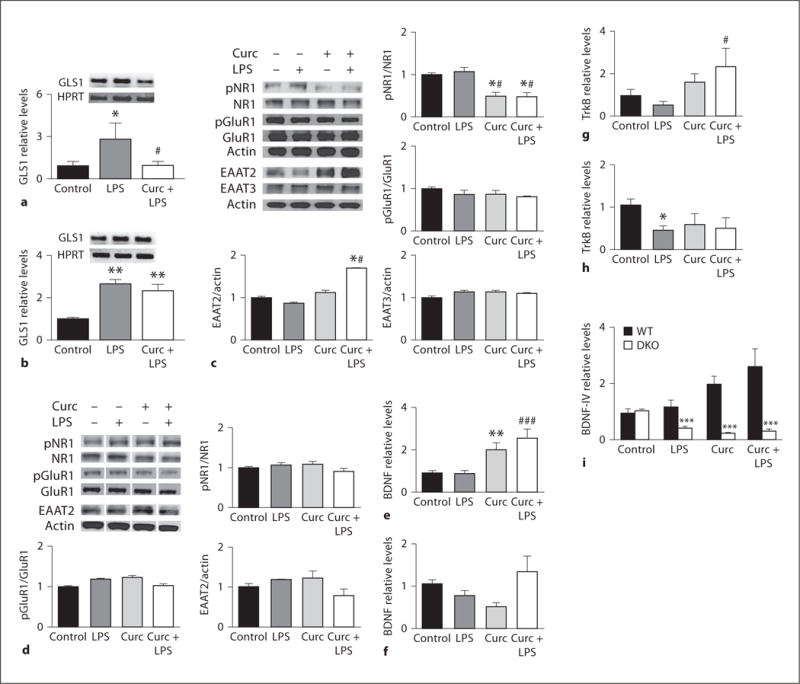
Curcumin modulates glutamate and BDNF signaling. Levels of glutaminase transcript were measured by quantitative RT-PCR in WT (a) and DKO (b) mice treated according to the experimental protocol and euthanized 4 h after LPS (n = 4–5). Results of immunoblot analysis of glutamate transporters (EAAT2 and EAAT3) and glutamate receptor subunits (GluR1 and NR1) in the hippocampus of WT mice (c) and DKO mice (d) (n = 3–10). Levels of mRNAs encoding BDNF and its receptor TrkB were measured by quantitative RT-PCR in samples from the hippocampus of WT mice (e, g) and DKO mice (f, h) (n = 3–6). * p < 0.05 versus control; ** p < 0.01 versus control; # p < 0.05 versus LPS; ### p < 0.001 versus LPS. i Relative levels of BDNF mRNA splice variants IV in hippocampal samples from WT and DKO mice. *** p < 0.001 compared to the same treatment in WT mice.
Excitatory amino acid transporters (EAATs) are responsible for the reuptake of glutamate from the synaptic cleft, thus playing a fundamental role in the termination of the glutamatergic signal and the preservation of the local integrity of excitatory synaptic transmission. We focused our analysis on EAAT2 and EAAT3. EAAT2 is almost exclusively expressed in glia, and is responsible for 90% of forebrain glutamate reuptake [42]. EAAT3 is the predominant post- and presynaptic neuronal transporter accounting for about 40% of hippocampal glutamate transport [43]. Levels of EAAT3 were unchanged, while levels of EAAT2 were significantly upregulated in WT mice that received curcumin or curcumin plus LPS (fig. 4c). In contrast, EAAT2 levels were not affected by curcumin in DKO mice (fig. 4d). EAAT2 levels were comparable in all groups and strains by day 7 (data not shown). The α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) subunit GluR1 and the N-methyl-D-aspartate (NMDA) subunit NR1 play important roles in learning and memory processes [44]. In our experimental model, neither LPS nor curcumin significantly affected hippocampal levels of GluR1 (fig. 4c), NR1 (fig. 4c), or NR2B (control = 1.0 ± 0.09; LPS = 1.0 ± 0.13; curcumin = 0.89 ± 0.08; curcumin + LPS = 0.98 ± 0.09). However, in WT mice, the phosphorylation of NR1 on Ser897 was reduced by more than 50% in all mice that received curcumin (fig. 4c). Changes in phosphorylation were specific for NR1 as demonstrated by the lack of changes in GluR1 phosphorylation levels (fig. 4c), and were absent in DKO mice (fig. 4d). Similarly to EAAT2, levels of phosphorylated NR1 returned to basal values by day 7 (data not shown). The phosphorylation of Ser897 of NR1 is associated with increased receptor sensitivity to glutamate and Ca2+ currents [45]. In WT mice, curcumin pretreatment may thus result in decreased amplitude of NMDA-driven calcium influx and possibly reduced neuronal dysfunction. In DKO mice, the combined increase of glutaminase levels, with normal EAAT2 levels, and NMDA and AMPA receptor activity suggests a scenario in which release of glutamate from the presynaptic terminal during the learning process may be exaggerated and possibly deleterious.
Curcumin Upregulates BDNF Signaling
BDNF plays an important role in learning and memory, anxiety behaviors and neuroprotection [46]. In our model, we found that in WT mice, the relative levels of BDNF mRNA were upregulated in response to curcumin treatment (fig. 4e), while TrkB mRNA levels were significantly elevated only in the curcumin + LPS group (fig. 4g). In DKO mice, total levels of BDNF mRNA were similar in all groups (fig. 4f), and there was a statistically significant reduction in TrkB mRNA levels in LPS-treated mice (fig. 4h). The transcriptional regulation of BDNF is quite complex and generates various transcripts from which an identical BDNF protein is produced. In the rodent brain, promoter IV accounts for the majority of the activity-dependent BDNF expression [47]. Interestingly, the most striking difference between WT and DKO mice concerned BDNF-IV, which exhibited opposite changes in expression in response to curcumin in WT mice (upregulation) and DKO mice (downregulation) (fig. 4i). Notably, BDNF-IV is also the only transcript that has been shown to be modulated by TNF-α [48].
Curcumin Modulates TNFR2 Levels, and TNFR2 Activation Mediates an Excitoprotective Action of Curcumin
TNF-α plays complex roles in the CNS depending upon which type of receptor is engaged [49]. Activation of TNFR1 signaling pathways is generally linked to apoptosis, and in microglia it promotes glutamate release facilitating excitotoxicity [39]. On the other hand, TNF-α signaling via TNFR2 has been shown to be neuroprotective and neurotrophic [50–52]. Curcumin failure to protect against LPS-induced changes in DKO mice prompted us to measure levels of TNFR1 and TNFR2 in the hippocampus of WT mice. Notably, TNFR2 was significantly upregulated early on in mice receiving curcumin + LPS (fig. 5a), returning to normal levels 1 week after LPS administration. To understand how curcumin-induced TNFR2 upregulation may impact neurons we moved to a cell culture system. Similarly to what was observed in vivo, TNFR1 levels were unaffected while TNFR2 levels were upregulated within 1 h in cells treated with curcumin and glutamate (fig. 5b). Curcumin pretreatment resulted in a significant decrease of glutamate-induced cell death in both hippocampal (fig. 5c, e) and cortical neurons (fig. 5d, f). To assess the role played by TNFR2 in curcumin-induced cell protection, we used two different approaches, soluble recombinant TNFR2 (rT2; a fusion protein containing the TNFR2 extracellular domain fused to the Fc of IgG, which acts as a decoy receptor to decrease levels of soluble TNF-α) and a TNFR2-neutralizing antibody. The addition of rT2 to the culture medium before glutamate exposure prevented neuronal cell death to an extent similar to curcumin (fig. 5g). On the other hand, the use of an antibody against the extracellular domain of TNFR2 exacerbated glutamate toxicity and completely nullified the protection afforded by curcumin (fig. 5h). These results are consistent with previous reports showing that TNFR2 signaling promotes neuronal survival [49, 52].
Fig. 5.
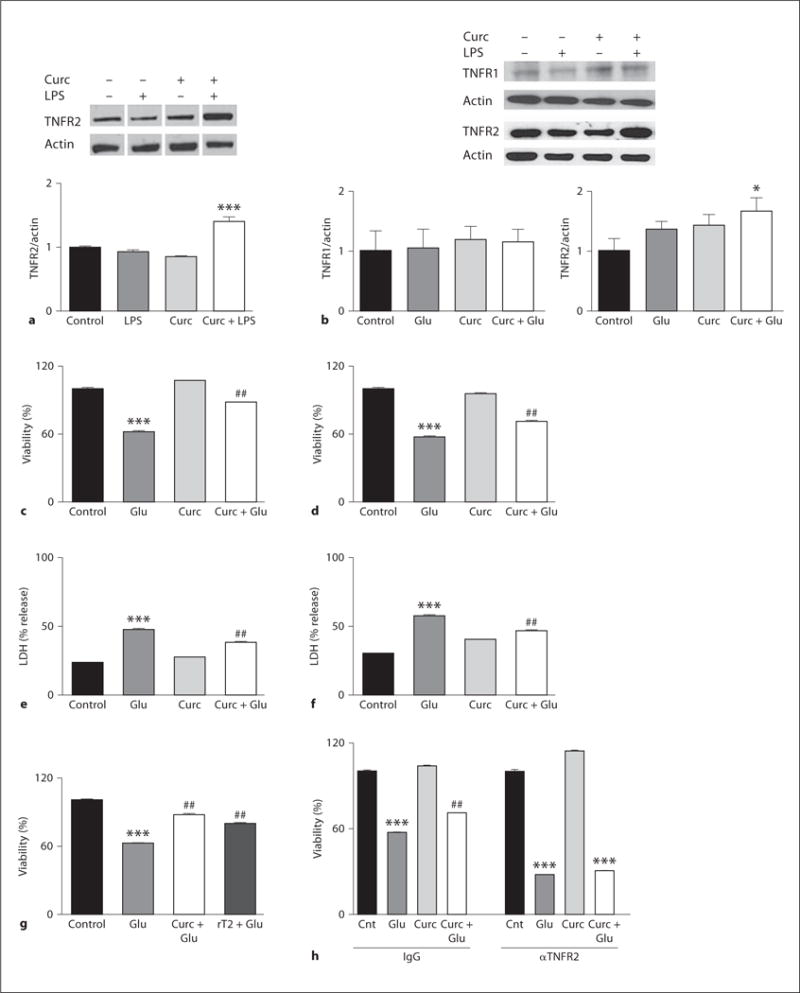
Curcumin upregulates TNFR2 and prevents glutamate-induced excitotoxicity. a Relative expression of TNFR2 in hippocampi from WT mice treated with LPS. b Primary neuronal cultures were incubated with vehicle or 10 μM curcumin (Curc) for 24 h, then challenged with 200 μM glutamate (Glu). Levels of TNFRs were measured 1 h after glutamate addition. Cell viability assessed by MTS assay (c, hippocampal and d, cortical), and lactate dehydrogenase (LDH) release assay (e, hippocampal and f, cortical). Survival of hippocampal neurons incubated with recombinant TNFR2 (rT2) (g) or anti-TNFR2 and IgG control (h) before glutamate challenge. All data are mean and SEM of 3–5 separate experiments. * p < 0.05 versus control; *** p < 0.001 versus control; ## p < 0.01 versus glutamate.
Discussion
Cognitive decline is associated with many neurodegenerative disorders, and neuroinflammation is believed to play a prominent role in the underlying synaptic dysfunction and neuronal degeneration [1]. However, in view of recent findings highlighting physiological roles for cytokines in the establishment and maintenance of learning and memory, the use of broad-spectrum anti-inflammatory therapies for chronic neurodegenerative disorders may be ineffective or even detrimental. An extensive array of health benefits attributed to curcumin has been identified in studies involving long-term supplementation (i.e., 4 weeks or longer) [53]. In contrast, there have been few studies to evaluate the prophylactic efficacy of short-term administration of curcumin. The data in the present study provide novel insight into the mechanism by which innate immune activation perturbs cognition, and how curcumin counteracts its adverse effects. Our findings suggest that: (1) cognitive impairment caused by activation of TLR4 is not mediated by TNF-α; (2) short-term administration of curcumin ameliorates cognitive deficits with a mechanism requiring TNFR signaling, and (3) curcumin-induced upregulation of TNFR2 is required to protect neurons from excitotoxicity.
Using LPS to elicit an immune response, we observed impairment of hippocampus-dependent memory which was efficiently prevented by a short-term supplementation with curcumin. The observed deficits were not the result of sickness behavior or learning differences amongst the experimental groups, but instead resulted from impaired memory consolidation. This result is in agreement with previous findings in rats, which the authors attributed to IL-1β upregulation [54]. However, our data strongly argue against an IL-1β-dependent neuroinflammatory mechanism. In fact, curcumin was able to normalize IL-1β levels in both WT and DKO mice, and hippocampus glial activation and cytokine levels were unchanged in DKO mice, in which LPS caused behavioral impairment even in the presence of curcumin. These observations suggest that LPS-induced cognitive impairment is not mediated by TNF-α signaling, and appears to involve a signaling pathway that does not require glial cell activation nor NF-κB-mediated cytokine production. TLRs are expressed in astrocytes, microglia and neurons wherein their activation can affect neuronal plasticity and survival [22, 26]. Specifically, TLR4 activation regulates neural progenitor cell self-renewal and neuronal differentiation [55], increases neuronal vulnerability to ischemia [22], regulates pain through the expression of nociceptin, and increases excitability of dorsal root ganglion neurons [56]. Also, TLR4 activation in parenchymal cells is alone sufficient to drive a typical acute-phase response to pathogens [57].
Our results highlight a new mechanism by which curcumin protects neurons against stress. We found that in the presence of a stressor curcumin induced the expression of TNFR2 both in vivo and in cultured neurons. This upregulation is fundamental for neuroprotection by curcumin because the ability of curcumin to ameliorate LPS-induced cognitive impairment and perturbations in the expression of glutaminase, NR1, EAAT2 and BDNF was absent in mice lacking TNFRs. Moreover, blockage of TNFR2 impaired curcumin-mediated neuroprotection, while addition of recombinant TNFR2 (which decreases soluble TNF-α/TNFR1 signaling) was as efficient as curcumin itself in protecting neurons against glutamate toxicity. We therefore propose that curcumin protects neurons against innate immune activation-induced cognitive impairment by triggering a shift from TNFR1- to TNFR2-mediated TNF-α signaling (fig. 6). Our model is supported by the recent observation that neuronal levels of TNFR2 are decreased while TNFR1 levels and its signaling are increased in the brains of AD patients compared to nondemented subjects [58]. TNFR1-mediated signaling has been shown to impair learning and memory in neurodegenerative mouse models [59]. Moreover, lack of TNFR2 exacerbates cognitive dysfunction after traumatic brain injury, while TNFR1 ablation ameliorates the cognitive deficit [60]. In addition, deletion of TNFR1 has been shown to decrease fear conditioning response [61]. Furthermore, while TNF-α does not seem to play a major role in acute plasticity, as shown by equivalent long-term potentiation responses in DKO and WT mice [62], it is required for homeostatic synaptic scaling and stabilization of neuronal network function [63]. TNF-α and BDNF have been shown to be important for maintaining synapses in a plastic state in which scaling can be expressed. TNFR1-dependent TNF-α signaling ‘scales up’ excitatory synapses by increasing cell surface AMPA receptors and reducing inhibitory GABAA receptors [62, 64]. However, TNFR1-dependent signaling also modifies AMPA receptor stoichiometry making the channels Ca2+ permeable and so rendering neurons more susceptible to excitotoxicity [62]. On the other hand, BDNF has been shown to inhibit neuronal excitability [63], and affords neuroprotection in animal models of a range of neurodegenerative disorders [46]. We found that, in the presence of LPS, curcumin modulates factors underlying synaptic scaling (TNF-α and BDNF) resulting in amelioration of cognitive deficits and enhanced neuronal resistance to excitotoxic damage.
Fig. 6.
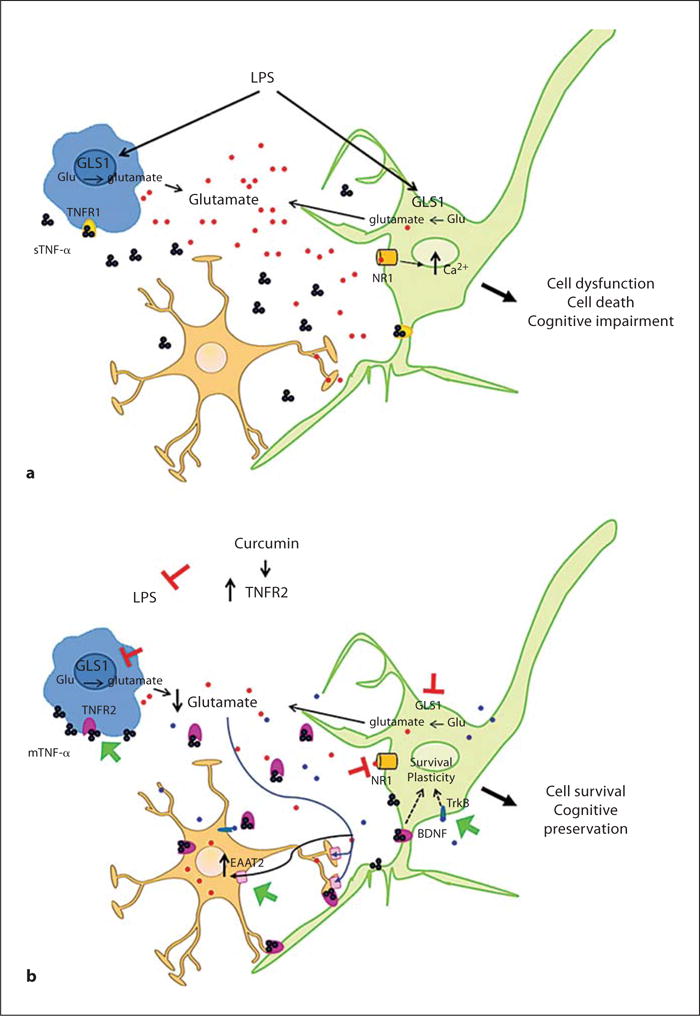
Schematic of the protective mechanisms of curcumin against innate immune-induced neuronal dysfunction. a LPS causes increased glutamate production and release (red dots) which may result in overstimulation of NMDA receptors, increased intracellular Ca2+ and neuronal dysfunction. b In the presence of curcumin, LPS induces an upregulation of TNFR2 (pink). TNFR2 may exert signaling via binding to transmembrane TNF-α (mTNF-α), and it can be shed resulting in decrease sTNF-α/TNFR1 (yellow) signaling. Changes in TNFR1/TNFR2 signaling are associated with a decrease of glutamatergic response through inhibition of glutaminase (GLS1) and NMDA receptor activity coupled with increased EAAT2 glutamate reuptake. Together with the enhancement of BDNF/TrkB (blue dots) signaling, these reactions appear to result in the preservation of neuronal functional integrity and cognitive performance. For colors, see online version.
Curcumin increased TNFR2 levels in the hippocampus of WT mice and protected against LPS-induced memory impairment, and these effects of curcumin were lost in DKO mice suggesting a fundamental role for a switch from TNFR1 to TNFR2 signaling to control neuronal network functionality in the mechanism of action of curcumin. TNF-α signaling is being pursued as a potential therapeutic target for neurodegenerative disorders. Among the different strategies developed to mod ulate TNF-α signaling are monomeric and dimeric (etanercept) TNFR2-Fc. Originally developed as TNF-α scavengers, it has become evident that their mechanism of action includes blockage of soluble TNF-α and induction of transmembrane TNF-α-TNFR2-mediated signaling [65]. Etanercept has been shown to increase transmembrane TNF-α [66], and to trigger transmembrane TNF-α reverse signaling that may induce cytokine suppression and/or other cellular events. While perispinal administration of etanercept improved cognitive performance in AD patients in two small nonrandomized studies [67], large controlled trials to test its efficacy have not yet been completed (www.clinicaltrials.org NCT01068353). Indirect evidence substantiating the validity of TNFR2-Fc in AD comes from a recent study demonstrating a 55% decreased incidence of AD in rheumatoid arthritis patients treated with etanercept versus those who received other drugs such as prednisone, sulfasalazine and rituximab [68]. While effective in several models of autoimmune disorders, a substantial drawback of the use of TNFR2-Fc is the high cost associated with the therapy.
Curcumin has been shown to ameliorate cognitive impairment in several animal models, and consumption of curry has been associated with lower risk of AD [69] and improved cognitive performance in elderly subjects [21]. However, large trials to determine the impact of curcumin on cognitive performance in neurodegenerative disorders have not yet been performed. The results of our study support the possible beneficial action of curcumin in treating cognitive impairment.
Supplementary Material
Acknowledgments
We thank E. Okun for advice on the behavioral testing procedures and S. Rothman for assistance in preparing the manuscript for publication. This research was supported by the Intramural Research Program of the National Institute on Aging of the National Institutes of Health, and by Program and Fundação de Amparo à Pesquisa do Estado de São Paulo-FAPESP.
References
- 1.Ownby RL. Neuroinflammation and cognitive aging. Curr Psychiatry Rep. 2010;12:39–45. doi: 10.1007/s11920-009-0082-1. [DOI] [PubMed] [Google Scholar]
- 2.Viviani B, Bartesaghi S, Corsini E, Galli CL, Marinovich M. Cytokines role in neurodegenerative events. Toxicol Lett. 2004;149:85–89. doi: 10.1016/j.toxlet.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 3.Anisman H, Merali Z, Hayley S. Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders. Prog Neurobiol. 2008;85:1–74. doi: 10.1016/j.pneurobio.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39:1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 5.McAfoose J, Koerner H, Baune BT. The effects of TNF deficiency on age related cognitive performance. Psychoneuroendocrinology. 2009;34:615–619. doi: 10.1016/j.psyneuen.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Yirmiya R, Goshen I. Immune modulation or learning, memory, neuronal plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Yirmiya R, Winocur G, Goshen I. Brain interleukin-1 is involved in spatial memory and passive avoidance conditioning. Neurobiol Learn Mem. 2002;78:379–389. doi: 10.1006/nlme.2002.4072. [DOI] [PubMed] [Google Scholar]
- 8.Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter-Levin G, Yirmiya R. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. 2003;13:826–834. doi: 10.1002/hipo.10135. [DOI] [PubMed] [Google Scholar]
- 9.Baier PC, May U, Scheller J, Rose-John S, Schiffelholz T. Impaired hippocampus-dependent and -independent learning in IL-6 deficient mice. Behav Brain Res. 2009;200:192–196. doi: 10.1016/j.bbr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Baune BT, Wiede F, Braun A, Golledge J, Arolt V, Koerner H. Cognitive dysfunction in mice deficient for TNF-α and its receptors. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1056–1064. doi: 10.1002/ajmg.b.30712. [DOI] [PubMed] [Google Scholar]
- 11.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 12.t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, Van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:515–521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- 13.Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imbimbo BP, Solfrizzi V, Panza F. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front Aging Neurosci. 2010 doi: 10.3389/fnagi.2010.00019. 2.pii:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, Assaid C, Nessly ML, Norman BA, Baranak CC, Reines SA, Rofecoxib Protocol 078 study group A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005;30:1204–1215. doi: 10.1038/sj.npp.1300690. [DOI] [PubMed] [Google Scholar]
- 16.ADAPT Research Group. Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65:896–905. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbaste M, Berke’ B, Dumas M, Soulet S, Delaunay JC, Castagnino C, Arnaudinaud V, Cheze C, Vercauteren J. Dietary antioxidants, peroxidation and cardiovascular risks. J Nutr Health Aging. 2002;6:209–223. [PubMed] [Google Scholar]
- 18.Gullett NP, Ruhul Amin AR, Bayraktar S, Pezzuto JM, Shin DM, Khuri FR, Aggarwal BB, Surh YJ, Kucuk O. Cancer prevention with natural compounds. Semin Oncol. 2010;37:258–281. doi: 10.1053/j.seminoncol.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Hatcher H, Planalp R, Cho J, Torti FM, Torti V. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci. 2008;65:1631–1652. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Son TG, Camandola S, Mattson MP. Hormetic dietary phytochemicals. Neuromolecular Med. 2008;10:236–246. doi: 10.1007/s12017-008-8037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng TP, Chiam PC, Lee T, Chua HC, Lim L, Kua EH. Curry consumption and cognitive function in the elderly. Am J Epidemiol. 2006;64:898–906. doi: 10.1093/aje/kwj267. [DOI] [PubMed] [Google Scholar]
- 22.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem. 2007;20:947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 24.Sparkman NL, Kohman RA, Scott VJ, Boehm GW. Bacterial endotoxin-induced behavioral alterations in two variations of the Morris water maze. Physiol Behav. 2005;86:244–251. doi: 10.1016/j.physbeh.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 25.Jagatha B, Mythri RB, Vali S, Bharath MM. Curcumin treatment alleviates the effects of glutathione depletion in vitro and in vivo: therapeutic implications for Parkinson’s disease explained via in silico studies. Free Radic Biol Med. 2008;44:907–917. doi: 10.1016/j.freeradbiomed.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Okun E, Griffioen K, Barak B, Roberts NJ, Castro K, Pita MA, Cheng A, Mughal MR, Wan R, Ashery U, Mattson MP. Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc Natl Acad Sci USA. 2010;107:15625–15630. doi: 10.1073/pnas.1005807107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, Moore TA, Luo W, Yu QS, Johnson DA, Johnson JA, Greig NH, Mattson MP. Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem. 2010;112:1316–1326. doi: 10.1111/j.1471-4159.2009.06552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camandola S, Cutler RG, Gary DS, Milhavet O, Mattson MP. Suppression of calcium release from nositol 1,4,5-trisphosphate-sensitive stores mediates the anti-apoptotic function of nuclear factor-kappaB. J Biol Chem. 2005;280:22287–22296. doi: 10.1074/jbc.M410923200. [DOI] [PubMed] [Google Scholar]
- 29.Jurenka JS. Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: a review of preclinical and clinical research. Altern Med Rev. 2009;14:141–153. [PubMed] [Google Scholar]
- 30.Ray B, Lahiri DK. Neuroinflammation in Alzheimer’s disease: different molecular targets and potential therapeutic agents including curcumin. Curr Opin Pharmacol. 2009;9:434–444. doi: 10.1016/j.coph.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 31.Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 2007;18:335–343. doi: 10.1016/j.cytogfr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 33.Patel A, Siegel A, Zalcman SS. Lack of aggression and anxiolytic-like behavior in TNF receptor (TNF-R1 and TNF-R2) deficient mice. Brain Behav Immun. 2010;24:1276–1280. doi: 10.1016/j.bbi.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gourine AV, Leon LR, Rudolph K, Tesfaigzi J, Kluger MJ. Cytokine cascade induced by endotoxin in TNF double receptor knockout mice: evidence supporting a role for IL-10 in mediating antipyretic action of TNF. J Therm Biol. 2000;25:21–27. [Google Scholar]
- 35.Reisel D, Bannerman DM, Schmitt WB, Deacon RM, Flint J, Borchardt T, Seeburg PH, Rawlins JN. Spatial memory dissociations in mice lacking GluR1. Nat Neurosci. 2002;5:868–873. doi: 10.1038/nn910. [DOI] [PubMed] [Google Scholar]
- 36.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1998;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 37.Huang WT, Niu KC, Chang CK, Lin MT, Chang CP. Curcumin inhibits the increase of glutamate, hydroxyl radicals and PGE2 in the hypothalamus and reduces fever during LPS-induced systemic inflammation in rabbits. Eur J Pharmacol. 2008;593:105–111. doi: 10.1016/j.ejphar.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 38.Holcomb T, Taylor L, Trohkimoinen J, Curthoys NP. Isolation, characterization and expression of a human brain mitochondrial gluataminase cDNA. Brain Res Mol Brain Res. 2000;76:56–63. doi: 10.1016/s0169-328x(99)00331-9. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- 40.Yawata I, Takeuchi H, Doi Y, Liang J, Mizuno T, Suzumura A. Macrophage-induced neurotoxicity is mediated by glutamate and attenuated by glutaminase inhibitors and gap junction inhibitors. Life Sci. 2008;82:1111–1116. doi: 10.1016/j.lfs.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 41.Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 43.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 44.Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Hugarin RL. Phosphorylation of the AMPA receptor GluR1 is required for synaptic plasticity retention of spatial memory. Cell. 2003;12:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 45.Maldve RE, Zhang TA, Ferrani-Kile K, Schreiber SS, Lippmann MJ, Snyder GL, Fienberg AA, Leslie SW, Gonzales RA, Morrisett RA. DARPP-32 and regulation of the ethanol sensitivity of NMDA receptors in the nucleus accumbens. Nat Neurosci. 2002;5:641–648. doi: 10.1038/nn877. [DOI] [PubMed] [Google Scholar]
- 46.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann NY Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tao X, West AE, Chen WG, Corfas G, Greenberg ME. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron. 2002;33:383–395. doi: 10.1016/s0896-6273(01)00561-x. [DOI] [PubMed] [Google Scholar]
- 48.Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol. 2006;1:212–222. doi: 10.1007/s11481-006-9020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontaine V, Mahand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 51.Yang F, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marchetti L, Schlett K, Pflzenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279:32869–32881. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- 53.Epstein J, Sanderson IR, MacDonald TT. Curcumin as a therapeutic agent: the evidence from in vitro, animal and human studies. Br J Nutr. 2010;103:1545–1557. doi: 10.1017/S0007114509993667. [DOI] [PubMed] [Google Scholar]
- 54.Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging. 2006;27:723–732. doi: 10.1016/j.neurobiolaging.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 55.Rolls A, Shechter RR, London A, Zv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 56.Wadachi R, Hargreaves KM. Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. J Dent Res. 2006;85:49–53. doi: 10.1177/154405910608500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen X, Libang Y, He P, Li R. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J Alzheimers Dis. 2010;19:621–630. doi: 10.3233/JAD-2010-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Longhi L, Ortolano F, Zanier ER, Perego C, Sticchetti N, De Simoni MG. Effect of traumatic brain injury on cognitive function in mice lacking p55 and p75 tumor necrosis factor receptors. Acta Neurochir Suppl. 2008;102:409–413. doi: 10.1007/978-3-211-85578-2_80. [DOI] [PubMed] [Google Scholar]
- 61.Simen BB, Duman CH, Simen AA, Duman RS. TNF alpha signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry. 2006;59:775–785. doi: 10.1016/j.biopsych.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 62.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 63.Turrigiano GG. The self-tuning of neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNF alpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 65.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonistic mechanism of action: a comprehensive review. Pharmacol Ther. 2008;117:244–279. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 66.Grattendick KJ, Nakashima JM, Feng L, Giri SN, Margolin SB. Effects of three anti-TNF-alpha drugs: etarnecept, infliximab and pirfenidone on release of TNF-alpha in medium and TNF-alpha associated with the cell in vitro. Int Immunopharmacol. 2008;8:679–687. doi: 10.1016/j.intimp.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 67.Tobinik EL, Gross H. Rapid improvement in verbal fluency and aphasia following perispinal etarnecept in Alzheimer’s disease. BMC Neurol. 2008;8:27. doi: 10.1186/1471-2377-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chou RC, Kane MA, Gautam S, Ghirmire S. Tumor necrosis factor inhibition reduces the incidence of Alzheimer’s disease in rheumatoid arthritis patients (abstract) ACR. 2010;640 [Google Scholar]
- 69.Chandra V, Pandav R, Dodge HH, Johnston JM, Belle SH, DeKosky ST, Ganguli M. Incidence of Alzheimer’s disease in a rural community in India: the Indo-US study. Neurology. 2001;57:985–989. doi: 10.1212/wnl.57.6.985. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.