

Abstract
Purpose
LY3022859 is an anti-TGFβRII IgG1 monoclonal antibody that inhibits receptor-mediated signaling activation. The primary objective of this phase I study was to determine a phase II dose in patients with advanced solid tumors. Secondary objectives were to assess safety and pharmacokinetics (PK).
Methods
LY3022859 was infused intravenously (IV) at 1.25 mg/kg over 1 hour every 2 weeks (Q2W) (cohort 1A) and at flat doses of 12.5 mg (cohort 1B) and 25 mg (cohort 2) over 3 hours Q2W.
Results
Fourteen patients were enrolled in cohorts 1A (n=2), 1B (n=5), and 2 (n=7). DLTs were experienced by both patients in cohort 1A (infusion-related reaction) and 2 patients in cohort 2 (cytokine release syndrome and infusion-related reaction). No MTD was determined. At the 25 mg dose level (cohort 2), after fifth infusion, LY3022859 had a short t1/2 (4.37–7.80 hours) and rapid clearance (CLss, 0.412 L/hr). Exposure increased 2-fold (from 28.5 μg·hr/mL to 60.2 μg·hr/mL) with increase in dose from 12.5 mg to 25 mg. No accumulation was observed after repeat administration.
Conclusions
The MTD for LY3022859 was not determined. Dose escalation beyond 25 mg was considered unsafe due to worsening symptoms (uncontrolled cytokine release) despite prophylaxis (corticosteroids, antihistamines).
Keywords: TGFβ, TGFβ receptor, monoclonal antibody, solid tumors, therapy resistance
Introduction
Ligand interactions with transforming growth factor β receptor type I (TGFβRI) and type II (TGFβRII) complexes modulate a variety of cellular processes and can therefore play a critical role in tumorigenesis [1–3]. In the premalignant state, transforming growth factor β (TGFβ) acts as a tumor suppressor and inhibitor of cell proliferation. Upon malignant progression, however, the TGFβ pathway’s tumor suppressor function is lost, and cancer cells become resistant to growth-inhibitory effects [4]. Aggressive tumors are typically associated with high ligand levels, which in turn are often closely associated with poor prognosis [5,6]. Ligand expression by tumor cells and stromal cells leads to autocrine and paracrine TGFβ signaling, which results in direct [1] and indirect [7] pro-tumor effects. Targeting the TGFβ pathway by inhibiting ligand receptor interactions may therefore allow therapeutic targeting of multiple aspects of a cancer.
Binding of TGFβ ligands (TGFβ 1, 2, or 3) to TGFβRII induces phosphorylation of the receptor’s serine/threonine kinase domain, a first step in activating the TGFβ signaling pathway [8]. Once bound to TGFβRII, the ligand-receptor complex forms a heterotrimeric phosphoprotein complex with TGFβRI that activates both canonical (ie, SMAD) and noncanonical (ie, MAPK) pathways [1,9]. These pathways modulate transcription of numerous target genes, resulting in various effects. Therefore, blockade of ligand binding to TGFβRII may be an effective way to inhibit the TGFβ pathway’s many effects on cancer progression.
LY3022859 (also referred to as TR1 and previously as IMC-TR1) is a fully human anti-TGFβRII immunoglobulin G, subclass 1 (IgG1) monoclonal antibody that blocks the ectodomain of TGFβRII, preventing the formation of the ligand-receptor complex and thus inhibiting receptor-mediated signaling activation. In preclinical experiments with LY3022859 or the murine surrogate MT1, TGFβ pathway inhibition significantly reduced direct and indirect TGFβ-mediated cancer cell growth and metastasis [10].
Agents targeting the TGFβ pathway have been well tolerated [11–18]. Published evidence of pathway modulation and antitumor activity [12,13,15] suggests that, at least in some contexts, tumors are more sensitive than normal tissue to TGFβ pathway inhibition.
In the present study, the anti-TGFβRII monoclonal antibody LY3022859 was evaluated for safety and tolerability in patients with advanced solid tumors who had failed standard therapy or for which no standard was available.. The primary objective was to determine a recommended phase II dose for LY3022859. Secondary objectives were to assess the safety and pharmacokinetic (PK) profiles of LY3022859 and determine the maximum tolerated dose (MTD).
Materials and Methods
Study Design and Patients
This was a phase I, multicenter, nonrandomized, open-label, dose-escalation study of LY3022859 in patients with advanced solid tumors.
Eligible patients were ≥18 years old and had a histologically or cytologically confirmed solid tumor that had failed standard therapy or for which no standard therapy was available, evidence of progressive disease, and an Eastern Cooperative Oncology Group performance status of ≤1.
Excluded were patients who had clinically significant cardiac disease, had a high risk of gastrointestinal bleeding, had received a systemic thrombolytic agent within 28 days before enrollment, had evidence of retinal pathology, relied on monocular vision, had received a solid organ transplant, or had a symptomatic central nervous system malignancy.
This study was conducted in accordance with principles of the Declaration of Helsinki and Good Clinical Practice guidelines and with local ethics committee approval and was registered (NCT01646203). Written informed consent was obtained from all patients.
Study Procedures
LY3022859 was infused intravenously (IV) over approximately 1 hour every 2 weeks (Q2W) in cohort 1A (1.25 mg/kg) and over 3 hours Q2W in cohort 1B (12.5 mg flat dose) and cohort 2 (25 mg flat dose), at a maximum rate of 25 mg/minute. Patients were observed for infusion-related or other adverse events (AEs) every 15 minutes during and every 30 minutes for 1 hour after each infusion.
Baseline evaluations included medical history; physical examination; vital signs; routine hematology, serum chemistry, and urine analyses; electrocardiography, echocardiography, and ophthalmology assessments; and radiographic tumor assessments. Patients were monitored throughout the study for AEs, dose-limiting toxicities (DLTs), ophthalmological abnormalities, performance status, weight, vital signs, laboratory parameters, and tumor status.
Blood samples for PK analysis were collected up to 336 hours after the end of infusion (before infusion; at end of infusion; and 1, 2, 4, 8, 24, 48, 96, 168, and 336 hours after end of infusion) following the first and fifth doses. Additional samples were collected before and 1 and 168 hours after the end of the second infusion, before and 168 hours after the end of the third infusion, and before the fourth infusion. At and after the seventh infusion, samples were collected before the first infusion of every cycle.
Serum samples obtained during this study were analyzed for LY3022859 using a validated enzyme-linked immunosorbent assay (ELISA) method. The lower and upper limits of quantification were 100 ng/mL and 2000 ng/mL, respectively. Samples above the limit of quantification were diluted to yield results within the calibrated range. The interassay accuracy (% relative error) ranged from −2.9% to 5.9%. The interassay precision (% coefficient of variation [CV]) during validation ranged from 1.9% to 9.3%.
Study Endpoints
Efficacy endpoints included disease control rate (best overall response of complete response [CR], partial response [PR], stable disease, or non-CR/non-progressive disease), response rate (best overall response of CR or PR). Radiographic disease response (per Response Evaluation Criteria in Solid Tumors [RECIST], version 1.1) was assessed at the end of every cycle (last week of the cycle, before the onset of the next cycle) where appropriate for patients without disease progression who continued to receive study therapy.
Safety endpoints included treatment-emergent AEs (TEAEs), deaths, DLTs, performance status, vital signs, electrocardiograms, laboratory tests, and concomitant medications. A DLT was defined as an AE occurring during Cycle 1 (ie, the first 6 weeks of treatment) that was considered at least possibly related to the study drug, was considered dose-dependent, and fulfilled any of the criteria (using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0 [NCI CTCAE v 4.0]) described in the Supplementary Materials. The MTD was defined as the highest dose level at which ≤33% of patients experienced a DLT during Cycle 1.
Noncompartmental PK parameters of LY3022859 were calculated after the first and fifth infusions.
Statistical Analysis
The planned statistical methods were primarily descriptive. Safety and toxicity were evaluated and summarized descriptively for all patients who received any study treatment (safety population).
The incidence and percentage of patients with AEs were determined, according to the most severe grade (using NCI-CTCAE v 4.0).
PK parameter for LY3022859 were calculated by standard noncompartmental analysis using Phoenix WinNonlin 6.3.
The planned enrollment of approximately 50 patients for the dose-escalation portion was based on the standard 3+3 method.
Efficacy and safety analyses were performed using SAS version 9.2.
Results
Patient Disposition
A total of 14 patients were enrolled from July 2012 to October 2014 at 3 study sites in the United States. Two patients were assigned to cohort 1A, 5 to cohort 1B, and 7 to cohort 2 (Figure 1). All 14 patients discontinued study drug for reasons that included progressive disease (n=7), AE (n=3), death (n=2), withdrawal of consent (n=1), and other (n=1). The 2 deaths were due to progressive disease and were not considered treatment related.
Fig. 1.

Patient disposition
At the end of cohort 2, the study team decided to stop further enrollment and terminate the study in the absence of any indication of clinical efficacy (all patients who discontinued had stable disease at best, with 5 of them completing at least 2 treatment cycles) and in the presence of safety concerns over major cytokine release, which prohibited further escalation to effective dose levels and thereby did not allow the study to reach the assumed minimal effective exposure.
Key Baseline Demographics and Clinical Characteristics
Patient demographics and baseline characteristics are presented in Table 1. The most common tumor types were colorectal carcinoma (n=3) and prostate carcinoma (n=2). Prior cancer-related therapy types included chemotherapy (85.7%), radiotherapy (71.4%), and hormonal therapy (14.3%). The majority of patients (64.3%) had previously undergone cancer-related surgery.
Table 1.
Patient demographics and baseline characteristics
| Characteristica | Cohort 1A (1.25 mg/kg Q2W) (N=2) | Cohort 1B (12.5 mg Q2W) (N=5) | Cohort 2 (25 mg Q2W) (N=7) | Total (N=14) |
|---|---|---|---|---|
| Sex | ||||
| Male | 2 (100) | 3 (60.0) | 6 (85.7) | 11 (78.6) |
| Female | 0 | 2 (40.0) | 1 (14.3) | 3 (21.4) |
| Age (years), median (range) | 65.5 (52.0–79.0) | 59.0 (53.0–75.0) | 58.0 (24.0–74.0) | 58.5 (24.0–79.0) |
| Race | ||||
| White | 2 (100) | 5 (100) | 7 (100) | 14 (100) |
| ECOG PS | ||||
| 0 | 0 | 1 (20.0) | 2 (28.6) | 3 (21.4) |
| 1 | 2 (100) | 4 (80.0) | 5 (71.4) | 11 (78.6) |
| Primary tumor type | ||||
| Colorectal carcinoma | 0 | 1 (20.0) | 2 (28.6) | 3 (21.4) |
| Esophageal carcinoma | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Prostate carcinoma | 2 (100) | 0 | 0 | 2 (14.3) |
| Sarcoma, bone | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Small cell lung carcinoma | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Other | ||||
| Adenoid cystic carcinoma, head | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Adenoid cystic carcinoma, breast | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Ewing's sarcoma | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Rectal cancer | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Unknown primary (epithelioid neoplasm) | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Uterine leiomyosarcoma | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Disease duration (months) | ||||
| N | 2 | 4 | 7 | 13 |
| Mean (SD) | 171(60.24) | 42.7 (8.83) | 53.8 (55.36) | 68.4 (62.82) |
| Median (range) | 171 (128–213) | 40.5 (35.0–55.0) | 41.2 (9.4–173) | 43.0 (9.4–213) |
| Metastatic disease site | ||||
| Bone | 1(50.0) | 0 | 0 | 1 (7.1) |
| Liver | 0 | 3(60.0) | 2 (28.6) | 5 (35.7) |
| Lung | 0 | 3 (60.0) | 5 (71.4) | 8 (57.1) |
| Lymph nodes | 2 (100) | 2 (40.0) | 4 (57.1) | 8 (57.1) |
| Peritoneal | 0 | 2 (40.0) | 1 (14.3) | 3 (21.4) |
| Pleura | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Soft tissue | 1 (50.0) | 0 | 1 (14.3) | 2 (14.3) |
| Other | 0 | 2 (40.0) | 4 (57.1) | 6 (42.9) |
| Prior therapy types | ||||
| Chemotherapy | 2 (100) | 4 (80.0) | 6 (85.7) | 12 (85.7) |
| Hormonal | 2 (100) | 0 | 0 | 2 (14.3) |
| Radiotherapy | 1 (50.0) | 3 (60.0) | 6 (85.7) | 10 (71.4) |
| Previous surgery | ||||
| Yes | 1 (50.0) | 3 (60.0) | 5 (71.4) | 9 (64.3) |
| No | 1 (50.0) | 2 (40.0) | 2 (28.6) | 5 (35.7) |
Abbreviations: ECOG PS = Eastern Cooperative Oncology Group performance status; SD = standard deviation; Q2W = every 2 weeks.
All data presented as n (%), except for age and disease duration as indicated.
Safety
Treatment Exposure
The starting dose in this study was based on body weight (1.25 mg/kg, cohort 1A) but was reduced to flat doses of 12.5 mg and 25 mg in later cohorts (cohorts 1B and 2, respectively). Both patients in cohort 1A were exposed to LY3022859 only once, and both had an infusion-related reaction that led to study drug interruption after 14 minutes and 50 minutes, respectively. In the subsequent cohorts, all patients received prophylactic therapy and had study drug infused over 3 hours (instead of the 1-hour infusion scheduled for patients in cohort 1A).
In cohort 1B, all patients received at least 1 full dose infusion, for a mean of 4 infusions and 1.6 cycles. The mean duration of treatment was 8 weeks; mean cumulative dose, 50 mg; mean dose intensity, 6.3 mg/week; and mean relative dose intensity, 100%.
Adverse Events
Overall, 13 patients (92.9%) experienced at least 1 TEAE (Table 2). DLTs were experienced by both patients in cohort 1A (infusion-related reaction in each) and by 2 patients in cohort 2 (cytokine release syndrome and infusion-related reaction). TEAEs that led to study drug discontinuation occurred in both patients in cohort 1A (infusion-related reaction in each), 1 patient in cohort 1B (intestinal obstruction), and 2 patients in cohort 2 (dehydration and cytokine release syndrome). The infusion-related reactions in cohort 1A and the cytokine release syndrome in cohort 2 that led to study drug discontinuation were considered treatment related.
Table 2.
Most common treatment-emergent adverse events (≥4 patients overall)
| Cohort 1A (1.25 mg/kg Q2W) (N=2) | Cohort 1B (12.5 mg Q2W) (N=5) | Cohort 2 (25 mg Q2W) (N=7) | Total (N=14) | |
|---|---|---|---|---|
| Any TEAEa | 2 (100) | 4 (80.0) | 7 (100) | 13 (92.9) |
| Nausea | 0 | 2 (40.0) | 3 (42.9) | 5 (35.7) |
| Fatigue | 0 | 2 (40.0) | 3 (42.9) | 5 (35.7) |
| Anemia | 0 | 1 (20.0) | 3 (42.9) | 4 (28.6) |
| Infusion-related reaction | 2 (100) | 0 | 2 (28.6) | 4 (28.6) |
| Tumor pain | 1 (50.0) | 1 (20.0) | 2 (28.6) | 4 (28.6) |
Abbreviations: Q2W = every 2 weeks; TEAE = treatment-emergent adverse event.
Patients reporting more than 1 TEAE within a preferred term were only counted once.
Two patients in cohort 2 had treatment-related TEAEs (infusion-related reaction in each case) that led to interruption but not discontinuation of study drug. Both events included symptoms of dyspnea/shortness of breath.
Ten patients (71.4%) had TEAEs considered by the investigator to be treatment related. The most common TEAEs (occurring in ≥4 patients overall) were nausea, fatigue, anemia, infusion-related reaction, and tumor pain. Overall, 8 patients (57.1%) experienced at least 1 TEAE of grade ≥3 (Table 3). Of these, 4 had a treatment-related grade ≥3 TEAE.
Table 3.
Grade ≥3 treatment-emergent adverse events
| Cohort 1A (1.25 mg/kg Q2W) (N=2) | Cohort 1B (12.5 mg Q2W) (N=5) | Cohort 2 (25 mg Q2W) (N=7) | Total (N=14) | |
|---|---|---|---|---|
| Any Grade ≥3 TEAEa | 2 (100) | 3 (60.0) | 3 (42.9) | 8 (57.1) |
| Anemia | 0 | 1 (20.0) | 1 (14.3) | 2 (14.3) |
| Dehydration | 0 | 0 | 2 (28.6) | 2 (14.3) |
| Infusion-related reaction | 2 (100) | 0 | 0 | 2 (14.3) |
| Abdominal pain | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Ascites | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Gamma-glutamyltransferase increased | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Hyponatraemia | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Hypotension | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Hypovolaemia | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Intestinal obstruction | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Lymphocyte count decreased | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Lymphocyte count increased | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Nausea | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Renal failure acute | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Small intestinal obstruction | 0 | 1 (20.0) | 0 | 1 (7.1) |
| Syncope | 0 | 0 | 1 (14.3) | 1 (7.1) |
| Troponin increased | 0 | 0 | 1 (14.3) | 1 (7.1) |
Abbreviations: Q2W = every 2 weeks; TEAE = treatment-emergent adverse event.
Patients reporting more than 1 TEAE within a preferred term were only counted once.
No treatment-emergent serious adverse events (SAEs) or deaths occurred in cohort 1A. Two patients in cohort 1B and 3 patients in cohort 2 had SAEs, but only 1 of these SAEs (infusion-related reaction in cohort 2) was considered to be treatment related.
Three patients died during the study or within 30 days of the last dose of study drug: 1 patient in cohort 1B and 2 patients in cohort 2. All 3 deaths were due to progressive disease.
Treatment Response Data
There were no responses observed in this study.
Pharmacokinetics
Table 4 summarizes the PK parameters of LY3022859 calculated after the first and fifth intravenous infusions at the 12.5 mg (cohort 1B) and 25 mg (cohort 2) dose levels. Figure 2 shows the mean serum concentration-time profiles of LY3022859 after the first and fifth infusions. The PK profile of LY3022859 was not characterized at the 1.25 mg/kg dose level (cohort 1A) because of the reported infusion interruptions.
Table 4.
Summary of pharmacokinetic parameters for LY3022859 following single and multiple intravenous infusions
| PK parameter | Geometric mean (CV%)a
|
||
|---|---|---|---|
| Cohort 1B (12.5 mg Q2W) (nPK=5) | Cohort 2 (25 mg Q2W) (nPK=5) | ||
| After first infusion | Cmax, μg/mL | 4.34 (9) | 5.10 (21) |
| tmax,b hr | 3.22 (3.00–4.00) | 3.30 (3.00–5.00) | |
| AUC(0-tlast), μg·hr/mL | 28.5 (18) | 60.2 (30) | |
| AUC(0-∞), μg·hr/mL | 32.8 (34)c | 63.9 (33) | |
| t1/2,d hr | 4.06 (1.88–5.59)e | 5.93 (5.04–8.12) | |
| CL, L/hr | 0.381 (34)c | 0.391 (33) | |
| Vz, L | 2.01 (25)c | 3.35 (19) | |
| After fifth infusion | Cmax, μg/mL | 3.305; 3.747f | 5.01 (28)c |
| tmax,b hr | 3.00; 3.83f | 3.12 (3.03–3.20)c | |
| AUCτ, μg·hr/mL | NA | 60.7 (35)c | |
| t1/2,d hr | 5.78g | 5.80 (4.37–7.80)c | |
| CLss, L/hr | NA | 0.412 (35)c | |
| Vss, L | NA | 3.81 (19)c | |
| RA,Cmax | 0.865; 0.879f | 1.12 (39)c | |
| RA,AUC | NA | 1.17 (67)c | |
Abbreviations: AUC(0-tlast) = area under the concentration versus time curve from the time zero to tlast; AUCτ = area under the concentration versus time curve during 1 dose interval (336 hours); CLss = clearance at steady state after intravenous administration; Cmax = maximum observed drug concentration; CV = coefficient of variation; NA = not applicable; nPK = number of PK observations; PK = pharmacokinetic; Q2W = every 2 weeks; RA,AUC = accumulation ratio calculated using AUC; RA,Cmax = accumulation ratio calculated using Cmax; t1/2 = terminal elimination half-life; tmax = time of maximum observed drug concentration; Vss = volume of distribution at steady state following intravenous administration; Vz = volume of distribution during the terminal phase.
Geometric mean and geometric CV% are provided for n≥3; otherwise, actual values are provided.
Median and range are provided for tmax.
n=3.
Geometric mean and range are provided for t1/2.
n=4.
n=2.
n=1.
Fig. 2.
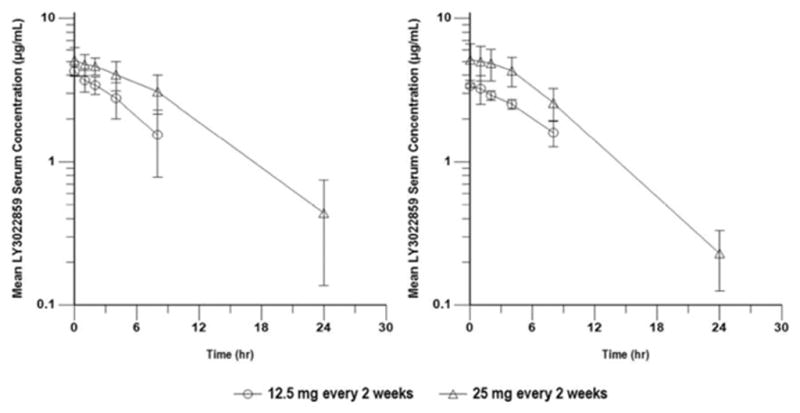
Serum concentration-time profiles of LY3022859 following first (left) and fifth (right) intravenous infusions at doses of 12.5 mg (cohort 1B) or 25 mg (cohort 2) over 3 hours every 2 weeks. Data are presented as arithmetic means (SD) on a semilogarithmic scale
At the 25 mg dose level (cohort 2), after fifth infusion, LY3022859 had a short t1/2 (4.37–7.80 hours) and rapid clearance (CLss, 0.412 L/hr; CV%, 35%). A 2-fold increase in exposure (from 28.5 μg·hr/mL to 60.2 μg·hr/mL) was observed when dose was increased from 12.5 mg to 25 mg. In addition, no accumulation was observed following repeated administration. Predose concentrations (Ctrough) following multiple infusions were either below the quantifiable limit or not available. No patient achieved the target trough levels required for efficacy based on data from nonclinical tumor xenograft models (based on xenograft models, the target serum concentration for LY3022859 was hypothesized to be one that would maintain trough concentrations at or above 37.6 μg/mL for antimetastatic activity, 188 μg/mL for immune-mediated antitumor activity, and 101–216 μg/mL for antiproliferative activity).
Discussion
In this phase I study of LY3022859 in patients with advanced solid tumors, cytokine release syndrome pre-empted dose escalation, and the MTD was not determined. Even with reduced dose levels, dose escalation beyond a dose of 25 mg was considered unsafe due to worsening symptoms (uncontrolled cytokine release) despite prophylaxis.
The starting dose in this study, which was based on body weight (1.25 mg/kg), was supported by preclinical toxicokinetic studies in cynomolgus monkeys and PK studies in tumor models (unpublished observations). This starting dose, however, resulted in infusion-related reactions and consequent interruption of the initial infusion in the first 2 patients exposed to LY3022859 (ie, cohort 1A). The study protocol was subsequently amended to manage the risk of infusion-related reaction by introducing mandatory prophylactic therapy consisting of diphenhydramine (or a similar histamine H1 antagonist), ranitidine (or a similar H2 antagonist), an intravenous corticosteroid, and acetaminophen; changing from a weight-normalized dose to a fixed (flat) dose; and extending the infusion time from 1 hour to 3 hours. Despite these measures, 2 more patients (both of whom received the 25 mg dose in cohort 2) experienced infusion-related reactions.
Together, there are two possible etiologies for the infusion reactions. One is that, theoretically, affected patients were reacting to infusion of the humanized monoclonal antibody, an event rarely seen with modern therapeutic antibodies and very unlikely since LY3022859 shares a backbone in common with a number of other antibodies that have not produced such reactions. Alternatively, these findings could be related to the study drug’s mechanism of action or an off-target (but drug-specific) activity; LY3022859 targets TGFβRII, which is ubiquitously expressed in the body, including on immune cells.
No PK profile could be obtained for LY3022859 administered at the 1.25-mg/kg dose level because of the above-mentioned infusion interruptions. However, the PK profiles obtained for cohorts 1B and 2 indicated that LY3022859 administered at the 25-mg flat dose had a short half-life, was rapidly cleared, and resulted in a 2-fold increase in exposure (from 28.5 μg·h/mL to 60.2 μg·h/mL) when compared with the 12.5-mg flat dose. In addition, accumulation of LY3022859 was not observed following repeated administration, and all predose concentrations (Ctrough) following multiple infusions were below the quantifiable limit. Hence, no patient achieved the target trough levels required for efficacy based on nonclinical tumor xenograft models (as discussed in the Results section above). Reaching targeted active dose levels of LY3022859 (37.6 μg/mL and 101–216 μg/mL) was considered not feasible, given the observed infusion-related safety profile despite prophylactic therapy at the highest dose (25 mg) administered in this study.
In conclusion, the primary objective of this study was not achieved since a safe dose without infusion-related reactions could not be determined at doses and concentrations that were biologically active.
Supplementary Material
Acknowledgments
Funding support
The study was sponsored by Eli Lilly and Company. Jude Richard (INC Research, Austin, TX) provided medical writing services on behalf of the authors, funded by the study sponsor. These included assisting with initial drafting of the manuscript and subsequent revision according to guidance from the authors.
Other contributors/acknowledgements
We thank David Schaer (Eli Lilly and Company) for useful discussions during the development of this manuscript.
Footnotes
Compliance with Ethical Standards
Conflict of interest
JC was an employee of Lilly during the conduct of the study but is now an employee of Sotio. EC has served on advisory boards for Lilly, Taiho, Bayer, EMD Serono, Amgen, Advaxis, Merrimack, Castle Biosciences, and Genentech. JK, ST, KD, RK, SRPK, and IG are employees of Lilly. The remaining authors declare no conflict of interest.
Ethical approval
This study was conducted in accordance with principles of the Declaration of Helsinki and Good Clinical Practice guidelines and with local ethics committee approval and was registered (NCT01646203).
Informed consent
Written informed consent was obtained from all patients.
clinicaltrials.gov Identifier: NCT01646203
References
- 1.Massagué J. TGFβ in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-β and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13:5262–5270. doi: 10.1158/1078-0432.CCR-07-1157. [DOI] [PubMed] [Google Scholar]
- 3.Elliott RL, Blobe GC. Role of transforming growth factor beta in human cancer. J Clin Oncol. 2005;23:2078–2093. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 4.Inman GJ. Switching TGFB from a tumor suppressor to a tumor promoter. Curr Opin Genet Dev. 2011;21:93–99. doi: 10.1016/j.gde.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Teicher BA. Transforming growth factor-beta and the immune response to malignant disease. Clin Cancer Res. 2007;13:6247–6251. doi: 10.1158/1078-0432.CCR-07-1654. [DOI] [PubMed] [Google Scholar]
- 6.Levy L, Hill CS. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10:554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prud'homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest. 2007;87:1077–1091. doi: 10.1038/labinvest.3700669. [DOI] [PubMed] [Google Scholar]
- 9.Massagué J, Gomis RR. The logic of TGFβ signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 10.Zhong Z, Carroll KD, Policarpio D, Osborn C, Gregory M, Bassi R, Jimenez X, Prewett M, Liebisch G, Persaud K, Burtrum D, Wang S, Surguladze D, Ng S, Griffith H, Balderes P, Doody J, Schwartz JD, Youssoufian H, Rowinsky EK, Ludwig DL, Witte L, Zhu Z, Wu Y. Anti-transforming growth factor β receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin Cancer Res. 2010;16:1191–1205. doi: 10.1158/1078-0432.CCR-09-1634. [DOI] [PubMed] [Google Scholar]
- 11.Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapartra AK, Suri A, Balasubramaniam A, Nair S, Oliushine V, Parfenov V, Poverennova I, Zaaroor M, Jachimczak P, Ludwig S, Schmaus S, Heinrichs H, Schlingensiepen KH Trabedersen Glioma Study Group. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol. 2011;13:132–142. doi: 10.1093/neuonc/noq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris JC, Shapiro GI, Tan AR, Lawrence DP, Olencki TE, Dezube BJ, Hsu FJ, Reiss M, Berzofsky JA. Phase I/II study of GC1008: a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody (MAb) in patients with advanced malignant melanoma (MM) or renal cell carcinoma (RCC) J Clin Oncol. 2008;26(15 suppl) doi: 10.1371/journal.pone.0090353. abstract 9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahnert J, Baselga J, Calvo E, Seoane J, Brana I, Sicart E, Gueorguieva I, Cleverly A, Lahn MMF, Pillay S, Holdhoff M, Blakeley JO, Carducci MA. First human dose (FHD) study of the oral transforming growth factor-beta receptor I kinase inhibitor LY2157299 in patients with treatment-refractory malignant glioma. J Clin Oncol. 2011;29(15 suppl) abstract 3011. [Google Scholar]
- 14.Lonning S, Mannick J, McPherson JM. Antibody targeting of TGF-β in cancer patients. Curr Pharm Biotechnol. 2011;12:2176–2189. doi: 10.2174/138920111798808392. [DOI] [PubMed] [Google Scholar]
- 15.Oettle H, Hilbig A, Seufferlein T, Tsianakas A, Luger T, Schmid RM, von Wichert G, Endlicher E, Garbe C, Kaehler KK, Hauschild A, Enk A, Kiessling P, Schmaus S, Heinrichs H, Schlingensiepen K. Phase I/II study with trabedersen (AP 12009) monotherapy for the treatment of patients with advanced pancreatic cancer, malignant melanoma, and colorectal carcinoma. J Clin Oncol. 2011;29(15 suppl) abstract 2513. [Google Scholar]
- 16.Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, Silliman N, Streisand J, Powell J, Akesson A, Coppock J, Hoogen Fv, Herrick A, Mayes MD, Veale D, Haas J, Ledbetter S, Korn JH, Black CM, Seibold JR Cat-192 Study Group; Scleroderma Clinical Trials Consortium. Recombinant human anti-transforming growth factor β1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007;56:323–333. doi: 10.1002/art.22289. [DOI] [PubMed] [Google Scholar]
- 17.Lahn M, Kloeker S, Berry BS. TGF-beta inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 2005;14:629–643. doi: 10.1517/13543784.14.6.629. [DOI] [PubMed] [Google Scholar]
- 18.Trachtman H, Fervenza FC, Gipson DS, Heering P, Jayne DR, Peters H, Rota S, Remuzzi G, Rump LC, Sellin LK, Heaton JP, Streisand JB, Hard ML, Ledbetter SR, Vincenti F. A phase 1, single-dose study of fresolimumab, an anti-TGF-β antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011;79:1236–1243. doi: 10.1038/ki.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.