

SUMMARY
The molecular mechanism of transmembrane proton translocation in rotary motor ATPases is not fully understood. Here we report the 3.5 Å resolution cryoEM structure of the lipid nanodisc-reconstituted Vo proton channel of the yeast vacuolar H+-ATPase, captured in a physiologically relevant, autoinhibited state. The resulting atomic model provides structural detail for the amino acids that constitute the proton pathway at the interface of the proteolipid ring and subunit a. Based on the structure and previous mutagenesis studies, we propose the chemical basis of transmembrane proton transport. Moreover, we discovered that the C-terminus of the assembly factor Voa1 is an integral component of mature Vo. Voa1’s C-terminal transmembrane α helix is bound inside the proteolipid ring, where it contributes to the stability of the complex. Our structure rationalizes possible mechanisms by which mutations in human Vo can result in disease phenotypes, and may thus provide new avenues for therapeutic interventions.
Keywords: Vacuolar H+-ATPase, Vo proton channel, cryoEM, lipid nanodisc, V-ATPase assembly, Voa1, membrane protein structure, proton pumping, reversible disassembly
ETOC BLURB
Here we report the 3.5 Å resolution cryoEM structure of lipid nanodisc-reconstituted yeast vacuolar H+-ATPase Vo proton channel. The resulting atomic model provides insight into the chemical basis of transmembrane proton transport. Moreover, we discovered that the C-terminus of the assembly factor Voa1 is an integral component of mature Vo.
INTRODUCTION
Vacuolar H+-ATPases (V-ATPases, V1Vo-ATPases) are membrane bound multi-subunit enzyme complexes that acidify subcellular compartments in every eukaryotic cell and the extracellular space in some animal tissues (Forgac, 2007). V-ATPase’s proton pumping activity is essential for pH and ion homeostasis (Kane, 2006), protein trafficking, endocytosis, mTOR (Zoncu et al., 2011) and Notch (Yan et al., 2009) signaling, hormone secretion (Sun-Wada et al., 2006), and neurotransmitter release (Vavassori and Mayer, 2014). While complete loss of V-ATPase is embryonic lethal (Inoue et al., 1999), aberrant (hypo- or hyper-) activity is associated with human diseases such as renal tubular acidosis (Smith et al., 2000), sensorineural deafness (Karet et al., 1999), osteoporosis (Thudium et al., 2012), diabetes (Sun-Wada et al., 2006), microbial infection (Wong et al., 2011), infertility (Brown et al., 1997), cancer (Sennoune et al., 2004), and AIDS (Geyer et al., 2002) making the enzyme a potential drug target (Kartner and Manolson, 2014).
Yeast V-ATPase, a well characterized model system for the mammalian enzyme, is composed of ~30 polypeptides that are organized in two sectors: a cytoplasmic V1-ATPase (subunits A3B3(C)DE3FG3H) and a membrane embedded Vo proton channel (ac8c′c″def (Oot et al., 2017); subunit c′ is not found in higher organisms) (Figure S1A). V-ATPase is a rotary motor enzyme, employing a catalytic mechanism shared with the F-, A- and A/V-type ATPases (Muench et al., 2011). In V-ATPase, ATP hydrolysis on V1 drives rotation of the rotor subcomplex made up of subunits DFd and the “proteolipid” subunits c, c′ and c″, which are arranged in a ten-membered ring (c8c′c″; otherwise known as the proteolipid- or c-ring). Each proteolipid subunit carries one lipid exposed glutamic acid residue that is essential for proton pumping (Hirata et al., 1997). Proton translocation occurs through two aqueous half-channels at the interface of the c-ring and the membrane-integral C-terminal domain of subunit a (aCT). During c-ring rotation, the essential glutamates are protonated from the cytosolic half-channel and, after rotating ~360°, the protons are released into the luminal half-channel. A strictly conserved arginine residue in aCT located mid-membrane across from the c-ring glutamates is essential for transmembrane proton transport (Kawasaki-Nishi et al., 2001).
V-ATPase is regulated in vivo by a unique mechanism referred to as “reversible disassembly” wherein V1-ATPase is released into the cytoplasm, leaving behind free Vo in the membrane (Kane, 1995; Sumner et al., 1995) (Figure S1A). Upon enzyme dissociation, both sectors become “autoinhibited”, that is V1 no longer hydrolyzes MgATP (Gräf et al., 1996; Oot et al., 2016; Parra, 2000) and unlike the free membrane sector of F-ATPases (Fo) (Schneider and Altendorf, 1985), Vo does not permit passive proton translocation (Zhang et al., 1992). While reversible disassembly has been well characterized on a cellular level in yeast (Parra and Kane, 1998), insect (Voss et al., 2007), and to some extent in higher animals (Bodzeta et al., 2017; Stransky and Forgac, 2015; Trombetta et al., 2003; Zoncu et al., 2011), little is known about the molecular mechanism of proton translocation, largely due to the lack of high-resolution structural information for the enzyme’s Vo proton channel. This lack of structural information has also limited our understanding of the disease-causing mechanisms of human V-ATPase mutations, many of which are found in subunit a.
Recently, a 3.9 Å resolution cryoEM structure of amphipol solubilized yeast Vo (Mazhab-Jafari et al., 2016) showed the proton carrying glutamate of c″ (E108) in contact with one of the essential arginines (R735) in aCT. However, as the complex was vitrified in the presence of the V-ATPase inhibitor Bafilomycin A1, it was uncertain whether the unique position of the c-ring relative to aCT was caused by inhibitor binding, or represented the native, autoinhibited conformation of the complex. Furthermore, resolution in that reconstruction was anisotropic, thus making it difficult to ascertain the reliability of the model in some regions such as the subunit interfaces implicated for proton translocation.
Here we report a cryoEM reconstruction of S. cerevisiae Vo reconstituted into lipid nanodiscs (VoND) determined at a resolution of 3.5 Å. Backbone and side chain densities were sufficiently resolved so that all subunits in the complex could be built as an all-atom model with high confidence. Furthermore, the map of VoND contained an α helical density within the cavity of the c-ring, which we were able to assign as the C-terminal transmembrane α helix (TMH) of the Vo assembly factor Voa1. Based on the structural data, we can deduce the mechanism of proton translocation at the aCT-c-ring interface in holo V-ATPase, discuss aspects of autoinhibition of free Vo, and identify the spatial locations of the amino acid residues implicated in various human diseases.
RESULTS AND DISCUSSION
CryoEM Reconstruction of Yeast Vo in Lipid Nanodiscs
Following starvation-induced dissociation of yeast V-ATPase (Figure S1A), the autoinhibited Vo was purified and reconstituted into lipid nanodiscs (VoND) using membrane scaffold protein MSP1E3D1 (Stam and Wilkens, 2017)(Figure S1B). CryoEM images of vitrified VoND revealed monodisperse and homogeneous particles with random orientations (Figure 1A, S1C). 2D class averages showed intact Vo complexes with clearly visible TMHs within a disc-like density contributed by the lipid bilayer, and a cytoplasmic domain extending above the membrane (Figure 1B). The ~150×120 Å2 nanodiscs are of sufficient size to accommodate the ~120×100 Å2 Vo, such that isotropic lipid density surrounds the transmembrane regions of the complex. CryoEM reconstruction was carried out to an overall resolution of 3.5 Å (Figure 1C, S1D, Table 1). The final map shows clear density for all known protein components of the Vo, including subunits a, d, e, f and the c-ring (Figure 1C-E; Movie S1). Furthermore, the map contained an α helical density inside the proteolipid ring, which we identified as the C-terminus of the assembly factor Voa1 based on biochemical analysis (see below). A comparison of our map of VoND to the recently reported 3.9 Å map of amphipol solubilized Vo (Mazhab-Jafari et al., 2016)(EMD-8409) revealed that, while both maps had overall similar features, our map VoND displayed higher, and more isotropic resolution, with prominent side chain densities for most of the complex (Movie S2). Notably, improvements were not limited to transmembrane regions, but also extended to the cytoplasmic domain of the complex (Figure S1E, F), including subunit d and the N-terminal half of subunit a (aNT), which were only partially resolved previously. In line with this observation, unlike the dataset of Vo in amphipol (Mazhab-Jafari et al., 2016), our dataset of VoND was homogeneous, without detectable subcomplexes based on the 3D classification analysis. Thus, the data suggest that lipid nanodisc reconstitution improves the overall stability of Vo, allowing for a 3D reconstruction in which the density of each subunit component is well resolved for model building.
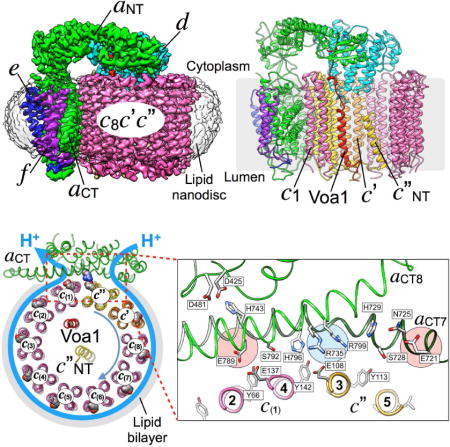
Figure 1. 3.5 Å resolution cryoEM map of VoND.
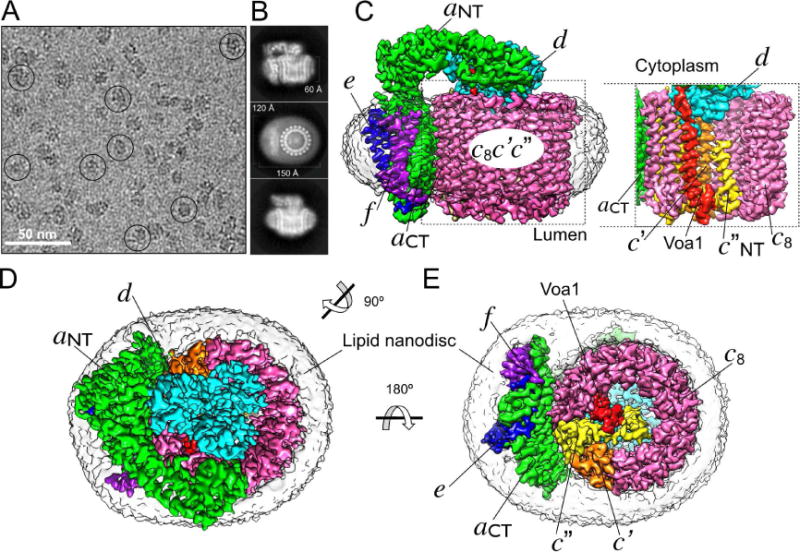
(A) Electron micrograph of ice-embedded VoND showing uniform particle distribution with representative particles highlighted by circles. (B) Averaged 2D projections showing elements of secondary structure surrounded by isotropic density of the 150×120×60 Å3 lipid nanodiscs. (C) VoND density map showing subunits a (green), d (cyan), e (blue), f (purple), and c-ring subunits c8 (pink), c′ (orange) and c″ (yellow). The image to the right shows the inside of the c-ring, highlighting the N-terminal α helix of c″ (c″NT), and the here identified integral component of Vo, the C-terminal TMH of the assembly factor Voa1 (red). (D) View from the cytoplasm towards the membrane showing the rotor subunit d atop the c-ring and next to aNT. (E) View from the lumen showing the position of Voa1 and subunits c′ and c″ relative to aCT. The low-pass filtered density of the lipid nanodisc is shown in transparent grey. The same color codes for different protein subunits are used throughout the remaining figures. See also Table 1, Figure S1 and Movies S1, S2.
Table 1.
| CryoEM map reconstruction parameters
| ||
|---|---|---|
| Data Collection/Image Processing | Instrument/Software/Settings | Specifics |
| Grid | UltrAuFoil 1.2/1.3 | Quantifoil |
| Cryo-specimen freezing | Leica EM GP | 1 blot; 90% RH; Blot time = 2~3 sec |
| Electron microscope | JEM3200FSC | 300 keV, in-column energy filter (25 eV) |
| Detector | Gatan K2 Summit | 4K × 4K, 5μm pixel size |
| Sampling interval | 0.615 Å/Pixel | Super-resolution, 1.23 Å/Pixel (2×2 binned) |
| Exposure rate on specimen | 5 e−/Å2/s | 5 frames/sec |
| Exposure time | 10 sec | Total dose 50 e−/Å2 |
| Drift correction | Unblur with exposure filter | 1,741 movies |
| Defocus range | 0.7 – 4 μm | |
| Defocus determination | CTFFIND4 | |
| Particles picked | RELION 1.4 | |
| Particle box size | 200 × 200 | |
| Number of particle used | 180,528 | out of 285,000 boxed particles |
| Initial map generation | EMAN 2.1 | |
| Map refinement | RELION 1.4 | |
| Resolution | 3.5 Å | Gold standard FSC at 0.143 |
| Modeling | Chimera, Coot and Phenix | De novo model building |
| Model refinement statistics | |||
|---|---|---|---|
| Non-hydrogen atoms | 22,110 | ||
| Protein residues modeled/expected | 2,866/3,000 | Excluding Voa1 | |
| All-Atom Contacts (Clash score*) | 4.44 | 100th percentile (N=37, 3Å - 9999Å) | |
|
| |||
| Protein Geometry | |||
| Poor rotamers | 14 | 0.60% | Goal: <0.3% |
| Favored rotamers | 2180 | 93.04% | Goal: >98% |
| Ramachandran outliers | 0 | 0.00% | Goal: <0.05% |
| Ramachandran favored | 2794 | 97.54% | Goal: >98% |
| MolProbity score | 1.32 | 100th percentile (N=342, 3.50Å ± 0.25Å) | |
| Cβ deviations >0.25Å | 0 | 0.00% | Goal: 0 |
| Bad bonds: | 0/22558 | 0.00% | Goal: 0% |
| Bad angles: | 0/30638 | 0.00% | Goal: <0.1% |
|
| |||
| Peptide Omegas | |||
| Cis Prolines: | 0/86 | 0.00% | Expected: ≤1 per chain, or ≤5% |
|
| |||
| Low-resolution Criteria | |||
| Cα BLAM outliers | 28 | 0.98% | Goal: <1.0% |
| Cα Geometry outliers | 6 | 0.21% | Goal: <0.5% |
|
| |||
| EmRinger Score (1603 residues) | 1.93 | ||
| Isotropic B factor Å2 (average) | 165.1 | ||
Atomic Model of Yeast Vo
The 3.5 Å resolution map of VoND was used to build an all-atom model for over 95% of the expected residues (Figure 2A), including the N-terminal domain of subunit a (aNT), subunits e, f, and large portions of subunit d (Figure 2, S2A, Movie S2). The final model of the entire complex displayed proper geometry (Molprobity score = 1.32; Table 1), and matched well with the experimental map (4 Å at 0.5 correlation; Figure S1D). Of note, our map contains several small, unmodeled densities observed in both half maps (Figure S2B), possibly representing stably bound lipids, or yet to be identified (non)-protein components of the Vo.
Figure 2. Atomic model of VoND.
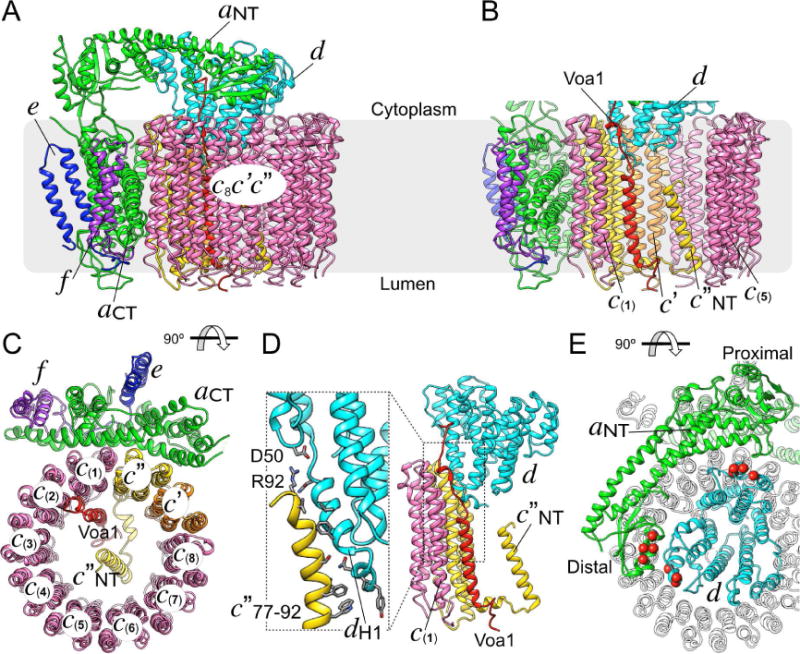
(A) Ribbon diagram of the all-atomic model of nanodisc reconstituted yeast Vo. (B) View inside the c-ring highlighting the arrangement of Voa1, the N-terminal TMH of c″ (c″NT) and subunit d. (C) The interface between the c-ring and aCT is formed by c(1) and c″. (D) The interaction between subunits d and c″ is mediated by hydrophobic contacts and a possible salt bridge between c″R92 and dD50. dH1 indicates subunit d’s N-terminal α helix. (E) The interaction between aNT and subunit d when viewed from the cytoplasm towards the membrane, with contact residues previously identified biochemically (Stam and Wilkens, 2017) highlighted by space fill Cαs shown in red. See also Table 1, Figure S2 and Movie S3.
The c-ring
Yeast V-ATPase c-ring contains three proteolipid variants c, c′, and c″ in the ratio of 8:1:1 (Figure 2A-C; from hereon, individual copies of the subunit c variant will be designated as c(1)-(8)). The c″ and c′ variants were identified unambiguously in our map by their longer luminal loops compared to c, and the extra TMH at the N-terminus of c″ (c″NT; residues 17-39). c″NT is in the center of the ring, with its N-terminus pointing towards the cytoplasm (Figure 2B, S2C). The proton binding sites on the c-ring are homologous to those found in the F-ATPase from S. platensis (Pogoryelov et al., 2009). However, the essential glutamates in our model appear to be in a more open conformation in all ten proteolipids, with each glutamate (except E137 in c(1)) in H-bond distance to a tyrosine residue contributed by the same or a neighboring subunit (Figure S2C, D). Importantly, unlike holo yeast V-ATPase, where the c-ring was seen in three rotational states relative to subunit a (Zhao et al., 2015), the c-ring in our map of inhibitor-free VoND was found to be in a single orientation. This indicates that upon V-ATPase dissociation, the c-ring assumes a unique “resting position” relative to aCT (Figure 2C), and that this resting conformation is an inherent feature of free, autoinhibited Vo not caused by inhibitor binding.
Specific interactions between subunit d and the c-ring
The role of subunit d is to link the c-ring rotor to the V1 motor. Our structure provides a detailed picture of the specific molecular contacts between d and the c-ring, which are largely mediated by subunit d’s N-terminus and the cytosolic loops of c″. The interaction buries a surface area of ~670 Å2 (calculated by PISA (Krissinel and Henrick, 2007)) that includes a salt bridge between dD50 and c″R92, hydrophobic contacts, and multiple residue pairs that are at distances suggesting H-bond formation (Figure 2D). Additional contacts of d are with the cytosolic loops of the adjacent c(1) and c′ subunits, with buried surface areas of ~400 and 250 Å2, respectively. This observation is consistent with previous mutagenesis experiments that showed defective enzyme assembly upon removal of d’s N-terminal residues (Owegi et al., 2006).
Subunit a and its interaction with subunit d
Subunit a is organized in N-terminal cytoplasmic (aNT) and C-terminal membrane integral (aCT) domains of ~400 residues each (Figure 2A). While subunit a plays a key role in proton transport across Vo (Toei et al., 2011), its atomic structure has not yet been resolved in detail. Here, we built an all-atom model for subunit a, except two unstructured loops (residues 153-183 and 657-705), for which there was no interpretable density in our map. Our model shows that aNT is folded as a hairpin containing two globular segments (proximal and distal subdomains) connected by a long coiled-coil (Figure 2E). The model also reveals unprecedented molecular details of the aNT-d interface, an interaction that is only seen in free Vo. The structure shows both proximal and distal subdomains of aNT in contact with subunit d (Figure 2E), with a total buried surface area of ~940 Å2. The interaction of aNT and d seen in free Vo is consistent with our recent study that defined the binding sites of the two polypeptides using site directed mutagenesis and isothermal titration calorimetry of recombinant proteins (Figure 2E) (Stam and Wilkens, 2017). These structural characteristics unique to free Vo may contribute to the stability of the complex in the autoinhibited state, and protect against loss of subunit d upon enzyme dissociation (Ediger et al., 2009). However, previous mutagenesis and functional assays have suggested that the interaction between a (part of the stator) and d (part of the rotor) is not solely responsible for forming the autoinhibition state of the eukaryotic Vo (Couoh-Cardel et al., 2015; Qi and Forgac, 2008). Of note, aCT and its interaction with the c-ring will be described in a separate section below.
The Assembly Factor Voa1 is a Component of Yeast Vo
Our map revealed two α-helical densities inside the central pore of the c-ring, one in the center, and one on the side, in contact with c″ and c(1) (Figure 1C). Equivalent helical densities were also observed in the map of amphipol solubilized Vo (Mazhab-Jafari et al., 2016) and in that study, both helical densities were modeled as part of subunit c″, with the N-terminus of c″ oriented towards the vacuolar lumen. However, this interpretation contradicts earlier biochemical studies that placed the N-terminus of c″ toward the cytoplasm (Flannery et al., 2004; Wang et al., 2007). By matching the amino acid sequence to the side chain densities, we confirmed that the central α helix belonged to c″, with its N-terminus pointing toward the cytoplasm, consistent with the previous biochemical studies (Figure 1C, S2B). When examining the other α-helical density that is in contact with c″ and c(1), we found that it extends from the lumen to the cytoplasm, where it contacts subunit d (Figure 3A, red density), suggesting that this α-helical density may belong to a separate and unknown polypeptide. To determine the identity of this structural component, we performed mass spectrometry of our VoND preparation. The analysis revealed multiple peptides that matched the open reading frame of Voa1 (YGR106C) (Figure S3A), a 265 residue polypeptide that functions in the assembly of Vo in the ER membrane (Figure S3B)(Ryan et al., 2008). While not required for assembly of a functional Vo, deletion of Voa1 causes synthetic lethality when combined with a mutation of the ER retention motif of another assembly factor (Vma21)(Ryan et al., 2008).
Figure 3. Voa1 is a component of Vo and contributes to the c-ring stability and subunit d association.
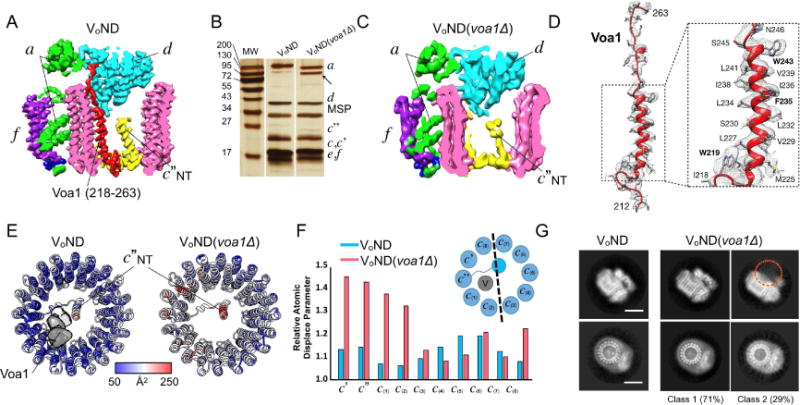
(A) Slice through cryoEM map of VoND, with density highlighted in red representing the C-terminal TMH of the assembly factor Voa1. (B) Silver stained SDS-PAGE of wild type VoND and VoND(voa1Δ). The arrow indicates partial degradation of subunit a in the VoND(voa1Δ) preparation. (C) The cryoEM density (red) assigned to Voa1 in wild type VoND (panel A) is missing in the map of VoND(voa1Δ). (D) Model of the C-terminal TMH of Voa1 within the corresponding map density highlighted in panel (A). (E, F) Atomic displacement parameter (ADP) display of the c-rings of VoND and VoND(voa1Δ) (E), and comparison of the normalized ADP for VoND and VoND(voa1Δ) (F). (G) 2D class averages of particles in different orientations from the VoND and VoND(voa1Δ) datasets. Deletion of Voa1 results in loss of subunit d in ~30% of the particles (as indicated by the orange circle in the 2D average shown at the top-right). Scale bars are 50 Å. See also Figure S3.
To assess whether the second α helical density inside the c-ring represented the C-terminal TMH of Voa1, we purified Vo from a yeast strain in which the gene for Voa1 was deleted, reconstituted the resulting Vo(voa1Δ) into lipid nanodiscs (VoND(voa1Δ)) (Figure 3B), and determined its cryoEM structure at 4.6 Å resolution (Figure 3C). While the map of VoND(voa1Δ) has a similar appearance compared to the map of wild type VoND (Figure 3A), inspection of the interior of the c-ring revealed that the α helical density next to c(1) (red density in Figure 3A) was missing in the map of VoND(voa1Δ) (Figure 3C), providing strong evidence that the density belongs to Voa1. Our assignment is further supported by the presence of side chain density, which allowed de novo building of Voa1’s C-terminal TMH (residues 222-244) (Figure 3D, S3C). It should be noted that there was no clear density that could account for the remainder of the protein (residues S26-D211). This suggests that the luminal domain of Voa1 is either flexible, or proteolytically removed upon trafficking of the complex to the vacuole, with the latter possibility supported by mass spectrometry analysis of both free Vo and holo V1Vo-ATPase (Figure S3D).
To examine the structural contribution of Voa1 to the mature Vo complex, we compared atomic displacement parameters (ADP)(Trueblood, 1996) of wild type and mutant complex (Figure 3E). While all c-ring components in wild type have similar ADP, c-subunits proximal to Voa1 displayed a significant increase of the relative ADP in the mutant complex (Figure 3F). This increase in ADP suggests a decreased stability of the complex upon loss of Voa1, which is also evident from increased degradation of subunit a in the VoND(voa1Δ) preparation (Figure 3B, see arrow). Moreover, Voa1 is located between c(1) and c″, where it forms a unique binding pocket for subunit d (Figure 3A), and in line with this observation, we noticed during the 3D classification of the VoND(voa1Δ) dataset that ~30% of mutant complexes lacked subunit d (Figure 3G). Thus, absence of Voa1 likely reduces the affinity between d and the c-ring such that d is either inefficiently recruited during assembly, or lost after assembly and/or during purification of the mutant complex. It has been shown that upon mutation of Voa1’s C-terminal ER retention motif, Voa1 is found on the vacuole (Ryan et al., 2008)). In our structure, subunit d is seen to bind to the C-terminus of Voa1 (Figure 2D). It is possible that this interaction masks Voa1’s ER retention motif, thereby serving as a quality control measure to ensure that only correctly assembled mature Vo complex can traffic to the vacuole, similar to what has been proposed for the trafficking of immunoglobulin E receptors in T cells (Letourneur et al., 1995). Our hypothesis that the interaction between Voa1 and subunit d plays a role in quality control during V-ATPase assembly is also supported by the observation that in a subunit d deletion strain, no Vo(Δd) subcomplex is found on the vacuolar membrane (Bauerle et al., 1993). To summarize, we have shown that the C-terminal TMH of Voa1 is an integral part of the mature yeast Vo complex, and that the assembly factor contributes to the stability of the V-ATPase membrane sector.
The Proton Path
According to current models of proton transport by rotary ATPases, two laterally offset aqueous half-channels (or “cavities”) in subunit a near the interface to the c-ring provide access for protons from the cytoplasm and lumen (or outside of the cell) to the sites of protonation and deprotonation of the c-ring’s essential glutamates (or aspartates) (Vik and Antonio, 1994). However, how and where protons are transferred during c-ring rotation, is still not fully understood. Our structure of VoND provides an atomic picture of how essential residues in subunit a and the c-ring interact to constitute the proton translocation pathway.
Aqueous access to cytoplasmic and luminal cavities in subunit a
aCT contains eight TMHs (TMH1-8; hereafter referred to as aCT1-8), with additional short α helical segments at the cytoplasmic and luminal sides (Figure 4A; TM helix numbering as in ref.(Toei et al., 2011)). Our map and model revealed two prominent aqueous cavities, one with access from the cytoplasmic, and another with access from the luminal side of the complex. The cytoplasmic cavity is formed by the cytoplasmic ends of aCT4, 5, 7, and 8, and lined by charged and polar residues including E721, N725 and H729 (Figure 4A). Aqueous access from the lumen is via the loops connecting aCT3, 4, 7, and 8, as well as the luminal loop of subunit f, with several polar residues inside the cavity including D425, D481 and H743 (Figure 4A). In contrast to the cytoplasmic cavity, aqueous access from the luminal cavity to the c-ring appears to be blocked by aCT7 and 8, reminiscent of what has been proposed for F-ATPase (Allegretti et al., 2015). Importantly, mutations of negatively charged residues in the cytosolic and luminal cavities have resulted in functional defects (Toei et al., 2011), suggesting that the negative charge distribution in the cavities is critical for proper proton transport.
Figure 4. Subunit a and the proton path.
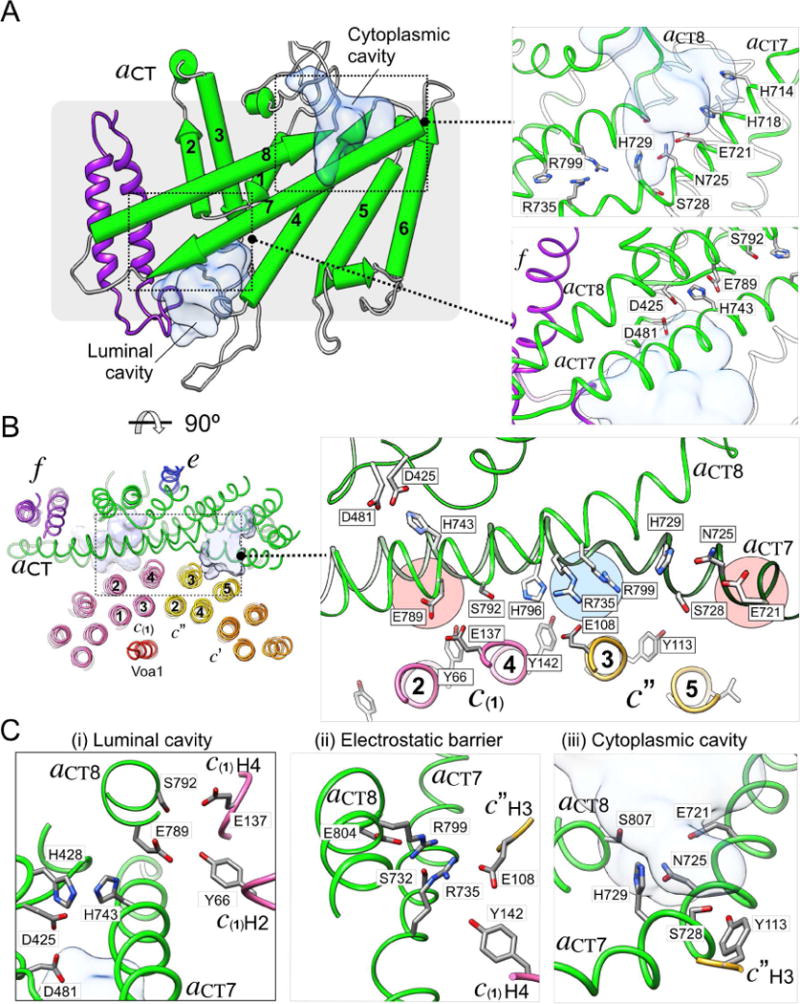
(A) Schematic of aCT showing TMHs 1-8 (aCT1-8), with the cytosolic and luminal aqueous access cavities shown in the top and bottom insets annotated with key amino acid residues, respectively. (B) Interface between aCT and the c-ring as seen from the cytoplasm. The interface buries aCT7 and 8 and the outer surface of c(1) and c″. Some of the residues that mediate the interaction between c-ring and aCT are shown in the zoomed-in view; (i) Residues near the luminal cavity, (ii) the electrostatic barrier (aR735 and aR799), and (iii), the cytosolic access cavity. See also Figure S4 and Movie S4.
Molecular contacts at the interface between aCT and the c-ring
In our structure of autoinhibited Vo, the aCT-c-ring interface is composed of aCT7 and 8, and the outer TMHs of c(1) and c″, with a total buried surface area of ~2100 Å2 (Figure 4B, left panel). It is noteworthy that the aCT-c-ring interface is the best resolved region in our map, suggesting that the interface is in a low energy state stabilized by multiple side chain interactions (Figure S1F, S2A). The side chain densities in the aCT-c-ring interface (Figure S4B, Movie S4) provide a picture of the residues involved in proton transport at a level of detail not seen in previous studies (Figure 4B, right panel). The nearly horizontal aCT7 and 8, a structural hallmark of rotary ATPase membrane sectors, contain numerous strictly conserved and polar residues (Figure S4A), many of which are essential for efficient proton pumping (Figure 4B, right panel, and Figure S4A) (Toei et al., 2011). Our structure shows that many of these residues are in close proximity to the essential glutamates of the c-ring (Figure 4B). Most notably, the two essential arginines of aCT (R735, R799) are located at the center of the interface, and, together with the neighboring H729 and H796, represent a physical and electrostatic barrier in the middle of the membrane that separates the cytoplasmic and luminal cavities (Figure 4B, blue highlight in right panel). This barrier may function to prevent direct transfer of protons between the cytosolic and luminal cavities (Figure 4B, red highlights in right panel, and Figure S4C, D), and thus ensure that protons travel along with the c-ring clockwise through the lipid bilayer (see Figure 5A below). Notable interactions resolved at the aCT-c-ring interface are between c(1)E137 and aCT’s S792 (Figure 4C(i)), and most prominently, a salt bridge between c″E108 and aCT’s essential R735 at the center of the interface (Figure 4C(ii)). Another interaction is between c″Y113, and aCTS728, which are seen at H-bond distance (Figure 4C(iii)). c″Y113 together with multiple other conserved tyrosines (Y66, Y142 in c(2)-(8), Y72 and Y150 in c′) constitute a circular, belt-like structure located at the same height on the outer TMHs of the c-ring (Figure S4E). The interaction of these tyrosines with polar residues in aCT7 and 8 likely function to maintain the proper proximity between c-ring and aCT during c-ring rotation.
Figure 5. Mechanistic model of proton pumping.
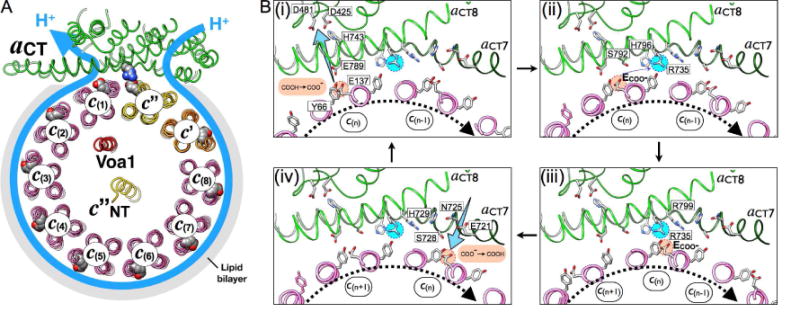
(A) View of our structure from the cytoplasm illustrating the proton path in Vo. (B) Mechanistic model of c-ring-aCT interactions upon c-ring clockwise rotation during V-ATPase driven proton pumping. One glutamate (E137) in one c-ring subunit (c(n)) is deprotonated by passing its H+ to E789 at the luminal cavity (i), and the H+ is delivered to the lumen through a sequence of polar residues (H743, D425 and D481 in aCT); (ii), (iii) The deprotonated E137 is stabilized by interactions with S792, H796 (ii), and attracted by the positively charged R735, forming a salt bridge (iii); (iv) Further rotation breaks the salt bridge and brings the glutamate into contact with the cytoplasmic cavity, where it is re-protonated from the cytoplasm near E721. See also Movie S5.
Mechanism of proton translocation in holo V-ATPase
Eukaryotic V-ATPase is a dedicated proton pump, with the free energy of ATP hydrolysis generated on V1 coupled to clockwise rotation of the c-ring past aCT (when viewed from the cytoplasm towards the membrane) (Figure 5A). Based on our structural observation (Figure 4) and the available mutagenesis data (Toei et al., 2011), we can begin to rationalize the mechanism of proton translocation across holo V-ATPase’s Vo membrane sector during ATP hydrolysis driven proton pumping. To illustrate the mechanism, we modeled several intermediate positions of the c-ring relative to aCT to highlight the steps of de-protonation, salt bridge formation, and re-protonation (Figure 5B(i)–(iv) and Movie S5). These three steps are described in more detail in the following paragraphs.
Deprotonation of the essential c-ring glutamic acid
During c-ring rotation, protonated c-ring glutamic acid residues (E-COOH; e.g. E137 on c(n)) emerge from the lipid bilayer and encounter the essential E789 on aCT8 (Figure 5B(i)). E789 is strictly conserved in eukaryotic V-ATPases, and appears well positioned for shuttling protons from the essential glutamates on the c-ring to the luminal cavity (Figure 4C(i). E137 and E789 have similar pKa values (~7.8; as predicted by PROPKA (Rostkowski et al., 2011)), a prerequisite for efficient proton transfer. However, the two residues in our model appear to be too distant for a direct interaction, and we therefore propose that proton transfer from E137 to E789 occurs via H-bonding to the tyrosine residue that is part of the proton binding site (e.g. Y66 in Figure 5B(i)). In this model, E789 would then deliver the proton to the luminal aqueous cavity through a network of critical residues, including H743, D425, and D481 (Figure 4A). H743 is on the luminal border in our structure, consistent with previous solvent accessibility experiments (Toei et al., 2011).
Movement of the essential c-ring glutamates between luminal and cytoplasmic cavities
After deprotonation at E789, charged c-ring glutamates must traverse the space in the aCT-c-ring interface that separates the luminal from the cytoplasmic cavities. While it is ionized, the glutamate can interact sequentially with aCT residues S792, H796 (Figure 5B(ii)), and finally R735 (Figure 5B(iii)). The c-ring glutamates’ consecutive interactions with S792 and H796 before engaging in a salt bridge with the positively charged barrier residue R735 ensure the deprotonation of the essential glutamates near the luminal cavity. The importance of the hydroxyl and imidazole groups of S792 and H796, respectively, is highlighted by previous mutagenesis experiments showing that replacement of these two residues with alanine or phenylalanine, respectively, results in significantly reduced enzyme activity (Toei et al., 2011). Moreover, it has been speculated that the negatively charged carboxylates are not only stabilized by opposing, polar residues of aCT (including S792 and H796 in yeast V-ATPase), but also by structural water molecules that may be part of the aCT-c-ring interface (Pogoryelov et al., 2010).
Re-protonation of the essential c-ring glutamates
ATP hydrolysis on V1 then provides the free energy to break the salt bridge between R735 and the c-ring glutamates so that the c-ring can continue the rotation (Figure 5B(iv)). Since aqueous access to the middle of the membrane is blocked by H729 and several bulky hydrophobic residues contributed by aCT and the c-ring, re-protonation of the essential c-ring glutamates then occurs upon contact with aCT’s S728 and E721 at the cytoplasmic cavity, with the essential E721 likely playing a key role (E-COO− → E-COOH; Figure 5B(iv)). After receiving a proton from the cytoplasmic access cavity, the now neutral c-ring glutamic acid is stabilized by H-bond formation with its neighboring tyrosine so that it can reenter the lipid bilayer to complete the transfer of one proton from the cytoplasm to the lumen. Our structure is consistent with previous accessibility experiments using chemical probes, which indicated that E721 is at the border to the cytoplasmic cavity (Toei et al., 2011). It is noteworthy that this glutamic acid at the cytoplasmic access cavity is highly conserved not only in V-ATPase but also more distantly related rotary motor ATPases, with the glutamate either 3 or 4 helical turns upstream of the essential arginine residue. For example, in E. coli F-ATPase, E196, which is 14 residues away from R210, the same distance as between E721 and R735 in yeast Vo, is the only residue in the cytoplasmic access channel of that enzyme identified to date to be essential for proton translocation (Vik et al., 1988). Though our structure does not have sufficient resolution to visualize water molecules, solvent accessibility prediction using PROPKA reveals that E721 is >95% exposed to solvent with a predicted pKa of ~6.5, suggesting that the residue is mostly ionized at the pH of the cytoplasm (~7), and therefore coordinated by water molecules most of the time. In summary, E721’s location at the bottom of the cytoplasmic aqueous cavity, its accessibility to solvent and essential nature as revealed by mutagenesis studies, and the residue’s level of conservation in related proton translocating rotary motor enzymes provide strong evidence that E721 plays a key role in the final steps of the re-protonation of the essential c-ring glutamates (Figure 4B(iii), 5B(iv)).
More recently, a 3.6 Å cryoEM structure of dimeric yeast mitochondrial F-ATPase was reported (Guo et al., 2017), in which the two proton carrying glutamates of the c subunits in the a-c-ring interface are also in an extended conformation. However, none of these two carboxyl groups is close enough to the essential arginine for salt bridge formation, suggesting that yeast Fo was captured in a functional state different from the autoinhibited Vo. As mentioned above, unlike free Vo, isolated Fo catalyzes passive proton transport, and the conformation visualized in the yeast Fo structure may therefore represent a snapshot of the ‘active’ state of the proton channel.
Interpretation of loss-of-function mutations in yeast and human
Yeast subunit a
Site directed mutagenesis studies with yeast V-ATPase identified highly conserved residues within aCT that are critical for ATPase driven proton pumping and/or stability of the complex (Toei et al., 2011)(Figure 6A). Based on our atomic model of yeast Vo, we can now begin to understand the mechanism by which mutagenesis of these essential residues leads to functional defects. Not surprisingly, a majority of the identified residues that are essential for proton pumping activity are located along the proton path identified in our model. In particular, mutations of S732, H796, R799, R735, E789, and E721 that face the essential glutamates of the c-ring displayed significant activity defects, consistent with our proposed mechanism of proton pumping. Interestingly, three other mutants H801, K536, and S740 do not seem to reside along the proton path. Therefore, these residues may contribute to the overall structural stability of subunit a through intra subunit interaction (Figure 6A). For example, H801 in aCT8 and T414 in aCT1, and K536 in aCT4 and S740 in aCT7, respectively, are in H-bond distance, consistent with above hypothesis. Strikingly, mutations of three polar residues (K538, S534, and K593) that are not part of the proton path are in direct contact with the density of unknown identity, which is present in both independently refined half maps, suggesting that this unknown component of Vo may be important for V-ATPase function or stability.
Figure 6. Mechanistic interpretation of loss-of-function mutations in subunit a of yeast and human V-ATPase.
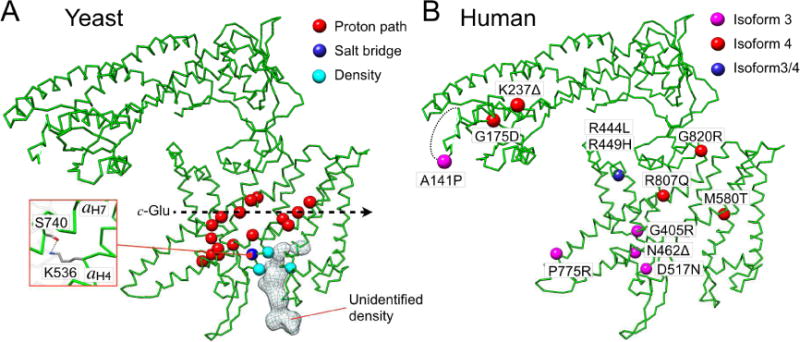
(A) Residues indicated by space fill Cαs (red circle) are implicated in V-ATPase activity based on mutagenesis and functional studies in yeast (Toei et al., 2011). Some of the residues are found to make contact with the unidentified density at the luminal side of aCT. S740 and K536 appear to interact via H-bond (box). (B) Disposition of the disease-causing mutations in isoforms 3 and 4 of human V-ATPase subunit a. See also Figure S5 and Table S1.
Human subunit a
The basic structure of the yeast V-ATPase complex is preserved in the enzyme from higher organisms, including human. There are four subunit a isoforms in human (a1-4), with a1 found in neurons, a2 in endothelial cells, a3 in osteoclast ruffled membrane and pancreatic β cells, and a4 in the kidney (Toei et al., 2010). Sequence identity between yeast and human subunit a isoforms is between 30-50% (up to 85% similarity), allowing reliable modeling of human subunit a isoforms using our model as template. Mutations in a3 and a4 have been associated with osteopetrosis and kidney disease, respectively (Smith et al., 2000; Sobacchi et al., 2001; Stover et al., 2002; Susani et al., 2004) (Table S1). Nine out of the reported twelve disease causing mutations are located in aCT, with five mutations in the putative proton pathway (Figure 6B). The mutations R444L (a3) and R449H (a4) (R462 in yeast) likely disrupt salt bridge formation with a neighboring E424L (a3) and E429H (a4) (E443 in yeast), which would result in a destabilization of aCT. Interestingly, G220R, K237Δ, and A131Δ are located at the distal domain of aNT (Figure 6B), which undergoes a large conformational change upon V-ATPase dissociation or (re)association (Couoh-Cardel et al., 2015). K237 interacts with subunit d in free Vo through an electrostatic interaction, and A131 is part of the disordered loop (153-183) that is in contact with peripheral stator EG3 in the holo V-ATPase (Figure S5). This suggests that the conformational change of aNT plays a role in activity regulation during reversible enzyme disassembly.
Voa1, the yeast homologue of mammalian Ac45
Our structure reveals that the assembly factor Voa1 is a structural component of the mature Vo complex. Voa1 has recently been recognized as the yeast ortholog of mammalian Ac45 (ATP6AP1), a ~45 kDa glycosylated polypeptide that is found to be associated with the V-ATPase in some tissues (Jansen et al., 2016; Smith et al., 2016). However, the evolutionary relationship between Voa1 and Ac45 is limited to the proteins’ C-terminal TMH, and it has been shown that the C-terminal TMH of Ac45 can complement the Voa1 deletion phenotype in yeast. Previous work has shown that Ac45 is present in both free Vo and holo V1Vo, and that the 45 kDa glycosylated protein is bound to V-ATPase’s Vo membrane sector on the luminal side of the complex via its C-terminal TMH (Wilkens and Forgac, 2001). Of note, the density in VoND here identified as belonging to Voa1’s C-terminus is also observed in holo V-ATPase (Mazhab-Jafari et al., 2016), indicating that the assembly factor does not prevent functional binding of V1 to Vo in vivo. Besides providing evidence for the homology of Ac45’s and Voa1’s C-termini, Jansen et al. also reported that certain mutations in Ac45 are associated with an immunodeficiency phenotype, including a mutation M428I (M225 in Voa1), that is predicted to be part of the C-terminal TMH (Jansen et al., 2016). Our results show that Voa1 is an integral part of Vo, and that the assembly factor contributes to the stability of the mature complex as well as the recruitment of subunit d (Figure 3). However, while the C-termini of Voa1 and Ac45 appear to fulfill similar tasks, further studies will be required to elucidate the function of the luminal domain of Voa1.
CONCLUSIONS
Here, we present the 3.5 Å resolution cryoEM structure of yeast Vo reconstituted into lipid nanodiscs. The isotropic resolution of the reconstruction highlights the potential of lipid nanodisc reconstitution of membrane protein complexes for high-resolution structure determination by cryoEM as previously demonstrated for the TRPV1 channel (Gao et al., 2016). We show that the assembly factor Voa1 is a part of the mature V-ATPase membrane sector and contributes to the structural stability of the proteolipid ring. We provide an atomic picture of the essential residues along the proton path and propose a mechanistic model of proton translocation that is well supported by a large body of available biochemical and functional data. Moreover, our structure provides a foundation for the study of V-ATPase related human diseases, information that may help with future development of therapeutic interventions.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for further information as well as resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephan Wilkens (wilkenss@upstate.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strain construction
Yeast VOA1Δ mutant in the BY4741 strain background was purchased as part of a yeast deletion mutant array from Open Biosystems (Pittsburgh, PA). voa1Δ::kanMX allele was PCR amplified from the mutant strain with oligonucleotides upstream and downstream of the locus: voa1-5′utr: GCA ACA GTA CGA TTA TTA CAC TGA CTA TGC TGC AG; voa1-3′utr: GGC CAT TGC AGC AGC TAA ACC TCC AC. The fragment was transformed into a Vo purification strain in the BY4741 background with C-terminal calmodulin binding peptide (CBP) tag on subunit a (vph1-CBP::ura3) (Couoh-Cardel et al., 2015). Transformants were selected on SD-ura+G418 media.
METHOD DETAILS
Purification of Vo from yeast microsomal membranes
Protein purification of native Vo and Vo in a voa1Δ background was performed as described previously (Stam and Wilkens, 2017). Briefly, cells were harvested during the second log phase of growth, broken by bead beating, and subjected to a differential centrifugation protocol to isolate microsomal membranes. Isolated membranes were solubilized using 0.6 mg of undecylmaltoside per mg of membrane protein and affinity purified via the CBP-tag with calmodulin sepharose (GE Life Sciences). Vo was reconstituted into lipid nanodisc as described previously (Stam and Wilkens, 2017). Briefly, detergent solubilized Vo, membrane scaffold protein (MSP1E3D1) and E. coli total lipid extract (Avanti Polar Lipids) were mixed in reconstitution buffer (20 mM Tris/HCl pH 7.4, 100 mM NaCl, 0.5 mM EDTA) at a molar ratio of 0.02:1:25, and subjected to detergent removal by Bio-Beads SM-2 (BioRad). Reconstitution was performed at 25 °C. Reconstituted samples were applied to calmodulin sepharose for removal of unfilled nanodisc, and subjected to size exclusion chromatography on a Superdex 200 column. Protein containing peak fractions were concentrated and stored at −80 °C until use. For the cryoEM image collection of the dataset presented here, frozen aliquots of VoND were thawed quickly and subjected to size exclusion chromatography on Superose 6GL (10 mm × 300 mm column) in 20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.5 mM EDTA, 0.5 mM TCEP) as a final polishing step. Peak fractions were taken straight for cryoEM sample preparation.
Purification of V1Vo from yeast vacuolar vesicles and mass spectrometry
Holo V-ATPase was isolated from a yeast strain expressing N-terminally FLAG tagged subunit G and reconstituted into lipid nanodiscs as described (Sharma and Wilkens, 2017). Briefly, a 12 l culture grown to an OD of 1 was converted to spheroplasts and lysed with 10 strokes in a Dounce homogenizer in 12% Ficoll containing buffer. The lysed spheroplasts were overlaid with 12% Ficoll containing buffer and centrifuged in a SW 32 Ti swinging bucket rotor at 24,000 rpm for 40 minutes. Vacuoles were scooped from the top, homogenized in 12% Ficoll containing buffer, overlaid with 8% Ficoll and centrifuged under the same conditions. Purified vacuoles had a Concanamycin A sensitive MgATPase activity of 1.6 U/mg. Vacuoles were solubilized with 1.2 mg/mg dodecyl maltoside, reconstituted with 1.5 mg/mg membrane scaffold protein, and detergent was removed with polystyrene beads (BioBeads SM-2). Nanodisc reconstituted V1Vo (V1VoND) was purified from total nanodisc reconstituted vacuolar membrane protein using 1 ml anti FLAG affinity resin and eluted with 0.1 mg/ml FLAG peptide. For mass spectrometry, V1VoND containing fractions were precipitated in presence of 10% TCA and precipitated protein was re-dissolved in 8 M urea, 100 mM Tris-HCl, pH 8, 5 mM DTT. Protein samples were heated at 65 °C for 10 minutes, alkylated with 15 mM iodoacetate and digested with mass spectrometry grade trypsin and chyomotrypsin according to the manufacturer’s (Promega) instructions. V1VoND samples were also resolved by SDS-PAGE to generate in-gel tryptic digests followed by mass spectrometry analysis on a Thermo LTQ Orbitrap mass spectrometer.
Cryo-EM specimen preparation and imaging
3 μl aliquots of VoND peak fractions from the Superose size exclusion chromatography run (0.3 mg/ml) were applied to glow-discharged gold grids (UltrAuFoil™ 1.2/1.3), blotted for 2~3 s, and then plunge-frozen in liquid ethane using a Leica EM GP (Leica). The grids were transferred into cartridges, loaded into an JEM3200FSC (300 KeV) electron-microscope with in-column omega filter (25e−V), and images were recorded at 0.7–3.5 m underfocus on a K2 summit direct electron detector (Gatan) with super-resolution mode at nominal 30K magnification corresponding to a sampling interval of 1.23Å/pixel (Super resolution 0.62 Å/pixel). Each micrograph was exposed for 10 sec with 5 e−/A2/sec dose rate (total specimen dose, 50 e−/A2), and ~50 frames were captured per specimen area using serial-EM.
Image processing
Each movie stack was initially binned by 2 and then corrected for drift and radiation damage using UNBLUR (Grant and Grigorieff, 2015) for total frames. From an initial data set, ~3,100 particles from 20 images were automatically picked by EMAN2 and the contrast transfer function (CTF) parameters were estimated internally based on the boxed particles (e2ctf.py)(Ludtke et al., 1999). Extracted particles were subjected to reference-free 2D classification and the resulting 2D class averages were used to generate an initial 3D model with C1 symmetry in EMAN2. For all the images, CTF was determined with CTFFIND4 (Rohou and Grigorieff, 2015). A total of 285,000 particles were automatically selected from 1,741 micrographs based on 2D references generated in EMAN2 using RELION1.4 (Scheres, 2012). After 2D class average and 3D classification, low-convergence classes were discarded and the remaining 193,646 particles were subjected to 3D auto-refinement in RELION. At this stage, the resulting map had a resolution of ~4.2 Å as determined by the gold-standard protocol implemented in RELION. The particles from 3D auto-refinement were then submitted to particle-based motion correction and radiation-damage weighting (Scheres, 2014). After another round of 3D classification using ‘polished particles’, 180,528 particles were then subjected to 3D auto-refinement and the resulting map had a resolution of 3.9 Å. To minimize potential errors from the signal of the lipid nanodisc, a solvent mask around the protein was used in additional iterations as focused refinement (Nguyen et al., 2015), and this focused refinement improved the resolution of the final map to 3.5 Å according to the gold-standard FSC at 0.143 correlation. The density map and resolution was validated by performing an additional round of reconstruction with original raw particle images after the Fourier phases were randomized beyond 7 Å using the same image processing protocol (Chen et al., 2013). The local resolution of the density map was assessed by ResMap (Kucukelbir et al., 2014).
Model building
To generate the molecular model, homology models of subunit d and the c-ring (Couoh-Cardel et al., 2016) and portions of previously published models (PDBIDs: 5TJ5, 3J9V; (Mazhab-Jafari et al., 2016; Zhao et al., 2015)) were fit into the density map using Chimera’s Fit in Map Tool (Pettersen et al., 2004). Subunit a had the largest amount of unknown density. At this resolution, its protein boundaries could be visually determined, even in regions lacking an associated model. A 30 Å color zone in Chimera was then used to segment density around the known modeled regions. Not all of subunit a had previously been modeled, and thus the extra-large segmentation could encompass the excess density. The de novo modeling pipeline (Baker et al., 2010) was then used to complete various regions that were not previously modeled, including the N-terminal domain of subunit a (aNT), subunits e, f, and large portions of subunit d (Figure 2, S2A, Movie S2). Here, the secondary structure prediction was assessed against predicted α helices in the density map, focusing on helix length, disorder and transmembrane regions; all predictions were based on the PHYRE2 Protein Fold Recognition Server (Kelley et al., 2015). Once the predicted α helices were positioned, large aromatic amino acids were used as anchors to register α helices within the density. Loop regions were then built connecting the assigned helices. Subunit a was then optimized against the segmented density alone, focusing on the de novo regions with Phenix.real_space_refine using default parameters plus simulated annealing (Adams et al., 2010). This quickly adjusted the fit to density for the individual subunit. The larger complex was then optimized with Phenix.real_space_refine (default parameters) using the complete 3.5Å density map as a constraint. Coot (Emsley and Cowtan, 2004) was then used to manually adjust loop regions that did not converge into the density. Ramachandran outliers were manually adjusted, in addition to amino acids with distortions in bond lengths and angles. Phenix.refine using structure factors generated by Phenix.map_to_structure_factors was then used to further optimize the model with stronger geometry restraints. Hydrogen atoms were included to improve geometry in later refinement cycles. Multiple iterations of reciprocal-space model optimization employing torsion-angle non-crystallographic symmetry (NCS) restraints, manual model building in Coot, and real-space optimization were carried out until no further improvement could be obtained. Model quality was assessed by analysis with Molprobity (Davis et al., 2004) and EMRinger (Barad et al., 2015), and the final map and model were used to model the atom positional uncertainty (atomic displacement parameters (ADP) (Roh et al., 2017; Trueblood, 1996)(Figure S2A, Movie S2). Buried surface areas were analyzed using PISA as implemented in the CCP4 program suite (Krissinel and Henrick, 2007). Aqueous cavities were analyzed using 3v (Voss and Gerstein, 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification, statistical analysis and validation are implemented in the software packages used for 3-D reconstruction and model refinement.
DATA AND SOFTWARE AVAILABILITY
The 3D cryoEM density map has been deposited in the Electron Microscopy Data Bank under the accession number EMD: EMD-7348. Coordinates have been deposited in the Protein Data Bank under the accession number PDB: 6C6L.
Supplementary Material
Near-atomic resolution density revealing both global and local features of VoND. Individual proteins have been segmented and colored based on the model. Unmodeled densities (in or around the complex) are shown in gray. These unmodeled densities may be attributed to lipid, potential ions, disordered protein, and/or noise. It should be noted that some of these unmodeled densities appear in both the even and odd density maps, which would indicate that they are actual map features.
Map and model of amphipol solubilized Vo (EMDB-8409; PDB5TJ5). Map densities are contoured at thresholds to demonstrate the match with reported models. These thresholds are reported in the EMDB with their associated map. Color designation of the subunits is the same as in Movie S1. Grey color refers to densities that are not modeled.
The following demonstrates the cryoEM map/model quality at both the global and local level. Density is colored based on a 2.5 Å radius around each coordinate that was assigned. Again, unmodeled density is in gray as in Movie S1.
aCT7 and 8 form an interface with the c-ring. Isolated experimental map density for the aromatic residues helped to anchor the model during refinement, ensuring that the helical register was modeled correctly. Important residues identified by activity defects upon mutagenesis (Toei et al., 2011) are highlighted, and either point towards the viewer, or towards the aCT-c-ring interface.
Essential c-ring glutamic acid residues (e.g. E137 on c(n)) are deprotonated in the chemical environment of the essential E789 in aCT8. As the c-ring rotates clockwise (as viewed from the cytosol towards the membrane), the deprotonated E-COO− is attracted to the electrostatic barrier between the offset luminal and cytoplasmic cavities, and forms a transient salt bridge with R735 in aCT7. Re-protonation of essential c-ring glutamates occurs upon entrance into the cytoplasmic cavity between S728 and E721. After receiving a proton in the chemical environment of E721, the neutralized c-ring glutamic acid can then re-enter the lipid bilayer.
Highlights.
-
–
Atomic cryoEM structure of yeast V-ATPase Vo proton channel sector in lipid nanodisc
-
–
Proton translocation pathway with atomic details
-
–
The C-terminal transmembrane α helix of the assembly factor Voa1 is part of the Vo
-
–
Functional interpretation of disease causing mutations in Human V-ATPase
Acknowledgments
This work has been supported by the National Institutes of Health grants GM058600 to SW and P41GM103832 and R01GM079429 to WC. CFH was supported by the pre-doctoral fellowship under the National Library of Medicine Training Program in Biomedical Informatics (Grant No. T15LM007093) awarded to the Keck Center of the Gulf Coast Consortia. The cryoEM data was collected at the National Center of Macromolecular Imaging and processed using the Computational and Integrative Biomedical Research Center (CIBR) at Baylor College of Medicine. We are grateful for the computing resources at the Texas Advanced Computer Center in University of Texas at Austin. We thank Drs. Steven J. Ludtke, Matthew L. Baker and Michael F. Schmid for helpful discussion on cryoEM data processing and modeling; Dr. Ebbing de Jong and SUNY Upstate’s mass spectrometry core facility for performing the mass spectrometry analysis; Dr. Rebecca Oot for help with multiple sequence alignment and critical reading of the manuscript, Stuti Sharma for help with purifying holo V-ATPase from vacuolar vesicles, and both Dr. Rebecca Oot and Stuti Sharma for many helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, W.C., and S.W.; Investigation, S.-H.R., N.J.S., C.F.H., S.C.-C., G.P., and S.W.; Writing, S.-H.R., N.J.S., C.H., G.P., W.C., and S.W.; Funding Acquisition, W.C. and S.W.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M, Klusch N, Mills DJ, Vonck J, Kühlbrandt W, Davies KM. Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature. 2015;521:237–240. doi: 10.1038/nature14185. [DOI] [PubMed] [Google Scholar]
- Baker ML, Baker MR, Hryc CF, Dimaio F. Analyses of subnanometer resolution cryo-EM density maps. Methods Enzymol. 2010;483:1–29. doi: 10.1016/S0076-6879(10)83001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad BA, Echols N, Wang RY, Cheng Y, DiMaio F, Adams PD, Fraser JS. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat Methods. 2015;12:943–946. doi: 10.1038/nmeth.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerle C, Ho MN, Lindorfer MA, Stevens TH. The Saccharomyces cerevisiae VMA6 gene encodes the 36-kDa subunit of the vacuolar H(+)-ATPase membrane sector. J Biol Chem. 1993;268:12749–12757. [PubMed] [Google Scholar]
- Bodzeta A, Kahms M, Klingauf J. The Presynaptic v-ATPase Reversibly Disassembles and Thereby Modulates Exocytosis but Is Not Part of the Fusion Machinery. Cell Rep. 2017;20:1348–1359. doi: 10.1016/j.celrep.2017.07.040. [DOI] [PubMed] [Google Scholar]
- Brown D, Smith PJ, Breton S. Role of V-ATPase-rich cells in acidification of the male reproductive tract. J Exp Biol. 1997;200:257–262. doi: 10.1242/jeb.200.2.257. [DOI] [PubMed] [Google Scholar]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, Henderson R. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013;135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couoh-Cardel S, Hsueh YC, Wilkens S, Movileanu L. Yeast V-ATPase Proteolipid Ring Acts as a Large-conductance Transmembrane Protein Pore. Sci Rep. 2016;6:24774. doi: 10.1038/srep24774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couoh-Cardel S, Milgrom E, Wilkens S. Affinity Purification and Structural Features of the Yeast Vacuolar ATPase Vo Membrane Sector. J Biol Chem. 2015;290:27959–27971. doi: 10.1074/jbc.M115.662494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ediger B, Melman SD, Pappas DL, Jr, Finch M, Applen J, Parra KJ. The tether connecting cytosolic (N terminus) and membrane (C terminus) domains of yeast V-ATPase subunit a (Vph1) is required for assembly of V0 subunit d. J Biol Chem. 2009;284:19522–19532. doi: 10.1074/jbc.M109.013375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Flannery AR, Graham LA, Stevens TH. Topological Characterization of the c, c′, and c″ Subunits of the Vacuolar ATPase from the Yeast Saccharomyces cerevisiae. J Biol Chem. 2004;279:39856–39862. doi: 10.1074/jbc.M406767200. [DOI] [PubMed] [Google Scholar]
- Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- Gao Y, Cao E, Julius D, Cheng Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature. 2016;534:347–351. doi: 10.1038/nature17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer M, Yu H, Mandic R, Linnemann T, Zheng YH, Fackler OT, Peterlin BM. Subunit H of the V-ATPase binds to the medium chain of adaptor protein complex 2 and connects Nef to the endocytic machinery. J Biol Chem. 2002;277:28521–28529. doi: 10.1074/jbc.M200522200. [DOI] [PubMed] [Google Scholar]
- Gräf R, Harvey WR, Wieczorek H. Purification and properties of a cytosolic V1-ATPase. J Biol Chem. 1996;271:20908–20913. doi: 10.1074/jbc.271.34.20908. [DOI] [PubMed] [Google Scholar]
- Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. eLife. 2015;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Bueler SA, Rubinstein JL. Atomic model for the dimeric FO region of mitochondrial ATP synthase. Science. 2017;358:936–940. doi: 10.1126/science.aao4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata R, Graham LA, Takatsuki A, Stevens TH, Anraku Y. VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+-ATPase. J Biol Chem. 1997;272:4795–4803. doi: 10.1074/jbc.272.8.4795. [DOI] [PubMed] [Google Scholar]
- Inoue H, Noumi T, Nagata M, Murakami H, Kanazawa H. Targeted disruption of the gene encoding the proteolipid subunit of mouse vacuolar H(+)-ATPase leads to early embryonic lethality. Biochim Biophys Acta. 1999;1413:130–138. doi: 10.1016/s0005-2728(99)00096-1. [DOI] [PubMed] [Google Scholar]
- Jansen EJ, Timal S, Ryan M, Ashikov A, van Scherpenzeel M, Graham LA, Mandel H, Hoischen A, Iancu TC, Raymond K, et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun. 2016;7:11600. doi: 10.1038/ncomms11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane PM. Disassembly and reassembly of the yeast vacuolar H(+)-ATPase in vivo. J Biol Chem. 1995;270:17025–17032. [PubMed] [Google Scholar]
- Kane PM. The where, when, and how of organelle acidification by the yeast vacuolar H+-ATPase. Microbiol Mol Biol Rev. 2006;70:177–191. doi: 10.1128/MMBR.70.1.177-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- Kartner N, Manolson MF. Novel techniques in the development of osteoporosis drug therapy: the osteoclast ruffled-border vacuolar H(+)-ATPase as an emerging target. Expert Opin Drug Discov. 2014;9:505–522. doi: 10.1517/17460441.2014.902155. [DOI] [PubMed] [Google Scholar]
- Kawasaki-Nishi S, Nishi T, Forgac M. Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc Natl Acad Sci U S A. 2001;98:12397–12402. doi: 10.1073/pnas.221291798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F, Hennecke S, Demolliere C, Cosson P. Steric masking of a dilysine endoplasmic reticulum retention motif during assembly of the human high affinity receptor for immunoglobulin E. J Cell Biol. 1995;129:971–978. doi: 10.1083/jcb.129.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Mazhab-Jafari MT, Rohou A, Schmidt C, Bueler SA, Benlekbir S, Robinson CV, Rubinstein JL. Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nature. 2016;539:118–122. doi: 10.1038/nature19828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muench SP, Trinick J, Harrison MA. Structural divergence of the rotary ATPases. Q Rev Biophys. 2011;44:311–356. doi: 10.1017/S0033583510000338. [DOI] [PubMed] [Google Scholar]
- Nguyen TH, Galej WP, Bai XC, Savva CG, Newman AJ, Scheres SH, Nagai K. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature. 2015;523:47–52. doi: 10.1038/nature14548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oot RA, Couoh-Cardel S, Sharma S, Stam NJ, Wilkens S. Breaking up and making up: The secret life of the vacuolar H+ -ATPase. Protein Sci. 2017;26:896–909. doi: 10.1002/pro.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oot RA, Kane PM, Berry EA, Wilkens S. Crystal structure of yeast V1-ATPase in the autoinhibited state. The EMBO journal. 2016;35:1694–1706. doi: 10.15252/embj.201593447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owegi MA, Pappas DL, Finch MW, Jr, Bilbo SA, Resendiz CA, Jacquemin LJ, Warrier A, Trombley JD, McCulloch KM, Margalef KL, et al. Identification of a domain in the V0 subunit d that is critical for coupling of the yeast vacuolar proton-translocating ATPase. J Biol Chem. 2006;281:30001–30014. doi: 10.1074/jbc.M605006200. [DOI] [PubMed] [Google Scholar]
- Parra KJ. The H Subunit (Vma13p) of the Yeast V-ATPase Inhibits the ATPase Activity of Cytosolic V1 Complexes. J Biol Chem. 2000;275:21761–21767. doi: 10.1074/jbc.M002305200. [DOI] [PubMed] [Google Scholar]
- Parra KJ, Kane PM. Reversible association between the V1 and V0 domains of yeast vacuolar H+-ATPase is an unconventional glucose-induced effect. Mol Cell Biol. 1998;18:7064–7074. doi: 10.1128/mcb.18.12.7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pogoryelov D, Krah A, Langer JD, Yildiz O, Faraldo-Gomez JD, Meier T. Microscopic rotary mechanism of ion translocation in the F(o) complex of ATP synthases. Nat Chem Biol. 2010;6:891–899. doi: 10.1038/nchembio.457. [DOI] [PubMed] [Google Scholar]
- Pogoryelov D, Yildiz O, Faraldo-Gómez JD, Meier T. High-resolution structure of the rotor ring of a proton-dependent ATP synthase. Nat Struct Mol Biol. 2009;16:1068–1073. doi: 10.1038/nsmb.1678. [DOI] [PubMed] [Google Scholar]
- Qi J, Forgac M. Function and subunit interactions of the N-terminal domain of subunit a (Vph1p) of the yeast V-ATPase. J Biol Chem. 2008;283:19274–19282. doi: 10.1074/jbc.M802442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh SH, Hryc CF, Jeong HH, Fei X, Jakana J, Lorimer GH, Chiu W. Subunit conformational variation within individual GroEL oligomers resolved by Cryo-EM. Proc Natl Acad Sci U S A. 2017;114:8259–8264. doi: 10.1073/pnas.1704725114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostkowski M, Olsson MH, Sondergaard CR, Jensen JH. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct Biol. 2011;11:6. doi: 10.1186/1472-6807-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M, Graham LA, Stevens TH. Voa1p functions in V-ATPase assembly in the yeast endoplasmic reticulum. Mol Biol Cell. 2008;19:5131–5142. doi: 10.1091/mbc.E08-06-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife. 2014;3:e03665. doi: 10.7554/eLife.03665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E, Altendorf K. All three subunits are required for the reconstitution of an active proton channel (F0) of Escherichia coli ATP synthase (F1F0) The EMBO journal. 1985;4:515–518. doi: 10.1002/j.1460-2075.1985.tb03658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennoune SR, Bakunts K, Martínez GM, Chua-Tuan JL, Kebir Y, Attaya MN, Martínez-Zaguilán R. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am J Physiol Cell Physiol. 2004;286:C1443–1452. doi: 10.1152/ajpcell.00407.2003. [DOI] [PubMed] [Google Scholar]
- Sharma S, Wilkens S. Biolayer interferometry of lipid nanodisc-reconstituted yeast vacuolar H+ -ATPase. Protein Sci. 2017;26:1070–1079. doi: 10.1002/pro.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–75. doi: 10.1038/79208. [DOI] [PubMed] [Google Scholar]
- Smith GA, Howell GJ, Phillips C, Muench SP, Ponnambalam S, Harrison MA. Extracellular and Luminal pH Regulation by Vacuolar H+-ATPase Isoform Expression and Targeting to the Plasma Membrane and Endosomes. J Biol Chem. 2016;291:8500–8515. doi: 10.1074/jbc.M116.723395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobacchi C, Frattini A, Orchard P, Porras O, Tezcan I, Andolina M, Babul-Hirji R, Baric I, Canham N, Chitayat D, et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet. 2001;10:1767–1773. doi: 10.1093/hmg/10.17.1767. [DOI] [PubMed] [Google Scholar]
- Stam NJ, Wilkens S. Structure of the Lipid Nanodisc-reconstituted Vacuolar ATPase Proton Channel: Definition of the interaction of rotor and stator and implications for enzyme regulation by reversible dissociation. J Biol Chem. 2017;292:1749–1761. doi: 10.1074/jbc.M116.766790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch ABS, Morton CC, Axon PR, Akil I, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky LA, Forgac M. Amino Acid Availability Modulates Vacuolar H+-ATPase Assembly. J Biol Chem. 2015;290:27360–27369. doi: 10.1074/jbc.M115.659128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner JP, Dow JA, Earley FG, Klein U, Jäger D, Wieczorek H. Regulation of plasma membrane V-ATPase activity by dissociation of peripheral subunits. J Biol Chem. 1995;270:5649–5653. doi: 10.1074/jbc.270.10.5649. [DOI] [PubMed] [Google Scholar]
- Sun-Wada GH, Toyomura T, Murata Y, Yamamoto A, Futai M, Wada Y. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J Cell Sci. 2006;119:4531–4540. doi: 10.1242/jcs.03234. [DOI] [PubMed] [Google Scholar]
- Susani L, Pangrazio A, Sobacchi C, Taranta A, Mortier G, Savarirayan R, Villa A, Orchard P, Vezzoni P, Albertini A, et al. TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum Mutat. 2004;24:225–235. doi: 10.1002/humu.20076. [DOI] [PubMed] [Google Scholar]
- Thudium CS, Jensen VK, Karsdal MA, Henriksen K. Disruption of the V-ATPase functionality as a way to uncouple bone formation and resorption - a novel target for treatment of osteoporosis. Curr Protein Pept Sci. 2012;13:141–151. doi: 10.2174/138920312800493133. [DOI] [PubMed] [Google Scholar]
- Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry. 2010;49:4715–4723. doi: 10.1021/bi100397s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toei M, Toei S, Forgac M. Definition of membrane topology and identification of residues important for transport in subunit a of the vacuolar ATPase. J Biol Chem. 2011;286:35176–35186. doi: 10.1074/jbc.M111.273409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science. 2003;299:1400–1403. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- Trueblood KN, Bürgi HB, Burzlaff H, Dunitz JD, Gramaccioli CM, Schulz HH, Shmueli U, Abrahams SC. Atomic Dispacement Parameter Nomenclature. Report of a Subcommittee on Atomic Displacement Parameter Nomenclature. Acta Crystallogr A. 1996;52:770–781. [Google Scholar]
- Vavassori S, Mayer A. A new life for an old pump: V-ATPase and neurotransmitter release. J Cell Biol. 2014;205:7–9. doi: 10.1083/jcb.201403040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vik SB, Antonio BJ. A mechanism of proton translocation by F1F0 ATP synthases suggested by double mutants of the a subunit. J Biol Chem. 1994;269:30364–30369. [PubMed] [Google Scholar]
- Vik SB, Cain BD, Chun KT, Simoni RD. Mutagenesis of the alpha subunit of the F1Fo-ATPase from Escherichia coli. Mutations at Glu-196, Pro-190, and Ser-199. J Biol Chem. 1988;263:6599–6605. [PubMed] [Google Scholar]
- Voss M, Vitavska O, Walz B, Wieczorek H, Baumann O. Stimulus-induced phosphorylation of vacuolar H(+)-ATPase by protein kinase A. J Biol Chem. 2007;282:33735–33742. doi: 10.1074/jbc.M703368200. [DOI] [PubMed] [Google Scholar]
- Voss NR, Gerstein M. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010;38:W555–562. doi: 10.1093/nar/gkq395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cipriano DJ, Forgac M. Arrangement of subunits in the proteolipid ring of the V-ATPase. J Biol Chem. 2007;282:34058–34065. doi: 10.1074/jbc.M704331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkens S, Forgac M. Three-dimensional structure of the vacuolar ATPase proton channel by electron microscopy. J Biol Chem. 2001;276:44064–44068. doi: 10.1074/jbc.M106579200. [DOI] [PubMed] [Google Scholar]
- Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 2011;108:19371–19376. doi: 10.1073/pnas.1109201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Denef N, Schupbach T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev Cell. 2009;17:387–402. doi: 10.1016/j.devcel.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Myers M, Forgac M. Characterization of the V0 domain of the coated vesicle (H+)-ATPase. J Biol Chem. 1992;267:9773–9778. [PubMed] [Google Scholar]
- Zhao J, Benlekbir S, Rubinstein JL. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature. 2015;521:241–245. doi: 10.1038/nature14365. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Near-atomic resolution density revealing both global and local features of VoND. Individual proteins have been segmented and colored based on the model. Unmodeled densities (in or around the complex) are shown in gray. These unmodeled densities may be attributed to lipid, potential ions, disordered protein, and/or noise. It should be noted that some of these unmodeled densities appear in both the even and odd density maps, which would indicate that they are actual map features.
Map and model of amphipol solubilized Vo (EMDB-8409; PDB5TJ5). Map densities are contoured at thresholds to demonstrate the match with reported models. These thresholds are reported in the EMDB with their associated map. Color designation of the subunits is the same as in Movie S1. Grey color refers to densities that are not modeled.
The following demonstrates the cryoEM map/model quality at both the global and local level. Density is colored based on a 2.5 Å radius around each coordinate that was assigned. Again, unmodeled density is in gray as in Movie S1.
aCT7 and 8 form an interface with the c-ring. Isolated experimental map density for the aromatic residues helped to anchor the model during refinement, ensuring that the helical register was modeled correctly. Important residues identified by activity defects upon mutagenesis (Toei et al., 2011) are highlighted, and either point towards the viewer, or towards the aCT-c-ring interface.
Essential c-ring glutamic acid residues (e.g. E137 on c(n)) are deprotonated in the chemical environment of the essential E789 in aCT8. As the c-ring rotates clockwise (as viewed from the cytosol towards the membrane), the deprotonated E-COO− is attracted to the electrostatic barrier between the offset luminal and cytoplasmic cavities, and forms a transient salt bridge with R735 in aCT7. Re-protonation of essential c-ring glutamates occurs upon entrance into the cytoplasmic cavity between S728 and E721. After receiving a proton in the chemical environment of E721, the neutralized c-ring glutamic acid can then re-enter the lipid bilayer.