

Abstract
The acetylcholine-activated inward rectifier potassium current (IKACh) is constitutively active in persistent atrial fibrillation (AF). We tested the hypothesis that the blocking of IKACh with the small molecule chloroquine terminates persistent AF. We used a sheep model of tachypacing-induced, persistent AF, molecular modeling, electrophysiology, and structural biology approaches. The 50% inhibition/inhibitory concentration of IKACh block with chloroquine, measured by patch clamp, was 1 μM. In optical mapping of sheep hearts with persistent AF, 1 μM chloroquine restored sinus rhythm. Molecular modeling suggested that chloroquine blocked the passage of a hydrated potassium ion through the intracellular domain of Kir3.1 (a molecular correlate of IKACh) by interacting with residues D260 and F255, in proximity to I228, Q227, and L299. 1H 15N heteronuclear single-quantum correlation of purified Kir3.1 intracellular domain confirmed the modeling results. F255, I228, Q227, and L299 underwent significant chemical-shift perturbations upon drug binding. We then crystallized and solved a 2.5 Å X-ray structure of Kir3.1 with F255A mutation. Modeling of chloroquine binding to the mutant channel suggested that the drug’s binding to the pore becomes off centered, reducing its ability to block a hydrated potassium ion. Patch clamp validated the structural and modeling data, where the F255A and D260A mutations significantly reduced IKACh block by chloroquine. With the use of numerical and structural biology approaches, we elucidated the details of how a small molecule could block an ion channel and exert antiarrhythmic effects. Chloroquine binds the IKACh channel at a site formed by specific amino acids in the ion-permeation pathway, leading to decreased IKACh and the subsequent termination of AF.—Takemoto, Y., Slough, D. P., Meinke, G., Katnik, C., Graziano, Z. A., Chidipi, B., Reiser, M., Alhadidy, M. M., Ramirez, R., Salvador-Montañés, O., Ennis, S., Guerrero-Serna, G., Haburcak, M., Diehl, C., Cuevas, J., Jalife, J., Bohm, A., Lin,Y.-S., Noujaim, S. F. Structural basis for the antiarrhythmic blockade of a potassium channel with a small molecule.
Keywords: potassium inward rectifier, atrial fibrillation, IKACh
The acetylcholine-activated inward rectifier potassium current (IKACh) is remodeled in humans with persistent atrial fibrillation (AF) and in animal models of persistent AF (1–5). IKACh flows through tetrameric sarcolemmal channels formed by the Kir3.1 and Kir3.4 proteins. IKACh activity is minimal in baseline physiology (6). Upon parasympathetic stimulation, acetylcholine binds to the G-protein-coupled muscarinic receptor (6). Subsequently, the Gβγ subunit docks directly into its binding sites on the channel, leading to its activation (7–9). As IKACh is important for heart rate modulation on a beat-to-beat basis, the on/off switching of the current is tightly regulated. However, in persistent AF, IKACh is constitutively active, irrespective of parasympathetic stimulation (3), leading to a net increase in the background inward rectifier current. Previous reports showed that an increase of the inward rectifier current results in shortening of the action potential duration (APD) and the subsequent formation of stable electrical rotors, which activate the myocardium at high frequencies and lead to fibrillation (10, 11). Therefore, the blocking of IKACh in the setting of chronic/persistent AF may be antiarrhythmic (12, 13). We aimed to elucidate the structural basis of how chloroquine blocks IKACh and whether it can terminate persistent AF. In sheep hearts with persistent AF, we compared the electrophysiological effects of chloroquine with those of tertiapinQ—a 21 residue peptide blocker of IKACh (14), initially isolated from the European honeybee venom—on the dynamics of the arrhythmia. We also used protein NMR spectroscopy, X-ray crystallography, and molecular modeling to investigate the structural details of how a small molecule (chloroquine) blocks a potassium current (IKACh).
MATERIALS AND METHODS
Persistent AF sheep model
All animal procedures followed institutional guidelines. A dual chamber pacemaker (St. Jude Medical, St. Paul, MN, USA) was implanted in male sheep (30–40 kg) with a bipolar pacing lead (St. Jude Medical) attached to the right atrial (RA) appendage under general anesthesia, as described previously (15, 16). A subcutaneous loop recorder (insertable loop recorder, Reveal XT; Medtronic, Minneapolis, MN, USA) was inserted parasternally in apposition to the left atrium (LA). After a 4 wk period of recovery from surgery, the RA was paced with an algorithm consisting of 30 s pacing at 20 Hz, followed by 10 s sensing. The pacemaker resumed pacing only if AF stopped, and sinus rhythm was detected. This algorithm reliably generated tachypacing-induced, self-sustained, persistent AF after 2 mo of pacing (16). Persistent AF was defined as episodes lasting >7 d without reversal to sinus rhythm.
Optical mapping
Hearts were removed via thoracotomy and connected to a Langendorff perfusion system with recirculating oxygenated (95% O2, 5% CO2) Tyrode’s solution at a constant flow rate of 240 ml/min (36.5–37.5°C, pH 7.4). The Tyrode’s solution composition (in millimolars) was the following: NaCl 130, KCl 4.0, MgCl2 1.0, CaCl2 1.8, NaHCO3 24, NaH2PO4 1.2, glucose 5.6, and albumin 0.04 g/l. Blebbistatin 10 μM (Enzo Life Sciences, Farmingdale, NY, USA) was used to reduce motion. After an atrial trans-septal puncture, all of the vein orifices were sealed, except for the inferior vena cava, which was connected to a digital sensor and to an open-end cannula to control the intra-atrial pressure. The pressure was kept at 5 cm H2O, resembling the diastolic LA pressure. Three custom-made bipolar electrodes were placed on the top of RA appendage, LA appendage, and the roof of LA. All recordings were bipolar signals obtained with a sampling rate of 1.0 kHz (DA100C; Biopac Systems, Goleta, CA, USA). The voltage-sensitive dye, di-4-ANEPPS (100–200 mg), was administered before imaging. [The imaging setup is drawn (see Fig. 2A).] The emitted fluorescence (645 nm) on the epicardial surfaces of the LA free wall and RA free wall was captured by 2 charge-coupled device (CCD) cameras (80 × 80 pixels, 600 frames/s; LittleJoe; SciMeasure Analytical Systems, Decatur, GA, USA). A third CCD camera was coupled to a rigid borescope (90° view angle), introduced through an incision in the left ventricle, passed across the mitral valve, and focused on the endocardial surface of the posterior LA (PLA). This is similar to what has been done before (15, 16). Five second movies of AF were recorded every 2 min during a 30 min period of baseline AF. Afterwards, we tested, for 15 min, the effects of 1 μM chloroquine or 0.1–0.2 μM tertiapinQ on the dynamics of persistent AF in 5 and 6 hearts, respectively.
Figure 2.

Effect of chloroquine on persistent AF in isolated Langendorff-perfused hearts. A) Ex vivo epicardial and endocardial mapping setup includes 3 synchronized CCD cameras: cameras 1 and 2 for the epicardial imaging of the LA and RA and camera 3 coupled to an endoscope advanced through the left ventricle into the LA for endocardial PLA imaging. B) APD75 and APD50 at constant 2.5 Hz pacing in PLA, LA, and RA, before and after the application of 1 µM chloroquine (n = 5). C) Representative DF maps of the PLA, LA, and RA in baseline AF and 2 and 4 min after chloroquine 1 µM perfusion. After 4 min, the heart spontaneously reverted to a 1.2 Hz SR. D) Time course of the average DF in the 5 hearts, 30 min before and after chloroquine perfusion. The black × marks indicate the time of spontaneous AF cardioversion in each of the hearts. E) Average maximum DF right before chloroquine application at baseline and immediately before AF termination. *P < 0.05, **P < 0.01.
APD and dominant frequency analyses in the isolated sheep heart
Five second optical movies were acquired during constant pacing or during sustained AF. Under constant pacing, action potentials were recorded, and the APD was computed at 50 and 75% repolarization (APD50 and APD75) at the LA, PLA, and RA, before and after drug application. During AF, dominant frequency (DF) maps were generated from the optical movies by performing fast Fourier transform on the fluorescence signal time series, as detailed earlier (15, 16).
Kir3.1 intracellular domain expression, purification, and 1H 15N HSQC NMR spectroscopy
The DNA sequence for the Kir3.1 intracellular domain consisting of residues 41–64 and 190–371, studied earlier with X-ray crystallography and NMR (17–19), was synthesized and cloned into the polyhistidine-tobacco etch virus-ligation-independent cloning vector. Protein expression was induced with 0.4 mM isopropyl-β-d-(−)-thiogalactopyranoside for 8 h at 25°C. To obtain the Kir3.1 protein in a uniform 1H/15N background, as detailed in earlier NMR studies of the Kir3.1 intracellular domain (17, 20), transformed BL21(DE3)pLysS-competent cells were grown to an optical density with a wavelength of 600 nm of 0.9 at 37°C in M9 minimal medium. The M9 medium contained 15NH4Cl (1 g/L), [U-2H] glucose (2 g/L), and [2H/15N] Celtone base powder (1 g/L) in 99% 2H2O (Cambridge Isotope Laboratories, Tewksbury, MA, USA). Cells were harvested by centrifugation and frozen at −80°C. The pellets were suspended in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 0.1 mM DTT, leupeptin, and aprotinin and lysed by sonication. The lysates were then centrifuged, and the supernatant was purified using the Ni affinity column (Qiagen, Germantown, MD, USA) and eluted in lysis buffer containing 300 mM imidazole. The polyhistidine-tag was then cleaved by tobacco etch virus protease, and the cleaved protein was purified on a size-exclusion chromatography column (HiLoad Superdex 200 pg; GE Healthcare, Waukesha, WI, USA), pre-equilibrated in 10 mM HEPES-NaOH, pH 7.0, 50 mM KCl, and 1 mM DTT. As in earlier studies (17, 19, 20), the chromatograms indicated that our protein traveled as a tetramer in solution. The NMR samples were concentrated to ∼0.25 mM tetrameric Kir3.1 in a buffer containing 10 mM HEPES-NaOH, pH 7.0, 50 mM KCl, and 1 mM DTT in 90% H2O/10% 2H2O. 1H 15N two-dimensional spectra of Kir3.1 were collected in the absence and presence of 1 mM chloroquine (Sigma-Aldrich, St. Louis, MO, USA) on a Burker DRX-600 with a cryoprobe using a transverse relaxation optimized spectroscopy (TROSY)-heteronuclear single-quantum correlation (HSQC) sequence. Spectra were processed with nmrPipe software (21). The processing scheme consisted of a solvent-suppression filter, cosine square-bell apodization function, 0 filling to double number of points, and phase correction in both dimensions: baseline correction in the first dimension and linear prediction in second dimension. Spectra were analyzed using CcpNmr software (22). Assignments were transferred from the published chemical shifts for Kir3.1 (20) (Biological Magnetic Resonance Data Bank Accession Number: 11067). Out of 187 backbone amides with published 1H 15N chemical shifts, 156 peaks could be picked in the Kir3.1 protein-alone spectrum above noise level. One residue (F46) was outside of the 1H spectral width. Assignments were then transferred from the Kir3.1 protein-alone spectrum to the Kir3.1 + chloroquine spectrum, where the closest peaks in the ligand-bound spectrum were assumed to be the same residues, as in the Kir3.1-alone spectrum. One hundred thirty peaks could be transferred from the Kir3.1 protein alone to the chloroquine-bound spectrum. Most of the peaks missing are likely a result of a low signal-to-noise ratio except for Q360, Q363, M364, M367, S368, and S369, which are likely a result of ligand-induced intermediate chemical exchange, as they are located in the very distal C-terminal helix of Kir3.1.
Chemical shift perturbations (CSPs) were calculated for peaks in the Kir3.1 alone and chloroquine-bound spectra using CcpNmr, according to the following equation:
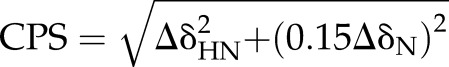 |
where ΔδHN and ΔδN are the changes in chemical shift of 1H and 15N during complex formation measured in parts per million. Transferred chemical shifts are reported in Supplemental Table 1.
Patch clamp and immunofluorescence
Patch pipettes were pulled from borosilicate glass tubes (World Precision Instruments, Sarasota, FL, USA), 2–3 MΩ resistance. Gigaohm seal and whole-cell recordings were performed at room temperature. The bath solution contained the following (in millimolars): 90 NaCl, 50 KCl, 1 CaCl2, 2 MgCl2, 10 HEPES, 10 glucose, pH 7.4, with NaOH. The pipette internal solution contained the following (in millimolars): K-aspartate 100, NaCl 10, KCl 40, Mg-ATP 5, EGTA 2, GTP-Tris 0.1, HEPES 10, pH 7.4. IKACh currents in human embryonic kidney 293 (HEK293) cells, transfected with Kir3.1 and Kir3.4 in the presence and absence of different doses of chloroquine (Sigma-Aldrich) or tertiapinQ (Tocris), were evoked in response to a 2 s ramp from −140 to 30 mV from a holding potential of 0 mV and subtracted from the residual current remaining after addition of 1 mM BaCl2 to the bath solution. Currents were also evoked in HEK293 cells transfected with either Kir3.1 and Kir3.4 or F255A or D260A mutant Kir3.1 (introduced via QuikChange mutagenesis and verified by sequencing) and Kir3.4 in the presence and absence of chloroquine. For fluorescence imaging, cells were plated on a coverslip and stained for 10 s with tertiapinQ-ATTO-488 (Alomone Laboratories, Jerusalem, Israel), and then images were acquired using an Olympus FV1000 multiphoton laser-scanning microscope.
Cloning, overexpression, and purification of the F255A Kir3.1 mutant
The Kir3.1 construct used for crystallization was the same as that used for the NMR experiment described above. The F255A mutation was introduced via QuikChange mutagenesis and verified by sequencing. The plasmid containing Kir3.1 F255A was transformed into Escherichia coli BL21 (DE3) cells. The cells were grown at 37°C in 2 × YT media containing ampicillin. At an optical density at 600 nm of ∼0.5, the temperature was lowered to 18°C. After 30 min at 18°C, isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 100 μM. Cells were harvested 16–20 h after induction by centrifugation at 4°C for 20 min. Cell pellets from 6 L culture were resuspended in 100 ml lysis buffer (20 mM Tris, pH 8.5, 150 mM NaCl, 1 mM PMSF, 1 μg/ml aprotinin) and stored at −80°C until ready for processing. All protein purification was performed on ice or at 4°C. The cells were lysed with 3 passes through an EmulsiFlex-C5 microfluidizer (Avestin, Ottawa, ON, Canada). The lysate was spun down at 35,000 rpm in an ultracentrifuge Ti70 rotor. The supernatant was loaded onto the Ni affinity column (Qiagen), pre-equilibrated in buffer (20 mM Tris, pH 8.5, 10 mM imidazole, 150 mM NaCl, 0.1 mM PMSF, 1 μg/ml aprotinin). After sufficient washing in the previous buffer, the bound protein was eluted (20 mM Tris, pH 8.5, 300 mM imidazole, pH 8.0, 150 mM NaCl, 0.1 mM PMSF, 1 μg/ml aprotinin). The Kir3.1 F255A fractions were analyzed by SDS-PAGE and pooled. The pooled fractions were diluted to a final NaCl concentration of 100 mM and loaded onto an anion exchange column (Source 15Q; GE Healthcare), pre-equilibrated in buffer QA (20 mM Tris, pH 8.0, 0.1 mM PMSF, 1 mM EDTA, 0.1% 2-ME). The protein was eluted using a gradient from buffer QA to QB (QA + 1 M NaCl; no EDTA). The peak fractions were analyzed by SDS-PAGE, and Kir3.1 F255A fractions were pooled. The purified protein was concentrated, buffer exchanged into storage buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 0.1% 2-ME, 5% glycerol), flash frozen in liquid nitrogen, and stored at −80°C.
Crystallization of Kir3.1 F255A and X-ray data collection
The protein was concentrated to 10 mg/ml and crystallized at 4°C in Linbro trays using the vapor-diffusion method by mixing equal amounts of protein and reservoir solution (25 mM Na/K phosphate, pH 5.0, 40 mM NaCl, 35% polyethylene glycol 400). The mixture was allowed to equilibrate over 1 ml reservoir solution. Crystals of Kir3.1 F255A were harvested and transferred to a cryogenic solution (reservoir + 10–15% ethylene glycol), flash frozen in liquid nitrogen, and stored in liquid nitrogen until ready for data collection. X-ray data were collected at 100K at a wavelength of 1.0003 Å at the Advanced Light Source Beamline 8.2.1 (Lawrence Berkeley National Laboratory, Berkeley, CA, USA). X-Ray data were reduced with the programs HKL2000 and MOSFLM (23). The crystals grew in the space-group P4212 with a unit cell of 80.07, 80.07, 85.04 Å; 90, 90, 90°, and 1 monomer in the asymmetric unit.
Structure determination and refinement
The Kir3.1 F255A structure was solved by molecular replacement using the program Phaser (24) within the CCP4 suite (25) of crystallographic programs. The wild-type (WT) Kir3.1 structure (18) [Protein Data Bank identification (PDB ID): 1N9P] was used as a search model. A large negative-difference density peak confirmed the F255A mutation. Refmac5 (26) and Phenix (27) were used to refine the structure at different stages. The molecular graphics program Coot (28) was used for manual rebuilding between successive rounds of refinement. PDB_REDO (29) was used to check for model errors. The final model contains aa 43–63 and 190–370. The final RWork and RFree were 22.39 and 26.68, respectively. F255A mutant Kir3.1 coordinates were deposited into the PDB (PDB ID: 5UM4). PyMOL (PyMOL Molecular Graphics System, v1.7.4; Schrödinger, Cambridge, MA, USA) was used for visualization and generation of molecular figures. The mutant was compared with WT structures (PDB IDs: 1N9P and 1U4E) using the program PDBeFOLD (30).
Molecular docking of chloroquine to Kir3.1
The intracellular domain of Kir3.1 (PDB ID: 1U4E) (19) and the Kir3.1 F255A structure obtained here were used for the docking simulations of the WT and mutant channels, respectively. The chloroquine SMILES string was obtained from DrugBank (31) (Accession Number: DB00608). The proteins and chloroquine were prepped using the Protein Preparation Wizard and LigPrep in Schrodinger’s Maestro (32). The protonation state of chloroquine was generated at a target pH of 7.0 using Epik (33, 34). The ligand was energy minimized using the OPLS_2005 forcefield (35). Before docking, the nonpolar hydrogens on both the protein and ligand were merged. To prepare the proteins for docking, a set of grids was created mapping the protein topology. The grids were generated using 60 × 60 × 60 points in the x, y, and z directions and centered around the aqueous channel with a spacing of 0.375 Å. Subsequently, the Lamarckian genetic algorithm was used for docking. The population size was set to 150, the number of energy evaluations was set to 2,500,000, and the number of generations was set to 27,000. One thousand runs were performed using AutoDock 4.2 (36). The lowest energy poses from molecular docking were analyzed. Hydrogen bond analysis was performed using Visual Molecular Dynamics (37).
Estimation of blocking ability of chloroquine
The atomic coordinates of tetrameric WT or F255A mutant Kir3.1 bound to chloroquine were transformed into a voxel grid. In this grid, each voxel was a cube of 0.2 × 0.2 × 0.2 Å. Each atom filled a number of cubes corresponding to its van der Waals radius (38). The use of the discrete grid allowed for a probe to search the space in the aqueous ion-permeation pathway. The probes used were voxel-approximated spheres with radii ranging from 1.4 Å (corresponds to the ionic radius of a bare K+ ion) to >3.9 Å (3.0 Å is the radius of a solvated K+ ion up to its first hydration shell) (39) in 0.1 Å increments. The probes were started toward the extracellular opening of the channel and were pushed longitudinally through the channel toward the opposite side. If, at any point in the pathway, the probe was unable to proceed further, then the probe’s lateral position was adjusted so as to search all possible paths through the channel. If the probe was physically unable to move past chloroquine (i.e., no path was found), then the channel was considered blocked at that probe radius.
RESULTS
Dose-response curve of IKACh block by chloroquine and tertiapinQ
In HEK293 cells stably transfected with Kir3.1 and Kir3.4, we measured the 50% inhibition/inhibitory concentration (IC50) of IKACh block by chloroquine and tertiapinQ. Figure 1A is a confocal image, showing the membrane staining of IKACh proteins by tertiapinQ conjugated to the fluorescent tag ATTO-488 in blank and in Kir3.1- and Kir3.4-expressing HEK293 cells. Figure 1B patch-clamp traces are currents in response to a 2 s voltage ramp (from −140 to +30 mV), showing BaCl2-sensitive IKACh. The current decreased by ∼50% upon addition of 1 μM chloroquine or 0.1 μM tertiapinQ. Figure 1C is the dose-response relationship for the block of IKACh by tertiapinQ and chloroquine. The IC50 for chloroquine is 1.02 μM, R2 = 0.96, and that of tertiapinQ is 0.07 μM, R2 = 0.94.
Figure 1.

Determination of the IC50 for chloroquine and tertiapinQ block of IKACh in HEK293 cells stably transfected with Kir3.1 and Kir3.4. A) Confocal microscopy. Fluorescence staining of IKACh proteins. Upper: Live HEK293 cells transfected with mCherry. TertiapinQ (TPQ) ATTO-488 does not show staining. Lower: HEK293 cells, transfected with mCherry and stably expressing Kir3.1/Kir3.4, show robust staining of the cell membranes by tertiapinQ ATTO-488. Laser and detector settings were identical in both cases. B) IKACh recorded in HEK293 cells expressing Kir3.1/3.4 in response to a ramp in the absence and the presence of 1 μM chloroquine (CQ) or 0.1 μM tertiapinQ. C) Dose-response curve of IKACh block by chloroquine, IC50 = 1.02 μM, R2 = 0.96; TertiapinQ, IC50 = 0.07 μM, R2 = 0.94.
Chloroquine terminates persistent AF in the sheep heart
The cardiac IKACh is a heterotetrameric channel formed by Kir3.1 and Kir3.4 proteins (6). Our earlier study suggested that chloroquine terminates cholinergic AF by blocking IKACh (40). However, it is not known whether chloroquine can terminate persistent AF. The effects of chloroquine in persistent AF were investigated using optical mapping in isolate Langendorff-perfused hearts. AF was easily reinduced by electrical pacing in all ex vivo hearts (n = 5) and was sustained throughout the 30 min baseline recording. Before and after the application of 1 μM chloroquine, 2.5 Hz constant pacing was applied to assess the effect of the drug on the APD. As shown in Fig. 2B, 1 μM chloroquine significantly prolonged APD75 in LA epicardium (LAepi; 125.7 ± 9.9 ms before chloroquine, 159.4 ± 10.7 ms after chloroquine, P < 0.01), RA epicardium (RAepi; 184.1 ± 8.0 ms before chloroquine, 208.4 ± 9.7 ms after chloroquine, P < 0.01), and PLA endocardium (PLAendo; 158.3 ± 7.2 ms before chloroquine, 180.6 ± 9.3 ms after chloroquine, P < 0.01). APD50 was also prolonged in LAepi (86.7 ± 8.0 ms before chloroquine, 114.4 ± 6.8 ms after chloroquine, P < 0.01), RAepi (139.6 ± 7.9 ms before chloroquine, 152.3 ± 7.7 ms after chloroquine, P < 0.01), and PLAendo (123.5 ± 9.2 ms before chloroquine, 137.5 ± 8.7 ms after chloroquine, P < 0.01). Representative maps of DF at baseline and during chloroquine perfusion showed that in the presence of chloroquine, DF was decreased in PLA, LA, and RA compared with baseline AF, until AF spontaneously terminated (Fig. 2C). The time course of maximal DF in all 5 experiments is shown in Fig. 2D. Before the administration of chloroquine, the DF was 8.4 ± 0.7 Hz in PLAendo and 7.6 ± 0.8 Hz in LAepi, higher than in RAepi (6.9 ± 0.9 Hz). Upon 1 μM chloroquine perfusion, maximal DF significantly decreased to 5.3 ± 0.7, 5.3 ± 0.7, and 5.3 ± 0.7 Hz in PLAendo, LAepi, and RAepi (P < 0.05), just before resumption of sinus rhythm (SR). In 5/5 hearts, AF terminated, and SR resumed after 8.4 ± 1.9 min of drug perfusion.
Effects of tertiapinQ on persistent AF in the sheep heart
To confirm further that the antiarrhythmic effects of chloroquine are, in part, as result of IKACh block, we tested 0.1–0.2 μM tertiapinQ in 6 isolated Langendorff- perfused sheep hearts with persistent AF. Sustained AF was easily inducible by pacing in all ex vivo hearts. Initially, 0.1 μM tertiapinQ was added to the perfusate, and after 15 min, if persistent AF was still present, then the peptide’s concentration was increased to 0.2 μM for another 15 min. Four out of six hearts (67%) reverted to SR with tertiapinQ application (0.1 μM: 1 heart, 0.2 μM: 3 hearts). Two hearts did not spontaneously convert to SR at 0.2 μM tertiapinQ; AF was cardioverted with a direct current shock. As depicted Fig. 3A, the 4 hearts that converted to SR (n = 4), pacing at 2.5 Hz, showed that tertiapinQ significantly prolonged APD75 in LAepi (106.0 ± 10.1 ms before tertiapinQ, 137.8 ± 17.3 ms after tertiapinQ, P = 0.015), RAepi (154.5 ± 17.6 ms before tertiapinQ, 196.3 ± 10.5 ms after tertiapinQ, P = 0.018), and PLAendo (126.3 ± 13.5 ms before tertiapinQ, 173.5 ± 18.9 ms after tertiapinQ, P < 0.01). APD50 was also prolonged in LAepi (77.3 ± 9.5 ms before tertiapinQ, 100.1 ± 12.9 ms after tertiapinQ, P < 0.01), RAepi (109.3 ± 7.9 ms before tertiapinQ, 148.3 ± 9.3 ms after tertiapinQ, P < 0.01), and PLAendo (86.8 ± 10.6 ms before tertiapinQ, 122.1 ± 15.5 ms after tertiapinQ, P < 0.01). Representative examples of DF maps during tertiapinQ perfusion showed that DF decreased compared with baseline AF and that AF spontaneously converted to SR (Fig. 3B). The time course of maximal DF is shown in Fig. 3C. DF in PLA, LA, and RA at baseline was 9.3 ± 0.8, 8.8 ± 0.5, and 7.7 ± 0.4 Hz before administration of tertiapinQ. The peptide significantly decreased maximal DF to 6.7 ± 0.9, 6.4 ± 0.8, and 5.2 ± 0.6 Hz (P < 0.05), respectively, immediately before AF termination (Fig. 3D; n = 4). In the 2 hearts where AF did not terminate, tertiapinQ tended to prolong the APD and slow down the DF of the arrhythmia (Supplemental Fig. 1), however, to a lesser extent than in the cases where AF terminated.
Figure 3.

Effect of tertiapinQ on persistent AF in isolated Langendorff-perfused hearts. A) APD at APD75 and APD50 at constant 2.5 Hz pacing in PLA, LA, and RA, before and after the application of 0.2 µM tertiapinQ (n = 4). B) Representative DF maps of the PLA, LA, and RA in baseline AF and 2 and 4 min after 0.1 µM tertiapinQ perfusion. After 4 min, the heart spontaneously reverted to a 0.8 Hz SR. C) Time course of the average DF in 4 hearts, 30 min before and after TertiapinQ perfusion. The black × marks indicate the time of spontaneous AF cardioversion in each of the hearts. D) Average maximum DF right before TertiapinQ application at baseline and immediately before AF termination. *P < 0.05, **P < 0.01.
Docking of chloroquine in the tetrameric Kir3.1 intracellular domain
The cardiac IKACh is a heterotetrameric channel formed by Kir3.1 and Kir3.4 proteins (5). The structure of Kir3.4 has not been elucidated; however, that of the Kir3.1 intracellular domain has been studied with X-ray crystallography and NMR spectroscopy (17, 18). Chloroquine docking into the Kir3.1 intracellular aqueous channel is shown in Fig. 4. One thousand docking runs were performed. The 2 lowest energy poses were analyzed. Figure 4A, top, is a magnified view of the tetrameric Kir3.1 channel, represented by gray ribbons, with docked chloroquine in cyan (lowest energy pose). The amine nitrogen of chloroquine formed a hydrogen bond (red line) with the side chain of residue D260 on 1 subunit, whereas the aminoquinoline ring underwent an aromatic–aromatic interaction with residue F255 from the adjacent subunit. The drug extended through the horizontal plane of the aqueous channel at the level of the D260/F255 residues. Figure 4B, top, is a view of the tetrameric Kir3.1 channel, represented by gray ribbons, with docked chloroquine in cyan (second-lowest energy pose). The amine nitrogen of chloroquine formed a hydrogen bond (red line) with the carbonyl oxygen chain of residue F255 on 1 subunit, whereas the aminoquinoline ring underwent an aromatic–aromatic interaction with residue F255 from the opposing subunit. In Fig. 4, the drug extended through the horizontal plane of the aqueous channel at the level of the D260/F255 residues. The van der Waals representations of the Kir3.1:chloroquine complex are shown from intracellular (middle) and extracellular (bottom) bird’s-eye views, with the ion-permeation pathway in the middle and chloroquine in cyan.
Figure 4.

Docking of chloroquine in the ion-permeation pathway of the Kir3.1 channel. A, B) Two lowest energy poses. Top: magnified view of the binding poses of chloroquine (cyan sticks) in Kir3.1. The D260 and F255 residues from each of the 4 Kir3.1 subunits are shown in green and orange sticks, respectively. A) The amine nitrogen of chloroquine forms a hydrogen bond (red line) with the side chain of D260 in 1 subunit, whereas the aminoquinoline ring of chloroquine is involved in an aromatic–aromatic interaction with the phenylalanine ring of F255 in the adjacent subunit. B) The amine nitrogen of chloroquine hydrogen bonds (red line) the carbonyl oxygen of F255 in 1 subunit, whereas the aminoquinoline ring of chloroquine is involved in an aromatic–aromatic interaction with the phenylalanine ring of F255 in the opposing subunit. Middle, bottom: van der Waals representations of the channel bound to chloroquine (cyan), viewed from the intracellular and extracellular sides, respectively.
Kir3.1 interaction with chloroquine studied with 1H 15N HSQC NMR
To validate the modeling data and delineate the binding site of chloroquine, we studied the interaction of chloroquine with the purified intracellular domain of Kir3.1 using NMR spectroscopy. We acquired 1H 15N HSQC spectra of the Kir3.1 tetramer at a concentration of 0.25 mM and then added 1 mM chloroquine and acquired the spectra once again. Spectral analysis of the hydrogen and nitrogen dimensions identified backbone residues with significant CSPs >0.04 ppm as a result of chloroquine binding. The transferred chemical shifts are reported in Supplemental Table 1. In total, 11 backbone residues underwent a significant difference in chemical shifts after chloroquine binding to the channel. They were (largest to smallest CSP) the following: L246, Q227, Q344, V47, K208, H57, S235, L299, I228, L251, and G51. The F255 peak in the spectrum of Kir3.1 alone was apparent above noise level. After addition of chloroquine, at noise level, there was a discernible peak corresponding to F255, which was significantly shifted, suggesting that this residue’s chemical environment has changed as a result of chloroquine binding. Residue D260 did not undergo significant CSP. However, this residue is linked via hydrogen bonding to residue I228, which underwent CSP. It should be noted that the TROSY-HSQC NMR sequence, which was used here, is an indirect method that does not provide actual structural restraints. The chemical shifts suggest the general location of the binding pocket. Figure 5A shows examples of backbone resonances, with and without bound chloroquine. The resonance contours of Kir3.1 alone are in black, whereas those of Kir3.1, in the presence of chloroquine, are in red. Residues W323 and K49 are examples of amino acid resonance contours that were unaffected by chloroquine. I228 and L299 are examples of residues that were affected by the drug. We subsequently mapped the backbone residues with significant CSP onto the tetrameric structure of Kir3.1 (PDB ID: 1U4E) and highlighted them in red (Fig. 5B). V47 and G51 are located adjacent to one another in a loop belonging to the N-terminal region. H57 is located at the end of the N-terminal cytoplasmic region, and K208 is located at the start of the β-strand βC close to the distal C-terminal α helix αB region of the Kir3.1 structure that contains the residues Q360, Q363, M364, M367, S368, and S369 connected to each other via hydrogen bonding. F255 and Q227 are located in the aqueous pore region, and I228 is in the β-sheet βD, along with Ser235, adjacent to the aqueous channel. I228 and L299 are connected through a hydrogen-bond network with L299, located at the base of the G-loop in βH, a region important for gating of the channel. Importantly, residues Q227, I228, F255, and L299 form a pocket in the channel’s aqueous vestibule. Figure 5C shows the channel’s longitudinal view of the van der Waals representation, with the 2 front subunits removed, to expose this binding pocket (circled in white). We then proceeded to combine the NMR experimental results, with the molecular docking shown in Fig. 4B. The binding pose of chloroquine from Fig. 4B placed the drug into the pocket, suggested by the 1H 15N HSQC NMR spectra (Fig. 5D–F). The binding pose of chloroquine from Fig. 4A also places the drug into the pocket, suggested by the 1H 15N HSQC NMR spectra (data not shown).
Figure 5.

Chloroquine binding to the isolated and purified Kir3.1 intracellular domain studied with 1H 15N HSQC NMR. A) Examples of resonance peaks of residues W323, K49, I228, and L299 obtained by a 1H 15N NMR TROSY-HSQC sequence. The black contours are of the Kir3.1 protein alone, and the red contours are after addition of chloroquine. The resonance peaks of W323 and K49 did not undergo significant shifts, suggesting that these residues are not involved in the binding of chloroquine to the channel. The peaks of I228 and L229 underwent significant shifts, indicating that the chemical environment of these residues have changed upon chloroquine binding to the channel. B) An intracellular bird’s-eye view of the tetrameric Kir3.1 is shown in gray ribbons, whereas the residues that underwent significant CSPs in the NMR experiments are shown as red sticks. C) A longitudinal representation of the tetrametric channel, with the front 2 subunits removed, exposing the amino acids (Q227, I228, F255, and L299) that form a binding pocket for chloroquine (white circle) in the aqueous vestibule. D) Placement of docked chloroquine in the binding pocket suggested by NMR. Longitudinal view of chloroquine (cyan sticks) docked into the Kir3.1 channel from Fig. 4 with the front subunits removed, and the residues that underwent significant shifts in the NMR experiment are shown in red. E) Magnified view of the boxed area in D showing agreement between the molecular docking of chloroquine and the binding pocket suggested by NMR, with chloroquine sitting in the NMR-suggested binding site. F) Chloroquine in cyan sticks is shown with the amino acids that underwent CSPs in NMR, depicted in red sticks.
X-Ray crystallography and structure determination of the Kir3.1 intracellular domain with F255A mutation
We explored the binding of chloroquine to Kir3.1 in further structural detail. We mutated residue F255 into an alanine (F255A) and proceeded to express, purify, crystallize, and solve a high-resolution X-ray structure of the F255A channel. X-Ray data were collected at the Advanced Light Source Beamline 8.2.1 (Lawrence Berkeley National Laboratory). The Kir3.1 protein with the F255A mutation crystallized in the P4212 space group with 1 monomeric Kir3.1 subunit in the asymmetric unit. A 2.5 Å resolution structure was determined. The data collection and refinement statistics are in Supplemental Table 2. The mutation introduced minimal changes into the overall structure of the channel compared with the 2 previously solved WT structures (PDB IDs: 1N9P and 1U4E) (18, 19). After molecular replacement with PDB ID 1N9P as a search model (18), the electron density map for the Kir3.1 F255A tetrameric channel was generated. The difference (Fo-Fc) electron density map, around the area of residue 255, contoured in red at −3 σ, is shown in Fig. 6A. The magnified view of the residue 255 region from 1 subunit in Fig. 6A, right, highlights the red negative-density blob, which indicates the absence of phenylalanine at position 255. Subsequently, we repeated the molecular modeling experiments similarly to Fig. 4, by docking chloroquine in the F255A mutant Kir3.1 channel. One thousand runs were performed, and the lowest energy pose was analyzed. Figure 6B is a magnified view of the tetrameric F255A Kir3.1 channel, represented by gray ribbons, with docked chloroquine in cyan. The amine nitrogen of chloroquine formed a hydrogen bond with the side chain of D260. The drug binding is off centered and tucked between 2 subunits. The van der Waals representation of the F255A mutant channel bound to chloroquine (cyan) is viewed from the intracellular side of Fig. 6B, right.
Figure 6.

X-Ray crystallography structure of Kir3.1 with F255A mutation at 2.5 Å resolution and docking of chloroquine in the mutant channel structure. A) Left: electron density map for Kir3.1 F255A tetrameric channel after molecular replacement. The difference (Fo-Fc) electron density map, around the area of residue 255, is contoured in red at −3 σ. Right: magnified view of the residue 255 region from 1 subunit highlighting the red negative-density blob, which indicates the absence of residue F255. B) Docking of chloroquine into the ion-permeation pathway of the F255A mutant Kir3.1 channel. Left: magnified view of the binding pose of chloroquine (cyan sticks) in Kir3.1. Chloroquine binding is off centered, with the amine nitrogen of chloroquine forming a hydrogen bond with residue D260. The F255 residues from each of the 4 Kir3.1 subunits are shown in red sticks. Right) van der Waals representation of the F255A mutant channel bound to chloroquine (cyan), viewed from the intracellular side.
Modeling and quantification of chloroquine block in WT and F255A mutant Kir3.1
Figure 7A is a magnified view of chloroquine docked in WT and mutant Kir3.1, where the drug’s ability to block the channel might be reduced by the absence of residue F255. Consequently, we quantified chloroquine’s blocking ability of the WT and mutant channels. Figure 7B is the voxelated ion-permeation pathway in the WT channel bound to chloroquine. It was found that a probe of radius ≥2.5 Å was blocked by the drug, and probes with radii ≤2.5 Å passed through. Figure 7C is the voxelated ion-permeation pathway in the F255A mutant channel bound to chloroquine. The drug was unable to block a probe of radius 2.5 Å, and the minimal radius of block occurred at 3.9 Å. We validated the numerical and structural data in patch-clamp experiments. HEK293 cells were transfected with WT Kir3.1 or F255A or D260A Kir3.1 and with Kir3.4. The mutant IKACh channels were expressed at the cell membrane (Fig. 8A). We then constructed dose-response curves of chloroquine block of the WT and mutant currents elicited by a ramp from −140 to 30 mV. The current-voltage relationships are in Fig. 8B, where the mutations reduced the ability of 1 μM chloroquine to block the BaCl2-sensitive current. Figure 8C, left, shows dose-response curves for the effect of chloroquine on the BaCl2-sensitive inward current, measured at −120 mV, where the IC50 for WT was significantly lower (0.8 μM, R2 = 0.81, n = 11) than F255A (3.2 μM, R2 = 0.75, n = 10) and D260 (2.8 μM, R2 = 0.9, n = 10; P < 0.01). Figure 8C, right, dose-response curves for the effect of chloroquine on the BaCl2-sensitive outward current measured at +20 mV, where the IC50 for WT was significantly lower (1.1 μM, R2 = 0.83, n = 7) than F255A (4.2 μM, R2 = 0.65, n = 7) and D260 (3.8 μM, R2 = 0.89, n = 7; P < 0.01). Additionally, we investigated if the F255A mutation interferes with the ability of tertiapinQ to block IKACh. Figure 8D is a compilation of 5 WT and 5 F255A mutant Kir3.1-expressing cells, showing that the ability of tertiapinQ to block IKACh was not affected by the F255A mutation. TertiapinQ (70 nM) blocked 46.2 ± 5.1% of WT IKACh and 52.3 ± 11.3% of the F255A mutant IKACh (P = 0.6). This could be a result of the fact that chloroquine binds the channel in the intracellular domain, whereas the tertiapinQ binding site is far away, in the extracellular loop region (41).
Figure 7.

Estimation of chloroquine’s ability to block the WT and F255A mutant Kir3.1 channel using voxelation. A) Intracellular, magnified view of the WT (left) and F255A mutant (middle) channel’s aqueous pore with docked chloroquine in cyan from Figs. 4 and 6, respectively. Right: A longitudinal view of the channel’s ribbon structure, with the orange box indicating the area of the channel represented by the voxelation experiments of B and C. B) Voxelated WT channel’s ion-permeation pathway in gray, with the front subunit removed for clarity. Chloroquine is in cyan. The different snapshots show a spherical voxelated probe colored in purple, with a radius = 2.5 Å traveling through the channel, starting toward the extracellular portion and getting blocked by chloroquine. C) The voxelated F255A mutant ion-permeation pathway is in gray, with the front subunit removed for clarity. Chloroquine is in cyan. The different snapshots show a spherical, voxelated probe colored in purple, with a radius = 2.5 Å traveling down the channel, starting toward the extracellular portion, without being affected by chloroquine.
Figure 8.

F255A and D260A mutations reduce the ability of chloroquine to block IKACh. A) Confocal microscopy. Fluorescence staining of IKACh proteins. Top) Live HEK293 cells transfected with mCherry, WT Kir3.1, and Kir3.4. Middle (bottom): cells transfected with mCherry, Kir3.1 F255A or D260A, and Kir3.4, where tertiapinQ ATTO-488 showed robust staining of the cell membranes. B) BaCl2-sensitive IKACh currents elicited in WT Kir.31/Kir3.4-, F255A Kir3.1/Kir3.4-, or D260A Kir3.1/Kir3.4-transfected cells, in response ramps from −140 to +30 mV, before and after addition of 1 μM CQ. C, left: Dose-response curves for the effect of chloroquine on the BaCl2-sensitive inward current measured at −120 mV. IC50 for WT: 0.8 μM, R2 = 0.81, n = 11; F255A: 3.2 μM, R2 = 0.75, n = 10; and D260: 2.8 μM, R2 = 0.9, n = 10. P < 0.01 for WT vs. F255A and D260. Right: dose-response curves for outward current measured at +20 mV. IC50 for WT: 1.1 μM, R2 = 0.83, n = 7; F255A: 4.2 μM, R2 = 0.65, n = 7; and D260: 3.8 μM, R2 = 0.89, n = 7. P < 0.01 for WT vs. F255A and D260. D) TertiapinQ (70 nM) block of WT (46.2 ± 5.1%, n = 5) and F255A (52.3 ± 11.3%, n = 5; P = 0.6) Kir3.1/ Kir3.4 currents at −120 mV.
Modeling of chloroquine’s interaction with the transmembrane domain of Kir3.1
We explored with modeling whether chloroquine binds the transmembrane domain of the channel. To do so, a homology model for Kir3.1, using the PDB of G protein-gated inwardly rectifying potassium channel 2 (PDB ID: 3SYO) (42), was created (Fig. 9A). Then, docking simulations were performed, where the chloroquine docking search space encompassed the entire aqueous region of the transmembrane domain. The simulations suggested that chloroquine (in cyan) binds the transmembrane domain of the channel at residue D173. The image in Fig. 9A (right) is the lowest binding-affinity pose, shown as a bird’s-eye view from the intracellular side, whereas chloroquine’s amine group hydrogen bonds (red line) the negative side chain of D173. As it has been demonstrated that the chloroquine pore block of Kir2.1 channels is intracellular (43), we explored in Fig. 9B–D if chloroquine can travel through the channel; traverse the narrowest portion at the G-loop formed by aa T306, G307, and M308; and reach the transmembrane binding site at residue D173. We calculated a voxelated version of the channel (depicted in red) overlaying the Kir3.1 ribbon structure (gray; Fig. 9B). Figure 9C shows the voxelated channel (red) with the corresponding z-axis scale along the length of the channel axis (starting at 0 Å at the intracellular entrance of the intracellular aqueous vestibule). Subsequently, in Fig. 9D, we calculated the aqueous vestibule’s cross-sectional area as a function of the z axis (the channel axis). The narrowest region occurs at position 37.25 Å, at the level of the G-loop, with a cross-sectional area of 8.25 Å2. Figure 10A is a slice of the voxelated channel at 37.25 Å (shown in red beads), along with the surrounding corresponding residues T306, G307, and M308. These residues are represented as sticks (Fig. 10A, left) and as van der Waals radii (Fig. 10A, right). Subsequently, we calculated the cross-sectional area of the chloroquine rings. It was 46.6 Å2, larger than the 8.25 Å2 area of the G-loop pore. In Fig. 10B, the voxelated chloroquine is shown in blue, on top of the G-loop. Therefore, the model suggests that it is unlikely that chloroquine will physically pass through the G-loop region and advance from the intracellular region to the transmembrane domain of the aqueous vestibule. This is in agreement with mutagenesis experiments in Kir2.1 that showed that D172 (equivalent to D173 in Kir3.1) is not involved in the chloroquine block of Kir2.1 channels (43).
Figure 9.

Modeling the binding of chloroquine to the Kir3.1 transmembrane domain. A) Chloroquine docking in the aqueous region of the transmembrane domain in the homology model for Kir3.1 using the PDB of Kir3.2 (PDB ID: 3SYO). Chloroquine (in cyan) binds the transmembrane domain of the channel at residue D173. Right: the lowest binding-affinity pose viewed from the intracellular side. Chloroquine’s amine group hydrogen bonds (red line) the negative side chain of D173. B) Voxelated version of the channel (in red) overlaying the Kir3.1 ribbon structure (gray). C) Voxelated channel (red) with the corresponding z-axis scale along the length of the channel axis (starting at 0 Å at the intracellular entrance of the intracellular vestibule). D) Aqueous vestibule’s cross-sectional area calculated as a function of the channel axis. The narrowest region is at 37.25 Å, with a cross-sectional area of 8.25 Å2.
Figure 10.

A) Slice of the voxelated channel at 37.25 Å (red beads) with the surrounding corresponding residues T306, G307, and M308 as sticks (left) and as van der Waals radii (right). B) Voxelated chloroquine (cross-sectional area 46.6 Å2) in blue, on top of the G-loop.
DISCUSSION
Altogether, the results demonstrate that chloroquine terminates persistent AF and restores SR, at least in part, by blocking IKACh via binding at a site formed by specific amino acids in the intracellular ion-permeation pathway. Specifically, molecular modeling of Kir3.1 intracellular domain interaction with chloroquine suggested that the drug binds the aqueous vestibule of the channel via hydrogen bonding and aromatic–aromatic interactions at aa F255 and D260, in proximity to I228, Q227, and L299. 1H 15N HSQC NMR of the purified Kir3.1 intracellular domain confirmed the molecular modeling predictions. The NMR experiments showed that I228, F255, Q227, and L299, in addition to 8 other amino acids located at the entrance of the vestibule and by the subunit/subunit interface, underwent significant resonance shifts, indicating that the local environment of these residues changed in response to chloroquine’s interaction with the channel. As molecular modeling and NMR experiments pointed to a role for F255 in the binding of chloroquine to the channel, we mutated that residue to alanine and crystallized and solved a 2.5 Å resolution X-ray structure of the mutant channel. Subsequently, we performed molecular modeling on the mutant channel to gain further insights into how a small molecule blocks the ionpermeation pathway of an inward rectifier channel. Docking of chloroquine into the F255A Kir3.1 channel showed an off-centered interaction of chloroquine with the mutant protein, hampering the drug’s ability to block potassium flow through the vestibule compared with the WT channel. Patch-clamp results confirmed that F255A and D260A mutations reduced chloroquine’s block of IKACh.
Our earlier molecular-modeling studies suggested that chloroquine binds the intracellular domain of Kir3.1 (40). However, the details of this binding were not fully explored. In the previous work, 10 docking runs were used. In this study, we performed extensive simulations with 1000 runs. In general, the suggested binding site is similar in the previous (40) and the current study.
We attempted to cocrystallize Kir3.1 in the presence of chloroquine, but we were unable to grow any crystals that contained the ligand. In addition, numerous X-ray dataset soaks of chloroquine into Kir3.1 crystals were performed, but no clear ligand electron density was observed from these experiments. Therefore, we combined molecular modeling and 1H 15N HSQC NMR to probe the structural basis of IKACh block by a small molecule. Comparison of the residues affected by chloroquine binding in this work with those of the proposed binding site for spermine from an earlier NMR study (17) indicates that chloroquine binds to the same region. 1H 15N shifts for backbone are mostly sensitive to changes in hydrogen-bond patterns and local changes in secondary structure. From our NMR study, it can be inferred that the binding of chloroquine induces minor changes in the secondary structure of Kir3.1, as a result of changes in the packing of side chains of amino acids located away from the binding pocket.
A limited clinical trial in 1958 suggested that chloroquine, a widely used 4-aminoquinoline antimalarial, could be antiarrhythmic in patients with AF (44). Oral administration of chloroquine terminated AF in ∼50% of the participants. However, the antiarrhythmic mechanism was not understood (44). Chloroquine has been shown to block the sodium, L-type calcium and delayed rectifier currents at concentrations higher than used in this study (45). We used 1 μM, which is consistent with the plasma concentration of the drug in patients on an antimalarial regimen (46). Nevertheless, it is likely possible that the observed antiarrhythmic actions of chloroquine are also a result of its ability to block other currents, such as IK1 (47). Therefore, we compared the electrophysiological effects of chloroquine with those of tertiapinQ on the dynamics of persistent AF in the sheep heart. TertiapinQ is a potent blocker of IKACh (14), and it was shown to prolong the APD in atrial myocytes from patients with chronic AF (2, 3, 5, 48). In our experiments, tertiapinQ slowed down and terminated AF and prolonged the APD similarly to chloroquine. This suggests that the antiarrhythmic effects of chloroquine can be attributed, at least in part, to IKACh block; however, involvement of other currents cannot be ruled out. For instance, the effects of chloroquine on the dynamics of AF were faster than those of tertiapinQ. In addition, 5/5 hearts reverted to SR with chloroquine, whereas 4/6 hearts terminated with tertiapin. It has been shown that IK1 is remodeled in persistent AF (49), and thus, it is possible that chloroquine’s action could also be a result of blocking IK1 as well. The IC50 for chloroquine block of IK1 and IKACh is very close as shown earlier (40). In Kir3.1, chloroquine interacts with aa D260, and F255 in the ion-permeation pathway, whereas in Kir2.1, it interacts with equivalent aa F254 and D259, in addition to E224 (40). Because of the similarities in the binding of chloroquine to Kir3.1 and Kir2.1, the IC50 for chloroquine block of IK1 and IKACh is similar.
It has been reported that NTC-801 (50), a benzopyrene derivative, and AZD2927 (51), a benzamide-related compound, can selectively inhibit IKACh at submicromolar concentrations. However, the drugs failed to revert paroxysmal AF and atrial flutter, respectively, in patients (50, 51). The results are not surprising, given the fact that IKACh is not constitutively active in patients with paroxysmal AF (52), let alone in atrial flutter whose mechanism differs from that of AF (53). Therefore, efforts aimed at finding novel IKACh blockers should focus on agents capable of inhibiting IKACh, but equally as important, they should target the correct subset of AF patients where the current has been established to play a role. For instance, it has been argued that in clinical trials of IKACh-blocking therapies for AF, selection of the appropriate patient population is crucial (54). Patients with a high burden of paroxysmal self-terminating AF (50) are most likely to have abnormal ectopic activity as an AF mechanism (54); therefore, an IKACh blocker designed to terminate reentrant AF would not be useful (54).
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank Dr. Kevin Nash (University of South Florida) for his help with creation of mutant constructs. This work was supported, in part, by an American Heart Association postdoctoral fellowship to Y.T.; U.S. National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI), R21 HL138064 and R01HL129136 to S.F.N.; Tufts start-up and the Knez Family Investment Funds for Y-S.L.; and NIH NHLBI R01HL122352, the Leducq Foundation: Transatlantic Network of Excellence Program on Structural Alterations in the Myocardium and the Substrate for Cardiac Fibrillation, and University of Michigan Health System–Peking University Health Science Center Joint Institute for Translational and Clinical Research to J.J. HEK293 cells, stably transfected with Kir3.1 and Kir3.4, were a kind gift from the laboratory of Dr. Douglas Bayliss (University of Virginia Charlottesville, VA, USA). The authors declare no conflicts of interest.
Glossary
- AF
atrial fibrillation
- APD
action potential duration
- APD50/75
action potential duration at 50 and 75% repolarization
- CCD
charge-coupled device
- CSP
chemical shift perturbation
- DF
dominant frequency
- HEK293
human embryonic kidney 293
- HSQC
heteronuclear single-quantum correlation
- IC50
50% inhibition/inhibitory concentration
- IKACh
acetylcholine-activated inward rectifier potassium current
- LA
left atrium
- LAepi
left atrial epicardium
- PDB ID
Protein Data Bank identification
- PLA
posterior left atrium
- PLAendo
posterior left atrial endocardium
- RA
right atrium
- RAepi
right atrial epicardium
- SR
sinus rhythm
- TROSY
transverse relaxation optimized spectroscopy
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
C. Diehl, A. Bohm, Y-S. Lin, and S. F. Noujaim designed research; Y. Takemoto, D. P. Slough, G. Meinke, C. Katnik, Z. A. Graziano, C. Bujjibabou, M. Reseir, M. M. Alhadidi, R. Ramirez, O. Salvador-Montanes, S. Ennis, G. Guerrero, M. Haburcak, C. Diehl, A. Bohm, Y.-S. Lin, and S. F. Noujaim performed research; J. Cuevas and J. Jalife contributed reagents and analytic tools; Y. Takemoto, D. P. Slough, G. Meinke, C. Diehl, A. Bohm, J. Jalife, Y.-S. Lin, and S. F. Noujaim wrote the paper.
REFERENCES
- 1.Dobrev D., Friedrich A., Voigt N., Jost N., Wettwer E., Christ T., Knaut M., Ravens U. (2005) The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706 10.1161/CIRCULATIONAHA.105.575332 [DOI] [PubMed] [Google Scholar]
- 2.Wakili R., Voigt N., Kääb S., Dobrev D., Nattel S. (2011) Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968 10.1172/JCI46315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makary S., Voigt N., Maguy A., Wakili R., Nishida K., Harada M., Dobrev D., Nattel S. (2011) Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ. Res. 109, 1031–1043 10.1161/CIRCRESAHA.111.253120 [DOI] [PubMed] [Google Scholar]
- 4.Dobrev D., Carlsson L., Nattel S. (2012) Novel molecular targets for atrial fibrillation therapy. Nat. Rev. Drug Discov. 11, 275–291 10.1038/nrd3682 [DOI] [PubMed] [Google Scholar]
- 5.Voigt N., Trausch A., Knaut M., Matschke K., Varró A., Van Wagoner D. R., Nattel S., Ravens U., Dobrev D. (2010) Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ Arrhythm Electrophysiol 3, 472–480 10.1161/CIRCEP.110.954636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., Kurachi Y. (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 10.1152/physrev.00021.2009 [DOI] [PubMed] [Google Scholar]
- 7.Kurachi Y., Ishii M. (2004) Cell signal control of the G protein-gated potassium channel and its subcellular localization. J. Physiol. 554, 285–294 10.1113/jphysiol.2003.048439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanina T., Rishal I., Varon D., Mullner C., Frohnwieser-Steinecke B., Schreibmayer W., Dessauer C. W., Dascal N. (2003) Mapping the Gbetagamma-binding sites in GIRK1 and GIRK2 subunits of the G protein-activated K+ channel. J. Biol. Chem. 278, 29174–29183 10.1074/jbc.M304518200 [DOI] [PubMed] [Google Scholar]
- 9.Logothetis D. E., Lupyan D., Rosenhouse-Dantsker A. (2007) Diverse Kir modulators act in close proximity to residues implicated in phosphoinositide binding. J. Physiol. 582, 953–965 10.1113/jphysiol.2007.133157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noujaim S. F., Pandit S. V., Berenfeld O., Vikstrom K., Cerrone M., Mironov S., Zugermayr M., Lopatin A. N., Jalife J. (2007) Up-regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J. Physiol. 578, 315–326 10.1113/jphysiol.2006.121475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atienza F., Almendral J., Moreno J., Vaidyanathan R., Talkachou A., Kalifa J., Arenal A., Villacastín J. P., Torrecilla E. G., Sánchez A., Ploutz-Snyder R., Jalife J., Berenfeld O. (2006) Activation of inward rectifier potassium channels accelerates atrial fibrillation in humans: evidence for a reentrant mechanism. Circulation 114, 2434–2442 10.1161/CIRCULATIONAHA.106.633735 [DOI] [PubMed] [Google Scholar]
- 12.Dobrev D., Nattel S. (2010) New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 375, 1212–1223 10.1016/S0140-6736(10)60096-7 [DOI] [PubMed] [Google Scholar]
- 13.Dobrev D., Nattel S. (2011) New insights into the molecular basis of atrial fibrillation: mechanistic and therapeutic implications. Cardiovasc. Res. 89, 689–691 10.1093/cvr/cvr021 [DOI] [PubMed] [Google Scholar]
- 14.Ramu Y., Klem A. M., Lu Z. (2004) Short variable sequence acquired in evolution enables selective inhibition of various inward-rectifier K+ channels. Biochemistry 43, 10701–10709 10.1021/bi049125x [DOI] [PubMed] [Google Scholar]
- 15.Takemoto Y., Ramirez R. J., Yokokawa M., Kaur K., Ponce-Balbuena D., Sinno M. C., Willis B. C., Ghanbari H., Ennis S. R., Guerrero-Serna G., Henzi B. C., Latchamsetty R., Ramos-Mondragon R., Musa H., Martins R. P., Pandit S. V., Noujaim S. F., Crawford T., Jongnarangsin K., Pelosi F., Bogun F., Chugh A., Berenfeld O., Morady F., Oral H., Jalife J. (2016) Galectin-3 regulates atrial fibrillation remodeling and predicts catheter ablation outcomes. JACC Basic Transl. Sci. 1, 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martins R. P., Kaur K., Hwang E., Ramirez R. J., Willis B. C., Filgueiras-Rama D., Ennis S. R., Takemoto Y., Ponce-Balbuena D., Zarzoso M., O’Connell R. P., Musa H., Guerrero-Serna G., Avula U. M., Swartz M. F., Bhushal S., Deo M., Pandit S. V., Berenfeld O., Jalife J. (2014) Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation 129, 1472–1482 10.1161/CIRCULATIONAHA.113.004742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osawa M., Yokogawa M., Muramatsu T., Kimura T., Mase Y., Shimada I. (2009) Evidence for the direct interaction of spermine with the inwardly rectifying potassium channel. J. Biol. Chem. 284, 26117–26126 10.1074/jbc.M109.029355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishida M., Cadene M., Chait B. T., MacKinnon R. (2007) Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 26, 4005–4015 10.1038/sj.emboj.7601828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pegan S., Arrabit C., Zhou W., Kwiatkowski W., Collins A., Slesinger P. A., Choe S. (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 8, 279–287 10.1038/nn1411 [DOI] [PubMed] [Google Scholar]
- 20.Yokogawa M., Muramatsu T., Takeuchi K., Osawa M., Shimada I. (2009) Backbone resonance assignments for the cytoplasmic regions of G protein-activated inwardly rectifying potassium channel 1 (GIRK1). Biomol. NMR Assign. 3, 125–128 10.1007/s12104-009-9156-6 [DOI] [PubMed] [Google Scholar]
- 21.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 10.1007/BF00197809 [DOI] [PubMed] [Google Scholar]
- 22.Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 24.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joosten R. P., Long F., Murshudov G. N., Perrakis A. (2014) The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220 10.1107/S2052252514009324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 31.Wishart D. S., Knox C., Guo A. C., Cheng D., Shrivastava S., Tzur D., Gautam B., Hassanali M. (2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36, D901–D906 10.1093/nar/gkm958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sastry G. M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 10.1007/s10822-013-9644-8 [DOI] [PubMed] [Google Scholar]
- 33.Greenwood J. R., Calkins D., Sullivan A. P., Shelley J. C. (2010) Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 24, 591–604 10.1007/s10822-010-9349-1 [DOI] [PubMed] [Google Scholar]
- 34.Shelley J. C., Cholleti A., Frye L. L., Greenwood J. R., Timlin M. R., Uchimaya M. (2007) Epik: a software program for pK( a ) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 21, 681–691 10.1007/s10822-007-9133-z [DOI] [PubMed] [Google Scholar]
- 35.Shivakumar D., Williams J., Wu Y., Damm W., Shelley J., Sherman W. (2010) Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 6, 1509–1519 10.1021/ct900587b [DOI] [PubMed] [Google Scholar]
- 36.Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 38.Iijima H., Dunbar J. B., Jr., Marshall G. R. (1987) Calibration of effective van der Waals atomic contact radii for proteins and peptides. Proteins 2, 330–339 10.1002/prot.340020408 [DOI] [PubMed] [Google Scholar]
- 39.Mähler J., Persson I. (2012) A study of the hydration of the alkali metal ions in aqueous solution. Inorg. Chem. 51, 425–438 10.1021/ic2018693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noujaim S. F., Stuckey J. A., Ponce-Balbuena D., Ferrer-Villada T., López-Izquierdo A., Pandit S., Calvo C. J., Grzeda K. R., Berenfeld O., Chapula J. A., Jalife J. (2010) Specific residues of the cytoplasmic domains of cardiac inward rectifier potassium channels are effective antifibrillatory targets. FASEB J. 24, 4302–4312 10.1096/fj.10-163246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li D., Chen R., Chung S. H. (2016) Molecular dynamics of the honey bee toxin tertiapin binding to Kir3.2. Biophys. Chem. 219, 43–48 10.1016/j.bpc.2016.09.010 [DOI] [PubMed] [Google Scholar]
- 42.Whorton M. R., MacKinnon R. (2011) Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208 10.1016/j.cell.2011.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodríguez-Menchaca A. A., Navarro-Polanco R. A., Ferrer-Villada T., Rupp J., Sachse F. B., Tristani-Firouzi M., Sánchez-Chapula J. A. (2008) The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc. Natl. Acad. Sci. USA 105, 1364–1368 10.1073/pnas.0708153105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burrell Z. L., Jr., Martinez A. C. (1958) Chloroquine and hydroxychloroquine in the treatment of cardiac arrhythmias. N. Engl. J. Med. 258, 798–800 10.1056/NEJM195804172581608 [DOI] [PubMed] [Google Scholar]
- 45.Sánchez-Chapula J. A., Salinas-Stefanon E., Torres-Jácome J., Benavides-Haro D. E., Navarro-Polanco R. A. (2001) Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J. Pharmacol. Exp. Ther. 297, 437–445 [PubMed] [Google Scholar]
- 46.Walker O., Dawodu A. H., Adeyokunnu A. A., Salako L. A., Alvan G. (1983) Plasma chloroquine and desethylchloroquine concentrations in children during and after chloroquine treatment for malaria. Br. J. Clin. Pharmacol. 16, 701–705 10.1111/j.1365-2125.1983.tb02244.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noujaim S. F., Stuckey J. A., Ponce-Balbuena D., Ferrer-Villada T., López-Izquierdo A., Pandit S. V., Sánchez-Chapula J. A., Jalife J. (2011) Structural bases for the different anti-fibrillatory effects of chloroquine and quinidine. Cardiovasc. Res. 89, 862–869 10.1093/cvr/cvr008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voigt N., Rozmaritsa N., Trausch A., Zimniak T., Christ T., Wettwer E., Matschke K., Dobrev D., Ravens U. (2010) Inhibition of IK,ACh current may contribute to clinical efficacy of class I and class III antiarrhythmic drugs in patients with atrial fibrillation. Naunyn Schmiedebergs Arch. Pharmacol. 381, 251–259 10.1007/s00210-009-0452-6 [DOI] [PubMed] [Google Scholar]
- 49.Ferrari R., Bertini M., Blomstrom-Lundqvist C., Dobrev D., Kirchhof P., Pappone C., Ravens U., Tamargo J., Tavazzi L., Vicedomini G. G. (2016) An update on atrial fibrillation in 2014: from pathophysiology to treatment. Int. J. Cardiol. 203, 22–29 10.1016/j.ijcard.2015.10.089 [DOI] [PubMed] [Google Scholar]
- 50.Podd S. J., Freemantle N., Furniss S. S., Sulke N. (2016) First clinical trial of specific IKACh blocker shows no reduction in atrial fibrillation burden in patients with paroxysmal atrial fibrillation: pacemaker assessment of BMS 914392 in patients with paroxysmal atrial fibrillation. Europace 18, 340–346 [DOI] [PubMed] [Google Scholar]
- 51.Walfridsson H., Anfinsen O. G., Berggren A., Frison L., Jensen S., Linhardt G., Nordkam A. C., Sundqvist M., Carlsson L. (2015) Is the acetylcholine-regulated inwardly rectifying potassium current a viable antiarrhythmic target? Translational discrepancies of AZD2927 and A7071 in dogs and humans. Europace 17, 473–482 10.1093/europace/euu192 [DOI] [PubMed] [Google Scholar]
- 52.Voigt N., Friedrich A., Bock M., Wettwer E., Christ T., Knaut M., Strasser R. H., Ravens U., Dobrev D. (2007) Differential phosphorylation-dependent regulation of constitutively active and muscarinic receptor-activated IK,ACh channels in patients with chronic atrial fibrillation. Cardiovasc. Res. 74, 426–437 10.1016/j.cardiores.2007.02.009 [DOI] [PubMed] [Google Scholar]
- 53.Olshansky B. (2004) Combining ablation of atrial fibrillation with ablation of atrial flutter: are we there yet? J. Am. Coll. Cardiol. 43, 2063–2065 10.1016/j.jacc.2004.03.020 [DOI] [PubMed] [Google Scholar]
- 54.Nattel S., Dobrev D. (2016) Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat. Rev. Cardiol. 13, 575–590 10.1038/nrcardio.2016.118 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.