

Abstract
The RecA/LexA axis of the bacterial DNA damage (SOS) response is a promising, yet non-traditional drug target. The SOS response is initiated upon genotoxic stress, when RecA, a DNA damage sensor, induces LexA, the SOS repressor, to undergo autoproteolysis, thereby de-repressing downstream genes that can mediate DNA repair and accelerate mutagenesis. As genetic inhibition of the SOS response sensitizes bacteria to DNA damaging antibiotics and decreases acquired resistance, inhibitors of the RecA/LexA axis could potentiate our current antibiotic arsenal. Compounds targeting RecA, which has many mammalian homologs, have been reported; however, small-molecules targeting LexA autoproteolysis, a reaction unique to the prokaryotic SOS response, have remained elusive. Here, we describe the logistics and accomplishments of an academic-industry partnership formed to pursue inhibitors against the RecA/LexA axis. A novel fluorescence polarization assay reporting on RecA-induced self-cleavage of LexA enabled the screening of 1.8 million compounds. Follow-up studies on select leads show distinct activity patterns in orthogonal assays, including several with activity in cell-based assays reporting on SOS activation. Mechanistic assays demonstrate that we have identified first-in-class small molecules that specifically target the LexA autoproteolysis step in SOS activation. Our efforts establish a realistic example for navigating academic-industry partnerships in pursuit of anti-infective drugs, and offer starting points for dedicated lead optimization of SOS inhibitors that could act as adjuvants for current antibiotics.
Keywords: SOS response, LexA, RecA, antimicrobial resistance, anti-virulence, high-throughput screening
Graphical Abstract
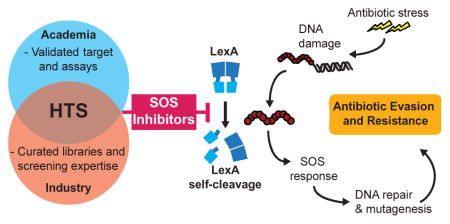
The bacterial SOS response is best known as the coordinated network of genes involved in DNA repair and mutagenesis in response to DNA damage (Figure 1A).1 The response is initiated by the interaction of two proteins, RecA, a DNA damage sensor, and LexA, a dual-functional repressor-protease. In the absence of stress, intact LexA binds to the promoters of SOS genes and blocks their transcription. Upon DNA damage, RecA polymerizes along exposed single-stranded DNA, forming activated nucleoprotein filaments (referred to as RecA*). RecA* functions as an allosteric effector, that promotes a latent serine protease activity within LexA that results in LexA self-cleavage (autoproteolysis). Autoproteolysis causes LexA to lose its repressor function, leading to the induction of genes involved in a host of adaptive processes, including nucleotide excision repair, error-prone translesion DNA replication, activation of mobile genetic elements, and cell division arrest.2,3 Expression of these SOS genes enables bacteria to survive and adapt to various genotoxic stresses in the environment, such as UV radiation, reactive oxygen species, and, importantly, antimicrobials.
Figure 1. Targeting LexA and RecA to inhibit the bacterial SOS response.

(A) Intact LexA (blue) acts as the DNA binding repressor for the SOS response. In the normal stress response, exposure to antibiotic stress triggers RecA polymerization along exposed, damaged single-stranded DNA, forming RecA* (red ovals). Formation of RecA* induces the autoproteolysis of the LexA repressor-protease, leading to the induction of SOS genes. Inhibition of the SOS response can increase antibiotic susceptibility by decreasing the proficiency of DNA repair or by preventing the development of antibiotic tolerant states. Inhibition of the response can also suppress acquired antibiotic resistance by inhibiting pro-mutagenic DNA repair mechanism or decreasing horizontal gene transfer (HGT). (B) Design of the FlAsH-LexA HTS assay. Structural images shown here are derived from existing E. coli LexA crystal structures (Full-length LexA: PDB 1JSO; Truncated LexA: PDB 1JHE). The N-terminal DNA binding domain of LexA was replaced with the short hexapeptide motif CCPGCC, which specifically binds to FlAsH-EDT2. (C) Incubation of 100 nM FlAsH-LexAEc with 300 nM RecA* induces autoproteolysis, resulting in the release of the small labeled peptide and an associated FP signal change (top panel). Mutation of the catalytic serine (S119) to an alanine abrogates self-cleavage (bottom panel). Data points represent the average values of five independent measurements and the error bars represent standard deviation.
In recent years, studies demonstrating a strong association between the SOS response and antimicrobial evasion have reinvigorated interest in this historically well-studied system.4–6 Numerous classes of antimicrobials, particularly DNA damaging agents, can trigger the SOS pathway.7–10 Genetically inactivating the RecA*/LexA axis can attenuate the SOS response and has been shown to result in both decreased antibiotic-associated mutagenesis and increased activity of DNA damaging antibiotics (decreased MIC), with recent evidence even demonstrating re-sensitization of resistant strains.7–12 Furthermore, tempering the SOS response can compromise multiple adaptive phenotypes, including persistence and biofilm formation, the activation of integron genes that mediate horizontal gene transfer of resistance elements, and the expression of resistance elements, such as qnrB fluoroquinolone-resistance genes (Figure 1A).13–17
Given these genetic precedents, inhibiting the SOS response has been proposed as a therapeutic strategy to potentiate current and future antibiotics.4,5 Small molecule SOS antagonists have been pursued both as tools to probe the SOS response or as intermediates toward potential antibiotic adjuvants. To date, reported inhibitors of the SOS response are confined to RecA antagonists: compounds that prevent the formation of ssDNA/RecA filaments or the protein’s ATP-dependent activities have been isolated as natural products or from screening based approaches.18–24 From a therapeutic perspective, however, specifically targeting RecA in bacteria poses a challenge due to the homology of the protein to the large and essential eukaryotic Rad51 family.25 By contrast, LexA does not possess eukaryotic homologs, and no prior in vitro screening efforts with LexA autoproteolysis as an endpoint have been reported in the literature. The absence of small molecules inhibiting LexA may be related in part to the challenges posed by this target, including the intramolecular nature of self-cleavage and the lack of insights into the interface between LexA and RecA*.
To broaden the pursuit of small molecule antagonists targeting the RecA/LexA axis, we considered the potential virtues of a different model for screening and inhibitor discovery: an academic-industry partnership. Academic institutions can provide in-depth knowledge of a given area of investigation and offer fresh paradigms for targets, while industry offers the infrastructure and expertise needed for large-scale library screening and translational efforts that can be important with non-traditional targets or those where a low hit-rate is anticipated. While such partnerships are increasingly common,26–29 their logistics are less commonly reported in the literature, despite the need for the description of precedents on issues ranging from work-flow to intellectual property to help template future efforts. Here, we report our collaborative effort with GlaxoSmithKline (GSK) to screen for inhibitors of the RecA*/LexA axis. We describe the identification of first-in-class inhibitors of LexA autoproteolysis and offer our experience as an example of navigating academic-industry partnerships to address the rising tide of antibiotic resistant bacteria.
RESULTS AND DISCUSSION
Design of the HTS FlAsH assay
Since LexA self-cleavage has been classically tracked by low-throughput, gel-based analysis, we (the UPenn team) first aimed to apply our biochemical understanding of LexA to devise an assay to directly monitor autoproteolysis in a manner amenable to screening. Many protease inhibitor discovery campaigns have employed synthetic peptide substrates containing fluorophore-quencher pairs flanking the cleavable peptide bond in their screening assays.30 However, the lack of LexA cleavage activity on peptide substrates in trans required us to devise an alternative strategy.
LexA exists as a homodimer in solution, with each monomer consisting of an N-terminal repressor domain (NTD) that binds DNA and a C-terminal domain (CTD) with serine protease activity. LexA autoproteolysis occurs within this CTD when a large conformational change positions the scissile bond (Ala84-Gly85 in E. coli LexA) for attack by the catalytic Ser (S119).31,32 A direct interaction between the activated RecA nucleoprotein filaments (RecA*) is believed to stabilize the cleavable conformation, thereby ensuring that LexA cleavage occurs rapidly in the presence of DNA damage. The in vivo cleavage reaction can be reconstituted in vitro with purified LexA and RecA* (RecA*-dependent cleavage). Importantly, in the absence of RecA*, a very low level of basal LexA hydrolysis also occurs at physiological pH (RecA*-independent cleavage), and this reaction can be accelerated by activation of the protein’s serine-lysine catalytic dyad at alkaline pH in vitro.33,34
Early work on the LexA of E. coli (LexAEc) suggested that tryptic removal of large portions of the NTD has no impact on the self-cleavage ability of the protease domain.35 Building on this observation and using existing LexA crystal structures as a guide,31,32 we further truncated the NTD to Gly75 for LexAEc (or Gly81 in the LexA from Pseudomonas aeruginosa) and attached an N-terminal tag containing a tetracysteine CCPGCC motif, which was specifically labeled with the biarsenical fluorophore, FlAsH-EDT2 (Figure 1B).36 Upon stimulation of autoproteolysis by filamentous RecA (RecA*), release of the short FlAsH-containing N-terminal peptide from this FlAsH-LexA construct resulted in a significant drop in fluorescence polarization (FP), allowing cleavage to be monitored either continuously or as an endpoint measurement after quenching with EDTA (Figure 1C); changing the catalytic Ser to Ala abolishes cleavage. The truncated LexA constructs mirror the behavior of the full-length protein in that alkaline-mediated cleavage occurs at comparable rates (Figure S1).
To develop the screening approach, we performed a small-scale screen of the Microsource Spectrum library (2560 compounds) using the FlAsH-LexA of P. aeruginosa (FlAsH-LexAPa). We screened compounds on two separate days at a fixed concentration of 16 μM in a single endpoint assay, according to standard screening procedures at our local screening facility. Across measurements, the average Z′-factor relative to the control reactions lacking RecA* was 0.77 on Day 1 and 0.65 on Day 2 (Figure S2). The independent replicates across two days showed a strong linear correlation (R2 = 0.93). While one of our goals was to focus on the inhibition of LexA autoproteolysis as the endpoint, we note that the design of our assay could identify compounds that target LexA, RecA, or their interface. In line with this possibility, one of the compounds identified in this pilot screen was suramin, which had been identified in prior RecA-focused screening efforts,37,38 but also shows concerning promiscuity as can often emerge from commercial library screening.39–41 Overall, the pilot screen indicated that the assay was robust and well-suited for advancing to high-throughput screening.
Engagement of collaboration with GSK
With a robust HTS assay available, but with screening capabilities limited to smaller, commercially-available libraries at our academic site, we considered whether a partnership with a pharmaceutical company could offer a means to utilize the HTS assay to its greatest effect. Such a partnership would permit access to a large, well-curated compound library and leverage drug discovery expertise – an important consideration since we anticipated that the intramolecular self-cleavage reaction of the LexA protease would likely present a difficult molecular target. To this end, we entered our concept of targeting LexA into the inaugural GSK Discovery Fast Track Challenge. The selection process proceeded in two stages. During the initial stage, we presented the therapeutic hypothesis without disclosure of confidential intellectual material, such as our FP-based HTS assay. Out of 140 applications, the LexA project was selected as one of 15 finalists to give a detailed presentation of the target background, availability of primary and secondary assays, and potential clinical applications. This finalist phase proceeded under a confidentiality disclosure agreement, which included a non-use clause prohibiting the unauthorized use of the disclosed material by GSK. At the end of the selection process, our group was among eight chosen for collaboration. Four of the eight groups selected involved anti-infective targets, highlighting the potential strengths of partnership approaches in this therapeutic area. The partnership terms stated that we would remain blinded to compound structures during the HTS process. Upon completion of the HTS screen, both we and the GSK team would have the right to decide whether to continue to advance the project. If we wished to proceed and GSK did not, GSK committed to releasing a limited number of compound structures (including their associated composition of matter rights), and both groups agreed to revenue sharing if leads advanced to therapeutics.
HTS screening campaign
The high-throughput screening campaign was designed to leverage the resources and expertise of both sides of the partnership (Figure 2A). Initial discussions resulted in the decision to use E. coli FlAsH LexA (FlAsH-LexAEc) in the screen (Figure S1). Although P. aeruginosa is a more pressing drug-resistant threat, both groups agreed that the genetic tools established in E. coli strains at UPenn would accelerate downstream validation steps (see below). We transferred assay reagents to the GSK team, who miniaturized the assay to a 1536-well format and performed a preliminary screen on ~9800 compounds in triplicate to independently validate the screen on the GSK platform (Figure S3). The statistics of the validation screen are summarized in Table S1.
Figure 2. Partnership between the University of Pennsylvania (UPenn) and GSK to screen for SOS inhibitors.

(A) Contributions of UPenn and GSK are colored with blue and red, respectively. The panels on the left summarize the major milestones in the partnership, while the right panels provide details on the HTS screen and the secondary screens. Despite the division of labor, methods and results were openly discussed between the two groups whenever appropriate. (B) Response distribution of the HTS screen. Plotted is a histogram of the responses (% activity) observed across 1.8 million compounds, grouped into 179 bins. The mean value of inhibition (0.99%) is noted and the 15.95% response cutoff is highlighted in purple, leading to 5544 statistical actives after primary screening.
Following validation, the screening proceeded to the full GSK discovery library of approximately 1.8 million compounds. Compounds were analyzed at a concentration of 10 μM per the screening practices of GSK. The screen (Table 1) was of high quality with an average Z′-factor of 0.72 and the average 3SD cutoff being ~16% inhibition and consistent results across batches (Figure 2B, Figure S4). Using these criteria, the screening campaign yielded 5,544 compounds selected for confirmation, corresponding to a hit rate of 0.31%. Re-testing of the initial hits identified 396 compounds with consistent activity in duplicate testing, following the removal of 111 auto-fluorescent compounds. The reconfirmation rate of 7.1% is consistent with many hits being close to the 3SD cutoff in initial screening. The 396 compounds were subjected to a dose-response analysis, which yielded a preliminary list of 245 compounds exhibiting >50% inhibition at the highest compound concentration of 200 μM. Analyzing the total fluorescence intensity values as a function of compound concentration and magnitude of inhibition enabled the GSK team to exclude 101 compounds with potential confounding fluorescence interference. Overall, the HTS earmarked 144 compounds for further biochemical studies. The GSK team clustered these compounds by visual inspection into seven chemotypes (families A-J) with multiple representatives and 12 singleton compounds (S1–S12). The singleton compounds and representatives from the different chemotypes – 32 compounds in total – were transferred via an MTA agreement to the UPenn team for further cell-based analysis. Though we remained blinded to the structures of the compounds during this screening and triage process, GSK shared descriptions of each compound’s physicochemical properties, potential reactive functionality, and other data obtained in the course of the HTS to foster collaborative decisions.
Table 1.
Screening statistics
| Total HTS compounds screened | 1,814,611 |
| Predicted hit rate, %a | 0.14 |
| Predicted 3SD cutoff, %a | 15.10 |
| No. primary 1536 assay plates | 1376 |
| Average Z′ (SD) | 0.72 (0.03) |
| Average robust 3SD cutoff, % | 15.95 |
| Average hit rate, % | 0.31 |
| Number robust actives in primary screen | 5544 |
| Statistical actives upon replicate screening | 396 |
| Potency bins (pIC50) earmarked compounds | |
| < 5 | 125 |
| 5 – 6 | 17 |
| 6 – 7 | 2 |
| >7 | 0 |
Based on data obtained in the validation set (see Table S1)
Secondary screening of candidates with cell-based assays
We then tested the transferred compounds for evidence of activity inside cells, using a well-established cell-based SOS reporter assay.8,42 In this assay, an E. coli strain with a deletion of the tolC efflux gene (ΔtolC) is transformed with a SOS-inducible reporter plasmid, which harbors GFP under the control of the recA promoter. Under ciprofloxacin stress (128 ng/mL), LexA repression on the recA promoter is relieved and a ~50-fold increase in GFP fluorescence over the unstressed control can be detected by flow cytometry at 2 hours (Figure 3A). Initially, compounds were assayed at a single dose (50 μM) for their ability to inhibit GFP expression (Figure 3B, S5, & S6A). Following this initial assay, we advanced those compounds that displayed at least 25% inhibition of GFP expression and/or exhibited the greatest decrease in GFP expression from each chemotype. Employing these criteria, we selected ten compounds (A3, B2, C1, D1, I2, J2, S2, S5, S10, and S12) for additional analysis, and observed dose-dependent inhibition of GFP expression (Figure 3C and S6B). To examine whether the alterations in GFP expression could be due to non-specific effects, we tested the most promising leads in two other conditions where GFP expression was either constitutively expressed or repressed. In the constitutively expressed condition, the ΔtolC strain was transformed with a reporter plasmid with a reversed LexA binding site, rendering it active independent of the SOS response; in the constitutively repressed condition, the regular reporter plasmid was introduced into a ΔtolC strain with the inactivating S119A mutation in the lexA gene. In both conditions, we observed minimal impacts on GFP expression up to 100 μM, with the exception of J2 (Figure S7). With J2, we observed a dose-dependent reduction in GFP fluorescence in both reporter strains, suggesting a direct interference with GFP expression or fluorescence. From these cell-based assays, the most promising compounds were earmarked for potential disclosure from GSK to UPenn, as described in the next section.
Figure 3. Cell-based SOS reporter assay.

(A) E. coli ΔtolC strains transformed with a GFP-based SOS reporter plasmid. Exposure of this strain to ciprofloxacin stress induces the SOS response and GFP expression. Inhibitors that block SOS induction would cause a reduction in GFP fluorescence. (B) Representative population histograms of the SOS reporter strain. Mid-log phase cell populations stressed by ciprofloxacin (+ FQ) are shown in blue, while non-stressed cells (No stress) are shown in red. Cells treated with 50 μM of compound are in green (+ FQ, + compound). Population histograms of all compounds are shown in Figure S4. (C) Dose response of compounds in the cell-based assay. The left panel shows a representative population histogram of MG1655 E. coli ΔtolC cells treated with increasing levels of compound C1. The right panel shows dose response curves of compounds C1, D1, and S5 in the reporter assay. The percent inhibition was calculated relative to fluorescence of cells not exposed to ciprofloxacin. Each data point represents the mean of two independent measurements and the error bars represent the range of values. Dose responses of all compounds are shown in Figure S5 and assay of C1 and D1 with WT MG1655 are shown Figure S12.
Decision to advance leads and disclosure of structures of select compounds
Upon completion of the HTS campaign and cell-based assay, both groups independently decided if advancement of the leads was desirable for their goals. Due to their micromolar potency and the projected challenges associated with optimizing the inhibitors, GSK decided not to proceed with the partnership and agreed to disclose compound structures to us to pursue independently. Both sides participated in joint meetings to review the combined data and select the best-suited compounds for disclosure. Although we remained blinded to compounds structures at this point, the GSK team provided opinions on the synthetic tractability of each compound to assist with our decision about which leads to select for disclosure. A significant criterion we used for selection was activity in the in vivo SOS reporter assay, as optimizing cell-based activity often serves as a major bottleneck in advancing lead compounds in the antibacterial area. With added consideration for synthetic tractability, we therefore selected compounds that showed sigmoidal dose-response activity in the cell-based assay, C1, D1, and S5; Furthermore, we chose A3 because it represented the most populous chemotype among the HTS hits. GSK disclosed one additional potential biochemical tool compound, S8, which did not show activity in cells, but did show activity in an assay focused on RecA polymerization only (described below). The structures of the compounds are shown in Figure 4 and their disclosed properties, including cytotoxicity in mammalian cells (see supplemental methods), are summarized in Table 2.
Figure 4. Structures of the compounds released after HTS.
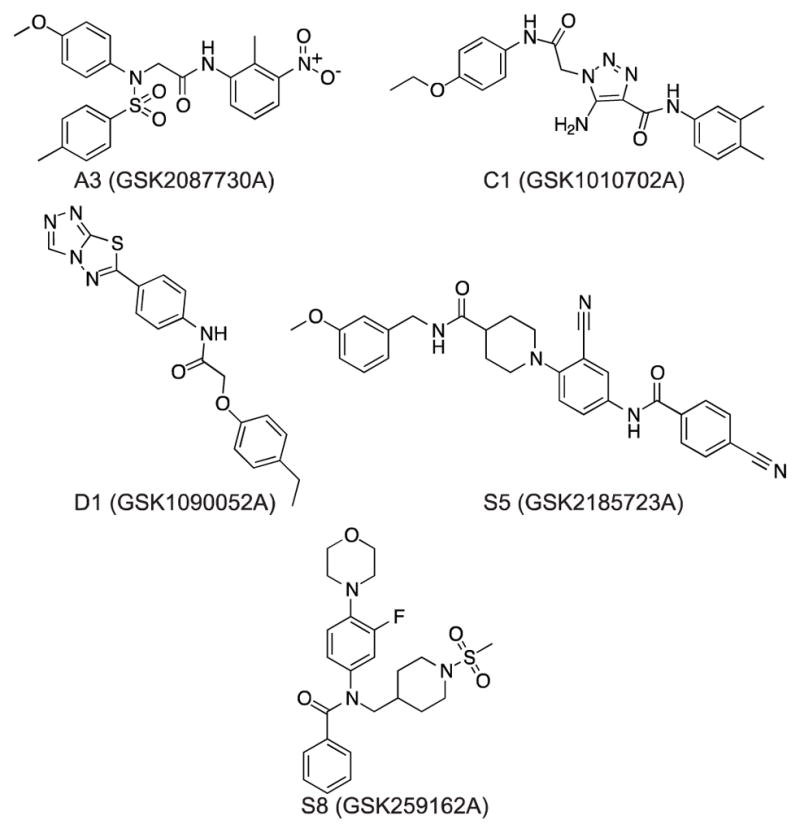
The five compounds were selected based on available biochemical and cell-based data, and subjected to additional validation and mechanistic assays.
Table 2.
Properties of the leads
| Compound | IC50 (μM) a | E. coli SOS reporter Assay (IC50, μM) | Cytotoxicity TC50 (μM) in L5178Y TK+/− (μM) | MW (g/mol) | IFI*** | ||||
|---|---|---|---|---|---|---|---|---|---|
| FlAsH-LexAEc Assay | RecA*/32P-LexAEc Assay | 32P-LexAEc only Assay | RecA Filament Assay | LexAPa Assays (RecA*/32P-LexAPa/LexAPa only) | |||||
| A3 | Not obtainable* | No activity | No activity | No activity | N/A | Active (37 ± 10) | 50 | 468.51 | 3/196, 2% |
| C1 | 32 (17–57) | 60 ± 2 | No activity | No activity | 150± 2/No activity | Active (31 ± 1) | 25 | 408.45 | 1/97, 1% |
| D1 | 20 (15–28) | 10 ± 1 | 13 ± 1 | No activity | 135± 2/63± 4 | Active (110 ± 7) | >100 | 379.44 | 0/179, 0% |
| S5 | 40 (26–51) | 9 ± 2 | 79 ± 1 | No activity | N/A | Active (46 ± 1) | >100 | 607.58 | 5/153, 3% |
| S8 | 105 (87–125) | Not obtainable** | No activity | 95 (74–120) | N/A | No activity | 100 | 475.58 | 29/230, 13% |
IC50 values represent compound concentration at which 50% inhibition is observed. The range of error given is from fit of duplicate data from dose response curves. Not obtainable indicates a plateau in maximal inhibition below 60%.
Total percent inhibition plateaus at 30%. Half of this maximal inhibition is observed at 0.54 μM (0.53–0.55).
Total percent inhibition plateaus at 57%. Half of this maximal inhibition is observed at 23 ± 2 μM.
Inhibition Frequency Index; how often >50% inhibition is observed out of the GSK screens conducted with the compound.
Characterization of leads with mechanistic assays
To build on results from the HTS screening, we next examined lead compounds using additional assays that could both validate their activities and reveal their mechanisms of action. We first assayed compounds using a non-fluorescence-based assay containing full-length E. coli LexA to account for fluorescence interference and perturbations caused by using a truncated reporter protein. In this assay, we monitored for dose-responsive inhibition of RecA*-stimulated cleavage of an N-terminally 32P-labelled LexA (Figure 5A & S8).33 Under these conditions, compounds C1, D1, and S5 inhibited LexA self-cleavage with IC50 values of 60, 10, and 9 μM, respectively (Table 2). Compound A3 did not display any detectable levels of inhibition. Compound S8 showed weak, incomplete inhibition in the RecA*/32P-LexA assay.
Figure 5. Orthogonal biochemical assays examining compound activity against LexA.

(A–B) Representative gels of compound D1 and dose response curves of compounds C1, D1, and S5 are shown across the two assays. Gel images and dose response curves of all five compounds are in Figure S7. (A) The RecA*/32P-LexA cleavage assay. Radiolabeled LexA was incubated with compound and self-cleavage was stimulated by adding RecA*. (B) The RecA*-independent cleavage assay identifies compounds that inhibit on LexA autoproteolysis alone. Radiolabeled LexA was incubated with compound and the progress of basal self-cleavage at neutral pH was monitored over 4 hours. Inhibition can be observed based on the decreased accumulation of labeled LexA NTD as a function of increasing compound. Given the longer exposure time, a minor contaminating band can be seen running above the NTD in the RecA*-independent assay, but not the RecA*/32P-LexA cleavage assay. (C) The LexA/DNA binding assay. Representative gels of C1, D1, and S5 are shown. EMSA gels of all five compounds are found in Figure S8.
Despite its low throughput, a notable strength of using 32P-LexA is that the construct can be used to examine LexA autoproteolysis that occurs independent of RecA*. Traditionally, this has been examined by quantifying the first-order kinetics of LexA self-cleavage under alkaline conditions, which could perturb compound stability and reactivity.33,35 Given the sensitivity afforded by 32P-LexA, we repurposed the assay to report on LexA autoproteolysis under conditions analogous to those studied in the HTS assay (pH 7), but excluding RecA* (Figure 5B). 32P-LexA was incubated at 37 °C for 4 hours in the presence or absence of compound and self-cleavage was quantified. When tested with this assay, the lead compounds display distinct features. Both D1 and S5 exhibited dose-dependent inhibition of LexA self-cleavage, with D1 showing an IC50 of 13 μM and S5 an IC50 of 79 μM (Figure 5B & S8). Notably, D1’s potency in the RecA*-independent assay closely resembles its IC50 in the RecA*/32P-LexA assay, whereas S5’s potency in the RecA*-independent assay dropped by approximately 9-fold. The three remaining compounds A3, C1, and S8 did not show detectable levels of inhibition. Thus, these results suggest that D1 specifically targets LexA, while compounds C1 and S5, which show activity in the FlAsH-LexA and RecA*/32P-LexA assays, could potentially interfere with the RecA*/LexA interface.
To gain further support for specificity in the mode of action and demonstrate that our leads were not false-positive, pan-assay interference compounds (PAINS),43 we examined whether the compounds were specifically inhibiting LexA autoproteolysis. First, since proteolysis is mediated by the CTD, we reasoned that DNA binding activity, primarily mediated by the NTD, should be unaffected. Compounds were examined for any impact on LexA binding to operator DNA.32 To quantify DNA binding, we performed electrophoresis mobility shift assays that monitor LexA binding to fluorescently-labeled operator DNA in the presence or absence of compound (Figure 5C). In the presence of the five lead compounds at 50 μM, no significant change was observed in the ability of LexA to bind to operator DNA (Figure 5C & S9). To further examine selectivity, two compounds, C1 and D1, were selected to estimate the specificity for inhibition of LexA autoproteolysis over inhibition for other proteases. The compounds were tested against two serine proteases (HCV1a protease and DPP4), an aspartyl protease (renin) and a cysteine protease (caspase 3). No inhibition was observed with D1 for all four proteases. C1 showed no activity against renin and the HCV1a protease, and only 31% and 26% inhibition at the highest concentration analyzed (100 μM) for DPP4 and caspase3, respectively (Table S2). Given the data indicating that D1 could inhibit LexA alone, we also performed a thermal shift assay with this pair. In this assay, D1 displayed stabilization of the LexA melting temperature, indicative of a direct binding interaction between the compound and the protein (Figure S10), a shift not observed with C1. As a complement to our biochemical assays, GSK noted that the Inhibition Frequency Indexes (defined as how often a given compound exhibited >50% inhibition in GSK screens conducted prior to ours) for C1, D1, and S5 are 1, 0 and 3 percent, respectively, suggesting an overall low promiscuity of these compounds (Table 2). Thus, we conclude that these leads show specificity for targeting the RecA*/LexA axis.
To further establish the target protein, the impact of the compounds on RecA alone was also investigated. Following completion of the main HTS campaign, the transferred compounds were subjected to a previously validated assay, which measures disruption of pre-formed RecA* filaments on fluorescently labeled single-stranded DNA by fluorescence polarization.19 Using this RecA*-only assay, one compound, S8, inhibited RecA polymerization, with a potency that was similar to that observed in the full HTS assay (IC50 = 95 μM); none of the remaining 31 compounds, including the other four disclosed leads, displayed detectable activity (Figure S11). The accumulation of data from the mechanistic assays suggest that of the compounds selected, D1 likely acts on LexA alone, C1 and S5 interfere with the RecA*/LexA interface, S8 weakly targets RecA, and A3 may be an anomalous hit.
Potential for advancement of tool compounds
At the conclusion of our HTS efforts, in a forward-looking manner, we next sought to assess the barriers in translating these leads from tool compounds for probing the SOS pathway to potential therapeutics. In this regard, we focused on analysis of C1 and D1, the two most potent compounds in established assays and with significant potential for downstream optimization through medicinal chemistry efforts. We focused on characterizing C1 and D1 with regards to three potential barriers: permeability and efflux, breadth of activity against pathogens, and synergy with DNA damaging antibiotics. To first address permeability, we examined C1 and D1 using our PrecA-GFP reporter assay, now utilizing E. coli with an intact tolC efflux pump. The level of SOS induction was monitored after inducing DNA damage with ciprofloxacin in the presence of C1 and D1. Consistent with expectations, both compounds reduce SOS induction in the cells in a dose-dependent manner, although not as potently as seen in ΔtolC strains (Figure S12), indicating that they are subject to active efflux to some extent. To next assess activity against a different pathogen, we turned our attention back to LexA from P. aeruginosa (LexAPa). We expressed, purified, and labeled full-length PKA-tagged LexAPa and evaluated C1 and D1 under both RecA*-dependent and independent conditions using 32P-LexAPa (Figure S13). In RecA*-dependent cleavage, C1 and D1 display IC50’s of ~150 and 135 μM (Table 2). In RecA*-independent cleavage assays, C1 did not inhibit full-length LexAPa, while D1 exhibited an IC50 of 63 μM (Table 2). Thus, C1 and D1 both show activity against LexAPa but with lower potency than against LexAEc, and the pattern of inhibition of C1 and D1 in mechanistic assays remained consistent across the two LexA species. Finally, we tested whether C1 and D1 displayed antibiotic activity in combination with other DNA damaging agents. We subjected ΔtolC E. coli strains to increasing levels of ciprofloxacin in the presence of 200 μM of either C1 or D1 (Figure S14) and measured the number of surviving cells at 1 hour following treatment. In the absence of ciprofloxacin, neither C1 nor D1 affected the survival of ΔtolC E. coli, relative to the DMSO carrier, and minimal impact on survival was observed at lower ciprofloxacin concentration. At elevated levels of ciprofloxacin (between 125 and 250 ng/mL), while surviving cells were detectable with ciprofloxacin alone, these were consistently diminished in the presence of C1 or D1. This phenotype suggests that the two compounds are weakly active inside cells, and underscores the need for further medicinal chemistry efforts to improve the potency of these lead compounds.
In summary, by devising an HTS assay focusing on LexA cleavage as an endpoint we have identified novel inhibitors of the SOS response that displayed specific mechanisms of action. Our identification of D1 as a small molecule inhibitor active against LexA alone is significant, as to date, only high levels of non-specific, pan-protease inhibitors or lariat peptide inhibitors have been shown to inhibit LexA autoproteolysis.44,45 Both C1 and D1 also show inhibition against LexA of P. aeruginosa, albeit with higher IC50’s, suggesting that these compounds can be used to study the RecA*/LexA axis of different bacterial species. The altered potency has prompted ongoing current work exploring modifications that could enhance species-specific targeting or show broad spectrum anti-SOS activity.
Our in vivo reporter and survival assays suggest a need for further medicinal chemistry efforts to improving the cell permeability of both C1 and D1. Notably, genetic analysis using E. coli with a hypoactive LexA that reduced the rate of self-cleavage by ~5-fold showed that the MIC of E. coli to genotoxic agents (e.g. ciprofloxacin) was not altered; reduced viability only became evident only when the LexA was fully inactivated by an S119A mutation.8 Thus, as opposed to some more conventional targets where partial inhibition can lead to antibacterial effects, an added barrier to targeting the SOS response appears to be that compounds targeting the RecA/LexA axis must display a high level of in-cell potency and produce near complete SOS inhibition. C1 and D1 are amenable to serving as stepping stones for medicinal chemistry efforts aimed at generating compounds with higher efficacy, revealing structure-activity relationships (SAR), and improving activity in cells. For example, compound C1, with a central triazole ring linked to two flanking phenyl rings is accessible from fragments via a Huisgen Cycloaddition. Notably, the central 1,4-connected 1,2,3-triazole ring of the molecule has been reported to provide a geometry that is similar to that of the β-turn,46 and LexA’s cleavage loop is known to organize into a β-turn in the cleavable conformation. For D1, compounds with similar structures have been identified and crystalized with the EthR repressor, implicated in ethionamide resistance in Mycobacterium tuberculosis.47 With regard to planned SAR studies, one limitation of our collaboration was that the terms of the relationship excluded the disclosure of additional compounds to UPenn. SAR trends within the chemotypes could not be readily disclosed by GSK without effectively releasing data on additional compounds. However, GSK provided reassurance that evidence of SAR was apparent in the initial HTS with the chemotypes of greatest interest (C and D).
Discovery in the anti-infective realm via academic-industry partnerships
Addressing the problem of antimicrobial resistance requires both novel targets and different models for drug discovery. The scientific challenges and economic disincentives, spanning discovery and marketing processes, have been documented as barriers to antibiotic discovery in industry.48,49 This problem is further compounded by competition, as well as a so-called “herd-mentality” that focuses attention on well-trodden targets.50 On the academic side, studies in the past decades have highlighted the potential of non-classical targets that could help stem the tide of resistance, including quorum sensing, stress responses, and virulence factors.51 However, to date, these insights have not yet yielded therapeutics for patients. This result is likely due in part to the limited practical experience of academic laboratories with the requirements for drug development, and also due to the regulatory challenges of advancing agents as adjuvants. Partnership models that both encourage the pursuit of novel, less validated targets in industry and help academic laboratories navigate the drug discovery landscape can help stimulate the antimicrobial discovery field.26
Our experience offers examples of the strengths and challenges associated with one such model for both partners. From the industry perspective, the GSK Discovery Fast Track Challenge process helps to mitigate the risks and costs associated with identifying novel targets, such the RecA*/LexA axis of the SOS response. Partnerships can often benefit from having established systems for validation in academia, exemplified in our case by our various cell-based reporter assays or strains with specific perturbations in the SOS pathway. Although the cost and scale of the HTS we performed is significant, the volume of screening in industry helps distribute these costs. From the academic perspective, libraries maintained by industry, in addition to often being larger than those at academic centers, have been highly curated to remove PAINS and other compounds with high false positive rates.43 In our case, the low hit rate of our assay made the scale of the screening critical, and discussions with GSK helped make us cognizant of the downstream challenges in clinical trial design with pursuing an adjuvant strategy. Enthusiasm and trust fostered by our interactions helped in finding compatible solutions to questions of intellectual property and material transfers, but we also faced limitations by being blinded to compounds along the screening process and in the number of structures which could be ultimately disclosed.
In our study, we have isolated novel small molecule inhibitors of the SOS response, including “first-in-class” agents that show direct inhibition of LexA self-cleavage. These compounds offer critical, and needed, tool compounds to probe the relevance of the SOS response to antibiotic evasion and starting points for ongoing medicinal chemistry efforts geared towards achieving drug-like potency and properties in cell-based assays. Having navigated our partnership towards this stated goal, we suggest that continued innovation in the models for relationships between academia and industry are highly justified, not only in lead identification but also in the many different phases needed to discover and develop new anti-infectives.
METHODS
Protein Constructs
The FlAsH-LexAEc construct was made by appending the following in sequence: an N-terminal maltose binding protein, a TEV-protease recognition site, a FLAsH-binding sequence (CCPGCC), a truncated version of E. coli LexA (residues 75–202), and a C-terminal poly-His tag in a pET41 expression vector backbone. An analogous construct with LexA from P. aeruginosa was generated using LexAPa residues 81–204. Corresponding control constructs with a mutation of the catalytic serine residue (S119A for E. coli, S125A for P. aeruginosa) were also prepared. Expression and purification of FlAsH-LexA variants, full-length LexA, and RecA proteins are described in the supplemental methods, along with fluorescent and radioactive labeling methods.
LexA cleavage assay conditions and plate setup
The assay setup for the pilot screen with FlAsH-LexAPa is described in the supplemental methods. For the FlAsH-LexAEc HTS screen, reactions were optimized to volumes suited for 1536-well plates. Each well contained 50 nM FlAsH-LexAEc, 133 nM RecA, 5 μM ssDNA (SKBT25: 5′-GCG TGT GTG GTG GTG TGC-3′),52 5 μM ATPγS in 100 mM Tris-HCl, pH 6.5, 150 mM NaCl, 5 mM MgCl2, 0.01% (w/v) BSA, 0.1 mM TCEP, and 0.01% (w/v) Pluoronic-F127. The controls, utilized for the Z′ and S/B calculation, were located in the last two columns of the plate and included all components of the assay listed above except for the RecA. Compounds or DMSO carrier were stamped onto the plates with an Echo 555 Liquid Handler (LabCyte). Reaction components were added as 2 μL additions of RecA* solution (ATPγS, ssDNA, and +/− RecA) and 2 μL of FlAsH-LexAEc using a Combi liquid handler (Thermo-Fisher). Reactions were centrifuged for 1 minute at 500 rpm and incubated for 30 minutes at room temperature. Reactions were quenched with a 2 μL addition of 45 mM EDTA, pH 8.0, and plates were read on ViewLux imagers (PerkinElmer). The final assay conditions resulted in a 100–120 mP difference between the uncleaved and cleaved control wells, representing an approximately 60 percent cleavage of the FlAsH-LexAEc. For each plate, the Z′-score was calculated from the cleaved and uncleaved control wells according to standard procedures.
HTS validation set
To assess the robustness of the FlAsH-LexA assay, an established set of 9722 compounds (the GSK validation set) was tested at 10 μM in triplicate. The Z′ score, the standard deviation of the sample population, false positive/negative rates, and statistical active rate were determined using a potency cut-off defined as the mean plus three standard deviations (3SD). The results are summarized in Figure S3 and Table S1.
Full HTS campaign
The full GSK diversity library (~1.8 million compounds) was tested at 10 μM in a 1536-well format. Batch screening was standardized to a volume of 96 plates (~135K compounds) and performed once or twice a day for a 2-week period, with a total of 1,376 plates tested. Compounds that displayed a percent inhibition of greater than or equal to 16% were selected for retesting in duplicate. Retested compounds that displayed auto-fluorescent properties were eliminated. The remaining compounds were tested in duplicate for dose-response in 2-fold serial dilutions with a top concentration of 200 μM and a total of 11 dose response points. Compounds that displayed dose-dependent changes in intrinsic fluorescence were also eliminated. The remaining compounds were then grouped by chemical family.
Cell-based SOS reporter assays
E. coli K12 MG1655 strain with or without the tolC transporter (ΔtolC)53 was transformed with a reporter plasmid where gfp expression is under the control of the recA promoter (pMS201 PrecA-GFP).42 To generate the S119A/ΔtolC strain, MG1655 strains with the S119A mutation in the lexA gene8 were transduced with a P1vir lysate derived from the tolC knockout strain of the KEIO collection.53 For the control strain with a constitutively repressed SOS pathway, the S119A/ΔtolC strain was transformed with PrecA-GFP plasmid. For the control strain with a constitutively expressed reporter, the MG1655 ΔtolC strain was transformed with a reporter plasmid containing GFP under the control of the recA promoter where the sequence of the LexA binding box in the recA promoter was reversed (pMS PrecA(rev)-GFP).8 Overnight cultures of the reporter strains were diluted 100-fold in M9 minimal media supplemented with 50 μg/mL of kanamycin and grown at 37 °C with agitation to an OD595 of ~0.6. 100 μL of culture were then added to a dilution series of compound in 96-well deep-well plates and 100 μL of 256 ng/mL ciprofloxacin (in M9 media with kanamycin) was added to the wells (final conditions: OD595 ~0.3 and ciprofloxacin 128 ng/mL). Cultures were incubated at 37 °C with agitation for 2 hours, after which the cells were fixed by adding 200 μL of phosphate buffer saline containing 4% paraformaldehyde. Fixed cells were analyzed using flow cytometry (BD FACSCalibur, Ex/Em: 488 nm/530 nm) and the mean fluorescence of 20,000 cells in each condition was recorded. Population histograms were analyzed with the FlowJo software. Additional details for the control cell-based SOS reporter assays are provided in the supplemental material.
32P-LexA cleavage assay
LexA was engineered with a RRXS phosphorylation site for protein kinase A on the N-terminus of full-length LexA. The construct was purified and labeled with 32P as described in the supplemental material. Labeled LexA was incubated with a 2-fold dilution series of compound, with the highest concentration being 200 μM. Cleavage assays were performed under the following conditions: 100 nM 32P-LexA, 200 nM of RecA, 66 nM of SKBT25 ssDNA, 10 μM of ATPγS, 5 mM MgCl2, 70 mM Tris-HCl, pH 7.0. Reactions were incubated for 15 minutes at room temperature, stopped with the addition of 2X Laemmli Buffer, and separated using 15% SDS-PAGE gels. For the RecA*-independent cleavage assay, 32P-LexA was subjected to the identical compound concentrations and buffer conditions as above, except without RecA, ssDNA, and ATPγS. Reaction aliquots were removed at 0, 2, and 4 hours. For both assay approaches, the protein bands were visualized with phosphorimaging and the compound IC50 was determined.
LexA electrophoresis mobility shift assay
Full-length, catalytically-inactive LexAEc-S119A was purified according to previously published protocols.8,33 Increasing concentrations (0 to 1000 nM) were mixed with 10 nM of SOS operator DNA dual-labeled with Cy3 and Cy5 in EMSA running buffer (70 mM Tris-HCl pH 7.5, 10 mM MgCl2, 150 mM NaCl, 5 mM DTT, 0.1 mg/ml BSA, 10 ng/μL ssDNA, 5% glycerol, 0.04% bromophenol blue) in the presence of 50 μM of compound or DMSO carrier. After incubation at room temperature for 30 minutes, 20 μL of each reaction was run on 6% native PAGE gels at room temperature. Gels were visualized on a Typhoon Imager using default fluorescence filter settings for Cy5. To obtain binding curves, gel bands were quantitated in ImageJ to determine the fraction of bound DNA for each LexA concentration. Data for each compound were fit to a variable-slope sigmoidal dose-response curve.
Protease assays
Compounds were submitted to BPS Bioscience to test their activities on dipeptidyl peptidase 4, caspase 3, HCV1a protease, and renin. Assays were performed in accordance with company practices.
Supplementary Material
Acknowledgments
We thank Jeffrey Faust (Wistar Institute) for help with the flow cytometry experiments and members of the Kohli laboratory for helpful discussions. We thank David Schultz of the High Throughput Screening Core at the Wistar Institute for his assistance with the pilot HTS screen with FlAsH-LexAPa. We also thank Manuela Roggiani and Mark Goulian for generously providing lysates for phage transduction. This work was supported by the National Institutes of Health (DP2-GM105444 to R.M.K.) as well as the Harrington Discovery Institute Scholar Innovator Award (to R.M.K.) and the Edward J. Mallinckrodt, Jr., Foundation. Fellowship training support was also provided from the National Institutes of Health (T32-GM7229 for C.Y.M.; T32-GM071399 for Z.M.H.; T32-AI060516 for J.M.K.; T32-AR007442 for M.J.C.)
ABBREVIATIONS
- LexAEc
LexA of E. coli
- LexAPa
LexA of P. aeruginosa
- RecA*
filamentous RecA
- CTD
C-terminal domain
- NTD
N-terminal domain
Footnotes
Author contributions. C.Y.M. and R.M.K. conceived of and designed the biochemical screen and cell-based reporter assays; C.Y.M. performed the initial small-scale validation screen at the University of Pennsylvania; C.Y.M. and R.M.K. wrote the proposal for the GSK Discovery Fast Track Challenge; A.J.J., P.M.K. and J.S. performed validation and main biochemical screens at GSK; A.Q. performed the RecA secondary screen; A.J.P. and K.L.M supervised the high-throughput screening campaign at GSK; T.S, M.J.C and C.Y.M. performed the radioactive cleavage assays; J.M.K., T.S. and C.Y.M performed the cell-based assays; Z.H. performed the EMSA studies; T.S. performed the thermal shift assays; all authors participated in the data analysis; C.Y.M and R.M.K wrote the manuscript, with input from all authors.
CONFLICTS OF INTEREST
A.J.J., P.M.K., A.J.P., A.Q., J.S., and K.L.W are employees of GlaxoSmithKline.
References
- 1.Radman M. SOS repair hypothesis: Phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci. 1975;5A:355–367. doi: 10.1007/978-1-4684-2895-7_48. [DOI] [PubMed] [Google Scholar]
- 2.Butala M, Zgur-Bertok D, Busby SJ. The bacterial LexA transcriptional repressor. Cell Mol Life Sci. 2009;66:82–93. doi: 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlacher K, Goodman MF. Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nat Rev Mol Cell Biol. 2007;8:587–594. doi: 10.1038/nrm2198. [DOI] [PubMed] [Google Scholar]
- 4.Culyba MJ, Mo CY, Kohli RM. Targets for combating the evolution of acquired antibiotic resistance. Biochemistry. 2015;54:3573–3582. doi: 10.1021/acs.biochem.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cirz RT, Romesberg FE. Controlling mutation: Intervening in evolution as a therapeutic strategy. Crit Rev Biochem Mol Biol. 2007;42:341–354. doi: 10.1080/10409230701597741. [DOI] [PubMed] [Google Scholar]
- 6.Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: From targets to networks. Nat Rev Microbiol. 2010;8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cirz RT, Chin JK, Andes DR, de Crecy-Lagard V, Craig WA, Romesberg FE. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005;3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mo CY, Manning SA, Roggiani M, Culyba MJ, Samuels AN, Sniegowski PD, Goulian M, Kohli RM. Systematically altering bacterial SOS activity under stress reveals therapeutic strategies for potentiating antibiotics. mSphere. 2016;1 doi: 10.1128/mSphere.00163-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thi TD, Lopez E, Rodriguez-Rojas A, Rodriguez-Beltran J, Couce A, Guelfo JR, Castaneda-Garcia A, Blazquez J. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J Antimicrob Chemother. 2011;66:531–538. doi: 10.1093/jac/dkq496. [DOI] [PubMed] [Google Scholar]
- 10.Kohanski MA, DePristo MA, Collins JJ. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell. 2010;37:311–320. doi: 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu TK, Collins JJ. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci U S A. 2009;106:4629–4634. doi: 10.1073/pnas.0800442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Recacha E, Machuca J, Diaz de Alba P, Ramos-Guelfo M, Docobo-Perez F, Rodriguez-Beltran J, Blazquez J, Pascual A, Rodriguez-Martinez JM. Quinolone resistance reversion by targeting the SOS response. MBio. 2017;8 doi: 10.1128/mBio.00971-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Da Re S, Garnier F, Guerin E, Campoy S, Denis F, Ploy MC. The SOS response promotes qnrB quinolone-resistance determinant expression. EMBO Rep. 2009;10:929–933. doi: 10.1038/embor.2009.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorr T, Lewis K, Vulic M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 2009;5:e1000760. doi: 10.1371/journal.pgen.1000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guerin E, Cambray G, Sanchez-Alberola N, Campoy S, Erill I, Da Re S, Gonzalez-Zorn B, Barbe J, Ploy MC, Mazel D. The SOS response controls integron recombination. Science. 2009;324:1034. doi: 10.1126/science.1172914. [DOI] [PubMed] [Google Scholar]
- 16.Gotoh H, Kasaraneni N, Devineni N, Dallo SF, Weitao T. SOS involvement in stress-inducible biofilm formation. Biofouling. 2010;26:603–611. doi: 10.1080/08927014.2010.501895. [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Jacoby GA, Mills DM, Hooper DC. SOS regulation of qnrB expression. Antimicrob Agents Chemother. 2009;53:821–823. doi: 10.1128/AAC.00132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alam MK, Alhhazmi A, DeCoteau JF, Luo Y, Geyer CR. RecA inhibitors potentiate antibiotic activity and block evolution of antibiotic resistance. Cell Chem Biol. 2016;23:381–391. doi: 10.1016/j.chembiol.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Lee AM, Wigle TJ, Singleton SF. A complementary pair of rapid molecular screening assays for RecA activities. Anal Biochem. 2007;367:247–258. doi: 10.1016/j.ab.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson EJ, Janzen WP, Kireev D, Singleton SF. High-throughput screening for RecA inhibitors using a transcreener adenosine 5′-O-diphosphate assay. Assay Drug Dev Technol. 2012;10:260–268. doi: 10.1089/adt.2011.0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sexton JZ, Wigle TJ, He Q, Hughes MA, Smith GR, Singleton SF, Williams AL, Yeh LA. Novel inhibitors of E. coli RecA ATPase activity. Curr Chem Genomics. 2010;4:34–42. doi: 10.2174/1875397301004010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wigle TJ, Sexton JZ, Gromova AV, Hadimani MB, Hughes MA, Smith GR, Yeh LA, Singleton SF. Inhibitors of RecA activity discovered by high-throughput screening: Cell-permeable small molecules attenuate the SOS response in Escherichia coli. J Biomol Screen. 2009;14:1092–1101. doi: 10.1177/1087057109342126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wigle TJ, Lee AM, Singleton SF. Conformationally selective binding of nucleotide analogues to escherichia coli RecA: A ligand-based analysis of the RecA ATP binding site. Biochemistry. 2006;45:4502–4513. doi: 10.1021/bi052298h. [DOI] [PubMed] [Google Scholar]
- 24.Bellio P, Di Pietro L, Mancini A, Piovano M, Nicoletti M, Brisdelli F, Tondi D, Cendron L, Franceschini N, Amicosante G, Perilli M, Celenza G. SOS response in bacteria: Inhibitory activity of lichen secondary metabolites against escherichia coli RecA protein. Phytomedicine. 2017;29:11–18. doi: 10.1016/j.phymed.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol. 2011;6:628–635. doi: 10.1021/cb100428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tralau-Stewart CJ, Wyatt CA, Kleyn DE, Ayad A. Drug discovery: New models for industry-academic partnerships. Drug Discov Today. 2009;14:95–101. doi: 10.1016/j.drudis.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Hall ZW, Scott C. University-industry partnership. Science. 2001;291:553. doi: 10.1126/science.1058411. [DOI] [PubMed] [Google Scholar]
- 28.Oduor RO, Ojo KK, Williams GP, Bertelli F, Mills J, Maes L, Pryde DC, Parkinson T, Van Voorhis WC, Holler TP. Trypanosoma brucei glycogen synthase kinase-3, a target for anti-trypanosomal drug development: A public-private partnership to identify novel leads. PLoS Negl Trop Dis. 2011;5:e1017. doi: 10.1371/journal.pntd.0001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mann J, Taylor PW, Dorgan CR, Johnson PD, Wilson FX, Vickers R, Dale AG, Neidle S. The discovery of a novel antibiotic for the treatment of Clostridium difficile infections: A story of an effective academic-industrial partnership. Medchemcomm. 2015;6:1420–1426. doi: 10.1039/c5md00238a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010;9:690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo Y, Pfuetzner RA, Mosimann S, Paetzel M, Frey EA, Cherney M, Kim B, Little JW, Strynadka NC. Crystal structure of LexA: A conformational switch for regulation of self-cleavage. Cell. 2001;106:585–594. doi: 10.1016/S0092-8674(01)00479-2. [DOI] [PubMed] [Google Scholar]
- 32.Zhang AP, Pigli YZ, Rice PA. Structure of the LexA-DNA complex and implications for SOS box measurement. Nature. 2010;466:883–886. doi: 10.1038/nature09200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mo CY, Birdwell LD, Kohli RM. Specificity determinants for autoproteolysis of LexA, a key regulator of bacterial SOS mutagenesis. Biochemistry. 2014;53:3158–3168. doi: 10.1021/bi500026e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slilaty SN, Rupley JA, Little JW. Intramolecular cleavage of LexA and phage lambda repressors: Dependence of kinetics on repressor concentration, pH, temperature, and solvent. Biochemistry. 1986;25:6866–6875. doi: 10.1021/bi00370a020. [DOI] [PubMed] [Google Scholar]
- 35.Little JW. Autodigestion of lexA and phage lambda repressors. Proc Natl Acad Sci U S A. 1984;81:1375–1379. doi: 10.1073/PNAS.81.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: Synthesis and biological applications. J Am Chem Soc. 2002;124:6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 37.Wigle TJ, Singleton SF. Directed molecular screening for RecA ATPase inhibitors. Bioorg Med Chem Lett. 2007;17:3249–3253. doi: 10.1016/j.bmcl.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nautiyal A, Patil KN, Muniyappa K. Suramin is a potent and selective inhibitor of mycobacterium tuberculosis RecA protein and the SOS response: RecA as a potential target for antibacterial drug discovery. J Antimicrob Chemother. 2014;69:1834–1843. doi: 10.1093/jac/dku080. [DOI] [PubMed] [Google Scholar]
- 39.Nok AJ. Arsenicals (melarsoprol), pentamidine and suramin in the treatment of human african trypanosomiasis. Parasitol Res. 2003;90:71–79. doi: 10.1007/s00436-002-0799-9. [DOI] [PubMed] [Google Scholar]
- 40.Albulescu IC, Kovacikova K, Tas A, Snijder EJ, van Hemert MJ. Suramin inhibits zika virus replication by interfering with virus attachment and release of infectious particles. Antiviral Res. 2017;143:230–236. doi: 10.1016/j.antiviral.2017.04.016. [DOI] [PubMed] [Google Scholar]
- 41.Naviaux RK, Curtis B, Li K, Naviaux JC, Bright AT, Reiner GE, Westerfield M, Goh S, Alaynick WA, Wang L, Capparelli EV, Adams C, Sun J, Jain S, He F, Arellano DA, Mash LE, Chukoskie L, Lincoln A, Townsend J. Low-dose suramin in autism spectrum disorder: A small, phase I/II, randomized clinical trial. Ann Clin Transl Neurol. 2017;4:491–505. doi: 10.1002/acn3.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, Liebermeister W, Surette MG, Alon U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods. 2006;3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
- 43.Aldrich C, Bertozzi C, Georg GI, Kiessling L, Lindsley C, Liotta D, Merz KM, Jr, Schepartz A, Wang S. The ecstasy and agony of assay interference compounds. ACS Infect Dis. 2017;3:259–262. doi: 10.1021/acscentsci.7b00069. [DOI] [PubMed] [Google Scholar]
- 44.Barreto K, Bharathikumar VM, Ricardo A, DeCoteau JF, Luo Y, Geyer CR. A genetic screen for isolating “lariat” peptide inhibitors of protein function. Chem Biol. 2009;16:1148–1157. doi: 10.1016/j.chembiol.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 45.Roland KL, Little JW. Reaction of LexA repressor with diisopropyl fluorophosphate. A test of the serine protease model. J Biol Chem. 1990;265:12828–12835. [PubMed] [Google Scholar]
- 46.Li H, Aneja R, Chaiken I. Click chemistry in peptide-based drug design. Molecules. 2013;18:9797–9817. doi: 10.3390/molecules18089797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flipo M, Willand N, Lecat-Guillet N, Hounsou C, Desroses M, Leroux F, Lens Z, Villeret V, Wohlkonig A, Wintjens R, Christophe T, Kyoung Jeon H, Locht C, Brodin P, Baulard AR, Deprez B. Discovery of novel N-phenylphenoxyacetamide derivatives as EthR inhibitors ethionamide boosters by combining high-throughput screening and synthesis. J Med Chem. 2012;55:6391–6402. doi: 10.1021/jm300377g. [DOI] [PubMed] [Google Scholar]
- 48.Renwick MJ, Brogan DM, Mossialos E. A systematic review and critical assessment of incentive strategies for discovery and development of novel antibiotics. J Antibiot (Tokyo) 2016;69:73–88. doi: 10.1038/ja.2015.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Booth B, Zemmel R. Prospects for productivity. Nat Rev Drug Discov. 2004;3:451–456. doi: 10.1038/nrd1384. [DOI] [PubMed] [Google Scholar]
- 50.Schachter B. Partnering with the professor. Nat Biotechnol. 2012;30:944–952. doi: 10.1038/nbt.2385. [DOI] [PubMed] [Google Scholar]
- 51.Dickey SW, Cheung GYC, Otto M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat Rev Drug Discov. 2017;16:457–471. doi: 10.1038/nrd.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tracy RB, Kowalczykowski SC. In vitro selection of preferred DNA pairing sequences by the Escherichia coli RecA protein. Genes Dev. 1996;10:1890–1903. doi: 10.1101/gad.10.15.1890. [DOI] [PubMed] [Google Scholar]
- 53.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.