

Abstract
Peptides often suffer from short in vivo half-lives due to proteolysis and renal clearance that limit their therapeutic potential in many indications, necessitating pharmacokinetic (PK) enhancement. D-peptides, composed of mirror-image D-amino acids, overcome proteolytic degradation but are still vulnerable to renal filtration due to their small size. If renal filtration could be slowed, D-peptides would be promising therapeutic agents for infrequent dosing, such as in extended-release depots. Here, we tether a diverse set of PK-enhancing cargoes to our potent, protease-resistant D-peptide HIV entry inhibitor, PIE12-trimer. This inhibitor panel provides an opportunity to evaluate the PK impact of the cargoes independently of proteolysis. While all the PK-enhancing strategies (PEGylation, acylation, alkylation and cholesterol conjugation) improved in vivo half-life, cholesterol conjugation of PIE12-trimer dramatically improves both antiviral potency and half-life in rats, making it our lead anti-HIV drug candidate. We designed its chemical synthesis for large-scale production (CPT31) and demonstrated that the PK profile in cynomolgous monkeys supports future development of monthly or less frequent depot dosing in humans. CPT31 could address an urgent need in both HIV prevention and treatment.
Keywords: HIV entry inhibition, D-peptide, PIE12-trimer, CPT31, pharmacokinetics, PK-enhancing cargo, proteolysis, renal clearance, PEGylation, acylation, alkylation, cholesterol, PrEP, HIV treatment, HIV prevention, extended-release depot
Graphical abstract
INTRODUCTION
Peptides are an increasingly important class of therapeutics. They have several advantages over small molecules, including increased target affinity and specificity, as well as an ability to disrupt protein-protein interactions, which are often considered “undruggable” by small molecules1. Peptides also have advantages over protein therapeutics in that they are chemically synthesized (allowing simplified, modular assembly and manufacturing), and their smaller size allows access to sterically restricted targets and deeper tissue penetration.
Despite these advantages, peptide therapeutics face considerable pharmacokinetic (PK) challenges since they can be rapidly degraded by proteases as well as cleared by the kidney, leading to short half-lives (e.g., the HIV entry inhibitor enfuvirtide/Fuzeon2). To overcome the problem of protease degradation, our group focuses on the development of D-peptides, which are chemically synthesized with D-amino acids and are the mirror images of natural L-peptides3. D-peptides are intrinsically protease-resistant due to their inability to interact with protease active sites4,5. Our most advanced D-peptide is a broadly potent inhibitor of HIV entry and provides a unique vehicle with which to test PK-enhancing strategies in the absence of proteolysis6.
Our D-peptide inhibitor, PIE12 (Pocket-specific Inhibitor of Entry), targets a highly conserved hydrophobic pocket on gp41, the HIV Env subunit responsible for mediating membrane fusion between the virus and host cell6,7. This pocket is exposed transiently during viral entry, highly conserved, and functionally critical. PIE12 was trimerized on a scaffold with three equivalent, short, and discrete polyethylene glycol (PEG) arms whose length was optimized to harness avidity when binding trimeric gp41. PIE12-trimer has sub-pM binding affinity to trimeric gp41 and blocks all major circulating clades of HIV-1 with mid-pM to low-nM potency. For attaching potential potency and PK-enhancing cargoes, we added a fourth arm with an orthogonal reactive group. Previously, to this fourth arm we attached the membrane-localizing cargoes cholesterol and alkyl chains that were predicted to localize the inhibitor to sites of viral entry and saw an increase in antiviral potency up to 160-fold6. Here, we comprehensively study the effect of the membrane-localizing and other cargoes on the in vivo pharmacokinetic properties of PIE12-trimer to determine the optimal combination of potency and pharmacokinetic enhancement.
Both preclinical and clinical research have highlighted a variety of promising PK-enhancing cargoes. For example, PEG conjugation improves in vivo drug half-life by increasing drug size and reducing renal filtration and is found in 15 FDA-approved products8–11. Recently, attachment of a 2 kDa PEG to anti-HIV enfuvirtide led to improved in vivo half-life12. Acylation extends half-life, primarily due to the interaction of the fatty acid moiety with human serum albumin (HSA), and is seen in the once-daily dosed diabetes therapies liraglutide and insulin degludec13. Additionally, acylation has been shown to increase the half-life and improve potency of enfuvirtide-like peptides14,15. As it is likely that the aliphatic chain of the fatty acid is responsible for the HSA interaction16, and alkanes only differ from fatty acids in the lack of the terminal carboxylic acid, we also explore the PK effect of alkane conjugation. Cholesterol conjugation is being investigated for both cancer therapeutics and siRNA delivery17, and has been shown to decrease in vivo clearance rates of siRNA 30-fold in mice, likely due to serum protein binding18. Specifically for targeting HIV, cholesterol conjugation has been shown to not only increase half-life, but also to increase antiviral potency as it delivers entry inhibitors to lipid rafts on cellular membranes, the site of viral entry6,19. We tested the effect of each of these strategies in the context of our HIV D-peptide inhibitor to determine the clearance-reducing potential endowed by PK-enhancing groups without the need to consider concurrent changes in sensitivity to proteolysis.
In this study, we were ultimately interested in identifying the cargo that best pairs potency enhancement and improved in vivo half-life of PIE12-trimer to produce an HIV drug conducive to infrequent dosing. An enduring global epidemic, HIV/AIDS claims over 1 million lives annually. Although modern antiretroviral therapy has managed this chronic infection in many patients, side effects and drug resistance are obstacles that still lead to poor treatment adherence and failure for many. The field is especially enthusiastic about the potential of long-acting therapies to improve patient compliance and outcomes20, and a combination of two long-acting drugs is currently in clinical trials21. Therefore, the future of anti-HIV therapy is expected to include the option for monthly or less frequent injections of multiple antiretrovirals, and a long-lasting PIE12-trimer entry inhibitor would be an ideal candidate for such regimens. Additionally, an infrequently dosed, highly potent anti-HIV agent such as PIE12-trimer could be an ideal preventative for use in pre-exposure prophylaxis (PrEP).
In this work we show that all of the tested modifications (PEGylation, acylation, alkylation, and cholesterol conjugation) are capable of enhancing the in vivo survival of a D-peptide drug in the absence of proteolysis. Cholesterol conjugation provides the combined benefits of highly improved potency (>100-fold) and half-life. After identifying cholesterolated PIE12-trimer as our final PK-enhanced candidate, we redesigned its synthesis for manufacturing scalability, resulting in our lead molecule, CPT31. Surprisingly, the small chemical change in the 4-arm scaffold of CPT31 compared to a previous compound led to an even further reduced clearance rate in rats. Importantly, we studied CPT31’s pharmacokinetics in a non-human primate model, and the results support the feasibility of extended-release (depot) formulation for monthly or less frequent dosing.
MATERIALS AND METHODS
Synthesis of PIE12-trimer and PIE12-trimer conjugates
PIE12 (Ac-HPCDYPEWQWLCELGK, all D-amino acids, except Gly) was synthesized using standard Fmoc methods by RS Synthesis (Louisville, KY) or in-house using a Prelude automated synthesizer (Gyros Protein Technologies). PIE12-trimer andmaleimide-PEG24-PIE12-trimer were synthesized as previously described6. PIE12-trimers were conjugated to membrane-anchoring cargoes in a similar manner by reacting maleimide-PEG24-PIE12-trimer (3 mM) with 4.5 mM thiocholesterol (Sigma Aldrich #136115), 1-hexadecanethiol (Sigma Aldrich #52270), or 1-octadecanethiol (Sigma Aldrich #01858) in dimethylacetamide (DMAC) with Et3N (200 mM) for 60 min at RT, then purified by RP-HPLC to produce cholesterol-PIE12-trimer (Chol-PIE12-trimer), C16-PIE12-trimer, andC18-PIE12-trimer, respectively. Palmitate-conjugated PIE12-trimer was synthesized by first reacting maleimide-PEG24-PIE12-trimer (3 mM) with D-cysteine (4.5 mM) in DMAC with Et3N (200 mM) for 60 min at RT, then purified by RP-HPLC. The resulting product, Cys-PEG24-PIE12-trimer (2 mM), was then reacted with palmitic acid NHS ester (5 mM, Sigma Aldrich #P1162) in DMAC with Et3N (500 mM) for 45 min at RT, then purified by RP-HPLC. Chol-PEG5K-PIE12-trimer was synthesized by reacting Cys-PEG24-PIE12-trimer (2 mM) with maleimide-PEG5K-NHS (3 mM, JenKem #A5003) followed by thiocholesterol (4.5 mM), then purified by RP-HPLC. Forty kDa PEG-PEG24-PIE12-trimer (PEG40-PIE12-trimer) was synthesized by reacting Cys-PEG24-PIE12-trimer (2 mM) with excess NHS-PEG5-NHS (ChemPep #281903), purified by RP-HPLC, reacted with 2.5 mM 40 kDa Y-branched PEG-amine (JenKem, A0010), then purified by RP-HPLC. Peptides and all final products, except PEG40-PIE12-trimer, were verified by mass spectrometry (Sciex 3000 triple quadrupole): PIE12 (calculated mass 2290.58 Da, observed mass 2290.2 Da), PIE12-trimer (calculated mass 7154.07 Da, observed mass 7156.0 Da), Chol-PIE12-trimer (calculated mass 8837.22 Da, observed mass 8839.4 Da), C16-PIE12-trimer (calculated mass 8693.25 Da, observed mass 8694.3 Da), C18-PIE12-trimer (calculated mass 8722.3 Da, observed mass 8724.6 Da), Palm-PIE12-trimer (calculated mass 8794.4 Da, observed mass 8795.0 Da), Chol-PEG5K-PIE12-trimer (calculated mass 13986.74 Da, observed mass 13943.6 Da, due to the polydispersity of the PEG5K). Polydispersity in the PEG40 conjugate prevented obtaining a mass for this product.
Synthesis of Fmoc-PEG27-triNHS, the heterotetrameric scaffold for CPT31
Fmoc-PEG27-COOH (Polypure, #15137-2790, 10 mmol) and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxidhexafluorophosphate (HATU, Chempep, 120801, 10 mmol) were suspended in 40 ml dimethylformamide (DMF). To this was added N,N-diisopropylethylamine (Sigma Aldrich, D125806, 10 mmol), followed by aminotriester (Frontier Scientific, #NTN1963, 20 mmol). This solution was stirred for 3 hours before purification by flash chromatography (Biotage Zip column) using a gradient of ethanol in dichloromethane (DCM). The product, Fmoc-PEG27-triester, was dried by rotary evaporation to yield a viscous amber oil at 90% yield. This material was dissolved in DCM (5 ml/g) and placed on ice with stirring. To this solution, 20 equivalents of trifluoroacetic acid (TFA) was added dropwise, and the reaction was stirred for 30 min before warming to RT. After 1 h, the reaction was purified by reverse-phase chromatography (Biotage C18 flash column) using an acetonitrile gradient in water, at a yield of 95%. The product was lyophilized, then dried repeatedly from toluene. The resulting Fmoc-PEG27-triacid was suspended in acetonitrile to a concentration of 500 mM, to which N,N’-disuccinimidyl carbonate (Sigma Aldrich, #225827) was added to 1650 mM, followed by triethylamine to 400 mM. The reaction was stirred for 45 min at 45°C, then purified using flash chromatography (Biotage ZIP column) using a gradient of ethanol in DCM at a yield of 90%, for a final yield of 75%. Intermediate and final products were verified by mass spectrometry (Agilent 6130B single quadrupole): Fmoc-PEG27-triester (calculated mass 1942.35 Da, observed mass 1942.5 Da), Fmoc-PEG27-triacid (calculated mass 1772.94 Da, observed mass 1773.2 Da), Fmoc-PEG27-triNHS (calculated mass 2063.99 Da, observed mass 2064.5 Da).
Synthesis of Cholesteryl-PEG4-NHS, for cholesterol attachment to CPT31
Fmoc-PEG4-COOH (ChemPep, #280109) was suspended in DCM to a concentration of 200 mM. To this, 5 equivalents of N,N-Diisopropylethylamine (DIPEA, Sigma Aldrich) were added, then the solution was added to 2-chlorotrityl chloride resin (Aapptec, #RTZ001). The mixture was agitated with argon gas for 2 h, then washed with DCM (3×) followed by DCM:methanol:DIPEA (17:2:1), then DCM (3×). To this, a solution of DMF:DCM:piperidine (1:1:1) was added to remove the Fmoc protecting group, and the reaction was agitated with argon gas for 40 min before being washed sequentially with DMF, DMF:DCM (1:1), and DCM. Two equivalents of cholesteryl chloroformate (Sigma Aldrich, #C77007) and 3 equivalents of DIPEA in DCM were added to the resin. The reaction was agitated with argon gas for 12 h, then washed with DCM. Cleavage of the cholesteryl-PEG4-COOH was carried out in 100 ml 5% TFA in DCM with agitation for 2 h. The resulting solution was dried by rotary evaporation to yield a viscous oil, then purified by flash chromatography (Biotage ZIP Sphere column) using a gradient of ethanol in DCM at a yield of 68%.
Cholesteryl-PEG4-COOH was then dissolved in acetonitrile to a concentration of 800 mM before adding 1.1 equivalents of N,N’-disuccinimidyl carbonate (DSC, Sigma Aldrich, #225827) followed by 0.8 equivalents of triethylamine. The solution was heated to 45°C and stirred for 60 min before purification by flash chromatography (Biotage ZIP sphere column) using a gradient of ethanol in DCM. The resulting product was dried extensively by rotary evaporation to yield a viscous yellow oil. Final product yield was 56%. Intermediate and final product were verified by mass spectrometry (Agilent 6130B single quadrupole): Cholesteryl-PEG4-COOH (calculated mass 677.95 Da, observed mass 677.5 Da) and Cholesteryl-PEG4-NHS (calculated mass 775.02 Da, observed mass 774.5 Da).
Synthesis of CPT31
PIE12-2 monomer (Ac-HPCDYPEWQWLCELG-PEG4-K) was synthesized by Ambiopharm (North Augusta, SC) using all D-amino acids (except Gly). PIE12-2 was suspended in DMAC buffered with triethylamine (150 mM) to a concentration of 20 mM. To this, Fmoc-PEG27-triNHS was added to a concentration of 6.06 mM. The reaction proceeded for 2 h at RT before piperidine was added to 30%, and the reaction was mixed for 40 min to remove the Fmoc group. NH2-PEG27-PIE12-trimer was then purified by RP-HPLC (Waters X-Bridge C18). This product (10 mM) was reacted with cholesteryl-PEG4-NHS (12 mM) in DMAC buffered by triethylamine (150 mM) for 90 min and purified by RP-HPLC (Waters X-Bridge C18) to generate CPT31 (cholesterol-PIE12-trimer with PEG31 fourth-arm spacer). Peptides, intermediates, and final product were verified by mass spectrometry (Agilent 6130B single quadrupole): PIE12-2 (calculated mass 2041.89 Da, observed 2041.9 Da), NH2-PEG27-PIE12-trimer (calculated mass 8369.53 Da, observed mass 8368.73 Da), and CPT31 (calculated mass 9029.46 Da, observed mass 9029.7 Da).
Pseudovirion Entry Assay
Pseudovirion assays in Table 1 were performed as previously described7,22. Briefly, a six-point dilution series of each inhibitor was generated in quadruplicate on HOS-CD4-CCR5 monolayers in 96 well plates, after which JRFL luciferase reporter pseudovirions were added. After 2 days, cells were lysed using GloLysis buffer (Promega), and BrightGlo luciferase substrate (Promega) was added. Luminescence was read on a PolarStar Optima (BMG) plate reader and normalized to uninhibited controls. Inhibition curves were plotted and fit to a standard IC50 equation for normalized data [(1 − c/(IC50 + c)], weighting each point by its standard error (with a minimum 1% error allowed) using KaleidaGraph (Synergy Software). Reported IC50 values are the average of at least two independent quadruplicate assays, and the standard error of the mean (SEM) is reported.
Table 1.
Antiviral Potency of Conjugates
| Compound | JRFL(nM) |
|---|---|
| PIE12-trimer | 2.1 ± 0.28* |
| PEG40-PIE12-trimer | 71 ± 8.5 |
| Palm-PIE12-trimer | 0.54 ± 0.029 |
| C16-PIE12-trimer | 0.11 ± 0.012* |
| C18-PIE12-trimer | 0.087 ± 0.012* |
| Chol-PIE12-trimer | 0.022 ± 0.0018 |
| Chol-PEG5k-PIE12-trimer | 0.030 ± 0.0010 |
| CPT31 | 0.015 ± 0.0062 |
(from ref.6)
Rodent Pharmacokinetics
Initial in-life rodent studies were performed at Navigen (Salt Lake City, UT). For each study, three Sprague Dawley rats (0.22-0.44 kg) were dosed as shown in Table 2. At each timepoint, plasma was obtained using lithium heparin. For Chol-PIE12-trimer, Chol-PEG5k-PIE12-trimer, and CPT31 data presented in Table 3, in-life studies were conducted at Calvert Laboratories (Scott Township, PA). Male rats per route were dosed with either Chol-PIE12-trimer (n=3), Chol-PEG5k-PIE12-trimer (n=3), or CPT31 (n=2) formulated at 2 mg/mL in 50 mM HEPES (pH 7.4). For both subcutaneous (SC, lower lumbar region) and intravenous (IV, bolus injection into lateral tail vein) administration, a dose of 1 mg/kg was delivered, and plasma (K2EDTA) samples were collected via jugular vein catheter at time points from 5 min to 24 h for the IV group and 15 min to 48 h for the SC group. Plasma samples were stored at −80 °C and shipped to Navigen on dry ice for bioanalysis. Compounds evaluated were stable in plasma at the storage and shipment conditions.
Table 2.
Median and (range) IV and SC plasma PK parameters of PIE12-trimer and conjugates in rats
| Compound | Route of Admin | Dose (mg/kg) |
T1/2 (hr) | Tmax (hr) |
C0 or Cmax (nM) |
AUC (0-inf) (hr* nM) |
Vd (obs) (mL/kg) |
Cl (obs) (mL/hr/kg) |
F* (%) |
|---|---|---|---|---|---|---|---|---|---|
| PIE 12-trimer | IV | 1.0 | 0.6 (0.5-0.8) |
NA | 275 (267-392) |
168 (137-221) |
704 (668-771) |
835 (634-1020) |
NA |
| SC | 1.0 | 0.8 (0.7-0.9) |
0.5 (0.5-1.0) |
80 (48-114) |
208 (161-264) |
NA | NA | 124 (73-193) |
|
| Palm-PIE12-trimer | IV | 1.2 | 1.8 (1.4-1.9) |
NA | 2242 (2180-2460) |
2241 (2210-2510) |
144 (127-171) |
61 (55-62) |
NA |
| SC | 1.2 | 2.2 (1.6-3.3) |
1.0 (1.0-2.0) |
585 (582-615) |
2313 (2010-2570) |
NA | NA | 103 (80-116) |
|
| C16-PIE12-trimer | IV | 1.0 | 0.9 (0.9-1.0) |
NA | 1875 (898-2170) |
1155 (746-1190) |
137 (128-206) |
100 (97-154) |
NA |
| SC | 1.0 | 1.2 (1.1-1.6) |
1.0 (1.0-1.0) |
192 (191-252) |
442 (435-606) |
NA | NA | 38 (37-81) |
|
| C18-PIE12-trimer | IV | 1.0 | 1.1 (1.0-1.1) |
NA | 900 (500-1230) |
760 (743-1140) |
227 (143-243) |
150 (100-153) |
NA |
| SC | 1.0 | 1.4 (1.2-1.4) |
2.0 (1.0-2.0) |
196 (150-221) |
713 (457-922) |
NA | NA | 94 (40-124) |
|
| Chol-PIE12-trimer | IV | 1.0 | 1.8 (1.5-2.0) |
NA | 1112 (953-1230) |
2394 (1710-2470) |
134 (117-138) |
47 (46-66) |
NA |
| SC | 1.0 | 2.7 (2.5-4.2) |
4.0 (2.0-4.0) |
304 (203-347) |
1748 (1280-2140) |
NA | NA | 73 (52-125) |
n=3, NA=not applicable, T1/2=elimination half-life, Tmax=time at maximum plasma concentration, Co=initial plasma drug concentration (IV injection), Cmax=maximum plasma drug concentration (SC injection), AUC=area under curve, Vd=apparent volume of distribution, Cl(obs)=observed total body clearance of the drug from plasma, F=bioavailability,
range reflects the largest/smallest possible ratios among all animals tested
Table 3.
Median IV and SC plasma PK parameters of cholesterol conjugates of PIE12-trimer in rats
| Compound | Route of Admin | Dose (mg/kg) |
T1/2 (hr) |
Tmax (hr) |
C0 or Cmax (nM) |
AUC (0-inf) (hr* nM) |
Vd (obs) (mL/kg) |
Cl (obs) (mL/hr/kg) |
F (%) |
|---|---|---|---|---|---|---|---|---|---|
| Chol-PIE12-trimer* | IV | 1.0 | 1.6 (1.6-1.7) |
NA | 4530 (3050-4980) |
4660 (4390-5660) |
57 (49-61) |
24 (20-26) |
NA |
| SC | 1.0 | 3.9 (3.0-4.6) |
2 (2-2) |
395 (300-493) |
2390 (2140-2560) |
NA | NA | 51 (38-57) |
|
| Chol-PEG5k-PIE12-trimer* | IV | 1.0 | 5.6 (4.5-5.7) |
NA | 1100 (1100-1190) |
4580 (3380-4710) |
118 (115-129) |
15 (14-20) |
NA |
| SC | 1.0 | 7.2 (7.2-7.9) |
4 (4-8) |
89 (78-104) |
1560 (1500-1910) |
NA | NA | 34 (33-56) |
|
| CPT31† | IV | 1.0 | 3.3 (2.6-3.9) |
NA | 2950 (2670-3230) |
3840 (3780-3910) |
134 (105-163) |
29 (28-29) |
NA |
| SC | 1.0 | 5.4 (5.3-5.5) |
2 (2-2) |
261 (207-314) |
2110 (1810-2410) |
NA | NA | 55 (46-64) |
n=3,
n=2,
NA=not applicable, T1/2=elimination half-life, Tmax=time at maximum plasma concentration, Co=initial plasma drug concentration (IV injection), Cmax=maximum plasma drug concentration (SC injection), AUC=area under the curve, Vd=apparent volume of distribution, Cl(obs)=observed total body clearance of the drug from plasma, F=bioavailability* range reflects the largest/smallest possible ratios among all animals tested
Non-human Primate Pharmacokinetics
The in-life portion of this study was performed by Calvert Laboratories. Three male cynomolgus monkeys (3.4-3.9 kg) were administered CPT31 (2 mg/ml in 50 mM HEPES, pH 7.4) intravenously as a single bolus injection (saphenous vein) at a dose of 1 mg/kg (0.5 ml/kg). One ml blood samples were collected by venipuncture of a femoral vein at 0.083, 0.167, 0.25, 0.5, 1, 2, 4, 8, 16 and 24 h post-dose into chilled tubes containing K2EDTA. Plasma was stored at −80 °C until bioanalysis.
Following a 13-day washout period, study animals were administered a single SC dose of CPT31 (10 mg/ml in 50 mM HEPES, pH 7.4) between the shoulder blades at a dose of 3 mg/kg. Plasma samples were collected pre-dose and 0.25, 0.5, 1, 2, 4, 8, 16, 24, 48 and 72 h post-dose and treated as described above. The pre-dose sample confirmed drug levels were below the lower limit of quantitation (5 nM). Compounds evaluated were stable in plasma at the storage and shipment conditions.
Quantitative Bioanalysis - PIE12-trimer conjugates (Navigen studies)
Samples were spiked with an internal standard (IS), then precipitated with two volumes of 98% acetonitrile/2% formic acid. Supernatants were analyzed by LC/MS/MS using an Agilent HPLC system (Waters X-Bridge BEH C18 column) paired to an AB Sciex API 3000 triple-quad mass spectrometer using MRM methods. Lipid conjugates required lower source temperatures (300°C vs 500°C) for improved reproducibility. For all studies, the column was regenerated after each group of three rats by running an isocratic gradient of 25% water/25% methanol/25% isopropanol/25% acetonitrile for 30 min.
Mass transitions and IS were as follows for each analyte: PIE12-trimer (1431.7/180.1 Da, IS: ΔHP-PIE12-trimer – a light version of PIE12-trimer where the N-terminal residues were deleted6), palmitate-PEG24-PIE12-trimer (1466.5/554.4 Da, IS: C8-PEG24-PIE12-trimer), C16-PEG24-PIE12-trimer (1450.1/453.4 Da, IS: C18-PEG24-PIE12-trimer), C18-PEG24-PIE12-trimer (1454.5/481.3 Da, IS: C16-PEG24-PIE12-trimer) and Chol-PIE12-trimer (1474.2/1694.9 Da, IS: Chol-PEG12-PIE12-trimer – an analog to Chol-PIE12-trimer, but synthesized using Malemide-PEG12-triNHS, Quanta BioDesign 10676 as previously described6).
Quantitative Bioanalysis - Chol-PIE12-trimer in rat plasma (Calvert study)
Fifty-microliter aliquots of plasma for each time point were precipitated with 3 volumes of ice-cold acetonitrile containing 2% formic acid (v/v) and 1.56 μM Chol-PEG12-PIE12-trimer (IS). Following centrifugation, 8 μL of supernatant were injected onto a Poroshell 300 SB-C8 column (2.1 × 75 mm, 5 μm) (Agilent). Analyte (Chol-PIE12-trimer) and IS were separated on an Agilent 1290 UHPLC system using a gradient consisting of 0.2% formic acid in 5 mM aqueous ammonium acetate buffer and 0.2% formic acid in acetonitrile/isopropanol (1:1) at a flow rate of 0.65 mL/min. The column temperature was maintained at 70 °C. Ions were formed by a dual electrospray source operated in positive-ion mode and detected on an Agilent quadrupole time-of-flight (Q-TOF) mass spectrometer (6540A). Extracted-ion chromatograms were processed with MassHunter Quantitative Analysis software (Agilent V. B.06). A m/z of 1476.7156 with a m/z window of 40 ppm was used to extract the peak area for Chol-PIE12-trimer. This ion corresponds to the second most abundant C13 isotope peak in the 6+ charge state cluster and represents the M+7 isotope of the (M+5H+NH4)6+ ion cluster. A m/z of 1662.5882 with a m/z window of 200 ppm was used to extract the peak area for Chol-PEG12-PIE12-trimer. This ion corresponds to the most abundant C13 isotope peak in the 5+ charge state cluster and represents the M+6 isotope of the (M+5H)5+ ion cluster. Plasma concentrations were determined from peak area ratio of analyte/IS compared against an 8-point calibration curve spanning a concentration range of 15.6 to 2,000 nM. Two quality control samples (100 and 400 nM) were prepared independently of the calibration curve and analyzed in triplicate to define limits of variability, with minimum acceptable determined concentrations ±20% of calculated concentration.
Chol-PEG5k-PIE12-trimer in Rat Plasma (Calvert Study)
Samples were processed as described above with subtle variations including: 2.5 volumes of acetonitrile containing 2% trifluoroacetic acid (v/v) was used for plasma protein precipitation, 370 nM Chol-PEG12-PIE12-trimer IS was used, 10 uL of supernatant was injected, and the UHPLC flow rate was 0.7 mL/min. Due to the polydispersity of the 5kDa PEG, three separate m/z ions of 1074.2127, 1157.9981 and 1159.3843 each with a m/z window of 200 ppm were used to extract the peak area for Chol-PEG5k-PIE12-trimer. These ions correspond to the 14+ and 13+ charge states. A m/z of 1662.5882 with a m/z window of 100 ppm was used to extract the peak area for Chol-PEG12-PIE12-trimer. This ion corresponds to the most abundant C13 isotope peak in the 5+ charge state cluster and represents the M+6 isotope of the (M+5H)5+ ion cluster. Plasma concentrations were determined from peak area ratio of analyte/IS compared against an 8-point calibration curve spanning a concentration range of 15.6 to 2,000 nM. Three quality control samples (40, 400, and 1,200 nM) were prepared independently of the calibration curve and analyzed in triplicate to define limits of variability, with minimum acceptable determined concentrations ±20% of calculated concentration.
CPT31 in Rat Plasma (Calvert Study)
Samples were processed as described above with subtle variations including: 5 volumes of acetonitrile containing 1% formic acid (v/v) was used for plasma protein precipitation, no internal standard was used or needed, 1 μL of supernatant was injected onto a Poroshell 120 EC-C8 column (2.1 × 5 mm, 2.7 μm), and a gradient consisting of 20 mM aqueous ammonium bicarbonate buffer and acetonitrile at a flow rate of 0.45 mL/min was used. The column temperature was maintained at 40 °C. A m/z of 1508.7473 with a m/z window of 40 ppm was used to extract the peak area for CPT31. This ion corresponds to the second most abundant C13 isotope peak in the 6+ charge state cluster and represents the M+7 isotope of the (M+5H+NH4)6+ ion cluster. Plasma concentrations were determined from the peak area of analyte compared against an 8-point calibration curve spanning a concentration range of 5 to 4,000 nM. Three quality control samples (15, 150 and 3,000 nM) were prepared independently of the calibration curve and analyzed in duplicate to define limits of variability, with minimum acceptable determined concentrations ±20% of calculated concentration.
CPT31 in Monkey Plasma (Calvert Study)
The internal standard, CPT31-IS was synthesized with an additional glycine on each PIE12-2 monomer (three in total), increasing the molecular mass by 171.1 Da. Plasma aliquots (200 μl) were spiked with CPT31-IS to a concentration of 60 or 150 nM, then precipitated with 2% NH4OH in acetonitrile (500 μl). Following centrifugation, the supernatant was applied to a strong anion exchange solid-phase extraction 96-well plate (SOLAμ SAX, 2 mg/ml 96-well plate). The anion exchange plate was first conditioned with 400 μl 2% NH4OH in methanol, followed by 400 μl of 2% NH4OH in water. The precipitated supernatant (500 μl) was then loaded into each well, followed by washing with 500 μl 2% NH4OH in water, then 500 μl methanol. Sample was eluted using two 50 μl aliquots of 2% formic acid in methanol.
LC-MS analysis was conducted using the Agilent instruments described above. Sample (1 μl) was injected at a flow rate of 0.45 ml/min on a Thermo Scientific Accupore 150 C4 column (2.1 × 50 mm, 2.6 μm), using a gradient of 20 mM ammonium bicarbonate (pH 7.9) in water and acetonitrile. Samples were analyzed against a standard curve of CPT31 from 5–2,000 nM. Three quality control samples (15, 150 and 1,500 nM) were prepared independently of the calibration curve and analyzed in duplicate to define limits of variability, with minimum acceptable determined concentrations ±20% of calculated concentration.
Pharmacokinetic Data Fitting
All bioanalytical data was fit using noncompartmental analysis with Phoenix edition v.6.4 WinNonlin (Pharsight, Cary, NC).
Animal Studies
All animal studies were performed in accordance with protocols approved by the IACUCs at the University of Utah or Calvert.
RESULTS
Conjugate Syntheses
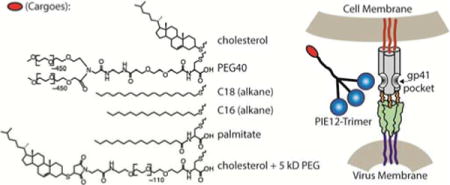
PIE12-trimer comprises three PIE12 monomers, each containing a unique primary amine (C-terminal Lys), coupled to a scaffold using a homobifunctional PEG4-NHS ester crosslinker. The 4th arm of our previously reported 4-arm scaffold6 is composed of a PEG24 spacer that terminates in a maleimide (thiol-reactive) group (Fig. 1A). The length of the 4th-arm PEG was empirically optimized to best bridge the distance between the membrane and the gp41 pocket drug target6. The orthogonal maleimide reactivity provides a convenient way to couple various cargoes to PIE12-trimer to explore their effect on potency and PK properties of the molecule. PEG, fatty acids, alkyl chains and cholesterol were selected from among clinical and promising preclinical PK-enhancing moieties (Fig 1B).
Figure 1.
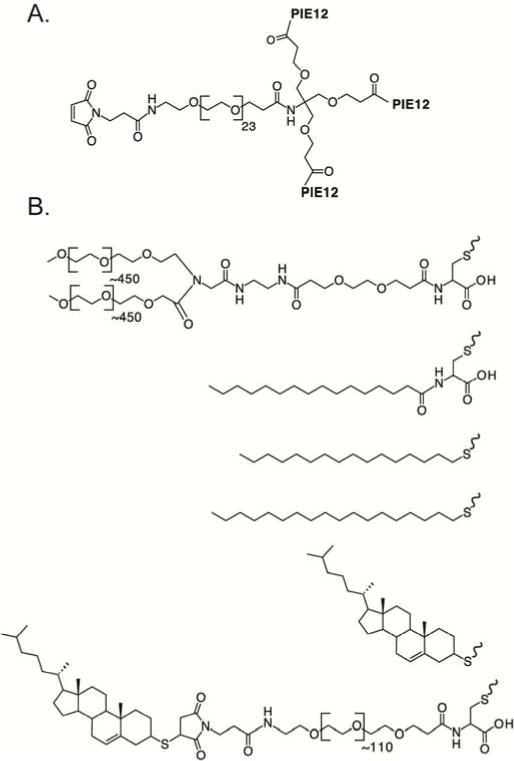
Schematic of PIE12-trimer and Conjugates: A) Maleimide-PEG24-PIE12-trimer, which is functionalized for conjugation with thiol-containing PK-enhancing cargoes, B) the PK-enhancing thiol cargoes used to make (from top to bottom): PEG40-PIE12-trimer, palm-PIE12-trimer, C16-PIE12-trimer, C18-PIE12-trimer, Chol-PIE12-trimer and Chol-PEG5k-PIE12-trimer.
For our PEGylation strategy, we coupled a 40 kDa Y-branched PEG (similar to the PEG in PEGASYS, a PEGylated interferon that slows human clearance of interferon 100-fold23) to PIE12-trimer’s 4th arm (PEG40-PIE12-trimer). For acylation, we attached palmitate (a fatty acid with 16 carbon atoms) using Cys as a bridge to generate the necessary reactivity with the 4-arm scaffold (Palm-PIE12-trimer). Alkanes with chain lengths of 16 and 18 carbons were conjugated to PIE12-trimer by reacting commercially available thio-alkanes with the maleimide on the scaffold (C16-PIE12-trimer and C18-PIE12-trimer). Finally, cholesterol conjugation was achieved by coupling thiocholesterol to the scaffold maleimide (Chol-PIE12-trimer; previously known as Chol-PEG24-PIE12-trimer from our study investigating the ideal PEG length for potency enhancement6).
Antiviral Potency
Previously, we demonstrated that membrane-localizing cargoes, such as alkanes and cholesterol, enhanced the anti-HIV potency of PIE12-trimer, presumably by concentrating the inhibitor at the membrane site of viral entry6. Since our ultimate goal is to produce an inhibitor for clinical use, it was important to determine the impact of each of our conjugates on antiviral potency. We determined the IC50 of viral entry for each conjugate using an HIV pseudovirus harboring a primary strain Env (JRFL) that is relatively insensitive to entry inhibitors. The majority of the conjugates (acylated, alkylated and cholesterol-conjugated; all of which are predicted to interact with lipid bilayers) improved antiviral potency, ranging from ~4-fold (for Palm-PIE12-trimer) to ~100-fold (for Chol-PIE12-trimer) (Table 1). The two alkane conjugates (C16-PIE12-trimer and C18-PIE12-trimer) similarly boosted potency by ~20-fold. The only conjugate that was detrimental to antiviral activity was PEG40-PIE12-trimer (>30-fold loss of activity, likely due to steric interference surrounding the gp41 pocket24,25).
Rat Pharmacokinetics
To investigate the pharmacokinetics of each of our conjugates, we dosed Sprague Dawley rats in-house with both subcutaneous (SC) and intravenous (IV) administrations of our compounds and analyzed the plasma drug concentration at various time points using a quantitative LC/MS/MS bioanalytical assay (Table 2). In addition to improving potency, all of the lipid-based conjugates (acylated, alkylated and cholesterol-conjugated) also extended half-life and reduced clearance rate. Specifically, Palm-PIE12-trimer and Chol-PIE12-trimer showed a similarly increased IV half-life (3-fold) and significantly reduced clearance (~14-18-fold). Both Palm-PIE12-trimer and Chol-PIE12-trimer were highly bioavailable (100% and 73%, respectively) upon SC dosing with ~3-fold extension of apparent half-life (based on terminal-phase elimination). The alkane conjugates (C16-PIE12-trimer and C18-PIE12-trimer) demonstrated modest increases in half-life upon IV or SC dosing (<2-fold) and moderately reduced clearance rates (5-8-fold), likely due to increased plasma protein binding.
Preliminary rat PK experiments with the 40 kDa PEG (PEG40) attached to PIE12-monomer showed a ~15-fold increase in circulating half-life compared to PIE12-monomer. We would expect a similar circulating half-life improvement in the context of PIE12-trimer. However, PEG40-PIE12-trimer was >3500-fold less potent than our best conjugate, Chol-PIE12-trimer (Table 1). This result, coupled with its increased mass, would lead to significantly increased depot dosing requirements, so this construct was not pursued further.
Engineering of Chol-PIE12-trimer
With a combination of the best improvement in potency and among the best improvements in PK, Chol-PIE12-trimer became our molecule of interest for further development. Therefore, our next steps were to attempt further optimization of its pharmacokinetic properties as well as its synthetic design to facilitate larger-scale production for continued preclinical and future clinical studies.
Hoping to harness the PK-enhancing effect of PEG without the steric consequences on potency, we added 5 kDa of linear polydisperse PEG to Chol-PIE12-trimer. In Chol-PEG5k PIE12-trimer, the linear PEG was inserted on the 4th scaffold arm, between PIE12-trimer and thiocholesterol. Previously, we demonstrated that increasing the length of the PEG spacer between thiocholesterol and PIE12-trimer had little effect on potency6. As expected from this trend, the potency of Chol-PEG5k-PIE12-trimer is similar to that of Chol-PIE12-trimer (Table 1).
As with our earlier conjugates, we investigated the pharmacokinetics of this new conjugate in rats with both SC and IV dosing, and analyzed the plasma drug concentration with quantitative LC/MS/MS (Table 3). The animal portion of the study was performed at Calvert Laboratories with a slightly modified protocol, and the bioanalytical assay was updated from the earlier study. Due to the changes, we repeated the Chol-PIE12-trimer experiments to have a more accurate side-by-side comparison. The repeat Chol-PIE12-trimer data are similar to the original except for Co and its derived parameters, due to earlier sampling times.
For both IV and SC administration, the added 5 kDa PEG resulted in a prolonged half-life (3.5-fold and 1.8-fold, respectively) when compared to Chol-PIE12-trimer (Table 3). However, the volume of distribution (Vd) is 2.1-fold higher for Chol-PEG5k-PIE12-trimer. The increased Vd partially offsets the benefit of slowed clearance. Furthermore, bioavailability was significantly decreased to 34%, suggesting that the added PEG mass is responsible for additional metabolism/sequestration in the subcutaneous space or lymphatic system. Taken together, the beneficial half-life effects of the added PEG were insufficient to warrant the added synthetic complexity associated with the 5 kDa PEG, which, unlike the original PEG24 4th arm, is polydisperse. Therefore, we did not pursue this molecule further.
With Chol-PIE12-trimer as our empirically determined lead candidate, it was important to design its chemical synthesis to facilitate production of material for toxicology, efficacy in non-human primates, and, ultimately, human clinical studies. Our original scaffold, with the fourth arm functionalized with a maleimide group for reaction with thiols, was efficient for rapidly testing our PK cargoes. However, this scaffold is not ideal as a drug substance because the maleimide-thiol reaction introduces a heterogeneous stereocenter. We redesigned the scaffold to avoid such a stereocenter while simultaneously simplifying synthesis, improving yield and scalability, and reducing cost (of both the scaffold and final product) (Fig 2).
Figure 2.
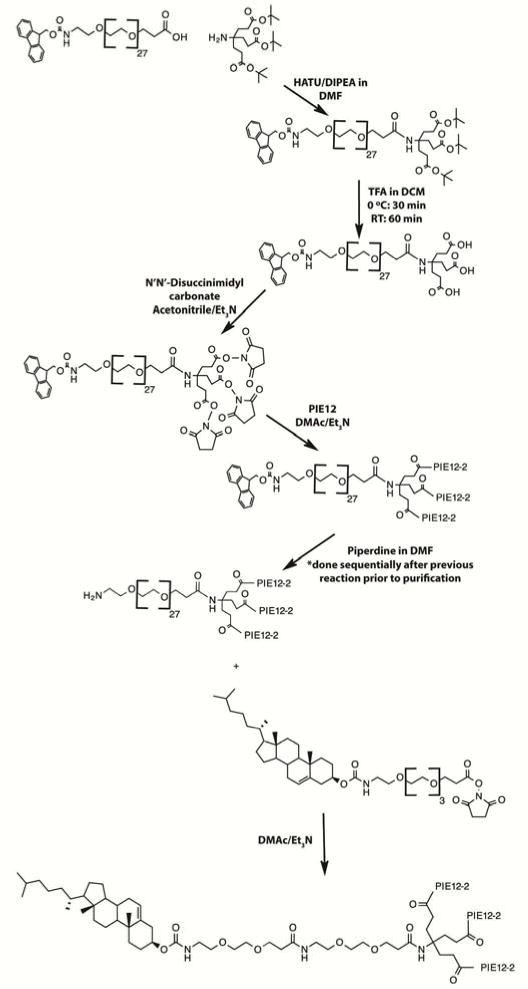
Streamlined Synthesis Strategy for CPT31. First, the PEG scaffold is assembled by adding the PEG27 spacer arm with an Fmoc-protected amine. The three carboxylic acids are then deprotected and activated to NHS esters, which are used to couple three PIE12-2 monomers. Finally, the Fmoc is removed to expose the amine and allow coupling to a cholesterol-PEG4-NHS ester to produce CPT31.
The revised scaffold comprises three short arms functionalized with NHS esters and a fourth arm (a high-quality monodisperse PEG27) terminating with an Fmoc-protected unique primary amine. After reaction of PIE12 monomer with the three NHS esters and removal of the Fmoc on the 4th arm, this trimer intermediate is purified by HPLC. Next, cholesterol-PEG4-NHS ester is conjugated to the primary amine on the fourth arm. Purification of the trimer intermediate simplifies synthesis since the main contaminant, PIE12-dimer (caused by competing hydrolysis of the NHS esters on the scaffold during trimerization), can be readily separated by HPLC purification prior to conjugation with cholesterol. After cholesterol conjugation, there is a dramatic shift to a later HPLC retention time, but much less separation between dimer and the correct trimer final product. Additionally, the location of the PEG linker on each of the three peptide arms was relocated from the Lys sidechain (PIE12GK-PEG4), which required orthogonal protection during solid-phase peptide synthesis, to the peptide backbone (PIE12G-PEG4-K, called PIE12-2 for simplicity), which was accomplished using a standard Fmoc-PEG4-acid reagent. The optimized molecule, CPT31 (Cholesterol-PIE12-trimer-PEG31), has a 4th arm that separates cholesterol from the trimer by 31 PEG units (vs. 24 in Chol-PIE12-trimer), lacks any heterogeneous stereocenters, and is easier and more efficient to produce. Like Chol-PIE12-trimer, CPT31 is soluble in standard aqueous buffers (e.g., PBS, HEPES) at physiological pH to ~40 mg/ml.
Somewhat surprisingly, the modest modifications of CPT31 compared to Chol-PIE12-trimer result in improved PK properties (Table 3). CPT31’s IV and SC half-lives in rats increase 2-fold and 1.4-fold, respectively, compared to Chol-PIE12-trimer. Additionally, the potency of CPT31 against the HIV JRFL strain is also slightly improved (Table 1).
NHP Pharmacokinetics
The ultimate goal for CPT31 is clinical use as an extended-release anti-HIV therapy (in combination with other extended-release antiretrovirals) and/or preventative. Therefore, it is critical to determine if its in vivo profile is compatible with infrequent dosing in humans. To pursue this question, we determined the PK profile of CPT31 in non-human primates (NHPs). These results will also inform future efficacy studies for preventing and treating SHIV in this definitive animal model. Three male cynomolgus monkeys were dosed IV at 1 mg/kg. After a 2-week washout period, these animals were dosed SC at 3 mg/kg (a high dose for evaluating therapeutic efficacy in NHPs). CPT31 found in plasma samples collected over a time course of 24 (IV study) and 72 (SC study) hours was quantitated by our bioanalytical assay. These data are shown in Fig. 3 and summarized in Table 4. CPT31 has slightly more favorable PK properties in NHPs than predicted from simple allometric scaling of the rat data, with longer IV and SC half-lives, increased bioavailability, and reduced clearance. Allometric scaling of the results are supportive of the feasibility of extended release human dosing. Although this study was not designed to assess toxicity, no adverse events were observed.
Figure 3.
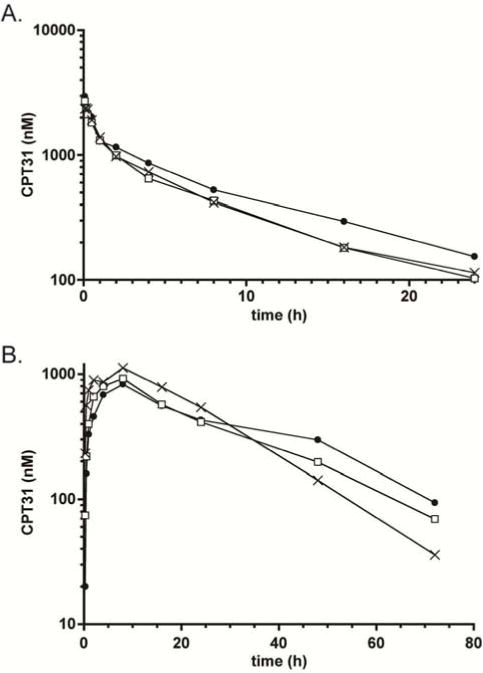
CPT31 Pharmacokinetics in Cynomolgous Monkeys. Characterization of single-dose PK curves for A) intravenous (1 mg/kg) and B) subcutaneous (3 mg/kg) dosing of CPT31 in three animals. The same animals were used for both studies with an intervening two-week washout period.
Table 4.
Median IV and SC plasma PK parameters of CPT31 in Male Cynomolgus Monkeys
| Compound | Route | Dose (mg/kg) |
T1/2 (hr) |
Tmax (hr) |
C0 or Cmax (nM) |
AUC (0-inf) (hr*nM) |
Vd(obs) (mL/kg) |
Cl (obs) (mL/hr/kg) |
F (%) |
|---|---|---|---|---|---|---|---|---|---|
| CPT31 | IV | 1.0 | 7.4 (7.1-9.4) |
NA | 3110 (2340-3940) |
11600 (11300-14900) |
97 (97-105) |
9.5 (7.4-9.8) |
NA |
| SC | 3.0 | 18.8 (12.3-23.0) |
8.0 (8.0-8.0) |
922 (828-1120) |
29400 (27200-30100) |
NA | NA | 80 (67-84) |
n=3, NA=not applicable, T1/2=elimination half-life, Tmax=time at maximum plasma concentration, Co=initial plasma drug concentration (IV injection), Cmax=maximum plasma drug concentration (SC injection), AUC=area under the curve, Vd=apparent volume of distribution, Cl(obs)=observed total body clearance of the drug from plasma, F=bioavailability
DISCUSSION
The therapeutic promise of peptides for potently and specifically blocking protein/protein interfaces has yet to be fully realized due to their in vivo instability caused by proteolysis and renal filtration. We are developing D-peptide therapeutics that inherently address protease instability. However, for long-term dosing, small D-peptides still require strategies to slow clearance via renal filtration. Using our most advanced D-peptide inhibitor, PIE12-trimer, that potently blocks all clinically relevant strains of HIV, we tested the effect of a panel of cargoes on proteolysis-independent in vivo clearance rates. These cargos included various lipid moieties (including palmitate, variable-length alkyl groups, and cholesterol) as well as a Y-branched 40 kDa PEG. The goal was to identify the best cargo to balance potency, pharmacokinetics, and scalability for manufacturing. A successful preclinical candidate will advance knowledge of PK-enhancing peptide modification as well as contribute to the nascent field of D-peptide-based therapeutics.
Acylation, and to a lesser extent the related alkylation, improved the PK properties of PIE12-trimer. Although these strategies were pursued primarily due to predicted HSA binding, other potential PK benefits of these modifications include self-association to prolong absorption from the subcutaneous space and interaction with cell membranes15,26,11. Based on the improvement in anti-HIV potency for each of these conjugates, at least transient interaction with the cell membrane, which would enrich the inhibitor at the site of viral entry, appears likely. Indeed, the additional hydrophobicity of C16-PIE12-trimer presumably increases membrane affinity, explaining the improved antiviral potency compared to Palm-PIE12-trimer. The significantly longer half-life of palmitoylated vs. thio-alkylated conjugates is surprising. Interestingly, the inhibitor containing the more hydrophobic alkane, C18-PIE12-trimer, also showed prolonged absorption from the subcutaneous space, but this effect did not increase the apparent terminal half-life upon SC dosing compared to palm-PIE12-trimer, since the latter had a lower clearance rate. Therefore, purely based on clearance, acylation appears to be the better choice for prolonging in vivo peptide survival.
Of the strategies we tested, PEGylation yields the greatest enhancement in half-life, but at the cost of a significant potency loss. PEG has a large hydrodynamic radius relative to its mass and primarily improves half-life by increasing drug size beyond the cut-off for effective renal filtration. For a target like the HIV gp41pocket that is sterically hindered24,25, the increased drug size is not compatible with effectively targeting the site of action. Interestingly, the potency of the gp41-targeting enfuvirtide is diminished by conjugation to even modest-sized PEG12. Therefore, although this modification is common in FDA-improved products and likely the superior option for less sterically hindered targets, we abandoned it for our purposes. Additionally, due to the results seen with the PEG conjugate, we did not test alternate PK-enhancing strategies such as conjugation to bulky HSA or Fc domains.
Conjugation to cholesterol yielded the best overall results for PIE12-trimer, with 100-fold improved potency and the slowest clearance of the lipid-based conjugates. Cholesterol has proven to be especially valuable for potency and PK enhancement in the context of viral entry inhibitors, even antibodies, as it likely enriches the inhibitors in lipid rafts where viral entry occurs and in which cholesterol is naturally enriched28–30. Additionally, it is possible that the cholesterol may also enhance the binding of the inhibitor to the prehairpin-intermediate target. Cholesterol is known to weakly interact with HSA31, and its affinity is lower than that of palmitate. Therefore, its interaction with cell membranes is likely to contribute to the PK enhancement. Although cholesterol conjugation is not yet utilized in any FDA-approved products, it is increasingly popular in preclinical research on cancer and siRNA therapeutics. Indeed, a cholesterol-conjugated miRNA to treat scleroderma is in early clinical trials (MRG-201) and will help establish the safety of this strategy32. Interestingly, in many of the current preclinical strategies utilizing cholesterol for target delivery, the conjugated product is a pro-drug with a labile linkage17. In CPT31, the cholesterol is crucial for delivery to the drug active site, and therefore our cholesterol conjugation strategy is designed for stability. Due to its successes in preclinical studies for a wide variety of therapeutic areas, including this study, cholesterol conjugation is likely to be an increasingly utilized strategy for enhancing peptide therapeutics.
All of our lipid-based conjugates demonstrate a decreased volume of distribution compared to Chol-PIE12-trimer. Currently, we cannot predict the significance of these findings because it is not clear which tissue compartments must be accessed for successful inhibition of HIV. Many anti-HIV drugs are highly HSA-bound33,34, but they remain effective in preventing and treating HIV (e.g., emtricitabine, a component of Truvada®). Ultimately, therapeutic efficacy in an animal model (such as SHIV inhibition in NHP) will be necessary to demonstrate the ability of CPT31 to access relevant viral reservoirs in vivo.
Importantly, once we decided on cholesterol as our PIE12-trimer conjugate, we optimized its chemical synthesis to produce our final drug candidate, CPT31. CPT31 incorporates design elements that simplify its synthesis, improve scalability, and eliminate heterogeneity compared to our original compound, Chol-PIE12-trimer. Somewhat unexpectedly, CPT31 has improved PK as well as slightly increased antiviral potency. A possible explanation for this observation is that the bulky maleimide group adjacent to thiochoesterol in Chol-PIE12-trimer hinders cholesterol insertion into the membrane and that the modified cholesterol linkage improves membrane association.
The future for injectable HIV drugs lies in extended-release dosing to remove the daily pill burden, increasing patient compliance and virologic control. Therefore, our minimum goal for CPT31 is to achieve monthly dosing via a sustained-release formulation. Such a formulation could be paired with promising long-acting HIV drugs such as cabotegravir (GSK744) from GlaxoSmithKline35–42 and rilpivirine (TMC278) from Tibotec38,39,43–46 for treatment or potentially used as monotherapy for PrEP (pre-exposure prophylaxis). Critically, the NHP pharmacokinetic evaluation described here supports the feasibility of this dosing goal. Allometric scaling of our NHP PK data to humans predicts a feasible CPT31 monthly depot dose of 42 mg (calculation assumptions: target in vivo drug level of 0.22 μg/mL (24 nM, 4X serum-adjusted IC90), projected human clearance rate of 3.0 mL/hr/kg (allometric scaling factor going from monkeys to humans is 0.3247), 70 kg weight, 30 days/month (720 h), and 80% bioavailability (Table 4)). CPT31’s low-mid pM potency in vitro, low clearance rate, and excellent bioavailability in non-human primates make it a very promising preclinical candidate for the treatment and/or prevention of HIV-1. Future studies will evaluate CPT31’s in vivo efficacy against SHIV in the NHP animal model and animal toxicology.
Acknowledgments
The authors would like to gratefully acknowledge Rebecca Macchione, Lyssa Lambert, and Haley Benzon for conducting the in vivo experiments at Navigen.
Funding Sources
This research was supported by NIH grants AI076168 to M.S.K., AI95172 to A.L.M., and GM082545 to D.M.E.
ABBREVIATIONS
- CPT31
Cholesterol-PIE12-trimer-PEG31
- FDA
Food and Drug Administration
- HIV
human immunodeficiency virus
- IS
internal standard
- LC/MS/MS
liquid chromatography, tandem mass spectrometry
- NHP
non-human primate
- PEG
polyethylene glycol
- PIE
pocket-specific inhibitor of entry
- PK
pharmacokinetics
- RP-HPLC
reversed-phase high performance liquid chromatography
- RT
room temperature
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.McGregor DP. Discovering and improving novel peptide therapeutics. Curr Opin Pharmacol. 2008;8(5):616–9. doi: 10.1016/j.coph.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Patel IH, Zhang X, Nieforth K, Salgo M, Buss N. Pharmacokinetics, pharmacodynamics and drug interaction potential of enfuvirtide. Clinical pharmacokinetics. 2005;44(2):175–86. doi: 10.2165/00003088-200544020-00003. [DOI] [PubMed] [Google Scholar]
- 3.Weinstock MT, Francis JN, Redman JS, Kay MS. Protease-resistant peptide design-empowering nature’s fragile warriors against HIV. Biopolymers. 2012;98(5):431–42. doi: 10.1002/bip.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milton RC, Milton SC, Kent SB. Total chemical synthesis of a D-enzyme: the enantiomers of HIV-1 protease show reciprocal chiral substrate specificity [corrected] Science. 1992;256(5062):1445–8. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 5.Zawadzke LE, Berg JM. A racemic protein. Journal of the American Chemical Society. 1992;114(10):4002–4003. [Google Scholar]
- 6.Francis JN, Redman JS, Eckert DM, Kay MS. Design of a Modular Tetrameric Scaffold for the Synthesis of Membrane-Localized d-Peptide Inhibitors of HIV-1 Entry. Bioconjug Chem. 2012 doi: 10.1021/bc300076f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J Virol. 2010;84(21):11235–44. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolate A, Baradia D, Patil S, Vhora I, Kore G, Misra A. PEG - a versatile conjugating ligand for drugs and drug delivery systems. J Control Release. 2014;192:67–81. doi: 10.1016/j.jconrel.2014.06.046. [DOI] [PubMed] [Google Scholar]
- 9.Corsetti M, Tack J. Naloxegol, a new drug for the treatment of opioid-induced constipation. Expert Opin Pharmacother. 2015;16(3):399–406. doi: 10.1517/14656566.2015.991306. [DOI] [PubMed] [Google Scholar]
- 10.English C, Aloi JJ. New FDA-Approved Disease-Modifying Therapies for Multiple Sclerosis. Clin Ther. 2015;37(4):691–715. doi: 10.1016/j.clinthera.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Wynn TT, Gumuscu B. Potential role of a new PEGylated recombinant factor VIII for hemophilia A. J Blood Med. 2016;7:121–8. doi: 10.2147/JBM.S82457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng S, Wang Y, Zhang Z, Lv X, Gao GF, Shao Y, Ma L, Li X. Enfuvirtide-PEG conjugate: A potent HIV fusion inhibitor with improved pharmacokinetic properties. European journal of medicinal chemistry. 2016;121:232–7. doi: 10.1016/j.ejmech.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar A. Insulin degludec/liraglutide: innovation-driven combination for advancement in diabetes therapy. Expert opinion on biological therapy. 2014;14(6):869–78. doi: 10.1517/14712598.2014.904851. [DOI] [PubMed] [Google Scholar]
- 14.Chong H, Wu X, Su Y, He Y. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS. 2016;30(8):1187–96. doi: 10.1097/QAD.0000000000001073. [DOI] [PubMed] [Google Scholar]
- 15.Ding X, Zhang X, Chong H, Zhu Y, Wei H, Wu X, He J, Wang X, He Y. Enfuvirtide (T20)-Based Lipopeptide Is a Potent HIV-1 Cell Fusion Inhibitor: Implications for Viral Entry and Inhibition. J Virol. 2017;91(18) doi: 10.1128/JVI.00831-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16(3):165–79. [PubMed] [Google Scholar]
- 17.Irby D, Du C, Li F. Lipid-Drug Conjugate for Enhancing Drug Delivery. Molecular pharmaceutics. 2017;14(5):1325–1338. doi: 10.1021/acs.molpharmaceut.6b01027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Rohl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vomlocher HP. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432(7014):173–8. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 19.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci USA. 2009;106(14):5801–6. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margolis DA, Boffito M. Long-acting antiviral agents for HIV treatment. Current opinion in HIV and AIDS. 2015;10(4):246–52. doi: 10.1097/COH.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, Eron JJ, Yazdanpanah Y, Podzamczer D, Lutz T, Angel JB, Richmond GJ, Clotet B, Gutierrez F, Sloan L, Clair MS, Murray M, Ford SL, Mrus J, Patel P, Crauwels H, Griffith SK, Sutton KC, Dorey D, Smith KY, Williams PE, Spreen WR. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet. 2017;390(10101):1499–1510. doi: 10.1016/S0140-6736(17)31917-7. [DOI] [PubMed] [Google Scholar]
- 22.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-Peptide Inhibitors of HIV-1 Entry. Proc Natl Acad Sci U S A. 2007;104(43):16828–33. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fishbum CS. The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008;97(10):4167–83. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- 24.Hamburger AE, Kim S, Welch BD, Kay MS. Steric accessibility of the HIV-1 gp41 N-trimer region. J Biol Chem. 2005;280(13):12567–72. doi: 10.1074/jbc.M412770200. [DOI] [PubMed] [Google Scholar]
- 25.Eckert DM, Shi Y, Kim S, Welch BD, Kang E, Poff ES, Kay MS. Characterization of the steric defense of the HIV-1 gp41 N-trimer region. Protein Sci. 2008;17(12):2091–100. doi: 10.1110/ps.038273.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Havelund S, Plum A, Ribel U, Jonassen I, Volund A, Markussen J, Kurtzhals P. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm Res. 2004;21(8):1498–504. doi: 10.1023/b:pham.0000036926.54824.37. [DOI] [PubMed] [Google Scholar]
- 27.Nauck M, Frid A, Hermansen K, Shah NS, Tankova T, Mitha IH, Zdravkovic M, During M, Matthews DR, Group L.-S Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care. 2009;32(1):84–90. doi: 10.2337/dc08-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lacek K, Urbanowicz RA, Troise F, De Lorenzo C, Severino V, Di Maro A, Tarr AW, Ferrara F, Ploss A, Temperton N, Ball JK, Nicosia A, Cortese R, Pessi A. Dramatic potentiation of the antiviral activity of HIV antibodies by cholesterol conjugation. The Journal of biological chemistry. 2014;289(50):35015–28. doi: 10.1074/jbc.M114.591826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pessi A. Cholesterol-conjugated peptide antivirals: a path to a rapid response to emerging viral diseases. Journal of peptide science: an official publication of the European Peptide Society. 2015;21(5):379–86. doi: 10.1002/psc.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urbanowicz RA, Lacek K, Lahm A, Bienkowska-Szewczyk K, Ball JK, Nicosia A, Cortese R, Pessi A. Cholesterol conjugation potentiates the antiviral activity of an HIV immunoadhesin. Journal of peptide science: an official publication of the European Peptide Society. 2015;21(9):743–9. doi: 10.1002/psc.2802. [DOI] [PubMed] [Google Scholar]
- 31.Peng L, Minbo H, Fang C, Xi L, Chaocan Z. The interaction between cholesterol and human serum albumin. Protein Pept Lett. 2008;15(4):360–4. doi: 10.2174/092986608784246542. [DOI] [PubMed] [Google Scholar]
- 32.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nature reviews Drug discovery. 2017;16(3):203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 33.Bocedi A, Notaril S, Narciso P, Bolli A, Fasano M, Ascenzi P. Binding of anti-HIV drugs to human serum albumin. IUBMB Life. 2004;56(10):609–14. doi: 10.1080/15216540400016286. [DOI] [PubMed] [Google Scholar]
- 34.Schon A, del Mar Ingaramo M, Freire E. The binding of HIV-1 protease inhibitors to human serum proteins. Biophys Chem. 2003;105(2–3):221–30. doi: 10.1016/s0301-4622(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 35.Spreen W, Min S, Ford SL, Chen S, Lou Y, Bomar M, St Clair M, Piscitelli S, Fujiwara T. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials. 2013;14(5):192–203. doi: 10.1310/hct1405-192. [DOI] [PubMed] [Google Scholar]
- 36.Andrews CD, Spreen WR, Mohri H, Moss L, Ford S, Gettie A, Russell-Lodrigue K, Bohm RP, Cheng-Mayer C, Hong Z, Markowitz M, Ho DD. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science. 2014;343(6175):1151–4. doi: 10.1126/science.1248707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spreen W, Ford SL, Chen S, Wilfret D, Margolis D, Gould E, Piscitelli S. GSK1265744 pharmacokinetics in plasma and tissue after single-dose long-acting injectable administration in healthy subjects. J Acquir Immune Defic Syndr. 2014;67(5):481–6. doi: 10.1097/QAI.0000000000000301. [DOI] [PubMed] [Google Scholar]
- 38.Spreen W, Williams P, Margolis D, Ford SL, Crauwels H, Lou Y, Gould E, Stevens M, Piscitelli S. Pharmacokinetics, safety, and tolerability with repeat doses of GSK1265744 and rilpivirine (TMC278) long-acting nanosuspensions in healthy adults. J Acquir Immune Defic Syndr. 2014;67(5):487–92. doi: 10.1097/QAI.0000000000000365. [DOI] [PubMed] [Google Scholar]
- 39.Margolis DA, Brinson CC, Smith GH, de Vente J, Hagins DP, Eron JJ, Griffith SK, St Clair MH, Stevens MC, Williams PE, Ford SL, Stancil BS, Bomar MM, Hudson KJ, Smith KY, Spreen WR, Team, L. A. I. S Cabotegravir plus rilpivirine once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15(10):1145–55. doi: 10.1016/S1473-3099(15)00152-8. [DOI] [PubMed] [Google Scholar]
- 40.Andrews CD, Bernard LS, Poon AY, Mohri H, Gettie N, Spreen WR, Gettie A, Russell-Lodrigue K, Blanchard J, Hong Z, Ho DD, Markowitz M. Cabotegravir long acting injection protects macaques against intravenous challenge with SIVmac251. AIDS. 2017;31(4):461–467. doi: 10.1097/QAD.0000000000001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrews CD, Yueh YL, Spreen WR, St Bernard L, Boente-Carrera M, Rodriguez K, Gettie A, Russell-Lodrigue K, Blanchard J, Ford S, Mohri H, Cheng-Mayer C, Hong Z, Ho DD, Markowitz M. A long-acting integrase inhibitor protects female macaques from repeated high-dose intravaginal SHIV challenge. Sci Transi Med. 2015;7(270):270ra4. doi: 10.1126/scitranslmed.3010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radzio J, Spreen W, Yueh YL, Mitchell J, Jenkins L, Garcia-Lerma JG, Heneine W. The long-acting integrase inhibitor GSK744 protects macaques from repeated intravaginal SHIV challenge. Sci Transi Med. 2015;7(270):270ra5. doi: 10.1126/scitranslmed.3010297. [DOI] [PubMed] [Google Scholar]
- 43.van’t Klooster G, Hoeben E, Borghys H, Looszova A, Bouche MP, van Velsen F, Baert L. Pharmacokinetics and disposition of rilpivirine (TMC278) nanosuspension as a long-acting injectable antiretroviral formulation. Antimicrobial agents and chemotherapy. 2010;54(5):2042–50. doi: 10.1128/AAC.01529-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson AG, Else LJ, Mesquita PM, Egan D, Back DJ, Karolia Z, Ringner-Nackter L, Higgs CJ, Herold BC, Gazzard BG, Boffito M. A compartmental pharmacokinetic evaluation of long-acting rilpivirine in HIV-negative volunteers for preexposure prophylaxis. Clin Pharmacol Ther. 2014;96(3):314–23. doi: 10.1038/clpt.2014.118. [DOI] [PubMed] [Google Scholar]
- 45.Verloes R, Deleu S, Niemeijer N, Crauwels H, Meyvisch P, Williams P. Safety, tolerability and pharmacokinetics of rilpivirine following administration of a long-acting formulation in healthy volunteers. HIV Med. 2015;16(8):477–84. doi: 10.1111/hiv.12247. [DOI] [PubMed] [Google Scholar]
- 46.McGowan I, Dezzutti CS, Siegel A, Engstrom J, Nikiforov A, Duffill K, Shetler C, Richardson-Harman N, Abebe K, Back D, Else L, Egan D, Khoo S, Egan JE, Stall R, Williams PE, Rehman KK, Adler A, Brand RM, Chen B, Achilles S, Cranston RD. Long-acting rilpivirine as potential pre-exposure prophylaxis for HIV-1 prevention (the MWRI-01 study): an open-label, phase 1, compartmental, pharmacokinetic and pharmacodynamic assessment. Lancet HIV. 2016;3(12):e569–e578. doi: 10.1016/S2352-3018(16)30113-8. [DOI] [PubMed] [Google Scholar]
- 47.Guidance for Industry: Estimating the Maximum Safe Starting Dose in Adult Healthy Volunteer. US Food and Drug Administration; Rockville, MD: 2005. [Google Scholar]