

Abstract
The intracellular multiprotein complex termed the inflammasome functions as a platform of pro-inflammatory cytokine production such as IL-1β and IL-18. Under certain conditions, however, the inflammasome produces non-canonical effects such as induction of cell death, pyroptosis and cell metabolism alterations. In mammalian cells, several types of inflammasomes were identified, but the most widely studied one is the inflammasome containing NOD-like receptor with pyrin domain 3 (NLRP3), which has recently been reported as a central pathogenic mechanism of chronic degenerative diseases. Many activators or danger factors exert their actions through activation of the NLRP3 inflammasome to produce a variety of functional changes in different cells including inflammatory, metabolic or survival responses. Several molecular signaling pathways are shown to mediate the activation of the NLRP3 inflammasome, and they are related to the modifications in K+ efflux, increased lysosome leakage and activation of cathepsin B or enhanced reactive oxygen species (ROS) production. In the kidney, inflammation is believed to mediate or promote the progression of glomerular sclerotic pathologies resulting in end-stage renal disease (ESRD). NLRP3 inflammasome activation may turn on glomerular inflammation and other cell damages, contributing to the onset of glomerular injury and ESRD. This inflammasome activation not only occurs in immune cells, but also in residential cells such as endothelial cells and podocytes in the glomeruli. This review briefly summarizes current evidence of NLRP3 inflammasome activation and related molecular mechanisms in renal glomeruli. The possible canonical and non-canonical effects of this inflammasome activation and its potential implication in the development of different glomerular diseases are highlighted.
Keywords: Inflammation, glomerulosclerosis, inflammasome, homocysteine, diabetes mellitus
Introduction
The characteristic feature of the innate immune system (IIS) is to detect and defend the cellular microenvironment from cellular disturbances such as invading pathogens and injury. As the IIS surveys for disruption in cell homeostasis, when exogenous or endogenous molecular patterns are recognized it initiates defense or inflammatory responses via activation of different pattern recognition receptors (PRRs); including C-type lectin receptors (CLRs), toll-like receptors (TLRs), retinoic inducible gene-I (RIG-I), hemopoetic interferon-inducible nuclear antigen with 200 repeats (HIN-200) and Nod-like receptors (NLRs). Instigating the classical inflammatory response is a distinctive trait of these PRRs; however, the NLR family of PPRs activates the inflammasome, a potent intracellular inflammatory machinery by proteolytic cleavage of inflammatory cytokines interleukin-1β or IL-18 into their active forms. The nucleotide-binding oligomerization domain Nod-like receptor containing pyrin domain 3 (NLRP3, also known as NALP3, CIAS1, cryopyrin, or PYPAF1)-centered inflammasome, namely, NLRP3 inflammasome has been well characterized and its activation is considered as ubiquitous in different organs and cells. Some investigators believed that this inflammasome activation may be the root of many chronic degenerative diseases. In the kidney, formation and activation of the NLRP3 inflammasome have been reported to occur not only in immune cells like dendritic cells [33] and infiltrating macrophages [8, 51], but also in other renal cells such as tubular epithelial cells [49, 90, 101] and podocytes [3, 97], signifying its role in a broad spectrum of glomerular and tublointerstital diseases as well as tubular injury and repair. This review will focus on the molecular mechanisms underlying NLRP3 inflammasome activation in renal glomeruli, its well-established and unconventional effects and the potential implication in the development of different glomerular diseases. Together with an analysis of the literature, we attempt to highlight the critical pathogenic role of the NLRP3 inflammasome during glomerular injury or disease and illustrate how this intracellular inflammatory machinery may serve as a potential therapeutic target for prevention and treatment of glomerular diseases and consequent end-stage renal disease (ESRD).
NLRP3 Inflammasomes in the Glomeruli
Both microbial and non-microbial inflammation has been reported to participate in glomerular inflammatory responses, which may represent a critical pathogenic mechanism responsible for glomerular injury and subsequent ESRD. In particular, the non-microbial inflammation is widely recognized as a key pathological mechanism that mediates or promotes the development of glomerular sclerotic pathology during ESRD associated with several common systemic diseases such as hypertension, hyperhomocysteinemia, diabetes mellitus, and erythematosus lupus [80, 97]. In this regard, activation of inflammatory responses in the residential renal cells may be critical in initiating glomerular injury and renal dysfunction. There is evidence that glomerular IL-1β is mainly generated from podocytes, but not other cell types. Increased IL-1β production from podocytes are critically involved in the progression of many non-proliferative forms of glomerulonephritis and other glomerular diseases induced by local inflammatory processes in humans and rodents [15, 73, 84]. Using different approaches, our recent studies have shown that 3 major NLRP3 components were indeed enriched in murine podocytes and that their assembling into a multiprotein complex in podocytes occurs during stimulations by various danger factors such as L-homocysteine (L-Hcys), uric acid crystals, and adipokine-visfatin [92, 97]. It has been proposed that the NLRP3 inflammasome activation may importantly contribute to the onset or gradual progression of glomerular injury under different pathological conditions [1, 13, 97].
NLRP3 inflammasome
Originally, NLRP3 inflammasomes were identified as a crucial player necessary for activation of caspase-1, and their primary function is to activate cysteine proteases that subsequently induces inflammatory responses in tissues or organs by generation of pro-inflammatory interleukins (ILs) [79]. It is known that there are 23 NLR genes in the human genome including the NLRP family that contains a pyrin domain and the NLRC family that contains a caspase recruitment domain. Among all NLRs, only a few can be assembled into the inflammasome to activate caspases [88], which are NLRP1, NLRC4, and NLRP3 inflammasomes. NLRP1 was the first identified inflammasome, and it was found to provide immunity against bacterial cell wall components, which directly activates caspase-5 without ASC [16, 29]. The presence of ASC, however, greatly accelerates NLRP1 inflammasome activity [29]. Another NLR-associated inflammasome is NLRC4, and its activation is only triggered by bacterial flagellin and endogenous cell cytoplasmic components [52, 68, 99].
The NLRP3 inflammasome has been well characterized and its activation strongly links to sterile inflammation in a variety of chronic degenerative diseases [17]. The NLRP3 gene was first found to have mutation in patients with familial cold urticara (FCU) and Muckle-Wells syndrome (MWS) [4]. Consequently, the over exuberant inflammatory response seen in these patients was attributed to an NLRP3-dependent mechanism that results in defective apoptosis and transcriptional regulation. Further studies in these MWS patients demonstrated that they spontaneously secrete active IL-1β, which is due to the interaction of NLRP3 with the ASC protein. This interaction was later recognized as the formation of NLRP3 inflammasomes [5]. Over the last 5 years, many bacterial and viral pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) released from damaged tissues or cells have been identified as activators of the NLRP3 inflammasome. Tremendous efforts have been made worldwide to address the possible role of this inflammasome in the initiation or development of different diseases and to find inducers or activators of the NLRP3 inflammasome under different pathological conditions, which may help identify therapeutic targets for treatment of related diseases.
NLRP3 inflammasome activators
There are a very diverse range of danger factors that initiate activation of the NLRP3 inflammasome through different mechanisms. It has been reported that microbes such as the influenza virus, adenoviruses, Staphylococcus aureus, E. coli, Neisseria gonorrhoe, and Candida albicans may stimulate NLRP3 inflammasome activation [27, 44, 47, 64, 71, 85]. One of the mechanisms activating the NLRP3 inflammasome during infections of these microorganisms may be associated with the formation of pannexin-1 pores that permits entry of these microbial toxins into the cytoplasm. In addition, monosodium urate (MSU) crystals, aluminum salts, silica or asbestos are also reported to stimulate the NLRP3 inflammasome [25, 41, 65] and as non-microbial materials they are phagocytosed into the cell leading to lysosomal damage, triggering NLRP3 inflammasome activation to produce IL-1β and other cytokines. Additionally, extracellular ATP and potassium were shown to induce NLRP3 assembly and activation, which may be mediated by the purinergic P2X7 receptor and pannexin-1 [74].
NLRP3 formation and activation have been originally attributed to autoinflammatory diseases and more than 20 such autoinflammatory diseases can be treated by inhibition of NLRP3 activation and by antagonizing the action of IL-1β. Recently, many studies found that the activation of theNRLP3 inflammasome also contributes to the development of various chronic metabolic diseases such as diabetes mellitus, gout, silicosis, and obesity. In addition, in acute myocardial infarction, atherosclerosis, glomerular sclerosis, Alzheimer’s disease and liver cirrhosis the NLRP3 inflammasome is also shown to be activated [21, 39, 61, 97]. In this regard, accumulation of the amyloid-β protein in the brain is considered as an activator of the NLRP3 inflammasome to promote local inflammation or cell injury leading to the development of Alzheimer’s disease [37]. In addition, the high levels of IL-1β has been detected in the brains of patients suffering from Alzheimer’s disease. During atherosclerosis, cholesterol crystals or even oxidative products of cholesterol may serve as a danger factor to cause phagolysosomal damage, activating the NLRP3 inflammasome and thereby promoting a pro-atherosclerotic phenotype in arteries [26]. In addition, trauma and excessive exercises may produce some chemokines that may activate NLRP3 inflammasomes. This process may be associated with hyaluronan-mediated cellular responses [94].
In a murine model of diabetes, hyperglycemia stimulated NLRP3 inflammasome activation, subsequently causing injury to pancreatic islet cells, glucose intolerance and insulin resistance [100]. Recent findings in our lab demonstrated that homocysteine (Hcys), an amino acid derived from protein catabolism involving methionine, if increased in blood or extracellular space, can stimulate NLRP3 inflammasome formation and activation in different types of cells. In endothelial cells, this NLRP3 inflammasome activation will lead to endothelial dysfunction and induce atherogenic effects. In podocytes, increased activation of the NLRP3 inflammasome results in local glomerular inflammation and podocyte injury or transformation, ultimately leading to glomerular sclerosis [97]. We have shown that inhibition of the NLRP3 inflammasome via genetic or pharmacological interventions prevented Hcys-induced podocyte injury and glomerular sclerosis [97]. We also demonstrated that the adipokine, visfatin instigates vascular inflammation and injury by activation of the NLRP3 inflammasome, which ultimately leads to atherosclerosis [93]. To our knowledge, increasing numbers of danger factors including PAMPs and DAMPs are reported as activators of the NLRP3 inflammasome under different pathological conditions. Therefore, the NLRP3 inflammasome is evolving as a new common pathogenic mechanism for different diseases, in particular, those chronic degenerative diseases associated with inflammatory pathology. Table 1 summarizes the exogenous and endogenous activators of the NLRP3 inflammasome as well as pharmacological inhibitors of this intracellular inflammatory machinery.
Table 1.
A list of NLRP3 inflammasome activators and inhibitors
| Activators | Regulators | |||
|---|---|---|---|---|
| Endogenous | Exogenous | Caspase-1 Inhibitors | K+ Efflux Inhibitors | NLRP3 Inflammasome Inhibitors |
| Homocysteine | Lipopolysaccharide | VX-765 | Glybenclamide | Parthenolide |
| Adenosine triphosphate | Asbestos | Ac-YVAD-cmk | Isoliquiritigenin | |
| Uric acid crystals | Silica | Z-VAD-FMK | ||
| Visfatin | Nanoparticles | |||
| Hyaluronan | Adjuvants | |||
| Cholesterol crystals | Nucleic acids | |||
| Amyloid-β | Muramyl dipeptide | |||
| Nigericin | ||||
Mechanisms mediating NLRP3 inflammasome activation
Detection of pathogenic microorganisms and sterile stressors, recruitment of ASC, caspase-1 cleavage and production of inflammatory molecules are main steps directing the pathological path to NLRP3 inflammasome activation. Given that more than 120 substrates were predicted for caspase-1, it is predictable that its maturation or activation will produce a variety of biological responses in addition to production of cytokines. However, IL-1β and IL-18 are well-studied products from the NLRP3 inflammasome. It is now known that the production of various cytokines via activation of caspase-1 triggers the inflammatory response and other cellular activities [53]. In phagocytes, the NLRP3 inflammasome activation follows a tightly regulated intracellular signaling and regulatory process, which include two important steps. Initially, NF-κB is involved which initiates the increased expression of genes encoding for inflammasome formation such as NLRP3, pro-caspase-1, pro-IL-1β and pro-IL-18 [58]. This initial step is referred to as “priming”, which is influenced by a cellular disturbance that activates PRRs. Signal II involves the detection of PAMPS or DAMPs by NLRP3, which promotes the recruitment of the inflammasome components to form the molecular complex [58]. Aggregation of these molecules together leads to the autocleavage of pro-caspase-1 into two subunits, p10 (10 kDa) and p20 (20 kDa), and active caspase-1 is the heterodimer [79]. Considerable evidence has indicated that the regulation of inflammasome activity can be controlled by transcription of NLRP3 via signaling cytokine receptors [31]. Additionally, tight transcriptional regulation through microRNA, miR-223, influences inflammasome activation by manipulating NLRP3 mRNA levels [12, 38]. In some other cells including endothelial cells, podocytes, neurons, epithelial cells and hepatic stellate cells, however, a sustained and small scale inflammasome activation may not require the priming step. Several signaling pathways have been reported to account for activation of the NLRP3 inflammasome. Among them, potassium (K+) efflux, reactive oxygen species (ROS) generation, and cathepsin B leakage into the cytosol due to phagolysosomal rupture are often studied [46]. For example, an intracellular surge of K+ ions into the cellular milieu is capable of triggering NLRP3 inflammasome activation. This model of activation occurs when extracellular ATP interacts with gated cation channels like P2X7R or bacterial toxins to produce pore formation [48, 64]. Researchers have shown experimentally that inhibition of high K+ concentrations diminishes the activation of the inflammasome, although this molecular mechanism has not been characterized in all K+ efflux inflammasome inducers [24, 34, 76]. It has been reported that many inflammasome activators stimulates production of ROS and these activators could be exogenous stimuli such as microbes or endogenously produced or secreted molecules including DAMPs, Hcys and uric acids [87]. Uptake of particulate and crystalline matter is reported to cause disruption of the lysosomal compartment causing cathepsin B release that may lead to NLRP3 inflammasome activation [28, 37, 41]. All these signaling pathways and mechanisms of NLRP3 inflammasome activation are summarized in Figure 1.
Figure 1.
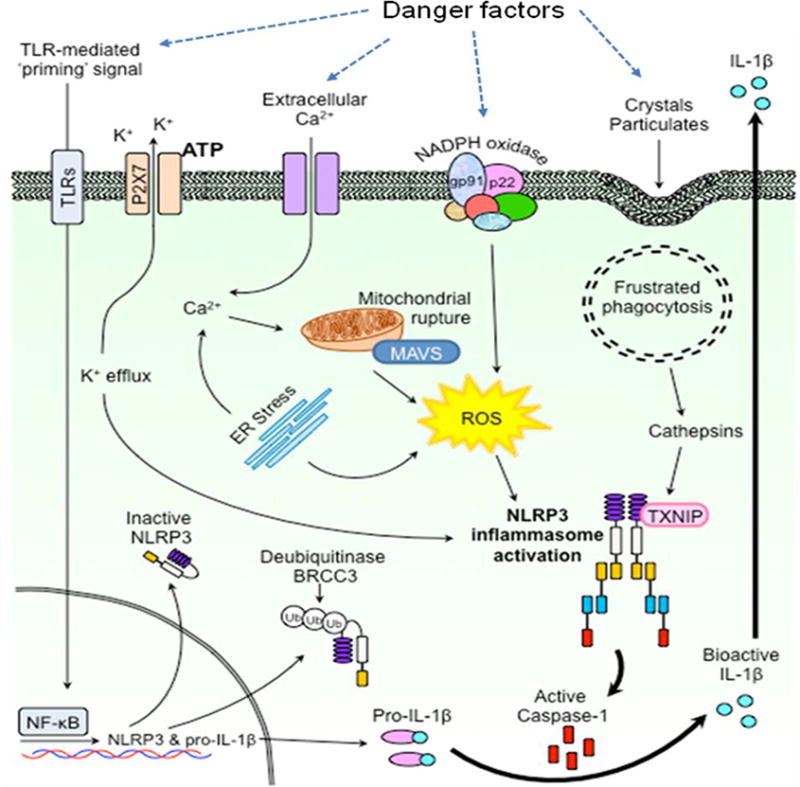
NLRP3 inflammasome formation and activation through different pathways including signal sensing, amplifying and mediating mechanisms.
In recent studies, we have demonstrated that during hyperhomocysteinemia (hHcys) podocyte injury and glomerular sclerosis are consequences of NLRP3 inflammasome activation [1, 3, 97]. We found that this activation is associated with NADPH oxidase (NOX)-derived superoxide (O2−) and related oxidants [2, 3]. Chemical scavengers and pharmacological inhibitors of ROS were used to confirm that elevated plasma Hcys may serve as a DAMP that activates the NLRP3 inflammasome in podocytes, which leads to podocyte dysfunction, glomerular injury, inflammation and ultimate glomerular sclerosis [1]. It has been suggested that DAMPs like Hcys, cholesterol, or visfatin may increase NOX activity via membrane raft clustering to form lipid raft redox signaling platforms in podocytes or glomerular endothelial cells, thereby producing O2−. NLRP3 can sense and monitor intracellular redox changes. If intracellular O2− increases, it dissociates from thioredoxin-interacting protein (TXNIP) and then binds to ASC and forms the NLRP3 inflammasome [1], where IL-1β, IL-18 and HMGB1 are produced. These factors, in particular, IL-1β act to recruit inflammatory cells to the glomeruli, in which O2− and additional cytokines are generated, creating a chronic sterile inflammatory cascade contributing to glomerular injury and sclerosis. Excessive amounts of active caspase-1 and IL-1β or other inflammasome products may reduce the level of podocyte-specific proteins, nephrin and podocin, which will result in slit diaphragm derangement and proteinuria. These inflammasome products may also directly induce podocyte pyroptosis, consequently reducing podocyte numbers and causing foot process effacement [3, 13, 97]. Figure 2 summarizes this new working model for NLRP3 inflammasome activation.
Figure 2.
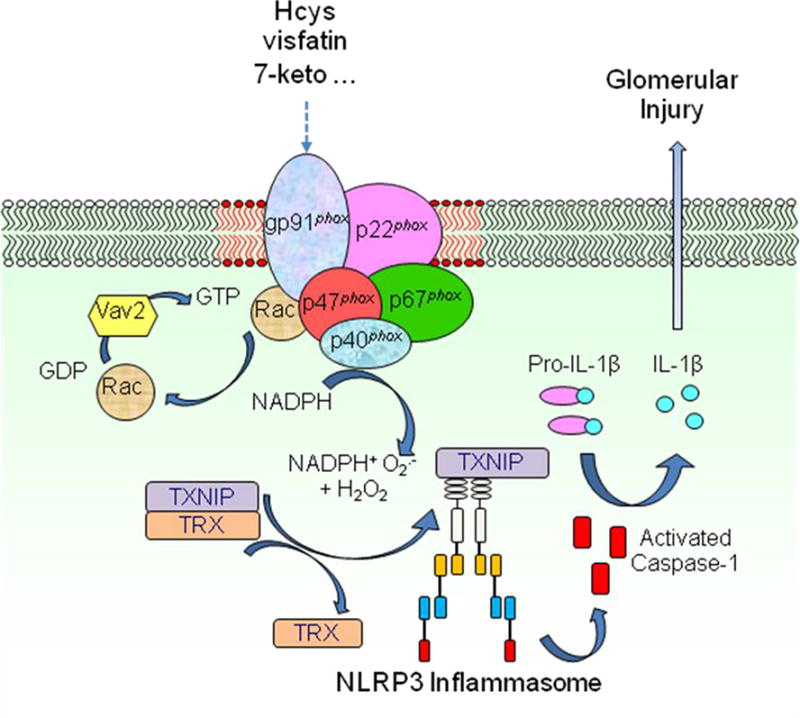
Membrane raft redox signaling platforms mediate activation of NLRP3 inflammasomes in podocytes in association with Rac1-NADPH oxidase activation, ROS production and interactions of NLRP3 with thioredoxin interacting protein.
Effector response of inflammasome activation
Activation of the NLRP3 inflammasome turns on inflammatory responses which may influence the progression to many chronic degenerative disease including chronic kidney diseases by eliciting damaging insults independent of inflammation [60]. NLRP3 inflammasome activation can cause both inflammatory and non-inflammatory effects [22]. In this regard, there is evidence that activated caspase-1 may act on more than 120 substrates and that pyroptosis, enhanced glycolysis and lipid metabolism, altered cell survival and many other direct effects during NLRP3 inflammasome activation may influence cell function and associated metabolism. It is believed that the physiological or pathological role of activated NLRP3 inflammasome extends far beyond inflammation [56].
With respect to the classical pathway, various cellular proteins such as cytokines or chemokines may be produced and secreted during NLRP3 inflammasome activation, which is associated with an internal signal or amino-terminal that direct their translocation from the endoplasmic reticulum (ER) lumen. These factors produced during inflammasome activation are then actively transported to the Golgi complex, packaged into vesicles, fusing with the plasma membrane and excreted out of the cells [59, 86]. Fibroblast growth factor 2 (FGF2), galectin 1 and 3 as well as mature IL-1β and the DAMPs including high mobility group box 1 (HMBG1) have been documented to be released during NLRP3 inflammasome activation [72]. It is these secreted factors that induce inflammatory, cell survival and repair reactions by activating cell surface receptors including FGF receptor-1, the IL receptors and the receptor for advanced glycation end-products (RAGE) [56]. Among these cytokines or chemokines, IL-1β and IL-18 are two common products. Both have extensive biological activities that result in the onset and development of inflammation as well as in the disturbance of cell functions. Moreover, IL-1α, may also contribute in the inflammatory process. IL-1α, is released from necrotic cells and further processing of this interleukin is not necessary because it descends from an active precursor [23]. Interestingly, some recent studies have indicated that almost all inflammasome activators are able to induce co-secretion of IL-1α with other interleukin molecules [35]. However, IL-α release is not directly from the NLRP3 inflammasome activation, but may be dependent upon intermediate reactions after inflammasome activation [58].
Pyroptosis, a specialized form of cell death serves as a non-inflammatory or non-canonical effector mechanism during NLRP3 inflammasome activation. It is caspase-1 mediated and proceeds independently of pro-inflammatory cytokine production. This form of cell death can enhance the immune responses by exposing foreign agents to the immune system’s surveillance [70]. Cytoplasmic swelling, osmotic lysis and release of intracellular components into the outside of the cell are all characteristic of pyroptosis [56]. Although attempts have been established to fill the gaps in knowledge of pyroptosis, characterization of its molecular features still remains under debate. A distinctive characteristic that may contribute to the pathogenesis of organ failure include the abnormal overexpression of collagen leading to fibrosis [7]. With the initial insult often unknown in fibrosis, this form of wound healing can cause high mortality rates and proceed independent of inflammation [7]. In addition to pyroptosis, many other non-canonical effects induced by activation of the NLRP3 inflammasome or consequently enhanced caspase-1 activity such as enhanced glycolysis, derangement of cellular lipid metabolism, disturbance of cell survival programs or altered cell membrane permeability may contribute to the pathogenic role of NLRP3 inflammasome activation.
NLRP3 Inflammasomes in Glomerular Diseases
Growing evidence reveal that pathological progression of many kidney diseases originates from inflammation as a result renal injury. Routes to resolving renal damage involves activation of transmembrane and intracellularly expressed PRRs as well as induction of transcriptional inflammatory mediators that are crucial in the innate immune response and cellular homeostasis [19, 82], in particular, the activation of the NLRP3 inflammasome. Here we highlighted some evidence showing the implications of this inflammasome in the onset or development of several different glomerular diseases.
Chronic glomerulonephritis (GN)
Chronic glomerulonephritis (GN) is referred to as inflammation of the glomeruli, which develops over several years with no or very few symptoms, but causes irreversible kidney damage, ultimately leading to ESRD. Infiltrating mononuclear phagocytes generate canonical NLRP3 inflammasome signaling, and the activation of NLRP3 inflammasome also occurred in some non-immune glomerular cells such as podocytes [15, 73, 84]. In addition, there is evidence that in a rodent model of nephrotoxic serum nephritis (NTN) interleukin (IL)-1 and tumor necrosis factor (TNF) are critical in promoting glomerular injury by interaction with the IL-1 receptor 1 (IL-1R1) [6]. Recent studies have demonstrated that inflammasome signaling components were upregulated during NTN, in particular, in renal dendritic cells, which led to increased production of mature IL-1β, suggesting the NLRP3 inflammasome was being formed and activated during NTN. Glomerular injury, renal leukocyte infiltration, and T-cell activation were significantly attenuated in Nlrp3 and Asc knockout mice having NTN. Interestingly, reduced secretion of active IL-1β was only observed in Asc knockout mice, but not in Nlrp3-deficient mice, suggesting another molecular mechanism to be involved independent of NLRP3 inflammasome activation. Additionally, NLRP3 may have non-canonical early inflammatory effects in NTN, which may be associated with glomerular release of the inflammatory protein, high-mobility group box 1 (HMGB1) in a NLRP3-mediated manner during glomerulonephritis. This HMGB1 is considered as one of the important mediators to promote the non-canonical functions of NLRP3/ASC inflammasome activation [6].
In addition, acute and chronic inflammatory responses are an important pathogenic mechanism of lupus nephritis (LN). Participation of the NLRP3 inflammasome may play a role in the progression of LN [98]. Recent studies have demonstrated that a selective inhibitor of NLRP3 inflammasome activity, Bay11-7082, prevented assembling and activation of the inflammasome, decreased proteinuria, blood urea nitrogen, and glomerular damage during LN. Accompanied with these beneficial effects, Bay11-7082 treatment also decreased renal immune complex deposition and the level of IL-1β, TNF-α and chemokine (C-C Motif) ligand 2 (CCL2) levels as well as infiltration of macrophages. However, some other studies have shown that active caspase-1 and renal secretion of pro-inflammatory cytokines such as IL-1β are not significant in acute heterologous NTN and other glomerulonephritis [62]. Obviously, more extensive research is needed to establish the contribution of this inflammasome in GN and other related molecular mechanisms.
Hyperhomocysteinemic nephropathy
Hyperhomocysteinemia (hHcys) is characterized as a homocysteine level that exceeds 15 μmol/L in the plasma of patients. Elevated homocysteine (Hcys) has been linked to the progression of many chronic metabolic diseases including hypertension, peripheral vascular disease, Alzheimer’s disease, diabetes and atherosclerosis [78]. In regard to renal disease, hHcys is considered an important pathogenic factor in the progression of ESRD, additionally it has been implicated in the development of cardiovascular complications related to ESRD [96]. Increasing evidence in studies using various animal models have confirmed that glomerular injury and ultimate sclerosis associated with chronic hHcys proceeds in a manner independent of hypertension. Accumulation of Hcys or hHcys in the blood induces pathological alterations in the glomeruli including extracellular matrix accumulation and podocyte injury. The inability of this toxic compound to be properly cleared or degraded from the body eventually leads to compromised renal function and glomerulosclerosis [45, 95]. Although the mechanism remains unclear on how Hcys causes cellular injury and sclerotic changes in many organs and tissues, some studies have revealed that inflammatory cytokines are upregulated during hHcys, suggesting the inflammasome may play a crucial role in these processes [20, 91]. Under some pathological conditions, Hcys-induced glomerular injury was found to activate local inflammatory responses by enhancing the production of monocyte chemoattractant protein 1 in glomerular mesangial cells and tubular epithelial cells [18]. However, inhibition of the inflammatory process significantly protected the kidney against hHcys-associated damage [18].
However, it remains poorly understood how the inflammation in glomeruli during hHcys is activated and whether this is associated with NLRP3 inflammasome formation and activation. In our studies, we determined that treatment of glomerular epithelial cells (podocytes) with Hcys induced the formation and activation of the NLRP3 inflammasome, contributing to the development of glomerular injury or sclerosis during hHcys. Moreover, there is evidence that genetic manipulation of the Nlrp3 gene resulted in protection of podocytes and glomeruli against injury or sclerosis as observed by decreased urinary protein excretion, glomerular damage and diminished expression of the podocyte-specific damage marker, desmin in a mouse model of hHcys. The Nlrp3 gene has been well documented as an essential component of NLRP3 inflammasomes, which serves as a sensor in the kidney for monitoring redox changes. This sensing function of NLRP3 is confirmed to be mediated by its association and dissociation with thioredoxin-interacting protein (TXNIP) [1].
Diabetic nephropathy
Diabetic nephropathy (DN) is the leading cause of end stage renal disease (ESRD) and remains a clinical concern due to high mortality and morbidity rates. Poor management of diabetes contributes to the loss of renal function and aside from the gradual changes in the diabetic kidney including thickening of the basement membrane, mesangial expansion, proteinuria and renal fibrosis. DN is different from other types of glomerular diseases that result in ESRD, it is generally categorized as a non-inflammatory glomerular disease. However, genome-wide transcriptome analysis were conducted and revealed that several inflammatory signaling pathways are present during DN. Although some reports have shown that hyperglycemia-induced cell death and the influx of immune cells to be an important inflammatory response in glomeruli, the precise mechanisms by which inflammation is activated during DN remains to be elucidated [77].
Evolving data now suggest that infiltration of inflammatory cells is crucial in the pathogenesis of DN [10]. Interleukin (IL)-1β and IL-18 secreted from immune cells and glomerular resident cells such as podocytes, endothelial cells or mesangial cells may possibly promote DN [81, 84, 97]. Initial studies from Shahzad et al [81] revealed that diabetic mice have upregulated expression of inflammasome molecules and pro-inflammatory cytokines in circulation when compared to their nondiabetic counterparts. It was demonstrated that morphological and progressive functional changes were also observed in these mice. When the authors utilized the db/db mouse model of diabetes, they discovered that after transplantation of Nlrp3 and caspase-1 deficient mice bone marrow, diabetic mice still experienced the same extent of damage as control db/db mice, suggesting that blocking of myeloid-lineage immune cells is insufficient in preventing the progression to DN. The production of mitochondrial reactive oxygen species has been shown to initiate NLRP3 inflammasome activation in diabetic conditions, further establishing the causative link between NLRP3 inflammasome activation and DN. Inhibition of NLRP3 or caspase-1 in the kidney led to protective outcomes seen by inactivation of the inflammasome. This provides a strong foundation in how the NLRP3 inflammasome serves as an important mediator for pro-inflammatory cytokine production by inducing caspase-1 activation and how inhibition of this complex may serve as a promising approach to treat DN. It has been reported that high glucose (HG) treatment in mice induced NADPH oxidase activity triggering NLRP3 inflammasome activation in glomerular podocytes and leading to podocyte injury during DN, however regulating thioredoxin-interacting protein (TXNIP) by shRNA and its inhibitors blocked this DN-induced inflammasome activation [32]. Additionally, in diabetic patients inflammasome activation was detected in other glomerular resident cells such as those localized in the endothelial.
Deletion of mouse Nlrp3 gene, antagonism of IL-1R and inhibition of mitochondrial ROS production all were shown to protect or even reverse DN in mice, suggesting that targeting the NLRP3 inflammasome in DN may serve as a beneficial strategy for treatment [81].
Glomerular injury and sclerosis in obesity
Chronic kidney disease (CKD) is now considered as one of the strongest risk factors for the morbidity and mortality in obese patients, with the prevalence of obesity increasing worldwide [14, 36]. Previous studies have identified that visceral fat generates bioactive substances that contribute to the pathophysiologic and structural changes leading to obesity-associated glomerular injury [11]. Mechanistically, obesity-induced glomerular sclerosis and ultimate ESRD is involved in chronic inflammation, abnormal vascular remodeling, rise in renal plasma flow, hyperfiltration and renal lipotoxicity [43].
Indeed, most recently, we have shown that the formation and activation of the NLRP3 inflammasome is implicated in the development of obesity and associated chronic glomerular injury and serves as an important initiating mechanism to activate glomerular inflammation leading to injury in obese mice [13]. Inhibition of the Asc gene significantly protected mice from high fat diet (HFD)-induced obesity, glomerular injury and podocyte damage. The associated molecular mechanism of HFD-induced inflammasome activation could be possibly due to increased production of fatty acid metabolites ceramide and palmitate, since their abundance in adipose tissue positively correlates with the development of obesity and type 2 diabetes. Vandanmagsar et al [89] showed that adding ceramide to adipose tissue explants leads to NLRP3-dependent caspase-1 activation and IL-1β production, suggesting that ceramide acts a danger signal to stimulate the NLRP3 inflammasome. In addition, we have shown that a HFD increased the glomerular ceramide production due to the activation of acid sphingomyelinase, a ceramide producing enzyme. The increased ceramide production induced the formation and activation of NLRP3 inflammasome in glomeruli and contributed to the obesity-induced glomerular injury. These results indicated that manipulation of the NLRP3 inflammasome formation or activation might be a possible strategy for treating obesity and related glomerular dysfunction and sclerosis.
Therapeutic Potential of Targeting the NLRP3 Inflammasome in Chronic Glomerular Diseases
The prolonged period of clinical silence in chronic glomerular disease or CKD leads to irreversible pathological damage including accumulation of extracellular matrix (ECM) proteins, proteinuria, renal hypertrophy, glomerular basement membrane thickening, mesangial expansion and glomerular fibrosis. It has been well known that early detection of these alterations and abnormalities in the impaired kidneys is necessary to facilitate therapeutics that can improve clinical outcomes. Management of risk factors such as hypertension, increase blood glucose level and albuminuria has been vital in slowing the progression to ESRD. Although current treatment targets are used in clinical practice, including combination therapies of angiotensin converting enzyme inhibitors (ACEi), angiotensin receptor blockers (ARB), mineralocorticoid receptor antagonists and statins [55], the morbidity and mortality of CKD patients still remains high. It is obvious that identification of new therapeutic targets and development of new strategies for treatment of ESRD are imperative there is a need to search for new pathways.
There is growing interest in administering anti-inflammatory therapies to halt renal and cardiovascular functional loss, for example, targeting oxidative stress and inflammatory responses. However, these therapies are met with a bit of skepticism and may only elicit partial improvements in the pathogenic process of CKD characterized by glomerular sclerosis. The root of glomerular dysfunction and other renal diseases previously associated with inflammation may potentially be eliminated by inhibiting NLRP3 inflammasome activation. Convincing evidence has shown that by blocking this complex there is a reduction in tissue and cellular inflammation, in addition the non-inflammatory or non-canonical damaging effects on cell function and metabolism during glomerular diseases are targeted.
In this regard, treatment with IL-1β antibodies has proven effective in patients with cyropyrin-associated periodic syndromes (CAPS) [66]. Additionally, activation of the NLRP3 inflammasome following MSU and other crystal-induced pathologies translate into the disease manifestation of gout or pseudogout [65]. There is also evidence that antagonism of the IL-1 signaling pathway immediately improved the clinical outcome of patients with these arthropathic diseases [67, 83]. However, whether crystal-associated nephropathies have the similar mechanisms has yet to be established. To our knowledge, approved inhibitors of IL-1β are commercially available for treatment of various inflammasome-mediated autoinflammatory diseases; however, they produce adverse effects at the injection site [30, 40]. Anakinra, a naturally occurring recombinant IL-1 receptor antagonist (IL-1Ra) was reported to act as a competitive inhibitor of IL-1 signaling [69]. A pilot clinical study reported that administration of anakinra improved the inflammatory response in hemodialysis patients [42]. In addition, glyburide, commonly used to treat type II diabetes, was found to block chloride and potassium channels in pancreatic β cells to regulate insulin release [9]. This compound is able to inhibit IL-1β release upon stimulation with LPS in human monocytes [75], but it does not block capsase-1 activation unless Nlrp3, NF-κB or P2X7 genes are knocked out in mice [50, 54, 57, 63]. It is expected that more therapeutic strategies will be forthcoming, which include the use of NLRP3 inhibitors, the P2X7 receptor blockers, IL-1 and IL-18 receptor blockers, caspase-1 inhibitors, alpha-1 antitrypsin, adenosine A2B receptor blockers, miRNAs, H2S donor Na2S, lysosome stabilizer, cathepsin-B inhibitors, and milk fat globule EGF-8 as an endogenous inhibitor of inflammasome-induced IL-1β production. These potential therapeutics may target different stages of NLRP3 inflammasome formation and activation, which may be selected for the use in prevention or treatment of ESRD and associated glomerular diseases.
Conclusion/Perspectives
There is indeed increasing evidence for an association between glomerular diseases and NLRP3 inflammasome activation, which leads to caspase-1-mediated IL-1β/IL-18 production. More knowledge is needed in this area to further assess the underlying relationship between NLRP3 inflammasome activation and the decline of glomerular function in various chronic kidney diseases, in particular, to define the temperospatial contribution of the activation of inflammasomes to the onset or development of glomerular disease and ultimate ESRD. Moreover, recognition and clarification of the non-canonical effects of NLRP3 inflammasome activation in glomeruli and alternative pathways to activate this inflammasome may be particularly interesting and important, because it may result in a combination of injurious actions independent of typical inflammation, which may cause direct damage to glomerular cells, interfere with synthesis of cell-specific proteins, increase cell membrane permeability and induce cell pyroptosis. These direct effects of NLRP3 inflammasome activation may suggest that targeting of the NLRP3 inflammasome may serve as a new therapeutic strategy for treatment and prevention of glomerular sclerosis and ESRD, which may be better than the approaches that just target the inflammatory pathways.
Acknowledgments
Many studies cited in this review from our laboratory were supported by grants HL57244, DK54927, HL091464 (to P. L.) and DK104031 (to K. M. B.) from the National Institutes of Health.
Abbreviations
- AIM2
absent in melanoma 2
- ASC
apoptosis-associated speck-like protein containing a CARD
- CARD
caspase recruitment domain
- CKD
chronic kidney disease
- CLRs
C-type lectin receptors
- DAMPs
damage-associated molecular patterns
- ESRD
end-stage renal disease
- ER
endoplasmic reticulum
- FCU
familial cold urticarial
- Hcys
homocysteine
- HFD
high fat diet
- hHcys
hyperhomocysteinemia
- HIN-200
hemopoetic interferon-inducible nuclear antigen with 200 repeats
- HMGB1
high mobility group box-1
- IL-1β
interleukin-1β
- IL-18
interlukin-18
- IPAF
interleukin-1β-converting enzyme protease-activating factor
- LOXs
lipoxygenases
- LR
lipid raft
- MDP
muramyl dipeptide
- miRNAs
microRNAs
- MSU
monosodium urate
- MWS
Muckle-Wells syndrome
- NLRP3
NOD-like receptor containing pyrin domain 3
- NLRs
nucleotide-binding domain leucine-rich repeats
- PAMPs
pathogen-associated molecular patterns
- PRRs
pattern recognition receptors
- RIGs-I
retinoic inducible gene-I
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- TLRs
toll-like receptors
- TXNIP
thioredoxin-interacting protein
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abais JM, et al. Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. J Biol Chem. 2014;289(39):27159–68. doi: 10.1074/jbc.M114.567537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abais JM, et al. Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free Radic Biol Med. 2014;67:211–20. doi: 10.1016/j.freeradbiomed.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abais JM, et al. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid Redox Signal. 2013;18(13):1537–48. doi: 10.1089/ars.2012.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aganna E, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 2002;46(9):2445–52. doi: 10.1002/art.10509. [DOI] [PubMed] [Google Scholar]
- 5.Agostini L, et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20(3):319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 6.Andersen K, et al. The NLRP3/ASC inflammasome promotes T-cell-dependent immune complex glomerulonephritis by canonical and noncanonical mechanisms. Kidney Int. 2014;86(5):965–78. doi: 10.1038/ki.2014.161. [DOI] [PubMed] [Google Scholar]
- 7.Artlett CM, Thacker JD. Molecular activation of the NLRP3 Inflammasome in fibrosis: common threads linking divergent fibrogenic diseases. Antioxid Redox Signal. 2015;22(13):1162–75. doi: 10.1089/ars.2014.6148. [DOI] [PubMed] [Google Scholar]
- 8.Asgari E, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013;122(20):3473–81. doi: 10.1182/blood-2013-05-502229. [DOI] [PubMed] [Google Scholar]
- 9.Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115(8):2047–58. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Awad AS, et al. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am J Physiol Renal Physiol. 2011;301(6):F1358–66. doi: 10.1152/ajprenal.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Axelsson J, Heimburger O, Stenvinkel P. Adipose tissue and inflammation in chronic kidney disease. Contrib Nephrol. 2006;151:165–74. doi: 10.1159/000095327. [DOI] [PubMed] [Google Scholar]
- 12.Bauernfeind F, et al. NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol. 2012;189(8):4175–81. doi: 10.4049/jimmunol.1201516. [DOI] [PubMed] [Google Scholar]
- 13.Boini KM, et al. Activation of inflammasomes in podocyte injury of mice on the high fat diet: Effects of ASC gene deletion and silencing. Biochim Biophys Acta. 2014;1843(5):836–45. doi: 10.1016/j.bbamcr.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boini KM, et al. Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim Biophys Acta. 2010;1801(12):1294–304. doi: 10.1016/j.bbalip.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boswell JM, et al. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J Immunol. 1988;141(9):3050–4. [PubMed] [Google Scholar]
- 16.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38(2):240–4. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 17.Cassel SL, Sutterwala FS. Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur J Immunol. 2010;40(3):607–11. doi: 10.1002/eji.200940207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung GT, Siow YL, O K. Homocysteine stimulates monocyte chemoattractant protein-1 expression in mesangial cells via NF-kappaB activation. Can J Physiol Pharmacol. 2008;86(3):88–96. doi: 10.1139/y08-002. [DOI] [PubMed] [Google Scholar]
- 19.Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27(8):352–7. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 20.de la Sierra A, Larrousse M. Endothelial dysfunction is associated with increased levels of biomarkers in essential hypertension. J Hum Hypertens. 2010;24(6):373–9. doi: 10.1038/jhh.2009.91. [DOI] [PubMed] [Google Scholar]
- 21.De Nardo D, De Nardo CM, Latz E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol. 2014;184(1):42–54. doi: 10.1016/j.ajpath.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denes A, Lopez-Castejon G, Brough D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012;3:e338. doi: 10.1038/cddis.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 24.Dostert C, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One. 2009;4(8):e6510. doi: 10.1371/journal.pone.0006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dostert C, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duncan JA, et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009;182(10):6460–9. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eisenbarth SC, et al. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453(7198):1122–6. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faustin B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25(5):713–24. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 30.Fleishmann RM. Safety of anakinra, a recombinant interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis and comparison to anti-TNF-alpha agents. Clin Exp Rheumatol. 2002;20(5 Suppl 27):S35–41. [PubMed] [Google Scholar]
- 31.Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol. 2009;183(2):792–6. doi: 10.4049/jimmunol.0900173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao P, et al. NADPH oxidase-induced NALP3 inflammasome activation is driven by thioredoxin-interacting protein which contributes to podocyte injury in hyperglycemia. J Diabetes Res. 2015;2015:504761. doi: 10.1155/2015/504761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghiringhelli F, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–8. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 34.Gross O, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459(7245):433–6. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 35.Gross O, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36(3):388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 36.Hall JE, et al. Is obesity a major cause of chronic kidney disease? Adv Ren Replace Ther. 2004;11(1):41–54. doi: 10.1053/j.arrt.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 37.Halle A, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haneklaus M, et al. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J Immunol. 2012;189(8):3795–9. doi: 10.4049/jimmunol.1200312. [DOI] [PubMed] [Google Scholar]
- 39.Heneka MT, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–8. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffman HM, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58(8):2443–52. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- 41.Hornung V, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9(8):847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hung AM, et al. IL-1beta receptor antagonist reduces inflammation in hemodialysis patients. J Am Soc Nephrol. 2011;22(3):437–42. doi: 10.1681/ASN.2010070760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hunley TE, Ma LJ, Kon V. Scope and mechanisms of obesity-related renal disease. Curr Opin Nephrol Hypertens. 2010;19(3):227–34. doi: 10.1097/MNH.0b013e3283374c09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ichinohe T, et al. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ingram AJ, et al. Activation of mesangial cell MAPK in response to homocysteine. Kidney Int. 2004;66(2):733–45. doi: 10.1111/j.1523-1755.2004.00795.x. [DOI] [PubMed] [Google Scholar]
- 46.Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol. 2010;30(5):628–31. doi: 10.1007/s10875-010-9440-3. [DOI] [PubMed] [Google Scholar]
- 47.Joly S, et al. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J Immunol. 2009;183(6):3578–81. doi: 10.4049/jimmunol.0901323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kahlenberg JM, Dubyak GR. Differing caspase-1 activation states in monocyte versus macrophage models of IL-1beta processing and release. J Leukoc Biol. 2004;76(3):676–84. doi: 10.1189/jlb.0404221. [DOI] [PubMed] [Google Scholar]
- 49.Kasimsetty SG, et al. Regulation of TLR2 and NLRP3 in primary murine renal tubular epithelial cells. Nephron Clin Pract. 2014;127(1-4):119–23. doi: 10.1159/000363208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kerur N, et al. TLR-independent and P2X7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Invest Ophthalmol Vis Sci. 2013;54(12):7395–401. doi: 10.1167/iovs.13-12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SM, et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am J Physiol Renal Physiol. 2015;308(9):F993–F1003. doi: 10.1152/ajprenal.00637.2014. [DOI] [PubMed] [Google Scholar]
- 52.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477(7366):592–5. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krishnan SM, et al. IL-1beta and IL-18: inflammatory markers or mediators of hypertension? Br J Pharmacol. 2014;171(24):5589–602. doi: 10.1111/bph.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuipers MT, et al. Ventilator-induced lung injury is mediated by the NLRP3 inflammasome. Anesthesiology. 2012;116(5):1104–15. doi: 10.1097/ALN.0b013e3182518bc0. [DOI] [PubMed] [Google Scholar]
- 55.Lambers Heerspink HJ, de Zeeuw D. Novel drugs and intervention strategies for the treatment of chronic kidney disease. Br J Clin Pharmacol. 2013;76(4):536–50. doi: 10.1111/bcp.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol. 2011;11(3):213–20. doi: 10.1038/nri2936. [DOI] [PubMed] [Google Scholar]
- 57.Lamkanfi M, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee MC, et al. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307. [DOI] [PubMed] [Google Scholar]
- 60.Li PL. Cardiovascular pathobiology of inflammasomes: inflammatory machinery and beyond. Antioxid Redox Signal. 2015;22(13):1079–83. doi: 10.1089/ars.2015.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, et al. Activation of Nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS One. 2014;9(1):e87552. doi: 10.1371/journal.pone.0087552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lorenz G, Darisipudi MN, Anders HJ. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol Dial Transplant. 2014;29(1):41–8. doi: 10.1093/ndt/gft332. [DOI] [PubMed] [Google Scholar]
- 63.Lottaz D, Beleznay Z, Bickel M. Inhibition of ATP-binding cassette transporter downregulates interleukin-1beta-mediated autocrine activation of human dermal fibroblasts. J Invest Dermatol. 2001;117(4):871–6. doi: 10.1046/j.0022-202x.2001.01451.x. [DOI] [PubMed] [Google Scholar]
- 64.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 65.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 66.Martinon F, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 67.Masters SL, et al. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGonagle D, et al. Successful treatment of resistant pseudogout with anakinra. Arthritis Rheum. 2008;58(2):631–3. doi: 10.1002/art.23119. [DOI] [PubMed] [Google Scholar]
- 69.Miao EA, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107(7):3076–80. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mitroulis I, Skendros P, Ritis K. Targeting IL-1beta in disease; the expanding role of NLRP3 inflammasome. Eur J Intern Med. 2010;21(3):157–63. doi: 10.1016/j.ejim.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 71.Monack DM, Detweiler CS, Falkow S. Salmonella pathogenicity island 2-dependent macrophage death is mediated in part by the host cysteine protease caspase-1. Cell Microbiol. 2001;3(12):825–37. doi: 10.1046/j.1462-5822.2001.00162.x. [DOI] [PubMed] [Google Scholar]
- 72.Muruve DA, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452(7183):103–7. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 73.Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009;10(2):148–55. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- 74.Niemir ZI, et al. Podocytes are the major source of IL-1 alpha and IL-1 beta in human glomerulonephritides. Kidney Int. 1997;52(2):393–403. doi: 10.1038/ki.1997.346. [DOI] [PubMed] [Google Scholar]
- 75.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25(21):5071–82. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perregaux DG, et al. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J Pharmacol Exp Ther. 2001;299(1):187–97. [PubMed] [Google Scholar]
- 77.Petrilli V, et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14(9):1583–9. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 78.Reidy K, et al. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124(6):2333–40. doi: 10.1172/JCI72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Robinson K, et al. Hyperhomocysteinemia confers an independent increased risk of atherosclerosis in end-stage renal disease and is closely linked to plasma folate and pyridoxine concentrations. Circulation. 1996;94(11):2743–8. doi: 10.1161/01.cir.94.11.2743. [DOI] [PubMed] [Google Scholar]
- 80.Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327(5963):296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 81.Segelmark M, Hellmark T. Autoimmune kidney diseases. Autoimmun Rev. 2010;9(5):A366–71. doi: 10.1016/j.autrev.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 82.Shahzad K, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87(1):74–84. doi: 10.1038/ki.2014.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 84.Terkeltaub R, et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis. 2009;68(10):1613–7. doi: 10.1136/ard.2009.108936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tesch GH, et al. Intrinsic renal cells are the major source of interleukin-1 beta synthesis in normal and diseased rat kidney. Nephrol Dial Transplant. 1997;12(6):1109–15. doi: 10.1093/ndt/12.6.1109. [DOI] [PubMed] [Google Scholar]
- 86.Thomas PG, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30(4):566–75. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annu Rev Cell Dev Biol. 2003;19:649–76. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- 88.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10(3):210–5. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 89.Turner CM, et al. Is the inflammasome a potential therapeutic target in renal disease? BMC Nephrol. 2014;15:21. doi: 10.1186/1471-2369-15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vandanmagsar B, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–88. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang J, et al. Involvement of endoplasmic reticulum stress in angiotensin II-induced NLRP3 inflammasome activation in human renal proximal tubular cells in vitro. Acta Pharmacol Sin. 2015;36(7):821–30. doi: 10.1038/aps.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Woo CW, Siow YL, O K. Homocysteine induces monocyte chemoattractant protein-1 expression in hepatocytes mediated via activator protein-1 activation. J Biol Chem. 2008;283(3):1282–92. doi: 10.1074/jbc.M707886200. [DOI] [PubMed] [Google Scholar]
- 93.Xia M, et al. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol. 2014 doi: 10.1016/j.ajpath.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xia M, et al. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol. 2014;184(5):1617–28. doi: 10.1016/j.ajpath.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yamasaki K, et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem. 2009;284(19):12762–71. doi: 10.1074/jbc.M806084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yi F, et al. Contribution of guanine nucleotide exchange factor Vav2 to hyperhomocysteinemic glomerulosclerosis in rats. Hypertension. 2009;53(1):90–6. doi: 10.1161/HYPERTENSIONAHA.108.115675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yi F, et al. Inhibition of ceramide-redox signaling pathway blocks glomerular injury in hyperhomocysteinemic rats. Kidney Int. 2006;70(1):88–96. doi: 10.1038/sj.ki.5001517. [DOI] [PubMed] [Google Scholar]
- 98.Zhang C, et al. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension. 2012;60(1):154–62. doi: 10.1161/HYPERTENSIONAHA.111.189688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao J, et al. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-kappaB activation. Int Immunopharmacol. 2013;17(1):116–22. doi: 10.1016/j.intimp.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 100.Zhao Y, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477(7366):596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 101.Zhou R, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 102.Zhuang Y, et al. Albumin impairs renal tubular tight junctions via targeting the NLRP3 inflammasome. Am J Physiol Renal Physiol. 2015;308(9):F1012–9. doi: 10.1152/ajprenal.00509.2014. [DOI] [PubMed] [Google Scholar]