

Abstract
Cutaneous melanoma (CM) is considered as a steroid hormone-related malignancy. However, few studies have evaluated the roles of genetic variants encoding steroid hormone receptor genes and their related regulators (SHR-related genes) in CM-specific survival (CMSS). Here, we performed a pathway-based analysis to evaluate genetic variants of 191 SHR-related genes in 858 CMSS patients using a dataset from a genome-wide association study (GWAS) from The University of Texas MD Anderson Cancer Center (MDACC), and then validated the results in an additional dataset of 409 patients from the Harvard GWAS. Using multivariate Cox proportional hazards regression analysis, we identified three independent SNPs (RORA rs782917 G>A, RORA rs17204952 C>T and DNMT1 rs7253062 G>A) as predictors of CMSS, with a variant-allele attributed hazards ratio (HR) and 95% confidence interval of 1.62 (1.25-2.09), 1.60 (1.20-2.13) and 1.52 (1.20-1.94), respectively. Combined analysis of risk genotypes of these three SNPs revealed a decreased CMSS in a dose-response manner as the number of risk genotypes increased (Ptrend < 0.001); however, no improvement in the prediction model was observed (area under the curve [AUC] = 79.6% to 80.8%, P = 0.656), when these risk genotypes were added to the model containing clinical variables. Our findings suggest that genetic variants of RORA and DNMT1 may be promising biomarkers for CMSS, but these results needed to be validated in future larger studies.
Keywords: cutaneous melanoma (CM), genome-wide association study (GWAS), single-nucleotide polymorphism (SNP), steroid hormone receptor, cutaneous melanoma-specific survival (CMSS)
Introduction
Cutaneous melanoma (CM) is the most lethal malignant tumor of the skin in the United States. In 2017, it is estimated that approximately 87,110 new cases of CM will be diagnosed in the United States, including: 52,170 males and 34,940 females. Of those diagnosed 9,730 individuals will die of this disease, including 6,380 males and 3,350 females1. Overall, the CM incidence is about 60% higher in males than in females, while females have an overall significant survival advantage than males2.
A number of clinicopathological variables have been reported to be associated with CM survival, such as tumor stage, Breslow thickness and ulceration; but the ability to accurately predict patient prognosis is still imperfect3. Sex differences in CM survival partly reflect the more common presence of unfavorable prognostic indicators in men. Interestingly, however, this sex difference disappears after the age of 65. Younger patients (<65) have a more favorable prognosis than older patients (≥65), a difference more prominent in women4. It has been reported that women aged 40-55 had a 7.55-fold of risk for pregnancy-associated CM than women aged 15-245. Currently, most of published studies showed a cumulative dose-dependent increased risk of CM with the use of estrogens, oral contraceptives and hormonal replacement therapy6, and the risk of premenopausal melanoma may be increased among women who are current oral contraceptive users, particularly among those with longer durations of use7. Women with a teenage acne history had a higher level of circulating testosterones, while men with a history of prostate cancer have been found to have an increased CM risk8, 9.
For these reasons, associations between steroid hormone receptors including estrogen receptors (ER), androgen receptors (AR) and progesterone receptors (PR) and CM risk have frequently been investigated. It has been shown that AR+/ER- human melanoma cell lines proliferate after incubation with estrogen10. The amount of ERs in the skin declines with age in women, whereas the positive feedback from estradiol decreases with decreasing plasma estradiol levels in the perimenopausal period11. In addition, clinical studies have showed that ERβ has been reported as a predominant ER subtype in melanoma as a most important prognostic factor, and its expression negatively correlate with melanoma growth and progression2. Furthermore, the loss of the ERβ protein in melanoma is directly proportional to Breslow thickness12. ERβ was more frequently expressed in the melanomas of women [both pregnant and non-pregnant] than in those of men13. Investigators have reported that progesterone can inhibit human melanoma cell growth in vitro14, while testosterone increased cell proliferation and reduced tyrosinase activity to increase melanoma tumor growth in a dose-dependent manner both in vivo and in vitro15. Furthermore, using the Xiphophorus fish melanoma model, following exposure to ultraviolet radiation (UV), levels of androgen rose while levels of estrogen dropped. Steroid hormone receptors may play an important role in CM development and progression. Thus, identification of genetic variants in steroid hormone receptor genes through hypothesis-based gene-set analysis, may reveal biomarkers that predict survival of CM patients, leading to novel cancer hormone therapies.
Materials and Methods
Study populations
All the patients in the two studies below provided a written informed consent under an Institutional Review Board-approved protocol.
The MDACC GWAS discovery dataset included 858 non-Hispanic white patients with CM, who were recruited between March 1993 and August 200816. All patients were followed using standardized methods and guidelines17. The GWAS dataset for genotype and clinical information, including: age, sex, Breslow thickness, metastasis, ulceration of tumor, mitotic rate and survival outcome, are available on dbGaP (accession: phs000187.v1.p1)18. Briefly, genomic DNA extracted from melanoma patient blood samples was genotyped with Illumina HumanOmni-Quad_v1_0_B array using the National Center for Biotechnology Information (NCBI) Database18, 19. Genome-wide imputation (imputation quality r2 ≥ 0.8) was conducted with the MACH software based on the 1000 Genomes Project CEU population (March 2010 release)20.
The Harvard GWAS replication dataset included 409 non-Hispanic white patients with invasive CM. The Harvard study consisted of CM patients from two cohort studies: Nurses’ Health Study (NHS) and Health Professionals Follow-up Study (HPFS). The GWAS dataset for genotypes and clinical information about age, sex, survival outcome and genotype data were available. Genotyping was performed using the Illumina HumanHap610 array. Genome-wide imputation (imputation quality r2 ≥ 0.8) was also performed using the MACH software based on the 1000 Genomes Project CEU population (March 2012 release)21, 22.
There were some differences in age and sex distributions between the two GWAS datasets. The proportion of young patients (≤ 50 years) was 43.24% in the MDACC dataset and 17.60% in the Harvard dataset. Also, the percentage of female participants was 42.19% and 66.26% in the MDACC and Harvard datasets respectively.
Gene and SNP extraction
The SHR-related genes were selected from the Molecular Signatures Database of GSEA website (http://software.broadinstitute.org/gsea/msigdb/search.jsp)23. Genotyped and imputed SNPs within these genes and their 2-kb flanking regions were extracted from the MDACC GWAS dataset following the outlined quality control criteria: (1) a minor allele frequency (MAF) ≥ 0.05, (2) a genotyping rate > 95%, and (3) Hardy-Weinberg equilibrium (HWE) P value > 1×10−5. We performed the linkage disequilibrium (LD) analysis by using HaploView 4.2 according to 373 Europeans from the 1000 Genomes Project21; pairwise r2 ≥ 0.8 was considered in high LD. Online bioinformatics tools were used to predict functions of the SNPs with a RegulomeDB24 score no more than 5 or a confirmatory functional prediction by SNPinfo and F-SNP25.
Statistical analysis
CMSS was the primary end-point used in the analysis, which was defined by the time of diagnosis to the last follow-up time or the CM-related death time. In the MDACC study, tumor stages were collapsed into two groups of stages I/II and stages III/IV, according to the presence or absence of regional/distant metastasis. Cox proportional hazards regression analysis was performed with adjustment for age, sex, Breslow thickness, tumor stage, ulceration and mitotic rate (in an additive genetic model). We estimated associations between SNPs in the SHR-related genes and CMSS by calculating hazards ratio (HR) and its 95% confidence interval (CI) by using the GenABEL package of R software. In the Harvard GWAS dataset, only age and sex were available for further analysis and adjusted for in the multivariate Cox regression analysis. The false-positive report probability (FPRP) method with a cut-off value of 0.20 was used for multiple testing corrections. FPRP was chosen, because the majority of SNPs included in the analysis were imputed and thus more or less in LD with other SNPs under investigation. The high prior probability of 0.20 could increase the possibility of identifying the significant SNPs through further validation. FPRP was calculated based on three factors: the observed P value, the prior probability of a true association of the tested genetic variant with a disease and the statistical power of the test. It is statistically less strict but more reasonable for the corrections among imputed SNPs in this hypothesis-based gene-set analysis different from the whole GWAS analysis, although it is likely to increases false positive results as well.
In the present study, we assigned a prior probability of 0.1 to detect an HR of 2.0 for an association with variant genotypes or minor alleles of the SNPs with P < 0.05. Then, we performed the multivariable stepwise Cox regression analysis including clinical variables and validated SNPs to identify independent representative SNPs in the MDACC dataset. A meta-analysis was followed by combining the results from both MDACC and Harvard studies by using PLINK 1.07. When no heterogeneity was found between two studies (Q-test P-value > 0.10 and I2 < 50.0%) a fixed-effects model was used; otherwise, a random-effects model was applied. Kaplan-Meier curve and log-rank tests were used to estimate the effects of risk genotypes on the cumulative probability of CMSS. Furthermore, we summarized and combined the risk genotypes to assess associations between the number of risk genotypes and CMSS. The heterogeneity test of associations between subgroups of each clinical variable was also evaluated by using the Chi-square-based Q-test in the stratified analyses, and P < 0.05 was considered significant for differences between the subgroups of each clinical variable. The receiver operating characteristic (ROC) curve was generated from the logistic regression model to assess the value of area under the curve (AUC) that predicts CMSS. A time-dependent AUC curve was generated with the survival ROC package of R software26. Haploview v4.227 was employed to construct Manhattan plots and LocusZoom28 was used to produce regional association plots. Finally, the expression quantitative trait locus (eQTL) analysis was performed by linear regression analysis using the R software, including additive, dominant and recessive models with the data of 373 Europeans from the 1000 Genomes project. The mRNA expression data from the lymphoblastic cell lines were derived from 373 Europeans available in the 1000 Genomes Project, which provides the needed evidence whether the effect of SNPs on the gene expression is genetically determined. We also queried the eQTL results in normal skin tissues from the GTEx Portal (http://www.gtexportal.org/home/)29. All statistical analyses were performed with SAS software (version 9.1.4; SAS Institute, Cary, NC, USA), if not specified otherwise.
Results
Basic characteristics of study populations
The baseline characteristics of the MDACC case-control and Harvard cohort studies were described previously16, 30-32. In the present analyses, 858 patients from the MDACC study and 409 patients from the Harvard study with GWAS data were included (Supplementary Table 1). The MDACC study had complete clinical information about age, sex, tumor stage, Breslow thickness, ulceration and mitotic rate and genotyping data, while the Harvard study only had age, sex, survival outcome and genotype data. The age of patients at diagnosis was younger in the MDACC study compared with that of the Harvard study, there were more men (496, 57.8%) than women (362, 42.2%) in the MDACC Study, and there were more patients with stages I/II (709, 82.6%) than stages III/IV (149, 17.4%) in the MDACC. The MDACC study also included cases with different stages of CM seen in the clinics, whereas the Harvard study consisted of new CM cases that were likely to be at an early stage identified from an active follow-up of healthy populations. We used 50 years of age as the cut-off value for age stratification, and the MDACC cases were younger with more late-stage diseases, while the Harvard cases were older with a more early-stage disease. For the above-mentioned reasons, the patients from the MDACC study had a relatively shorter median follow-up time (81.1 months), compared with that (179.0 months) for Harvard patients; however, the death rates of 95/858 (11.1%) and 48/409 (11.5%) in the MDACC versus Harvard studies during the follow-up period were similar.
Gene and SNP extraction from the MDACC dataset
There were 198 SHR-related genes selected from the Molecular Signatures Database23 (Supplementary Table 2), of which seven genes (AR, HDAC6, HSD17B10, MED12, MED14, NR0B1 and RHOXF1) on chromosome X were excluded from the gene list, because there are no standard statistics established for sex-specific analysis as females carry two copies of chromosome X and males are hemizygous for this chromosome. The flowchart of the study design is presented in Figure 1 to describe the analyses performed in the present study. As a result, a total of 4,177 genotyped and 22,062 imputed SNPs were extracted for 191 genes from the MDACC discovery dataset (Supplementary Figure 1). The QQ plot of the observed P values which showed a uniform distribution (Supplementary Figure 2). We then performed multivariate Cox regression analysis with adjustment for age, sex, Breslow’s thickness, regional/distant metastasis, ulceration and mitotic rate. After quality checks, 1,404 SNPs were found to be significantly associated with CMSS with P < 0.05, in which 660 SNPs with a false-positive report probability (FPRP) < 0.2 were extracted for further validation in the other independent dataset from the Harvard study.
Figure 1.
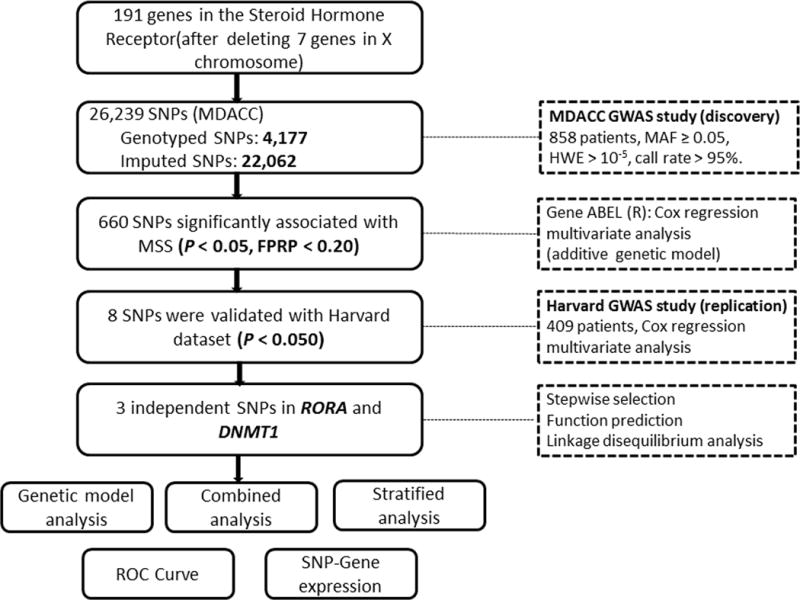
Research flowchart
Replication of the significant SNPs in the Harvard dataset
We validated the 660 SNPs by using the Harvard dataset. After Cox regression analysis with adjustment for age and sex, eight SNPs remained significantly associated with CMSS at P < 0.05, including seven SNPs (rs78293, rs782925, rs782915, rs782918, rs782917, rs17204952, and rs782919) in RORA, and one SNP (rs7253062) in DNMT1. The meta-analysis of these eight SNPs of both the MDACC and Harvard datasets showed that the same associations remained statistically significant, and no heterogeneity was observed for these eight SNPs between the two datasets (Table 1).
Table 1.
Meta-analysis of eight validated SNPs using two published melanoma GWAS datasets
| SNP | Allele1 | Gene | Chr | r2 | MDACC (n=858) | Harvard (n=409) | Meta-analysis | F-SNP6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| EAF | HR (95% CI)2 | P2 | EAF | HR (95% CI)3 | P3 | Phet4 | I2 | HR (95% CI) 5 | P5 | ||||||
| rs782923 | G/A | RORA | 15q22.2 | 0.993 | 0.21 | 1.52 (1.10–2.09) | 0.01 | 0.23 | 1.81 (1.19–2.77) | 0.006 | 0.52 | 0 | 1.62 (1.25–2.09) | 2.12E-04 | Transcriptional Regulation |
| rs782925 | C/T | RORA | 15q22.2 | 0.996 | 0.2 | 1.54 (1.11–2.14) | 0.009 | 0.21 | 1.73 (1.12–2.66) | 0.014 | 0.67 | 0 | 1.61 (1.24–2.09) | 3.78E-04 | Transcriptional Regulation |
| rs782915 | G/A | RORA | 15q22.2 | 0.998 | 0.2 | 1.54 (1.12–2.12) | 0.008 | 0.22 | 1.67 (1.07–2.58) | 0.023 | 0.77 | 0 | 1.58 (1.22–2.05) | 4.94E-04 | Transcriptional Regulation |
| rs782918 | C/T | RORA | 15q22.2 | genotyped | 0.2 | 1.55 (1.12–2.13) | 0.008 | 0.22 | 1.67 (1.07–2.58) | 0.023 | 0.79 | 0 | 1.59 (1.23–2.06) | 4.34E-04 | – |
| rs782917 | G/A | RORA | 15q22.2 | 0.998 | 0.21 | 1.61 (1.17–2.21) | 0.003 | 0.23 | 1.64 (1.06–2.52) | 0.026 | 0.95 | 0 | 1.62 (1.25–2.09) | 2.24E-04 | Transcriptional Regulation |
| rs17204952 | C/T | RORA | 15q22.2 | 0.986 | 0.17 | 1.56 (1.09–2.24) | 0.015 | 0.17 | 1.67 (1.05–2.66) | 0.03 | 0.82 | 0 | 1.60 (1.20–2.13) | 1.17E-03 | Transcriptional Regulation |
| rs782919 | C/T | RORA | 15q22.2 | 0.996 | 0.21 | 1.48 (1.07–2.05) | 0.017 | 0.22 | 1.61 (1.04–2.49) | 0.033 | 0.76 | 0 | 1.52 (1.18–1.98) | 1.45E-03 | Transcriptional Regulation |
| rs7253062 | G/A | DNMT1 | 19p13.2 | 0.932 | 0.4 | 1.44 (1.06–1.96) | 0.019 | 0.39 | 1.67 (1.13–2.48) | 0.01 | 0.56 | 0 | 1.52 (1.20–1.94) | 6.47E-04 | Transcriptional Regulation |
Abbreviations: SNP: single nucleotide polymorphism; GWAS: genome-wide association study; MDACC: The University of Texas M.D. Anderson Cancer Center; EAF: effect allele frequency; HR: hazards ratio; CI: confidence interval; Phet: P value for heterogeneity by Cochrane’s Q test.
Referring to reference allele/effect allele.
Adjusted for age, sex, Breslow thickness, distant/regional metastasis, ulceration and mitotic rate in Cox models of SNPs and melanoma-specific survival in MDACC study and all the tests of the proportional hazards assumption for the validated SNPs were not significant (P > 0.05);
Adjusted for age and sex in Harvard study;
Phet: P value for heterogeneity by Cochrane’s Q test;
Meta-analysis in the fix-effect model;
Independent representative SNPs
We further conducted a stepwise Cox regression analysis of selected clinical variables from the MDACC dataset plus the eight validated SNPs to identify SNPs as independent predictors of CMSS. Three SNPs (i.e., RORA rs782917, RORA rs17204952 and DNMT1 rs7253062) remained significant in the final model (Table 2). We then performed the LD analysis of RORA rs782917 and RORA rs17204952 and found that they were not in LD (r2 = 0). Functional prediction indicated that RORA rs782917, RORA rs17204952 and DNMT1 rs7253062 were putatively functional (Table 1), and these three SNPs were then subject to further analysis. For visual presentation, all genotyped and imputed SNPs in RORA and DNMT1 with an expansion of 100 kilobases in the flanking regions of the gene are shown in a regional association plot, in which these three independent SNPs are each labeled in purple (Supplementary Figure 3).
Table 2.
Independent predictors of CMSS as obtained from the stepwise Cox regression analysis of selected variables from the MDACC dataset
| Parameter1 | Category2 | Frequency | HR (95% CI) | P |
|---|---|---|---|---|
| Age | ≤50/>50 | 371/487 | 1.02 (1.01–1.04) | 0.005 |
| Sex | Female/Male | 362/496 | 1.34 (0.84–2.16) | 0.223 |
| Regional/distant metastasis | No/Yes | 709/149 | 4.22 (2.72–6.55) | <0.0001 |
| Breslow thickness(mm) | ≤1/>1 | 347/511 | 1.17 (1.10–1.24) | <0.0001 |
| Ulceration | No/Yes | 681/155 | 2.74 (1.77–4.23) | <0.0001 |
| Mitotic rate (mm2) | ≤1/>1 | 275/583 | 2.03 (0.99–4.17) | 0.052 |
| rs782917 G>A | GG/GA/AA | 542/276/40 | 1.58 (1.15–2.17) | 0.005 |
| rs17204952 C>T | CC/CT/TT | 590/244/24 | 1.60 (1.12–2.29) | 0.010 |
| rs7253062 G>A | GG/GA/AA | 297/430/131 | 1.41 (1.05–1.91) | 0.024 |
Abbreviations: CMSS, cutaneous melanoma-specific survival; MDACC, The University of Texas MD Anderson cancer Center; HR, hazards ratio; CI, confidence interval;
Stepwise analysis included age, sex, regional/distant metastasis, Breslow thickness, ulceration, mitotic rate and three SNPs in two genes (rs782917 and rs17204952 in RORA and rs7253062 in DNMT1);
The “category” was used as the reference.
Three independent SNPs as CM survival predictors in MDACC and Harvard studies
We further performed survival analysis with different genetic models for each of the three independent SNP. As shown in Table 3, we found that under an additive genetic model, RORA rs782917 A, RORA rs17204952 T and DNMT1 rs7253062 A variant alleles were associated with an increased death risk of CM, with a variant-allele attributed HR of 1.61 (95% CI = 1.17-2.21, P = 0.003), 1.56 (95% CI = 1.09-2.24, P = 0.015) and 1.44 (95% CI = 1.06-1.96, P = 0.019) in the MDACC study and 1.63 (95% CI = 1.06-2.52, P = 0.027), 1.67 (95% CI = 1.05-2.66, P = 0.030) and 1.67 (95% CI = 1.13-2.48, P = 0.010) in the Harvard study, as well as 1.62 (95% CI =1.25-2.09, P = 2.24E-04), 1.60 (95% CI =1.20-2.13, P = 1.17E-03) and 1.52 (95% CI =1.20-1.94, P = 6.47E-04) in a meta-analysis of the two datasets (Table 1).
Table 3.
Associations between three independent SNPs in the steroid hormone receptor-related genes and CMSS of patients in the MDACC study and Harvard study
|
Genotype |
MDACC
|
Harvard
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frequency | Univariate analysis | Multivariate analysis1 | Frequency | Univariate analysis | Multivariate analysis2 | |||||||
|
|
|
|||||||||||
| All | Death (%) | HR (95%CI) | P | HR (95%CI) | P | All | Death (%) | HR (95%CI) | P | HR (95%CI) | P | |
| RORA rs782917 G>A | ||||||||||||
| GG | 542 | 50 (9.2) | 1.00 | 1.00 | 243 | 24 (9.9) | 1.00 | 1.00 | ||||
| GA | 276 | 37 (13.4) | 1.49(0.98–2.28) | 0.065 | 1.67 (1.08–2.59) | 0.021 | 143 | 18 (12.6) | 1.33(0.72–2.45) | 0.364 | 1.39 (0.75–2.58) | 0.292 |
| AA | 40 | 8 (20.0) | 2012(1.01–4.48) | 0.048 | 2.46 (1.15–5.28) | 0.021 | 23 | 6 (26.1) | 2.92(1.19–7.14) | 0.019 | 3.11 (1.25–7.72) | 0.015 |
| GA+AA | 316 | 45 (14.2) | 1.58(1.05–2.36) | 0.027 | 1.77(1.17–2.69) | 0.007 | 166 | 24 (14.46) | 1.54(0.87–2.71) | 0.138 | 1.61(0.91–2.85) | 0.104 |
| Trend | 0.015 | 1.61 (1.17–2.21) | 0.003 | 0.039 | 1.63 (1.06–2.52) | 0.027 | ||||||
| RORA rs17204952 C>T | ||||||||||||
| CC | 590 | 56 (9.5) | 1.00 | 1.00 | 286 | 27 (9.4) | 1.00 | 1.00 | ||||
| CT | 244 | 34 (13.9) | 1.51 (0.98–2.31) | 0.060 | 1.43 (0.92–2.22) | 0.113 | 111 | 19 (17.1) | 1.91 (1.06–3.44) | 0.030 | 1.91 (1.06–3.44) | 0.030 |
| TT | 24 | 5(20.8) | 2.166 (0.99–6.15) | 0.053 | 3.09 (1.22–7.85) | 0.018 | 12 | 2(16.67) | 1.71 (0.41–7.19) | 0.465 | 1.98 (0.47–8.37) | 0.352 |
| CT+TT | 268 | 39 (14.6) | 1.58 (1.05–2.38) | 0.027 | 1.54 (1.01–2.35) | 0.044 | 123 | 21 (17.1) | 1.89 (1.07–3.35) | 0.028 | 1.92 (1.09–3.40) | 0.025 |
| Trend | 0.014 | 1.56(1.09 –2.24) | 0.015 | 0.042 | 1.67(1.05 –2.66) | 0.030 | ||||||
| DNMT1 rs7253062 G>A | ||||||||||||
| GG | 297 | 27 (9.1) | 1.00 | 1.00 | 156 | 12 (7.7) | 1.00 | 1.00 | ||||
| GA | 430 | 50 (11.6) | 1.23(0.77–1.96) | 0.389 | 1.41 (0.86–2.30) | 0.173 | 185 | 22 (11.9) | 1.62(0.80–3.27) | 0.179 | 1.59 (0.79–3.21) | 0.198 |
| AA | 131 | 18 (13.7) | 1.53 (0.84–2.78) | 0.163 | 2.09 (1.13–3.87) | 0.018 | 68 | 14 (20.6) | 3.06 (1.41–6.62) | 0.005 | 2.78 (1.28–6.06) | 0.010 |
| GA+AA | 561 | 68 (12.1) | 1.30 (0.83–2.03) | 0.254 | 1.55(0.97–2.47) | 0.067 | 253 | 36 (14.2) | 1.98 (1.03–3.81) | 0.040 | 1.90(0.99–3.66) | 0.055 |
| Trend | 0.161 | 1.44(1.06–1.96) | 0.019 | 0.005 | 1.67(1.13–2.48) | 0.010 | ||||||
| Number of risk genotypes3 | ||||||||||||
| 0 | 127 | 7 (5.51) | 1.00 | 1.00 | 66 | 5 (7.58) | 1.00 | 1.00 | ||||
| 1 | 379 | 36 (9.50) | 1.71 (0.76–3.83) | 0.197 | 2.35 (0.97–5.68) | 0.057 | 175 | 12 (6.86) | 0.94 (0.33–2.66) | 0.900 | 0.89 (0.31–2.52) | 0.819 |
| 2 | 290 | 40 (13.79) | 2.42 (1.08–5.39) | 0.031 | 3.54 (1.48–8.49) | 0.005 | 137 | 24 (17.52) | 2.55 (0.97–6.68) | 0.060 | 2.55 (0.97–6.68) | 0.058 |
| 3 | 62 | 12 (19.35) | 3.89 (1.53–9.89) | 0.004 | 5.57 (2.02– 15.40) | 0.001 | 31 | 7 (22.58) | 3.51 (1.11–11.06) | 0.032 | 3.39 (1.07– 10.69) | 0.038 |
| Trend | 0.001 | 1.66 (1.28–2.15) | <0.001 | <0.001 | 1.79 (1.27–2.52) | <0.001 | ||||||
| 0-1 | 506 | 43 (8.5) | 1 | 1 | 241 | 17 (7.05) | 1.00 | 1.00 | ||||
| 2-3 | 352 | 52 (14.8) | 1.73 (1.16–2.59) | 0.008 | 1.97(1.29–3.00) | 0.002 | 168 | 30 (18.45) | 2.85 (1.58–5.15) | <0.001 | 2.94 (1.63–5.33) | <0.001 |
Abbreviations: SNP, single nucleotide polymorphisms; CMSS, cutaneous melanoma-specific survival; GWAS, genome-wide association study; MDACC, The University of Texas MD Anderson cancer center; HR, hazards ratio; CI, confidence interval;
Adjusted for age, sex, Breslow thickness, distant/regional metastasis, ulceration and mitotic rate in Cox models of SNPs and CMSS in MDACC study;
Adjusted for age and sex in Harvard study;
Risk genotypes include RORA rs782917 GA+AA, RORA rs17204952 CT+TT and DNMT1 rs7253062 GA+AA.
Analysis of combined genotypes of the three independent SNPs
We further combined the risk genotypes of rs782917 GA+AA, rs17204952 CT+TT and rs7253062 GA+AA into a genetic score to assess the joint effect of these three independent SNPs on CMSS. First, we categorized all the patients into four groups with 0 to 3 risk genotypes (i.e., 0 to 3 genetic scores, Table 3). As a result, the trend test indicated that a risk-genotype dose-response effect, as in the effect on CMSS increased as the number of risk genotypes increased (Ptrend < 0.001) in both the MDACC and Harvard datasets after adjustments where appropriate (Table 3). After that, we further dichotomized all the patients into a low-risk group (0-1 risk genotypes) and a high-risk group (2-3 risk genotypes) because of the small number of subjects in some of the subgroups. Similar results were observed that the high-risk group had an increased risk of death in the MDACC study (HR =1.97, 95% CI = 1.29-3.00, P = 0.002) and the Harvard study (HR = 2.94, 95% CI = 1.63-5.33, P < 0.001), compared with the low-risk group. We also used Kaplan-Meier curves to illustrate the association between the number of unfavorable genotypes (NUGs) and CMSS (Figure 2A-D).
Figure 2.
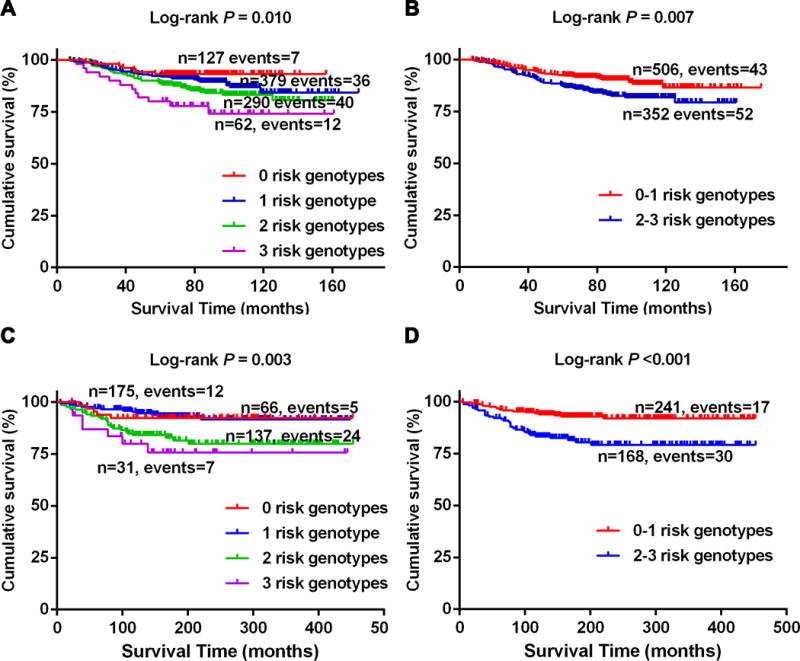
Kaplan-Meier survival curves for melanoma patients in the combined analysis of three risk genotypes in RORA and DNMT1 in both MDACC and Harvard studies. A and B: the MDACC study; C and D: the Harvard study.
Stratified analyses for the effect of combined risk genotypes on CMSS
We then conducted stratified analyses of the combined risk genotypes to evaluate whether there was a joint effect of the three risk genotypes as defined by the genetic score on CMSS, which may be modified by clinicalpathologic variables, including: age, sex, distant/regional metastasis, Breslow thickness, ulceration and mitotic rate in the MDACC dataset and age and sex in the Harvard dataset. We found that high-score risk genotypes were associated with: an increased risk of CM death in the age > 50 group in both MDACC and Harvard datasets, the male group in the MDACC dataset, and both the male and female groups in the Harvard dataset. No heterogeneity was observed in these two subgroups between the MDACC and Harvard datasets (Supplementary Table 3). These showed the number of genotypes influenced the prognosis of CM, although no differential sex effect was observed in the study populations. We also performed stratified analysis for the three SNPs by sex but no significant heterogeneity was found between males and females (Supplementary Table 4).
ROC curve estimators in the MDACC study
We used time-dependent AUC of the ROC curve in the MDACC dataset to assess the prediction effect of the three independent SNPs in the presence of other host and clinicalpathologic variables (i.e., age, sex, tumor stage, Breslow thickness, ulceration and mitotic rate). From the ROC curve, we found that the combination of clinicalpathologic variables and risk genotypes improved prediction performance of five-year CMSS by 1.2%, compared with the clinicalpathologic variables only (AUC = 80.8% to 79.6%, P = 0.656) (Supplementary Figure 4 A-B). We did not conduct ROC curve analysis in the Harvard dataset, because only clinical variables of age and sex were available.
Expression quantitative trait loci (eQTL) analysis
In the eQTL analysis to evaluate correlations between SNPs and mRNA expression levels of their corresponding gene in lymphoblastoid cell lines derived from 373 European descendants from the 1000 Genomes Project, we found that RORA rs17204952 genotypes were correlated with decreased mRNA expression levels of RORA in a recessive model (P = 0.015, Supplementary Figure 5 A-C). However, there was no significant correlation between rs782917 genotypes and RORA mRNA expression levels, nor between rs7253062 genotypes and DNMT1 mRNA expression levels. We have also queried the eQTL results (V7 release) in normal skin tissues from the sun-exposed lower leg and unexposed suprapubic from the GTEx Portal (http://www.gtexportal.org/home/), and found rs7253062 was correlated with the mRNA of expression DNMT1 with nominal P = 0.012 in normal skin tissues from sun-exposed lower leg29 (Supplementary Table 5).
Discussion
In the present study, we identified three significant SNPs (RORA rs782917 G>A, RORA rs17204952 C>T and DNMT1 rs7253062 G>A) as potential predictors of CMSS, which highlights the critical roles of these two genes, each encoding a member of steroid hormone receptors and their related regulators (RORA and DNMT1). In particular, there was a dose-response effect of their combined risk-genotypes on CMSS. Moreover, the RORA rs17204952 T allele was correlated with decreased mRNA expression levels of RORA in lymphoblastoid cell lines derived from 373 European descendants from the 1000 Genomes Project, rs782917 and rs17204952 may affect gene transcriptional regulation as histone modification enrichment region enhancer through assisting in the combination of transformation factor binding site and DNA enzymes, DNMT1 rs7253062 was correlated with the expression of DNMT1 mRNA in normal skin tissues from sun-exposed lower leg, and may play a role in gene transcription regulation. Our results suggested the potential biological roles of steroid hormone receptors and their related regulators in CM progression.
CM is known to be associated with altered steroid hormone receptor expression. Steroid hormones influence melanomagenesis through binding to the receptors and triggering downstream signaling that can lead to neoplastic proliferation33-37, they can also function as ligand-activated transcription factors. The nuclear receptor family of transcription factors including the closely related steroid hormone receptors (SHRs). SHRs are ligand-inducible transcription factors that exhibit potent effects on gene expression in living cells. AR and ER act as ligand-dependent transcription factors and are members of SHRs33. Their ligands bind to AR and ER non-specifically, regulating cell proliferation and differentiation, and genes in these pathways are commonly mutated in CM33. Previous study has showed that both males and females had an increasing risk of CM-specific death with an increasing age at diagnosis, this increase was more pronounced among females than males, especially in the higher stage groups38. The benefit is independent of other variables such as anatomic location of the tumor and histologic type of tumor. In addition, female patients have been found to have a significantly lower risk of both lymph node and distant metastases compared with male patients39. It has been reported that the incidence rates of melanoma increase steeply in women until about age 507, and women at 40-49 years of age showed higher rates of melanoma-related death and positive sentinel nodes40. These suggests that female hormones may have a protective role in the development of metastatic melanoma41, highlighting the critical role of the steroid hormone receptors and their related regulators in CM progression.
RORA, located at 15q22.2, encodes a protein called retinoic acid receptor-related orphan receptor A, which is a family member of the nuclear steroid hormone receptor superfamily42. RORA and ER share a consensus binding site on DNA (AGGTCA), and consequently they are the common target genes. Expression of RORA is inversely modulated by both male and female sex hormones. Dihydrotestosterone (DHT) and estradiol increase the binding of AR and ER to the RORA promoter region, and RORA in turn increases testosterone levels. The resultant negative feedback transcriptionally regulates aromatase, which converts male hormones to estrogens43. RORA is also known as a binding site of melatonin, which participates in anti-apoptotic and anti-inflammatory responses, suppressing neoplastic growth in a variety of SHRs-related tumors, including cancers of the breast, prostate, ovaries, and melanoma44. It has been demonstrated that RORA can reduce the invasive and migratory capacities of androgen-independent prostate cancer cells45. RORA transcript and protein expression levels increase significantly under the stress conditions of UV irradiation in breast epithelial cell lines, and thus it appears that RORA may be involved in human tumorigenesis42. RORA interacts with the ER and enhances ER transcriptional activity in breast cancer, and RORA expression has been shown to be required for activation of a subset of E2-mediated and upregulated genes that are associated with functions of lyase activity46. A research reported that RORα may also contribute to the effect of an enhancer through binding to and activating the Pcp-2(L7) gene promoter and structural gene in Purkinje cells47. Finally, one study found that RORA rs7164773 SNP was involved in a statistically significant interaction between breast cancer risk and menopausal status48. Taken together, these results suggest that RORA may be considered to function as a tumor suppressor gene, and its high expression may contribute to a good prognosis across multiple cancers, including CM.
DNMT1, located at 19p13.2, encodes an enzyme called DNA methyltransferase 1, which transfers methyl groups to cytosine nucleotides of genomic DNA. The deletion or reduction of DNMT1 leads to substantial genome-wide hypomethylation and chromosomal instability in human tumors49. Recent studies have identified associations between DNMT1 dysregulation, melanomagenesis, and melanocyte malignant transformation50-53. In addition, DNMT1 silencing is associated with reductions in the mesenchymal properties and invasive potential of melanoma cells50. Melanoma cells are characterized by an aberrant global DNA hypomethylation, ERβ activation induces global DNA methylation reprogramming in melanoma cells11. Previous studies have showed that changes in DNA methylation of cancer related genes can be an elementary process accounting for tumorigenesis. The polymorphisms of the DNMT1 gene rs2228611 contributes to favorable prognosis in gastric cancer54. The DNMT1 rs2228611 and rs759920 SNPs are associated with an increased risk of ovarian cancer development55. Finally, DNMT1 transient depletion is a causal factor of termed cancer-germline gene activation in vivo in melanoma56.
There are several limitations to the present study. First, the discovery and validation datasets had a different proportion of female patients with old age and disease stages, which may have introduced bias in the HR estimates. The validation dataset from the Harvard study lacks complete clinical data. Furthermore, neither of the two datasets we used had detailed information on hormone levels, history of acne, history of pregnancy and contraceptive drug use, tumor somatic mutation data, and systemic treatments and response. Second, because the number of patients was limited to the two datasets, particularly in stratification analysis by sex. So, there is limited study power to detect difference between males and females. Third, the patients in the present study were all non-Hispanic whites. Additional studies including multi-ethnic groups are needed. Fourth, function prediction websites have limitations in predicting the three identified SNPs, and thus more functional investigations are warranted to explore the precise mechanisms of the observed associations with CMSS. Fifth, we cannot exclude false positive findings as we used a relatively loose criterion to control for multiple testing and further population and functional validation are required.
Supplementary Material
Supplementary Figure 1. Manhattan plot of 26,239 SNPs of steroid hormone receptor-related genes in the MDACC study. The statistical values across the autosomes for associations between 26,239 SNPs and melanoma-specific survival were plotted as −log10 P values. The red horizontal line indicates P = 0.05, 1,404 SNPs are above the red line. The eight SNPs in RORA and DNMT1 were validated from the Harvard study, which are located at 15q and 19p, respectively. The other SNPs were not validated from the Harvard study.
Supplementary Figure 2. QQ plot of the null distribution of p-values.
Supplementary Figure 3. Regional association plots of the three independent SNPs in the steroid hormone receptor-related genes. The left-hand Y-axis shows the -log10 transformation of P values of individual SNPs, which is plotted against the chromosomal base-pair position with an extension of 100 KB in flanking region of the genes. The right-hand Y-axis shows the recombination rate estimated for European population from HapMap Data Rel 22/phase II. A. RORA rs782917; B. RORA rs17204952; C. DNMT1 rs7253062.
Supplementary Figure 4. Receiver operating characteristic (ROC) curve and time-dependent area under the ROC curve (AUC) estimation for prediction of melanoma-specific survival using the MDACC dataset. A. Five-year melanoma-specific survival rate; B. Time-dependent AUC estimation, based on age, sex, Breslow thickness, tumor stage, ulceration, mitotic rate and the risk genotypes of the two genes.
Supplementary Figure 5. The eQTL analysis of SNP rs17204952 by using the mRNA expression data from the lymphoblastic cell lines derived from 373 Europeans available in the 1000 Genomes Project. A. additive model, P = 0.989; B. dominant model, P = 0.286; C. recessive model, P = 0.015. (No eQTL results for rs782917 and rs7253062 in the 1000 Genomes Project). D-E: Annotations of rs782917, rs17204952 and rs7253062 in the steroid hormone receptor-related genes from ENCODE project data from the University of California Santa Cruz (UCSC, http://genome.ucsc.edu/). D. RORA rs782917 and rs17204952; E. DNMT1 rs7253062.
What’s new?
Steroid hormones and their receptors have been linked to cutaneous melanoma risk. Here, the authors identified three independent SHR-related genes SNPs (RORA rs782917 G>A, RORA rs17204952 C>T and DNMT1 rs7253062 G>A) as potential predictors of cutaneous melanoma-specific survival. Findings from this study highlight the possible role of steroid hormones and their receptors in the pathogenesis of cutaneous melanoma.
Acknowledgments
We would like to thank the John Hopkins University Center for Inherited Disease Research for conducting high-throughput genotyping for this study. We thank the participants and staff of the Nurses’ Health Study (NHS) and Health Professionals Follow-up Study (HPFS) for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. We also thank all the individuals who participated in this project. The authors assume full responsibility for analyses and interpretation of these data. The MD Anderson Study was support by NIH/NCI R01 CA100264, 2P50CA093459 and R01CA133996 as well as by The University of Texas MD Anderson Cancer Center Various Donors Melanoma and Skin Cancers Priority Program Fund; the Miriam and Jim Mulva Research Fund; the McCarthy Skin Cancer Research Fund and the Marit Peterson Fund for Melanoma Research. The Harvard Study was in part supported by NIH/NCI R01 CA49449, P01 CA87969, UM1 CA186107 and UM1 CA167552. Bo Li is sponsored by the China Scholarship Council for studying at the Duke University. Qingyi Wei was supported by start-up funds from Duke Cancer Institute, Duke University Medical Center and the P30 Cancer Center Support Grant (Grant ID: NIH CA014236).
Abbreviations
- CM
cutaneous melanoma
- CMSS
cutaneous melanoma-specific survival
- HR
hazards ratio
- CI
confidence interval
- SEER
Surveillance, Epidemiology, and End Results
- UV
ultraviolet
- GWAS
genome-wide association studies
- SNP
single-nucleotide polymorphisms
- MDACC
MD Anderson Cancer Center
- NHS
Nurses’ Health Study
- HPFS
Health Professionals Follow-up Study
- MAF
minor allelic frequency
- HWE
Hardy-Weinberg equilibrium
- FPRP
false-positive report probability
- ROC
receiver operating characteristic
- AUC
area under the curve
- eQTL
expression quantitative trait loci
Footnotes
Supplementary Material
Refer to Web version on PubMed Central for supplementary material.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Marzagalli M, Montagnani Marelli M, Casati L, Fontana F, Moretti RM, Limonta P. Estrogen Receptor beta in Melanoma: From Molecular Insights to Potential Clinical Utility. Front Endocrinol (Lausanne) 2016;7:140. doi: 10.3389/fendo.2016.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boland GM, Gershenwald JE. Principles of Melanoma Staging. Cancer Treat Res. 2016;167:131–48. doi: 10.1007/978-3-319-22539-5_5. [DOI] [PubMed] [Google Scholar]
- 4.Lasithiotakis K, Leiter U, Meier F, Eigentler T, Metzler G, Moehrle M, Breuninger H, Garbe C. Age and gender are significant independent predictors of survival in primary cutaneous melanoma. Cancer. 2008;112:1795–804. doi: 10.1002/cncr.23359. [DOI] [PubMed] [Google Scholar]
- 5.Bannister-Tyrrell M, Roberts CL, Hasovits C, Nippita T, Ford JB. Incidence and outcomes of pregnancy-associated melanoma in New South Wales 1994-2008. Aust N Z J Obstet Gynaecol. 2015;55:116–22. doi: 10.1111/ajo.12279. [DOI] [PubMed] [Google Scholar]
- 6.Koomen ER, Joosse A, Herings RM, Casparie MK, Guchelaar HJ, Nijsten T. Estrogens, oral contraceptives and hormonal replacement therapy increase the incidence of cutaneous melanoma: a population-based case-control study. Ann Oncol. 2009;20:358–64. doi: 10.1093/annonc/mdn589. [DOI] [PubMed] [Google Scholar]
- 7.Feskanich D, Hunter DJ, Willett WC, Spiegelman D, Stampfer MJ, Speizer FE, Colditz GA. Oral contraceptive use and risk of melanoma in premenopausal women. Br J Cancer. 1999;81:918–23. doi: 10.1038/sj.bjc.6690787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang M, Qureshi AA, Fortner RT, Hankinson SE, Wei Q, Wang LE, Eliassen AH, Willett WC, Hunter DJ, Han J. Teenage acne and cancer risk in US women: A prospective cohort study. Cancer. 2015;121:1681–7. doi: 10.1002/cncr.29216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li WQ, Qureshi AA, Ma J, Goldstein AM, Giovannucci EL, Stampfer MJ, Han J. Personal history of prostate cancer and increased risk of incident melanoma in the United States. J Clin Oncol. 2013;31:4394–9. doi: 10.1200/JCO.2013.51.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morvillo V, Luthy IA, Bravo AI, Capurro MI, Portela P, Calandra RS, Mordoh J. Androgen receptors in human melanoma cell lines IIB-MEL-LES and IIB-MEL-IAN and in human melanoma metastases. Melanoma Res. 2002;12:529–38. doi: 10.1097/00008390-200212000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Marzagalli M, Casati L, Moretti RM, Montagnani Marelli M, Limonta P. Estrogen Receptor beta Agonists Differentially Affect the Growth of Human Melanoma Cell Lines. PLoS One. 2015;10:e0134396. doi: 10.1371/journal.pone.0134396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Giorgi V, Gori A, Gandini S, Papi F, Grazzini M, Rossari S, Simoni A, Maio V, Massi D. Oestrogen receptor beta and melanoma: a comparative study. Br J Dermatol. 2013;168:513–9. doi: 10.1111/bjd.12056. [DOI] [PubMed] [Google Scholar]
- 13.Zhou JH, Kim KB, Myers JN, Fox PS, Ning J, Bassett RL, Hasanein H, Prieto VG. Immunohistochemical expression of hormone receptors in melanoma of pregnant women, nonpregnant women, and men. Am J Dermatopathol. 2014;36:74–9. doi: 10.1097/DAD.0b013e3182914c64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramaraj P, Cox JL. In vitro effect of progesterone on human melanoma (BLM) cell growth. Int J Clin Exp Med. 2014;7:3941–53. [PMC free article] [PubMed] [Google Scholar]
- 15.Allil PA, Visconti MA, Castrucci AM, Isoldi MC. Photoperiod and testosterone modulate growth and melanogenesis of s91 murine melanoma. Med Chem. 2008;4:100–5. doi: 10.2174/157340608783789185. [DOI] [PubMed] [Google Scholar]
- 16.Amos CI, Wang LE, Lee JE, Gershenwald JE, Chen WV, Fang S, Kosoy R, Zhang M, Qureshi AA, Vattathil S, Schacherer CW, Gardner JM, et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Human molecular genetics. 2011;20:5012–23. doi: 10.1093/hmg/ddr415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gershenwald JE, Ross MI. Sentinel-lymph-node biopsy for cutaneous melanoma. The New England journal of medicine. 2011;364:1738–45. doi: 10.1056/NEJMct1002967. [DOI] [PubMed] [Google Scholar]
- 18.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, et al. The NCBI dbGaP database of genotypes and phenotypes. Nature genetics. 2007;39:1181–6. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L, Lee M, Popova N, Sharopova N, Kimura M, Feolo M. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic acids research. 2014;42:D975–9. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: Using Sequence and Genotype Data to Estimate Haplotypes and Unobserved Genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–11. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biernacka JM, Tang R, Li J, McDonnell SK, Rabe KG, Sinnwell JP, Rider DN, de Andrade M, Goode EL, Fridley BL. Assessment of genotype imputation methods. BMC proceedings. 2009;3(Suppl 7):S5. doi: 10.1186/1753-6561-3-s7-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome research. 2012;22:1790–7. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic acids research. 2009;37:W600–5. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chambless LE, Diao G. Estimation of time-dependent area under the ROC curve for long-term risk prediction. Statistics in medicine. 2006;25:3474–86. doi: 10.1002/sim.2299. [DOI] [PubMed] [Google Scholar]
- 27.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 28.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Consortium G. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45:580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan H, Liu H, Liu Z, Zhu D, Amos CI, Fang S, Lee JE, Wei Q. Genetic variants in Hippo pathway genes YAP1, TEAD1 and TEAD4 are associated with melanoma-specific survival. International journal of cancer. 2015;137:638–45. doi: 10.1002/ijc.29429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin J, Liu H, Yi X, Wu W, Amos CI, Fang S, Lee JE, Han J, Wei Q. Genetic variants in the vitamin D pathway genes VDBP and RXRA modulate cutaneous melanoma disease-specific survival. Pigment Cell Melanoma Res. 2016;29:176–85. doi: 10.1111/pcmr.12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song F, Qureshi AA, Zhang J, Amos CI, Lee JE, Wei Q, Han J. Exonuclease 1 (EXO1) gene variation and melanoma risk. DNA repair. 2012;11:304–9. doi: 10.1016/j.dnarep.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitkov M, Joseph R, Copland J., 3rd Steroid hormone influence on melanomagenesis. Mol Cell Endocrinol. 2015;417:94–102. doi: 10.1016/j.mce.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 34.Meierjohann S, Schartl M. From Mendelian to molecular genetics: the Xiphophorus melanoma model. Trends Genet. 2006;22:654–61. doi: 10.1016/j.tig.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Cato AC, Peterziel H. The androgen receptor as mediator of gene expression and signal transduction pathways. Trends Endocrinol Metab. 1998;9:150–4. doi: 10.1016/s1043-2760(98)00039-3. [DOI] [PubMed] [Google Scholar]
- 36.Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266:510–8. [PubMed] [Google Scholar]
- 37.Zhu H, Dong D, Li F, Liu D, Wang L, Fu J, Song L, Xu G. Clinicopathologic features and prognostic factors in patients with non-cutaneous malignant melanoma: a single-center retrospective study of 71 cases. Int J Dermatol. 2015;54:1390–5. doi: 10.1111/ijd.12745. [DOI] [PubMed] [Google Scholar]
- 38.Enninga EAL, Moser JC, Weaver AL, Markovic SN, Brewer JD, Leontovich AA, Hieken TJ, Shuster L, Kottschade LA, Olariu A, Mansfield AS, Dronca RS. Survival of cutaneous melanoma based on sex, age, and stage in the United States, 1992-2011. Cancer Med. 2017 doi: 10.1002/cam4.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joosse A, de Vries E, Eckel R, Nijsten T, Eggermont AM, Holzel D, Coebergh JW, Engel J, Munich Melanoma G. Gender differences in melanoma survival: female patients have a decreased risk of metastasis. J Invest Dermatol. 2011;131:719–26. doi: 10.1038/jid.2010.354. [DOI] [PubMed] [Google Scholar]
- 40.Tellez A, Rueda S, Conic RZ, Powers K, Galdyn I, Mesinkovska NA, Gastman B. Risk factors and outcomes of cutaneous melanoma in women less than 50 years of age. J Am Acad Dermatol. 2016;74:731–8. doi: 10.1016/j.jaad.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 41.Kemeny MM, Busch E, Stewart AK, Menck HR. Superior survival of young women with malignant melanoma. Am J Surg. 1998;175:437–44. doi: 10.1016/s0002-9610(98)00070-1. discussion 44-5. [DOI] [PubMed] [Google Scholar]
- 42.Zhu Y, McAvoy S, Kuhn R, Smith DI. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene. 2006;25:2901–8. doi: 10.1038/sj.onc.1209314. [DOI] [PubMed] [Google Scholar]
- 43.Sarachana T, Xu M, Wu RC, Hu VW. Sex hormones in autism: androgens and estrogens differentially and reciprocally regulate RORA, a novel candidate gene for autism. PLoS One. 2011;6:e17116. doi: 10.1371/journal.pone.0017116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabrera J, Negrin G, Estevez F, Loro J, Reiter RJ, Quintana J. Melatonin decreases cell proliferation and induces melanogenesis in human melanoma SK-MEL-1 cells. J Pineal Res. 2010;49:45–54. doi: 10.1111/j.1600-079X.2010.00765.x. [DOI] [PubMed] [Google Scholar]
- 45.Moretti RM, Montagnani Marelli M, Motta M, Limonta P. Role of the orphan nuclear receptor ROR alpha in the control of the metastatic behavior of androgen-independent prostate cancer cells. Oncol Rep. 2002;9:1139–43. [PubMed] [Google Scholar]
- 46.Gu F, Hsu HK, Hsu PY, Wu J, Ma Y, Parvin J, Huang TH, Jin VX. Inference of hierarchical regulatory network of estrogen-dependent breast cancer through ChIP-based data. BMC Syst Biol. 2010;4:170. doi: 10.1186/1752-0509-4-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serinagaoglu Y, Zhang R, Zhang Y, Zhang L, Hartt G, Young AP, Oberdick J. A promoter element with enhancer properties, and the orphan nuclear receptor RORalpha, are required for Purkinje cell-specific expression of a Gi/o modulator. Mol Cell Neurosci. 2007;34:324–42. doi: 10.1016/j.mcn.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 48.Benna C, Helfrich-Forster C, Rajendran S, Monticelli H, Pilati P, Nitti D, Mocellin S. Genetic variation of clock genes and cancer risk: a field synopsis and meta-analysis. Oncotarget. 2017 doi: 10.18632/oncotarget.15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–92. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 50.Venza M, Visalli M, Catalano T, Biondo C, Beninati C, Teti D, Venza I. DNA methylation-induced E-cadherin silencing is correlated with the clinicopathological features of melanoma. Oncol Rep. 2016;35:2451–60. doi: 10.3892/or.2016.4618. [DOI] [PubMed] [Google Scholar]
- 51.Venza M, Visalli M, Catalano T, Fortunato C, Oteri R, Teti D, Venza I. Impact of DNA methyltransferases on the epigenetic regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in malignant melanoma. Biochem Biophys Res Commun. 2013;441:743–50. doi: 10.1016/j.bbrc.2013.10.114. [DOI] [PubMed] [Google Scholar]
- 52.Molognoni F, de Melo FH, da Silva CT, Jasiulionis MG. Ras and Rac1, frequently mutated in melanomas, are activated by superoxide anion, modulate Dnmt1 level and are causally related to melanocyte malignant transformation. PLoS One. 2013;8:e81937. doi: 10.1371/journal.pone.0081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venza M, Visalli M, Biondo C, Lentini M, Catalano T, Teti D, Venza I. Epigenetic regulation of p14ARF and p16INK4A expression in cutaneous and uveal melanoma. Biochim Biophys Acta. 2015;1849:247–56. doi: 10.1016/j.bbagrm.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 54.Jia Z, Wu X, Cao D, Wang C, You L, Jin M, Wen S, Cao X, Jiang J. Polymorphisms of the DNA Methyltransferase 1 Gene Predict Survival of Gastric Cancer Patients Receiving Tumorectomy. Dis Markers. 2016;2016:8578064. doi: 10.1155/2016/8578064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mostowska A, Sajdak S, Pawlik P, Lianeri M, Jagodzinski PP. DNMT1, DNMT3A and DNMT3B gene variants in relation to ovarian cancer risk in the Polish population. Mol Biol Rep. 2013;40:4893–9. doi: 10.1007/s11033-013-2589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cannuyer J, Van Tongelen A, Loriot A, De Smet C. A gene expression signature identifying transient DNMT1 depletion as a causal factor of cancer-germline gene activation in melanoma. Clin Epigenetics. 2015;7:114. doi: 10.1186/s13148-015-0147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Manhattan plot of 26,239 SNPs of steroid hormone receptor-related genes in the MDACC study. The statistical values across the autosomes for associations between 26,239 SNPs and melanoma-specific survival were plotted as −log10 P values. The red horizontal line indicates P = 0.05, 1,404 SNPs are above the red line. The eight SNPs in RORA and DNMT1 were validated from the Harvard study, which are located at 15q and 19p, respectively. The other SNPs were not validated from the Harvard study.
Supplementary Figure 2. QQ plot of the null distribution of p-values.
Supplementary Figure 3. Regional association plots of the three independent SNPs in the steroid hormone receptor-related genes. The left-hand Y-axis shows the -log10 transformation of P values of individual SNPs, which is plotted against the chromosomal base-pair position with an extension of 100 KB in flanking region of the genes. The right-hand Y-axis shows the recombination rate estimated for European population from HapMap Data Rel 22/phase II. A. RORA rs782917; B. RORA rs17204952; C. DNMT1 rs7253062.
Supplementary Figure 4. Receiver operating characteristic (ROC) curve and time-dependent area under the ROC curve (AUC) estimation for prediction of melanoma-specific survival using the MDACC dataset. A. Five-year melanoma-specific survival rate; B. Time-dependent AUC estimation, based on age, sex, Breslow thickness, tumor stage, ulceration, mitotic rate and the risk genotypes of the two genes.
Supplementary Figure 5. The eQTL analysis of SNP rs17204952 by using the mRNA expression data from the lymphoblastic cell lines derived from 373 Europeans available in the 1000 Genomes Project. A. additive model, P = 0.989; B. dominant model, P = 0.286; C. recessive model, P = 0.015. (No eQTL results for rs782917 and rs7253062 in the 1000 Genomes Project). D-E: Annotations of rs782917, rs17204952 and rs7253062 in the steroid hormone receptor-related genes from ENCODE project data from the University of California Santa Cruz (UCSC, http://genome.ucsc.edu/). D. RORA rs782917 and rs17204952; E. DNMT1 rs7253062.