

Abstract
Proteotoxic stress plays an important role in the pathogenesis of Huntington’s disease (HD). Autophagy is proposed as a compensatory mechanism to remove protein aggregates under proteotoxic stress by up-regulating p62 expression. In the present study, we investigated the molecular action of p62 to proteotoxic stress in HD cells. Using two different HD cellular models, STHdhQ7 and STHdhQ111 cells derived from wild type and HD knock-in mice and human fibroblasts from healthy and HD patients, we found that HD cells are more vulnerable to cell death under proteotoxic stress and during stress recovery. We further showed that P62 was up-regulated in both STHdhQ7 and STHdhQ111 cells in response to the stress with distinct subcellular localization patterns. While dispersed p62 puncti were found in STHdhQ7 cells, p62 bodies were initially present in the lysosomes and accumulated to the juxtanuclear regions of STHdhQ111 cells as MG132 incubation continued. Unlike in STHdhQ7 cells, p62 puncti were not associated with K48-linked polyubiquitinated protein aggregates or proteasomal components in STHdhQ111. Interestingly, addition of cysteine during MG132 incubation rescued cell death in STHdhQ111 cells caused by stress recovery and altered the subcellular distribution of p62. Our data suggest that aberrant positioning of p62 affects the proteasomal clearance of protein aggregates and may contribute to the increased vulnerability to proteotoxic stress-induced cell death in HD cells.
Keywords: Huntington’s disease, SQSTM1/p62, proteotoxicity, ubiquitin, protein aggregates
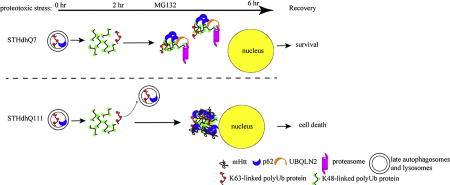
Graphical abstract
INTRODUCTION
Proper protein quality control is critical for neuronal functions due to the specialized structure and post-mitotic property of neurons. Failure to do so leads to accumulation of misfolded proteins, a hallmark present in various neurodegenerative diseases. Ubiquitin-proteasome system (UPS) and autophagy are the two major pathways for cellular protein degradations, which were traditionally viewed as two independent processes. Increasing evidence suggests that these two degradative pathways are closely related and several crosstalk mechanisms have been elucidated (Park and Cuervo 2013; Liebl and Hoppe 2016). While blocking UPS activity often leads to an up-regulation in autophagy (Pandey et al. 2007), blockage of autophagy leads to reduced proteasomal activities (Korolchuk et al. 2009). Therefore, the crosstalk between UPS and autophagy may serve as an important mechanism in compensating for impairments in degradative pathways. However, the crosstalk in the context of neurodegenerative diseases has been relatively unexplored.
P62, also known as sequestosome 1 (SQSTM1), functions as a signaling hub for diverse cellular events (Katsuragi et al. 2015). P62 polymerizes via the N-terminal Phox and Bem1p (PB1) domain and binds to ubiquitin through the C-terminal ubiquitin-binding domain (UBA). P62 also contains a microtubule-associated protein light chain 3 (LC3)-interacting domain (Pankiv et al. 2007). As an autophagy adaptor, p62 recognizes and shuttle ubiquitinated proteins to autophagosomes for degradation (Pankiv et al. 2007). As a result, p62 is normally degraded through autophagy (Bjørkøy et al. 2009). Under conditions that impair UPS activities, p62 expression was found to be increased (Kuusisto et al. 2001), indicating an up-regulation of the autophagic response. It is therefore implicated as a molecular link between UPS and autophagy. However, the molecular action of p62 in response to UPS impairment remains elusive.
Huntington’s disease (HD) is an inherited autosomal dominant neurodegenerative disease caused by polyQ expansion in the huntingtin (Htt) protein. A pathological hallmark of HD is the presence of intracellular mutant Htt (mHtt) aggregates. Proteomic analyses of the Htt interactome suggest that Htt has various roles in protein trafficking, proteomic homeostasis, energy metabolism and transcriptional regulation by interacting with different protein partners (Shirasaki et al. 2012; Ratovitski et al. 2012). Interestingly, it was reported that Htt serves as a scaffold protein to regulate autophagy (Rui et al. 2015). Htt directly interacts with p62 through its carboxyl region and positively regulates cargo recognition by facilitating the binding of p62 to ubiquitin (Ub)-K63-modified cargos and to LC3 (Rui et al. 2015). Additionally, Htt also interacts with unc-51 like autophagy activating kinase 1 (ULK1) to promote its activation and subsequent initiation of selective autophagy (Rui et al. 2015).
In the present study, we sought to investigate how mHtt affects p62 function in response to proteotoxic stress induced by MG132, a general proteasomal inhibitor. Using STHdhQ7 and STHdhQ111 striatal cell lines derived from wild type and mHtt knock-in mice (Trettel et al. 2000) and human fibroblasts from healthy and HD patients, we first demonstrate that HD cells are more vulnerable to cell death under prolonged proteotoxic stress or during recovery from the stress. p62 is up-regulated in both normal and HD cells in response to the UPS inhibition. Interestingly, we found a distinct difference in the subcellular localization of p62 in normal and HD cells in response to proteotoxic stress. STHdhQ7 but not STHdhQ111 cells exhibit dispersed p62 bodies that contain K48-linked polyubiquitinated (polyUb) protein aggregates and are associated with UPS components. Furthermore, we demonstrate that co-incubation of cysteine with MG132 rescues cell death in STHdhQ111 cells during stress recovery and prevents the formation of p62 macroaggregates. Taken together, these findings suggest that aberrant positioning of p62 in HD cells makes HD neurons more vulnerable to cell death during recovery from proteotoxic stress.
MATERIAL AND METHODS
Cell culture and drug treatment
STHdhQ7 (CH00097) and STHdhQ111 (CH00095) cells were obtained from Coriell Institute for Medical Research and cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum, 1% glutamine and 1% penicillin/streptomycin at 33°C. Primary fibroblasts, GM04693 from a HD individual (onset at age 41 years), GM05539 from a HD individual (onset at age 2 years) and AG02222 from a normal subject, were obtained from the Coriell Institute for Medical Research and cultured in DMEM supplemented with 20% FBS, 1% glutamine and 1% penicillin/streptomycin at 37°C. To induce proteotoxic stress, cells were incubated with 10 µM MG132 for various time as indicated. For stress recovery, cells were washed twice with PBS at the end of MG132 treatment and further incubated in the complete medium for 18 hours at 33°C. To block protein synthesis, STH cells were incubated with 10 µg/ml cycloheximide in the presence or absence of MG132 for 6 hours at 33°C. To inhibit autophagic influx, STH cells were incubated with 50 µM hydroxychloroquine (CQ) in the presence or absence of MG132 for various time as indicated at 33°C. For lactacystin treatment, STH cells were incubated with 5 µM lactacystin for 6 hours at 33°C. For cysteine treatment, 1 mM freshly made cysteine was added to the medium during MG132 incubation or during stress recovery. All these drugs were purchased from Sigma Aldrich (St. Louis, MO).
ATP assay
Cells were seeded in a 96-well plate at a density of 1 × 104 cells/well. After treatments, ATP content was measured with the CellTiter-Glo® luminescent cell viability assay (Promega, Madison, WI) as we previously described (Leon et al. 2010). The background luminescence from the culture medium was subtracted. Luminescence from the treated groups was normalized to their respective control groups.
Western blot
After treatments, cells were briefly washed with PBS, directly lysed in 1× SDS sample buffer and boiled for 5 minutes. About 20 µg of sample lysates were separated by SDS-PAGE and transferred to nitrocellulose membranes. Fluorescent western blotting was performed as previously described (Erie et al. 2015). The following primary antibodies were used: rabbit polyclonal anti-LC3B (1:500, Cat. #3868, Cell Signaling Techology, Danvers, MA), rabbit polyclonal anti-p62 (1:1000, Cat. #18420-1-AP, Proteintech, Rosemont, IL), mouse monoclonal anti-p62 (1:1000, Cat. #56416, Abcam, Cambridge, MA), mouse monoclonal anti-Ub (clone P4D1, 1:1000, Cat #14-6078-82, eBiosciences, San Diego, CA), Rabbit monoclonal anti-K48-linked Ub (clone Apu2, 1:1000, Cat. #05-1307, Millipore, Temecula, CA), Rabbit monoclonal, anti-K63-linked Ub (1:1000, Cat #179434, Abcam) and mouse monoclonal anti-actin [1:1000, Cat. #sc-47778, Santa Cruz Biotechnology (SCBT), Santa Cruz, CA]. Fluorescent signals were detected with a LI-COR Odyssey Fc system and bands were quantified by densitometric analysis using the provided Image Studio 2.0 software.
Immunofluorescence and image analysis
Cells were cultured on No. 1.5 glass coverslips coated with poly-L-lysine. After treatments, cells were fixed and permeablized in −20°C methanol for 1 min unless stated otherwise. Cells were then blocked and immunostained as described previously (Erie et al. 2015). The following primary antibodies were used: rabbit polyclonal anti-p62 (1:1000), mouse monoclonal anti-p62 (1:1000), mouse monoclonal anti-Ub (1:1000), Rabbit monoclonal anti-K48-linked Ub (1:1000), Rabbit monoclonal anti-K63-linked Ub (1:1000), mouse monoclonal ubiquilin-2 (1:1000. Cat. #NBP2-25164SS, Novus Biologicals, Littleton, CO), rabbit polyclonal anti-proteasome 20S α+β (1:1000, Cat. #22673, Abcam), rabbit monoclonal anti-huntingtin (1:500, Cat. #109115, Abcam), mouse monoclonal γ-tubulin (1:1000, GTU-88, Cat #T5326, Sigma) and rat monoclonal anti-lysosomal associated protein 1 (Lamp1, 1:500, 1D4B, Developmental Studies Hybridoma Bank, Iowa city, IA). Immunofluorescent signals were detected using the Zeiss LSM700 laser confocal microscope with a 63× oil Plan-Apochromat objective (numerical aperture: 1.4). In all presented images, DAPI was used as a nuclear counterstain.
Image analysis
Pixels with a protein expression level above the local mean value were designated as “punctate” and the remaining pixels as “diffuse”. The punctate sizes were measured using the Zen 2010 Software by outlining the aggresomes and measuring the longest axis. A co-localization analysis was performed using NIH ImageJ software at single-cell levels. Background was first subtracted from each channel using the “ROI/BG subtraction from ROI” plug-in. Manders’ coefficiency was calculated with the “JACoP” plug-in. A total of 6–15 cells from three independent images were analyzed per condition.
Quantitative RT-PCR analysis
P62 mRNA levels were measured with quantitative RT-PCR using the SYBR green method as we previously described (Leon et al. 2010). The forward and reverse primers for mouse p62 (NM_011018.3) are: 5’-GATGTGGAACATGGAGGGAAGA-3’ and 5’-GGAGTTCACCTGTAGATGGGT-3’. The forward and reverse primers for mouse actin (NM_007393) are: 5’-GGCTGTATTCCCCTCCATCG-3’ and 5’-CCAGTTGGTAACAATGCCATGT-3’.
Data analysis
All data were expressed as means ± S.E.M. To establish significance, data were subjected to unpaired student’s t-test, one-way or two-way ANOVA followed by the Tukey’s post-hoc analysis as indicated using the GraphPad Prism software statistical package 5.0 (GraphPad Software). The criterion for significance was set at p≤0.05.
RESULTS
HD cells are more vulnerable to cell death induced by proteotoxic stress
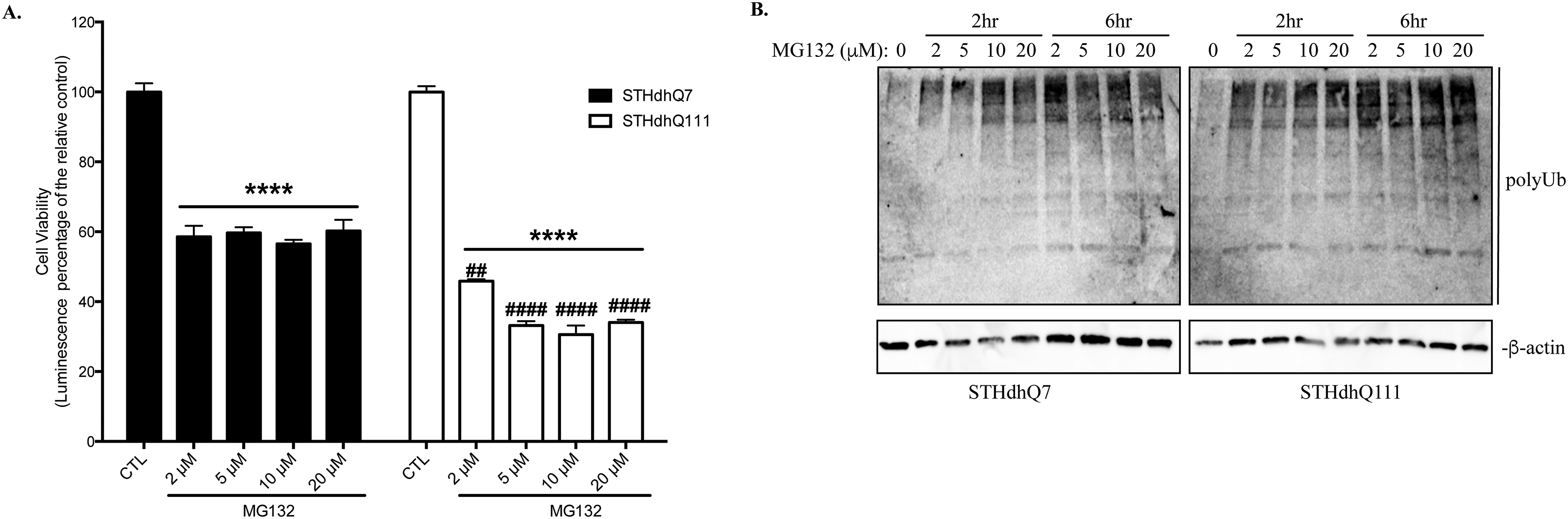
We first examined the survival response of STHdhQ7 and Q111 cells to proteotoxic stress induced by MG132 treatment, a general proteasomal inhibitor. Prolonged MG132 treatment at 10 µM for 16 hours caused significant cell death in both STHdhQ7 and Q111 cells (**** p<0.0001, compared to their relative controls, Fig. 1A). Interestingly, MG132 treatment is more toxic to STHdhQ111 cells than Q7 cells (40% survival in STHdhQ111 vs. 60% survival in STHdhQ7, #### p<0.0001, STHdhQ111 vs. STHdhQ7, Fig. 1A). Two-way ANOVA analysis showed a significant main effect for treatments, F(3, 15)=181.0, p<0.0001; a significant effect for cell types, F(1, 15)=19.74, p=0.0005; and a significant interaction between cell types and treatments, F(3, 15)=169.2, p<0.0001. In contrast, blocking autophagy flux by hydroxychloroquine (CQ) for 16 hours showed less toxicity in both cell lines (Fig. 1A). Co-incubation of CQ with MG132 did not cause further cell death compared to MG132 treatment alone (Fig. 1A). Western blot analysis demonstrated that MG132 but not CQ treatment induced a marked increase of polyUb proteins in both STHdhQ7 and Q111 cells (Fig. 1B–C), indicating that UPS is the primary degradation pathway. Noticeably, STHdhQ111 cells have a higher level of polyUb proteins at the basal level compared to that of STHdhQ7 cells (Fig. 1B–C, #p=0.03), indicating an impairment of UPS activity in STHdhQ111 cells at the basal level. We also tested the effect of different concentrations of MG132 on cell survival after 16 hours of incubation. STHdhQ111 cells are more sensitive to MG132 treatment at all concentrations we tested ranging from 2 to 20 µM (Supplementary Fig. 1A).
Figure 1. HD cells are more vulnerable to MG132-induced proteotoxicity.

A. Prolonged MG132 treatment causes more cell death in STHdhQ111 cells. STHdhQ7 and Q111 cells were incubated with 10 µM MG132 and/or 50 µM CQ for 16 hours as indicated. Cell viability was measured by ATP luminescence assay. ****p<0.0001; **p<0.01, *p<0.05, compared to their relative controls or as indicated. ####p<0.0001; ##p<0.01, STHdhQ111 compared to STHdhQ7 cells in the same treatment group. N=3 for each group. Two-way ANOVA with the Tukey’s post-hoc analysis. B. Representative Western blot from three independent experiments demonstrate that MG132 but not CQ treatment causes a significant accumulation of polyUb proteins in STHdhQ7 and Q111 cells. Both cells were treated with 10 µM MG132 or 50 µM CQ for 16 hours. Cell lysates were analyzed with antibodies against ubiquitin by Western blot. Actin was used as the loading control. C. Quantification of polyUb proteins in B by densitometry. *p<0.05, **p<0.01, compared to their relative controls. #p<0.05, STHdhQ111 vs. STHdhQ7, N=3 for each group. Student’s t-test. D. STHdhQ111 cells are more vulnerable to cell death after removal of proteotoxic stress. Cells were treated with 10 µM MG132 for 6 hours. After incubation, MG132 was removed and cells were further incubated in the complete medium for 18 hours. Cell viability was measured by ATP luminescence assay. ns, not significant; ****p<0.0001, compared to their respective MG132-treated groups. ####p<0.0001, STHdhQ111 compared to Q7 cells in the recovery group. N=7 for CTL and MG132 treated groups, N=10 for recovery groups. Two-way ANOVA with the Tukey’s post-hoc analysis. E. HD fibroblasts (GM04693 and GM05539) are more vulnerable to cell death after stress recovery or prolonged MG132 treatment. Cells were treated with 10 µM MG132 for the indicated time. At the end of the 6-hours incubation, MG132 was removed and cells were further incubated in the complete medium for 18 hours. Cell viability was measured by ATP luminescence assay. ns, not significant; **p<0.001, ***p<0.0001, compared to their respective control groups. ##p<0.001, HD fibroblasts compared to the healthy control in the same treatment group. N=4 for each group. Two-way ANOVA with the Tukey’s post-hoc analysis.
We next asked what happened to cells if we remove the proteotoxic stress. We noticed the presence of many floating cells after prolonged MG132 treatment. To reduce MG132-induced toxicity, we titrated MG132 concentration and incubation time, aiming for finding a point that causes little toxicity but significantly increases the level of polyUb proteins during incubation. While no cell death was detected when cells were incubated with various concentrations of MG132 for 6 hours (data not shown), polyUb proteins were markedly increased when cells were treated with 10 µM MG132 for 2 and 6 hours (Supplementary Fig. 1B). Cells were therefore treated with 10 µM MG132 for 6 hours and then returned to normal medium for 18 hours. While 6 hours of MG132 treatment did not cause a significant cytotoxicity in both cell lines, STHdhQ111 cells exhibited more cell death during recovery (~50% cell death in STHdhQ111 cells vs. 20% cell death in STHdhQ7 cells, #### p<0.0001, Fig. 1D). Two-way ANOVA analysis showed a significant main effect for treatments, F(2, 41)=75.64, p<0.0001; a significant effect for cell types, F(1, 41)=12.51, p=0.001; and a significant interaction between cell types and treatments, F(2, 41)=24.78, p<0.0001. This is also consistent with our microscopic observation that more floating cells were noticed in STHdhQ111 cells after recovery. Importantly, there was no difference in the recovery rate of UPS activity in these two cell lines (Supplementary Fig. S2A–C), ruling out the possibility that the increased cell death during recovery in STHdhQ111 cells is due to the impairment or slower recovery of UPS activity. To confirm our finding in an independent cell line, we further examined the effect of proteotoxic stress in primary fibroblasts from a healthy individual and HD patients. Similarly, there was more cell death in HD fibroblasts (GM04693 and GM05539) during stress recovery and after 24 hours of MG132 inhibition compared to normal fibroblasts (AG02222) (Fig. 1E). Two-way ANOVA analysis showed a significant main effect for treatments, F(3, 36)=11.22, p<0.0001; a significant effect for cell types, F(2, 36)=12.94, p<0.0001. Taken together, our data suggest that HD cells are more vulnerable to cell death during prolonged proteotoxic stress and recovery from the stress.
P62 is transcriptionally up-regulated in normal and HD cells under MG132 treatment
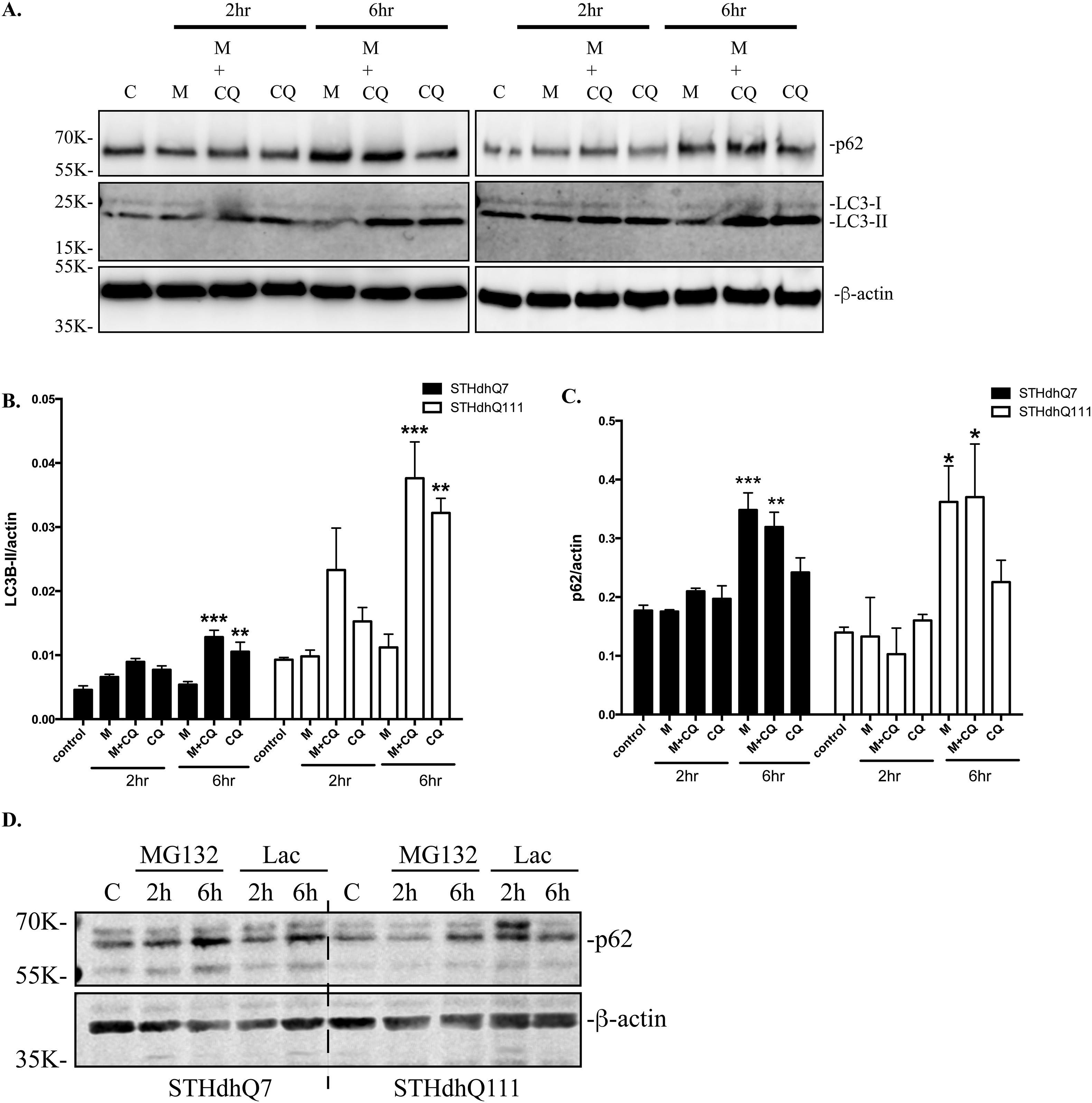
It was reported that proteasome inhibition induces autophagy as a compensatory response by increasing p62 levels (Kuusisto et al. 2001). We next analyzed the expression of p62 in STHdhQ7 and Q111 cells after MG132 treatments. Six hours of MG132 treatment caused a significant increase of p62 levels in both STHdhQ7 and Q111 cells (Fig. 2A–B). This up-regulation was blocked by cycloheximide (CHX), a general protein synthesis inhibitor, suggesting that p62 synthesis was increased (Fig. 2A). To further investigate whether changes in autophagy plays a role in regulating p62 levels, we measured autophagic influx in MG132-treatd cells. Using LC3, a specific autophagosomal marker, we showed that MG132 treatment did not affect LC3-II levels in STHdhQ7 and STHdhQ111 cells (supplementary Fig. S3A–B). In contrast, co-treatment of MG132 and CQ, especially for 6 hours, caused a significant increase in LC3-II levels (supplementary Fig. S3A–B). This increase is comparable to the group treated with CQ only, suggesting that MG132 treatment did not affect autophagy influx in STHdhQ7 and Q111 cells. In contrast, p62 levels were markedly increased in MG132 treated group compared to CQ-treated group in both cells (Supplementary Fig. S3A and S3C), further suggesting that the increase of p62 in MG132-treated group is not due to the blockage in autophagic influx. Consistently, p62 mRNA levels were significantly increased after MG132 treatment in both groups (Fig. 2C). Furthermore, we demonstrated that another proteasomal inhibitor, lactacystin, also caused a significant increase in p62 levels, indicating that the observed increase of p62 levels is not specific to MG132 treatment, but a general response to UPS impairment (Supplementary Fig. S3D). Together, our data suggest that p62 is significantly up-regulated at the transcriptional level in response to the proteotoxic stress and it is unlikely that this effect plays an autophagic role.
Figure 2. p62 is transcriptionally up-regulated in response to proteotoxic stress.

A. Representative Western blot analysis of p62 levels in response to MG132 treatment. Cells were treated with 10 µM MG132 in the presence or absence of 10 µg/ml cycloheximide (CHX) as indicated for 6 hours. Actin was used as the loading control. B. Quantification of p62 expression by densitometry. Cells were treated with 10 µM MG132 for 6 hours. * p<0.05; ** p<0.01, compared to their respective controls. N=9–10 for each group. Student’s t-test. C. Quantitative RT-PCR measurement of p62 mRNA. Cells were incubated with MG132 for 6 hours. ** p<0.01, Two-way ANOVA with the Tukey’s post-hoc analysis. There is a significant main effect for treatment, F(1, 12)=17.82, p=0.0012; no significant effect for cell types, F(1, 12)=1.537, p=0.2387 and no significant interaction between cell types and treatments, F(1, 12)=1.668, p=0.2208.
P62 displays distinct subcellular localization patterns in normal and HD cells under MG132 treatment
To further explore the functional indications of p62 up-regulation in response to proteotoxic stress in HD cells, we investigated the subcellular localization of p62. As an autophagic adaptor, p62 is normally associated with late autophagosomes or lysosomes labeled by Lamp1, a specific lysosomal marker (Fig. 3A and 3D, Supplementary Fig. S4A and B, left panels). Interestingly, p62 bodies were located outside of lysosomes in STHdhQ7 cells after 2-hr MG132 treatment. In contrast, most p62 puncti in STHdhQ111 cells were still sequestered in lysosomes (Fig. 3B vs. 3E). Co-localization analysis of p62 and Lamp1 further confirmed that MG132 treatment significantly reduced co-localization efficiency of p62 and Lamp1 in STHdhQ7 cells but not Q111 cells (Fig. 3G). Two-way ANOVA analysis showed a significant interaction between cell types and treatments, F(1, 19)=7.037, p=0.0157. Notably, MG132 treatment caused an increase in lysosomal size or swelling in STHdhQ111 cells (Fig. 3E, supplementary Fig. S4C). This is similar to what we observed in CQ treatment (Supplementary Fig. S4A–B, middle panels), which typically induces lysosomal swelling (King et al. 2016).
Figure. 3. P62 displays distinct subcellular localization patterns in response to MG132 treatment in STHdhQ7 and Q111 cells.

A–F. Representative confocal images of p62 and Lamp1 staining in STHdhQ7 (A–C) and Q111 (D–F) cells at basal conditions (A and D), after 2 hours (B and E) and 6 hours (C and F) of MG132 treatment. G. Manders’ co-localization efficiency analysis for the percentage of p62 puncti that are associated with Lamp1 after 2 hours of MG132 treatment in STHdhQ7 and Q111 cells. N=6–10 cells per group. **p<0.01, Two-way ANOVA with the Tukey’s post-hoc analysis. There is a significant interaction between cell types and treatments, F(1, 19)=7.037, p=0.0157. H. Quantitative analysis of the size of p62 puncti in STHdhQ7 and Q111 cells after 6 hours of MG132 treatment. A total of 20 puncti were analyzed per group. ****p<0.0001, student’s t-test. I. Quantitative analysis of the percentage of cells with p62 macroaggregates in STHdhQ7 and Q111 cells after 6 hours of MG132 treatment. A total of 10 different image fields (~300 cells) were analyzed per group. ****p<0.0001, student’s t-test. Right panel shows representative confocal images of p62 staining in STHdhQ7 and Q111 cells after 6 hours of MG132 treatment at a lower magnification.
When MG132 treatment was extended to 6 hours, p62 bodies accumulated to form juxtanuclear macroaggregates in STHdhQ111 cells while they were still largely dispersed in STHdhQ7 cells (Fig. 3C vs. 3F). We thus quantified the size of p62-positive macroaggregates in both cell lines and found a significant difference (0.553±0.022 µm in STHdhQ7 cells compared to 3.126±0.081 µm in STHdhQ111 cells, Fig. 3H). It is estimated that ~40% of STHdhQ111 cells had p62 macroaggregates (Fig. 3I). When MG132 treatment was extended to 16 hours, ~80–90% of STHdhQ111 but not STHdhQ7 cells had large, round macroaggregates (Supplementary Fig. S4A–B, right panels). These p62 aggregates co-localized with γ-tubulin, indicating that they were aggresomes (Supplementary Fig. S4D–E). Importantly, mHtt but not normal htt formed aggregates co-localized with p62 under MG132 treatment (Supplementary Fig. S4F). Collectively, these findings suggest that p62 responds differently to UPS inhibition in terms of subcellular localization in STHdhQ7 and Q111 cells.
P62 is not associated with K48-linked polyUb protein aggregates and proteasomal proteins in HD cells under proteotoxic stress
Using specific antibodies against K48- or K63-linked polyUb chain, we further explored the nature of the increased polyUb proteins. Consistent with the role of MG132 in UPS inhibition, a significant increase of K48-, but not K63-linked, polyUb proteins was observed in both cell lines at 2 hours of MG132 treatment and thereafter (Fig. 4A). We next investigated the subcellular distribution of K48-linked polyUb proteins. Compared to the punctate aggregates formation in STHdhQ7 cells, K48-linked polyUb proteins had a more diffuse appearance in STHdhQ111 cells (Fig. 4C vs. 4F, Supplementary Fig. S5). K48-linked polyUb protein aggregates were strongly correlated with p62 puncti in STHdhQ7 but not Q111 cells after 2 hours of MG132 treatment (Fig. 4C vs. 4F). Quantitative analysis using Manders’ co-localization coefficiency indicates that about 50% K48-positive puncti were associated with p62 in STHdhQ7 cells compared to that of 17% in Q111 cells (Fig. 4H, ## p<0.01). Further treatment with MG132 for 6 hours facilitated the formation of K48-linked polyUb protein aggregates as less diffused polyUb staining was observed in STHdhQ7 cells (Fig. 4D vs. 4C). This is also reflected by the increase in Manders’ colocalization coefficiency from 0.5 to 0.7 (Fig. 4H, **p<0.01). As MG132 treatment continued, K48-linked polyUb proteins and p62 co-localized to juxtanuclear macroaggregates in STHdhQ111 cells (Fig. 4G vs. 4F), which caused a marked increase in Manders’ coefficiency (Fig. 4H, ****p<0.0001). Two-way ANOVA analysis showed a significant interaction between cell types and treatments, F(1, 30)=21.67, p<0.0001. In both cell types, p62 puncti contained K63-linked polyUb proteins (Supplementary Fig. S6), consistent with the report that p62 has a higher affinity for K63-linked proteins.
Figure 4. Correlation of p62 with K48-linked polyUb proteins in response to MG132 treatment in STHdhQ7 and Q111 cells.

A. Representative Western blot analysis of the expression of K48-linked and K63-linked polyUb proteins in STHdhQ7 and Q111 cells after MG132 treatment. Actin was used as a loading control. M2, MG132 treatment for 2 hours; M6, MG132 treatment for 6 hours. B–G. Representative confocal images showing the localization of p62 puncti with K48-linked polyUb protein aggregates in STHdhQ7 (B–D) and Q111 (E–G) cells at basal conditions (B and E), after 2 (C and F) and 6 (D and G) hours of MG132 treatment. H. Manders’ co-localization coefficiency analysis for the percentage of K48 polyUb protein puncti that are positive with p62 staining in STHdhQ7 and Q111 cells after MG132 treatment. N=6–13 cells per group. **p< 0.01, ****p<0.0001, ## p<0.01 (STHdhQ111 2hr vs. STHdhQ7 2hr), two-way ANOVA with the Tukey’s post-hoc analysis.
We then investigated whether p62 targets K48-linked polyUb proteins to UPS or autophagosomes for degradation under MG132 treatment. It is difficult to determine whether the co-localization of p62 with K48-linked polyUb protein aggregates in STHdhQ111 cells after 6 hours of MG132 treatment has any functional indication (Fig. 4G). It is possible that this is just a precipitating effect as mHtt also formed aggregates with p62 (Supplementary Fig. S4F). We therefore focused on the interaction of p62 and proteasomal proteins at 2 hours of MG132 treatment. We did not detect a significant increase of LC3 expression after MG132 treatment by western blot (Supplementary Fig. S3A) or immunofluorescent staining (data not shown). Instead, p62 puncti were strongly associated with the 20S proteasomal subunit in STHdhQ7 but not Q111 cells after MG132 treatment (Fig. 5A vs. 5B). Quantitative analysis indicates a significant decrease in the percentage of p62 puncti positive for 20S proteasomal subunit in STHdhQ111 compared to Q7 cells (Fig. 5C). In line with this, p62 puncti were associated with ubiquilin-2 (UBQLN2), a proteasome shuttle factor in STHdhQ7 but not Q111 cells (Fig. 5D vs. 5E, 5F). Since p62 is associated with K48-linked polyUb protein aggregates in STHdhQ7 cells (Fig. 4B), it is expected that K48-linked polyUb protein aggregates are associated with proteasomes in STHdhQ7 but not Q111 cells (Supplementary Fig. S7). These data collectively suggest that p62 binds to K48-linked polyUb protein aggregates and target them to proteasomes in STHdhQ7 cells but failed to do so in Q111 cells upon UPS inhibition.
Figure 5. p62 puncti are associated with proteasomes in STHdhQ7 cells but not Q111 cells after MG132 treatment.

A–B. Representative confocal images showing that p62 puncti co-localize with the 20S proteasomal subunit in STHdhQ7 (A) but not Q111 (B) cells after 2 hours of MG132 treatment (right panels). The subcellular localization of p62 and 20S proteasomal subunit in untreated cells are also shown in the left panels. C. Manders’ co-localization coefficiency analysis for the percentage of p62 puncti that are positive for 20S proteasomal subunit. N=9 cells per group. ****p<0.0001, student’s t-test. D–E. Representative confocal images showing that p62 puncti co-localize with UBQLN2 in STHdhQ7 (D) but not Q111 (E) cells after 2 hours of MG132 treatment (right panels). The subcellular localization of p62 and UBQLN2 in untreated cells is also shown in the left panels. F. Manders’ co-localization coefficiency analysis for the percentage of p62 puncti that are positive for UBQLN2. N=10–11 cells per group. ****p<0.0001, student’s t-test.
Cysteine protects against stress recovery induced cell death and inhibits p62 macroaggregates formation in STHdhQ111 cells after MG132 treatment
It was reported that failure of amino acid homeostasis, especially the reduction of cysteine and aspartate in mammalian cells, causes cell death following proteasome inhibition (Suraweera et al. 2012). We therefore investigated whether cysteine can rescue cell death in STHdhQ111 cells during stress removal. Interestingly, we found that addition of cysteine during the MG132 incubation phase but not the recovery phase significantly rescued stress recovery induced cell death in STHdhQ111 cells (Fig. 6A, #### p<0.0001). The protective effect of cysteine on STHdhQ7 cells was not significant (Fig. 6A). Two-way ANOVA analysis showed a significant interaction between cell types and treatments, F(3, 65)=12.45, p<0.0001. Therefore, our data suggest that some molecular changes happened during MG132 treatment predispose STHdhQ111 cells to cell death after stress recovery and the protective effect of cysteine occurs during the MG132 incubation phase. We here further analyzed the effect of cysteine treatment on the subcellular localization of p62 in STHdhQ111 cells. Compared to STHdhQ111 cells after 6 hours of MG132 treatment (Fig. 6B), co-incubation with cysteine significantly reduced the p62 macroaggregates formation (Fig. 6C). Quantitative analysis indicates the percentage of STHdhQ111 cells having p62 macroaggregates reduced to 14% in the presence of cysteine (Fig. 6D).
Figure 6. Cysteine rescues proteotoxic stress recovery induced cell death in STHdhQ111 cells.

A. STHdhQ7 and Q111 cells were incubated with 10 µM MG132 in the presence or absence of 1mM cysteine for 6 hours. After incubation, drugs were removed and cells were further incubated in complete medium in the presence or absence of 1 mM cysteine for 18 hours. Cell viability was measured by ATP luminescence assay. ****p<0.0001, compared to their relative controls. N=6–11 for each group. Two-way ANOVA with the Tukey’s post-hoc analysis. B–C. Representative confocal images demonstrating the subcellular localization of p62 and K48-linked polyUb proteins in STHdhQ111 cells after 6 hours of MG132 treatment in the absence (B) or presence (C) of 1 mM cysteine. D. Quantitative analysis of the percentage of cells with p62 macroaggregates in STHdhQ111 cells after 6 hours of MG132 treatment in the absence or presence of 1 mM cysteine. A total of 3 different image fields (~300 cells) were analyzed per group. **p< 0.01, student’s t-test.
DISCUSSION
Various stresses, including oxidative stress (Niedzielska et al. 2016), proteotoxic stress (Morimoto 2008) and ER stress (Lindholm et al. 2006) play major roles in pathophysiological processes associated with neurodegenerative diseases and mental disorders. Neurons are particularly sensitive to the dysregulation of proteostasis. Protein misfolding is a common theme in many neurodegenerative diseases, including HD, leading to gradual proteasomal dysfunction and base-line accumulation of polyUb proteins. However, the effect of acute proteotoxic stress on HD cells is less investigated. Using MG132 to induce proteotoxic stress, our data collectively suggest that HD cells are more susceptible to MG132-induced cell death, especially during recovery from the stress. This finding is physiologically important as neurons are constantly facing repeated exposure to proteotoxic stress induced by various external stimuli. HD neurons may lose their capability to adapt to and recover from the stress, increasing the vulnerability of HD neurons to additional insults. Our cell viability test after stress recovery thus has further implications in screening for potential neuroprotective drug targets for HD treatment as a cell-based high-throughput assay. Since there is no difference in the recovery rate of UPS activity, it is likely that the molecular causes for the increased vulnerability of HD cells lie within the MG132 incubation phase and is therefore the focus of the present study. However, the molecular changes during stress recovery need to be further investigated.
Increasing evidence support the presence of a coordinated and complementary interaction between UPS and autophagy that becomes critical upon cellular stress (Nedelsky et al. 2008). P62 has been suggested as one of such regulators (Bjørkøy et al. 2005). In the present study, we show that p62 was up-regulated in response to proteasomal inhibition. This increase is not due to changes in autophagic influx as evidenced by minimal changes in LC3-II levels. This is in consistent with the report that proteasomal inhibition does not change LC3-II levels in primary neurons (Mitra et al. 2009). We observed an increase of p62 mRNA expression in normal and HD cells in response to UPS inhibition, indicating that p62 is transcriptionally up-regulated and the underlying mechanism is not affected in HD cells. In line with this, proteotoxic stress induced by blocking UPS activity transcriptionally up-regulates p62 in neuronal cells (Kuusisto et al. 2001). It is suggested that p62 is regulated at the transcriptional level possibly through activation of its transcriptional factor Nrf2 (NF-E2-related factor 2) (Jain et al. 2010).
The most significant finding in this study is the distinct subcellular localization of p62 in normal and HD cells upon UPS inhibition. P62 accumulates and forms larger aggresome-like structures in STHdhQ111 cells as proteasomal inhibition continues. These p62 bodies contain polyUb proteins and mutant huntingtin. It is not clear whether p62 plays a direct role in aggresome formation. It has been reported that knocking down p62 does not interfere with aggresome formation in Neuro2A cells after MG132 treatment, suggesting that p62 recruitment is secondary to aggresome formation and may cause a loss of its protective function (Nagaoka et al. 2004). Therefore, we focused on analyzing the nature of p62 bodies after 2 hours of MG132 treatment in the present study, when there were no p62 macroaggregates formed.
P62 interacts with LC3 and targets K63-linked polyUb proteins to autophagosomes for subsequent lysosomal degradation under physiological conditions (Pankiv et al. 2007). Recent studies demonstrate that p62 also acts as a shuttle protein for proteasomal degradation. For example, it was reported that p62 interacts with the 19S regulatory particle of the proteasome and may directly deliver K63-linked polyUb proteins for proteasomal degradation (Seibenhener et al. 2004), such as tau protein (Babu et al. 2005). P62 also binds K48-linked polyUb Chain, although it has a preference for K63-linked polyUb chain (Wurzer et al. 2015; Matsumoto et al. 2011; Seibenhener et al. 2004). However, the interaction of p62 with K48-linked polyUb proteins has not been well characterized. What is the significance of a marked increase of K48-linked polyUb protein with a concomitant increase of p62 in STHdhQ7 and Q111 cells upon UPS inhibition? Here we found that p62 bodies contain K48-linked polyUb protein aggregates in STHdhQ7 but not Q111 cells. We further demonstrate that p62 bodies are associated with ubiquilin-2 and the 20S proteasomal subunit in STHdhQ7 but not Q111 cells. It is thus reasonable to propose that K48-linked polyUb protein aggregates in STHdhQ7 cells are cleared by the UPS after recovery from proteotoxic stress, which leads to the survival of these cells. However, this process is impaired in STHdhQ111 cells, leading to increased cell death. Consistent with this idea, K48-linked polyUb proteins are usually targeted for proteasomal degradation (Clague and Urbé 2010) and protein aggregates caused by proteotoxic stress can be cleared through the UPS after removal of the stress, which is autophagy-independent and mediated by ubiquilin-2 (Hjerpe et al. 2016).
Studies of how mHtt interferes with p62 signaling are complicated by the fact that mHtt serves as both a substrate and a regulator of p62. Most studies have focused on the role of p62 in facilitating the autophagic clearance of mHtt aggregates. It was reported that endogenous or overexpressed p62 forms a shell-like structure around mHtt aggregates in HeLa cells expressing the N-terminus of mHtt and protects against mHtt-induced cell death (Bjørkøy et al. 2005). Moreover, overexpression of mHtt in HeLa cells induces p62 phosphorylation at Ser409, which in turn promotes the clearance of mHtt aggregates when autophagy is enhanced with rapamycin treatment (Lim et al. 2015). While these studies clearly demonstrate that p62 delivers mHtt aggregates for autophagic degradation, the actual clearance process has not been captured. On the other hand, Htt regulates selective autophagy by physically interacting with p62. The C-terminal region of normal Htt interacts with p62 and facilitates p62-mediated cargo recognition by promoting its association with LC3 and K63-linked polyUb substrates (Rui et al. 2015). mHtt may impair p62-mediated cargo recognition as supported by the evidence showing that “empty autophagosomes” are present in different HD cell types including the STHdhQ111 cell line (Martinez-Vicente et al. 2010). Interestingly, knocking down Htt expression by RNA interference reduced the formation of p62 bodies in HeLa cells upon MG132 inhibition (Rui et al. 2015), indicating that normal Htt is important for forming dispersed p62 bodies upon MG132 inhibition. In contrast, mHtt may facilitate the oligomerization of p62 through PB1 domain, which leads to the formation of non-functional p62 macroaggregates, as demonstrated by the co-localization of mHtt aggregates with p62 aggregates (Supplementary Fig. S4F). Histone deacetylase 6 (HDAC6) is important for aggresome formation by retrogradely transport protein aggregates to the perinuclear regions via microtubule (Kawaguchi et al. 2003; Ouyang et al. 2012). Interestingly, p62 interacts with HDAC6 and regulates its activity. Loss of p62 hyper-activates HDAC6 and impairs autophagic degradation (Yan 2014). Taken together, it is suggested that p62 function is comprised in STHdhQ111 cells despite the fact that aggresomes are formed in response to proteotoxic stress. As a result, these aggresomes cannot be effectively cleared out through autophagy even after removal of the stress. To support this notion, co-incubation of autophagic inducers, such as rapamycin, during the recovery period could not rescue the stress-induced cell death in STHdhQ111 cells (data not shown).
The molecular basis for aberrant p62 localization in HD cells upon UPS inhibition is yet to be determined. Our finding that co-incubation of cysteine with MG132 rescues cell death during stress recovery in HD cells is of particular interest. It was reported that the reduction of cysteine and aspartate caused by MG132 inhibition further initiates the integrated stress response and autophagy. However, the attempt to replenish the pool of intracellular amino acids is unsuccessful, leading to cell death (Suraweera et al. 2012). Notably, impaired cysteine homeostasis has been reported in HD. Cystathionine γ-lyase, the biosynthetic enzyme for cysteine, is severely depleted in HD, rendering SThdhQ111 cells more vulnerable to cysteine-depleted medium (Paul et al. 2014). R6/2 HD transgenic mice that were placed on a cysteine-rich diet had improved motor functions and longer life span (Paul et al. 2014). It was recently demonstrated that reduced cysteine in HD promotes oxidative stress and causes the dysfunction of activating transcription factor 4 (ATF4), the master regulator of amino acid metabolism (Sbodio et al. 2016). It is thus reasonable to speculate that cysteine levels are more affected in HD cells upon proteasome inhibition, which induces some unknown stress responses that affect the subcellular localization of p62.
CONCLUSIONS
In summary, our data suggest that HD neurons fail to maintain effective stress recovery mechanisms compared to normal neurons, which causes accumulation of cellular damages over time and eventually neurodegeneration. Further investigation of the underlying molecular mechanisms is important for therapeutic benefits.
Supplementary Material
Supplementary Figure S1. A. STHdhQ111 cells were more vulnerable to cell death when treated with different concentrations of MG132 for 16 hours. Cell viability was measured by ATP luminescence assay. ****p<0.0001, compared to their relative controls or as indicated. ####p<0.0001, ##p<0.01, STHdhQ111 compared to STHdhQ7 cells in the same treatment group. N=3 for each group. Two-way ANOVA with the Tukey’s post-hoc analysis. B. Dosage and time-dependent analysis of polyUb proteins by western blot as indicated. polyUb proteins were detected by anti-K48-linked Ub antibodies. Actin was used as the loading control.
{kind=link}
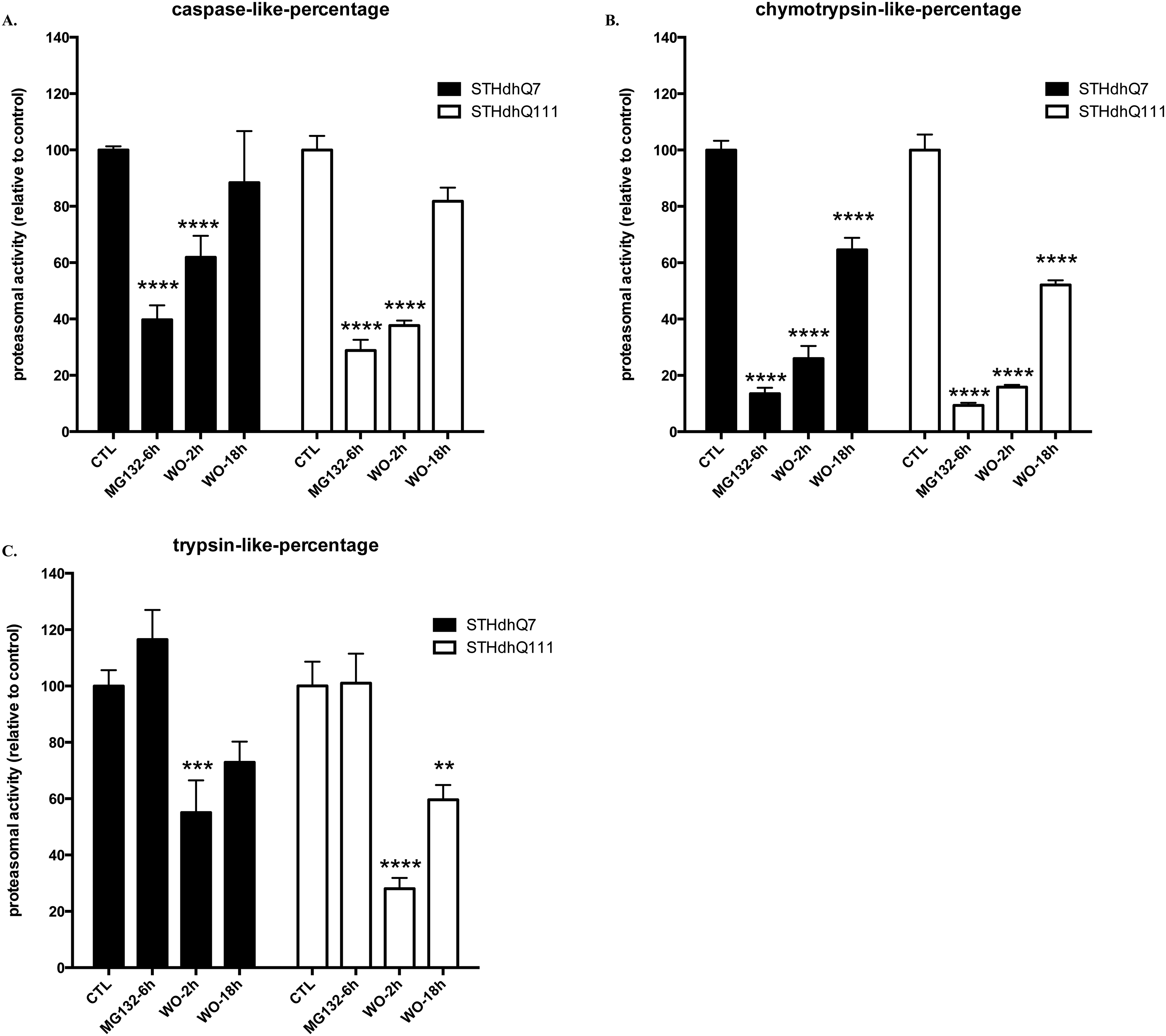
Supplementary Figure S2. A–C. Proteasomal activity assay. Caspase- (A), chymotrypsin- (B) and trypsin-(C) like activities of 20S proteasome were measured at different time intervals as indicated using substrates Suc-LLVY-aminomethycoumarin (AMC), Z-ARR-AMC and Z-LLE-AMC (Calbiochem) respectively, as we previously described (Leon et al. 2010). No difference in the recovery rate of proteasomal activity was observed in STHdhQ7 and Q111 cells after removal of MG132. N= 4–9 per group, ** p<0.01; *** p<0.001; **** p<0.0001 compared to their relative controls. Two-way ANOVA with the Tukey’s post-hoc analysis. For caspase-like activity, there is a significant main effect for treatments, F(3, 47)=83.17, p<0.0001; a significant effect for cell types, F(1, 47)=7.204, p=0.01; and no significant interaction between cell types and treatments, F(3, 47)=1.776, p=0.1646. For Chymotrypsin-like activity, there is a significant main effect for treatments, F(3, 52)=310.5, p<0.0001; a significant effect for cell types, F(1, 52)=7.217, p=0.0097; and no significant interaction between cell types and treatments, F(3, 52)=24.78, p=0.2768. For trypsin-like activity, there is a significant main effect for treatments, F(3, 52)=23.54, p<0.0001; a significant effect for cell types, F(1, 52)=4.849, p=0.0321; and no significant interaction between cell types and treatments, F(2, 52)=0.7930, p=0.5033.
{kind=link}
Supplementary Figure S3. A. Expression of p62 and LC3 in response to MG132 and CQ treatment in STHdhQ7 and Q111 cells. Both cells were treated with 10 µM MG132 and/or 50 µM CQ for 2 and 6 hours. Actin was used as the loading control. B. Quantification of LC3-II expression in A by densitometry. **p<0.01; ***p<0.001, compared to their respective controls. N=3 for each group. One-way ANOVA with the Tukey’s post-hoc analysis. C. Quantification of p62 expression in A by densitometry. *p<0.05; **p<0.01; ***p<0.001, compared to their respective controls. N=3 for each group. One-way ANOVA with the Tukey’s post-hoc analysis. D. Another proteasome inhibitor, lactacystin also induced up-regulation of p62 in both STHdhQ7 and Q111 cells. Cells were incubated with 5 µM lactacystin for the indicated time. P62 expression was analyzed by western blot. Actin was used as the loading control.
{kind=link}
Supplementary Figure S4. A–B. Representative confocal images showing the subcellular localization of p62 in response to CQ and MG132 treatment in STHdhQ7 (A) and Q111 (B) cells. Cells were treated with 50 µM CQ or 10 µM MG132 for 16 hours, fixed with methanol and co-stained with Lamp1 and p62 antibodies. C. Representative confocal images of p62 and Lamp1 staining in STHdhQ7 and Q111 cells after 2 hours of MG132 treatment. Note that lysosomes were swelled in STHdhQ111 cells. D–E. Representative confocal imaging showing the identification of p62 macroaggregates as aggresomes. p62 aggregates is co-localized to γ-tubulin, an aggresomal marker, in STHdhQ7 and Q111 cells treated with MG132 for 6 hours. F. Representative confocal imaging showing the co-localization of p62 macroaggregates with mHtt in STHdhQ111 but not Q7 cells. Cells were treated with MG132 for 6 hours and fixed in 4% paraformaldehyde and permeablized with 0.25% Triton X-100 and stained with anti-huntingtin and p62.
{kind=link}
Supplementary Figure S5. Representative confocal images showing the formation of K48-linked polyUb protein aggregates in STHdhQ7 (A) but not Q111 cells (B) after MG132 treatment for 2 hours.
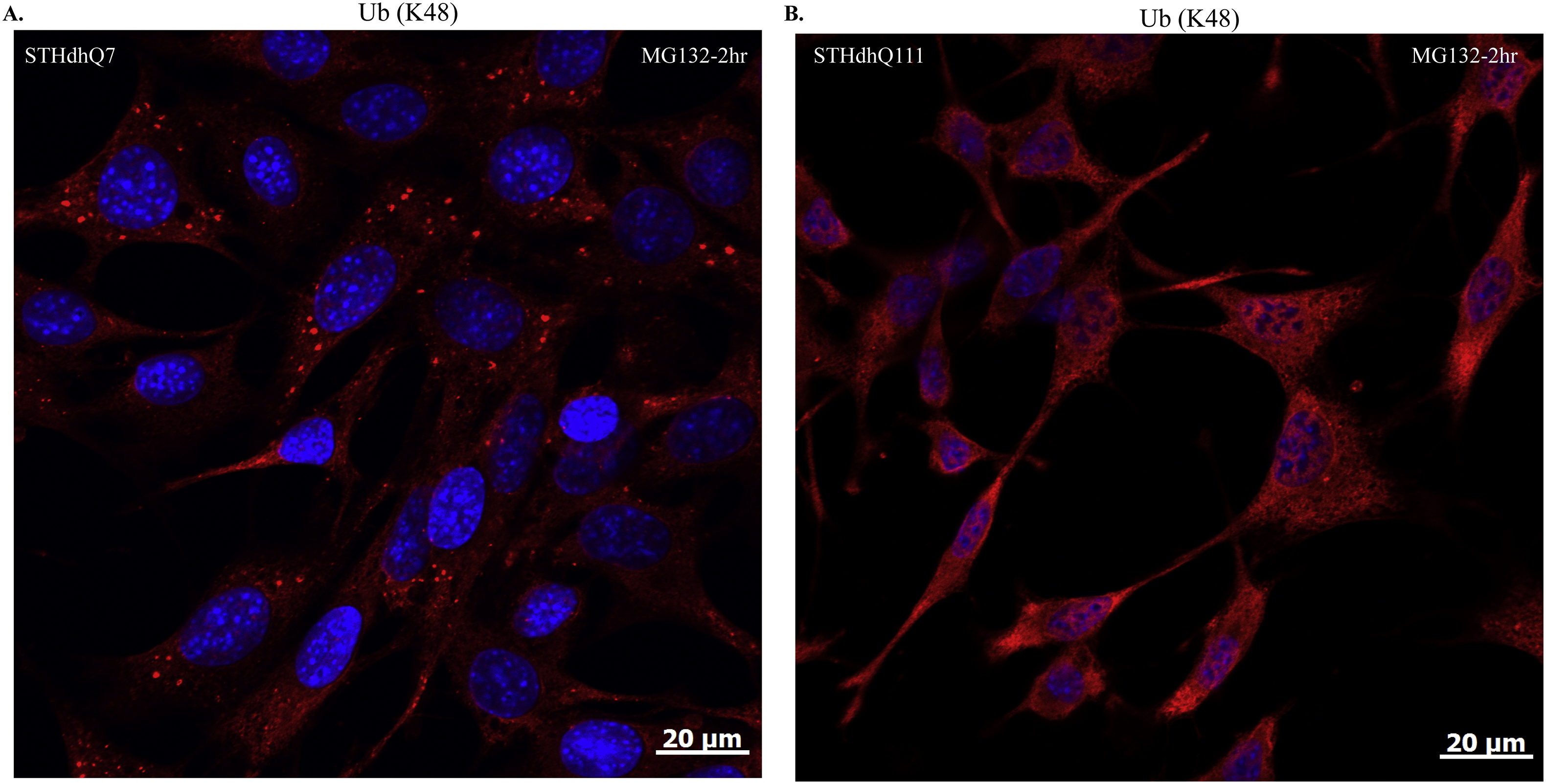{kind=link}
Supplementary Figure S6. Representative confocal images showing the co-localization of p62 with K63-linked polyUb proteins in STHdhQ7 (A–B) and Q111 (C–D) cells that were untreated (A and C) or after 2 hours of MG132 treatment (B and D).
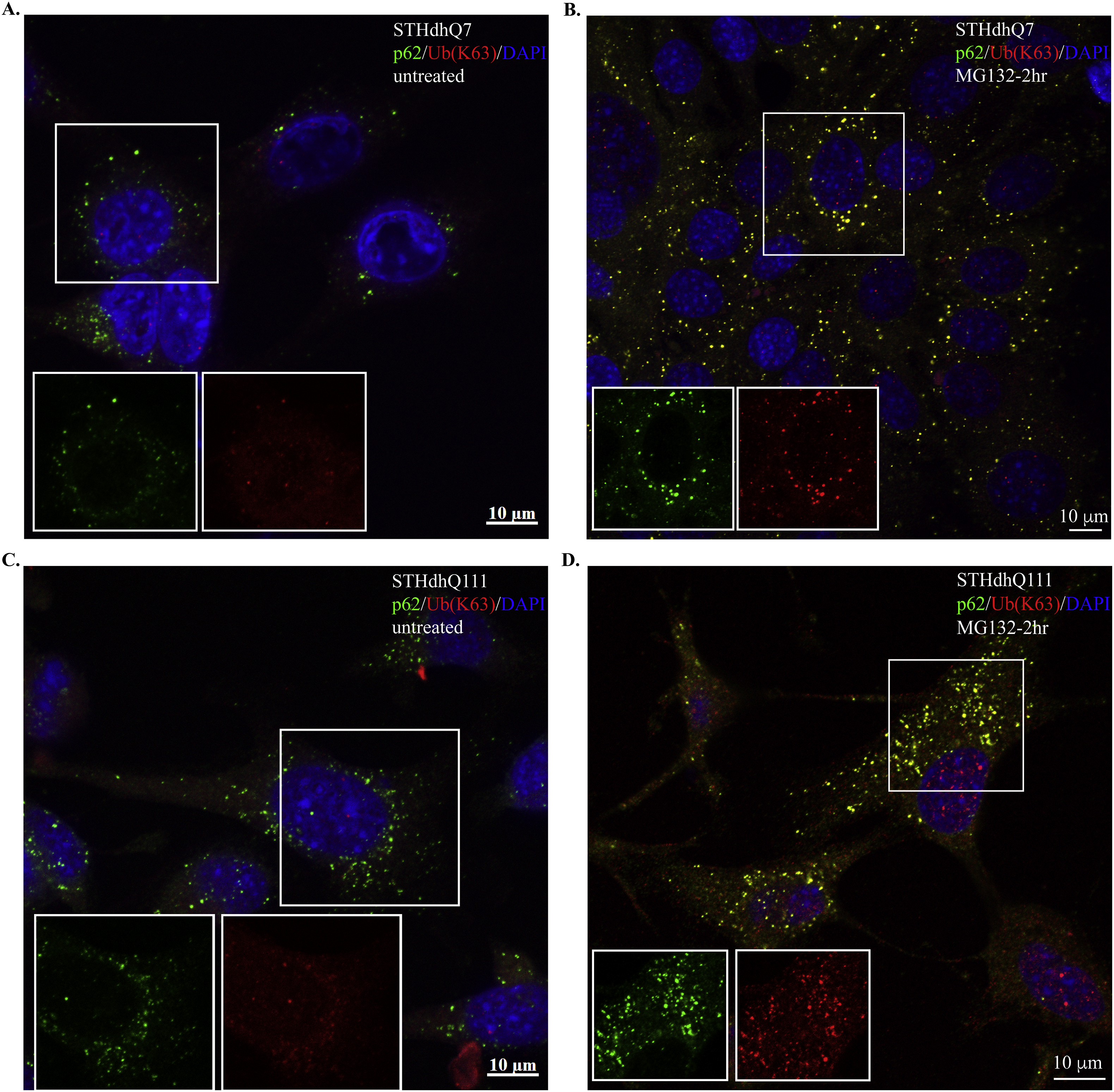{kind=link}
Supplementary Figure S7. Representative confocal images showing the subcellular localization of UBQLN2 and K48-linked polyUb proteins in untreated (A–B) and MG132-treated (C–D) STHdhQ7 and Q111 cells. Co-localization of UBQLN2 with K48-linked polyUb protein aggregates was detected in STHdhQ7 (C) but not Q111 (D) cells after 2 hours of MG132 treatment.
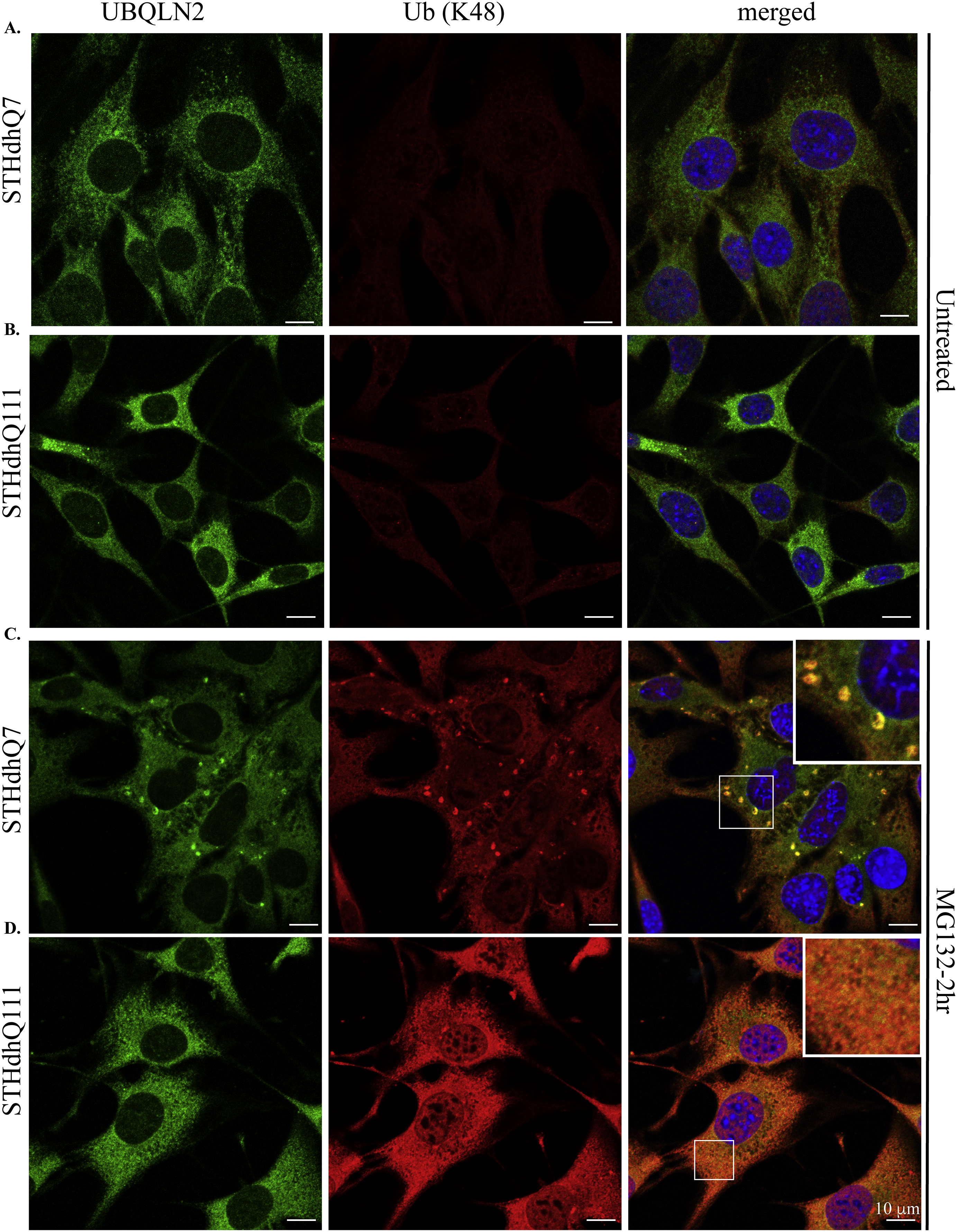{kind=link}
Highlights.
HD cells are more vulnerable to cell death induced by proteotoxic stress.
HD cells are more vulnerable to cell death during stress recovery.
P62 is up-regulated in response to proteotoxic stress
P62 displays distinct subcellular localization in normal and HD cells under stress
Cysteine protects against stress-induced cell death in HD cells
Acknowledgments
This work was supported by National Institute of Neurological Disorders and Stroke (NS066339-02 to J.W.).
Abbreviations
- ATF4
activating transcription factor 4
- BSA
bovine serum albumin
- CHX
cycloheximide
- CQ
hydroxychloroquine
- HD
Huntington’s disease
- HDAC6
histone deacetylase 6
- Htt
huntingtin
- mHtt
mutant huntingtin
- Lamp1
lysosomal associated membrane protein 1
- LC3
microtubule-associated protein light chain 3
- Nrf2
NF-E2-related factor 2
- PB1
Phox and Bem1p domain
- polyUb
polyubiquitinated
- SQSTM1
sequestosome 1
- Ub
ubiquitin
- UNQLN2
ubiquilin-2
- UBA
ubiquitin-binding domain
- ULK1
unc-51 like autophagy activating kinase 1
- UPS
ubiquitin-proteasome system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
There are no conflicts of interest to be reported.
References
- Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Meth. Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- Clague MJ, Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–685. doi: 10.1016/j.cell.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Erie C, Sacino M, Houle L, Lu ML, wei J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington's disease. Eur. J. Neurosci. 2015;42:1941–1951. doi: 10.1111/ejn.12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, Sahu I, et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell. 2016;166:935–949. doi: 10.1016/j.cell.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282:4672–4678. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao T-P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- King MA, Ganley IG, Flemington V. Inhibition of cholesterol metabolism underlies synergy between mTOR pathway inhibition and chloroquine in bladder cancer cells. Oncogene. 2016;35:4518–4528. doi: 10.1038/onc.2015.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuusisto E, Suuronen T, Salminen A. Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem. Biophys. Res. Commun. 2001;280:223–228. doi: 10.1006/bbrc.2000.4107. [DOI] [PubMed] [Google Scholar]
- Leon R, Bhagavatula N, Ulukpo O, McCollum M, wei J. BimEL as a possible molecular link between proteasome dysfunction and cell death induced by mutant huntingtin. Eur. J. Neurosci. 2010;31:1915–1925. doi: 10.1111/j.1460-9568.2010.07215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebl MP, Hoppe T. It's all about talking: two-way communication between proteasomal and lysosomal degradation pathways via ubiquitin. Am. J. Physiol., Cell Physiol. 2016;311:C166–78. doi: 10.1152/ajpcell.00074.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Lachenmayer ML, Wu S, Liu W, Kundu M, Wang R, Komatsu M, Oh YJ, Zhao Y, Yue Z. Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates. PLoS Genet. 2015;11:e1004987. doi: 10.1371/journal.pgen.1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat. Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Mitra S, Tsvetkov AS, Finkbeiner S. Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J. Biol. Chem. 2009;284:4398–4403. doi: 10.1074/jbc.M806269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka U, Kim K, Jana NR, Doi H, Maruyama M, Mitsui K, Oyama F, Nukina N. Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J. Neurochem. 2004;91:57–68. doi: 10.1111/j.1471-4159.2004.02692.x. [DOI] [PubMed] [Google Scholar]
- Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta. 2008;1782:691–699. doi: 10.1016/j.bbadis.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, Filip M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016;53:4094–4125. doi: 10.1007/s12035-015-9337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang H, Ali YO, Ravichandran M, Dong A, Qiu W, MacKenzie F, Dhe-Paganon S, Arrowsmith CH, Zhai RG. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 2012;287:2317–2327. doi: 10.1074/jbc.M111.273730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, Overvatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Park C, Cuervo AM. Selective autophagy: talking with the UPS. Cell Biochem. Biophys. 2013;67:3–13. doi: 10.1007/s12013-013-9623-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Sbodio JI, Xu R, Vandiver MS, Cha JY, Snowman AM, Snyder SH. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington's disease. Nature. 2014;509:96–100. doi: 10.1038/nature13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, Ross CA. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle. 2012;11:2006–2021. doi: 10.4161/cc.20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y-N, Xu Z, Patel B, Chen Z, Chen D, Tito A, David G, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015;17:262–275. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbodio JI, Snyder SH, Paul BD. Transcriptional control of amino acid homeostasis is disrupted in Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 2016;113:8843–8848. doi: 10.1073/pnas.1608264113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 Is a Polyubiquitin Chain Binding Protein Involved in Ubiquitin Proteasome Degradation. Mol. Cell. Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera A, Münch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell. 2012;48:242–253. doi: 10.1016/j.molcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, Martens S. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife. 2015;4:e08941. doi: 10.7554/eLife.08941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J. Interplay between HDAC6 and its interacting partners: essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 2014;33:567–580. doi: 10.1089/dna.2013.2300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. A. STHdhQ111 cells were more vulnerable to cell death when treated with different concentrations of MG132 for 16 hours. Cell viability was measured by ATP luminescence assay. ****p<0.0001, compared to their relative controls or as indicated. ####p<0.0001, ##p<0.01, STHdhQ111 compared to STHdhQ7 cells in the same treatment group. N=3 for each group. Two-way ANOVA with the Tukey’s post-hoc analysis. B. Dosage and time-dependent analysis of polyUb proteins by western blot as indicated. polyUb proteins were detected by anti-K48-linked Ub antibodies. Actin was used as the loading control.
Supplementary Figure S2. A–C. Proteasomal activity assay. Caspase- (A), chymotrypsin- (B) and trypsin-(C) like activities of 20S proteasome were measured at different time intervals as indicated using substrates Suc-LLVY-aminomethycoumarin (AMC), Z-ARR-AMC and Z-LLE-AMC (Calbiochem) respectively, as we previously described (Leon et al. 2010). No difference in the recovery rate of proteasomal activity was observed in STHdhQ7 and Q111 cells after removal of MG132. N= 4–9 per group, ** p<0.01; *** p<0.001; **** p<0.0001 compared to their relative controls. Two-way ANOVA with the Tukey’s post-hoc analysis. For caspase-like activity, there is a significant main effect for treatments, F(3, 47)=83.17, p<0.0001; a significant effect for cell types, F(1, 47)=7.204, p=0.01; and no significant interaction between cell types and treatments, F(3, 47)=1.776, p=0.1646. For Chymotrypsin-like activity, there is a significant main effect for treatments, F(3, 52)=310.5, p<0.0001; a significant effect for cell types, F(1, 52)=7.217, p=0.0097; and no significant interaction between cell types and treatments, F(3, 52)=24.78, p=0.2768. For trypsin-like activity, there is a significant main effect for treatments, F(3, 52)=23.54, p<0.0001; a significant effect for cell types, F(1, 52)=4.849, p=0.0321; and no significant interaction between cell types and treatments, F(2, 52)=0.7930, p=0.5033.
Supplementary Figure S3. A. Expression of p62 and LC3 in response to MG132 and CQ treatment in STHdhQ7 and Q111 cells. Both cells were treated with 10 µM MG132 and/or 50 µM CQ for 2 and 6 hours. Actin was used as the loading control. B. Quantification of LC3-II expression in A by densitometry. **p<0.01; ***p<0.001, compared to their respective controls. N=3 for each group. One-way ANOVA with the Tukey’s post-hoc analysis. C. Quantification of p62 expression in A by densitometry. *p<0.05; **p<0.01; ***p<0.001, compared to their respective controls. N=3 for each group. One-way ANOVA with the Tukey’s post-hoc analysis. D. Another proteasome inhibitor, lactacystin also induced up-regulation of p62 in both STHdhQ7 and Q111 cells. Cells were incubated with 5 µM lactacystin for the indicated time. P62 expression was analyzed by western blot. Actin was used as the loading control.
Supplementary Figure S4. A–B. Representative confocal images showing the subcellular localization of p62 in response to CQ and MG132 treatment in STHdhQ7 (A) and Q111 (B) cells. Cells were treated with 50 µM CQ or 10 µM MG132 for 16 hours, fixed with methanol and co-stained with Lamp1 and p62 antibodies. C. Representative confocal images of p62 and Lamp1 staining in STHdhQ7 and Q111 cells after 2 hours of MG132 treatment. Note that lysosomes were swelled in STHdhQ111 cells. D–E. Representative confocal imaging showing the identification of p62 macroaggregates as aggresomes. p62 aggregates is co-localized to γ-tubulin, an aggresomal marker, in STHdhQ7 and Q111 cells treated with MG132 for 6 hours. F. Representative confocal imaging showing the co-localization of p62 macroaggregates with mHtt in STHdhQ111 but not Q7 cells. Cells were treated with MG132 for 6 hours and fixed in 4% paraformaldehyde and permeablized with 0.25% Triton X-100 and stained with anti-huntingtin and p62.
Supplementary Figure S5. Representative confocal images showing the formation of K48-linked polyUb protein aggregates in STHdhQ7 (A) but not Q111 cells (B) after MG132 treatment for 2 hours.
Supplementary Figure S6. Representative confocal images showing the co-localization of p62 with K63-linked polyUb proteins in STHdhQ7 (A–B) and Q111 (C–D) cells that were untreated (A and C) or after 2 hours of MG132 treatment (B and D).
Supplementary Figure S7. Representative confocal images showing the subcellular localization of UBQLN2 and K48-linked polyUb proteins in untreated (A–B) and MG132-treated (C–D) STHdhQ7 and Q111 cells. Co-localization of UBQLN2 with K48-linked polyUb protein aggregates was detected in STHdhQ7 (C) but not Q111 (D) cells after 2 hours of MG132 treatment.