

Abstract
To overcome the chemical and metabolic stability issues of L-cystine dimethyl ester (CDME) and L-cystine methyl ester (CME), a series of L-cystine diamides with or without Nα-methylation was designed, synthesized, and evaluated for their inhibitory activity of L-cystine crystallization. L-Cystine diamides 2a–i without Nα-methylation were found to be potent inhibitors of L-cystine crystallization while Nα-methylation of L-cystine diamides resulted in derivatives 3b–i devoid of any inhibitory activity of L-cystine crystallization. Computational modeling indicates that Nα-methylation leads to significant decrease in binding of the L-cystine diamides to L-cystine crystal surface. Among the L-cystine diamides 2a–i, L-cystine bismorpholide (CDMOR, LH707, 2g) and L-cystine bis(N′-methylpiperazide) (CDNMP, LH708, 2h) are the most potent inhibitors of L-cystine crystallization.
Keywords: Cystine diamide, Cystinuria, Crystallization inhibition, Molecular imposter
Graphical Abstract
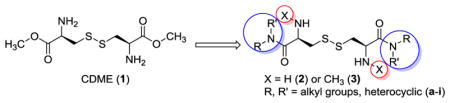
A series of L-cystine diamides with or without Nα-methylation was designed, synthesized, and evaluated for their inhibitory activity of L-cystine crystallization.
Cystinuria is an inherited disease caused by a defect in the reabsorption of cystine and dibasic amino acids in the renal proximal tubule.1–3 The transporter responsible for the reabsorption is a heterodimer consisting of rBAT and b0,+AT subunits. Mutations in either of the two subunits can result in abnormal transport of L-cystine and other dibasic amino acids from the luminal fluid of the renal proximal tubule and intestines, leading to elevated concentrations of these amino acids in the urine of affected individuals causing cystinuria.4. While dibasic amino acids such as lysine, ornithine, and arginine are completely soluble in urine, L-cystine has limited aqueous solubility, leading to its crystallization in urine and formation of L-cystine stones in the patient’s kidney, ureter, and bladder. Even though the incidence of L-cystine stones is much lower than that of calcium oxalate stones, L-cystine stones are larger, occur at a young age, recur more frequently, and are more likely to cause chronic kidney disease.5
Clinical treatment of cystinuria has not changed over the last 30 years. Current approaches are aimed at reducing the concentration of free L-cystine in urine and increasing its solubility.6 A high fluid intake of around 4–5 liters a day and urine alkalinization with citrate or bicarbonate salts can suppress but may not completely prevent stone formation. For severe cases, drug therapy can be used, utilizing the chemical reaction of D-penicillamine or α-mercaptopropionylglycine with L-cystine to generate more soluble mixed disulfides.5, 7 These drugs have side effects including loss of taste, fever, proteinuria, serum sickness-type reactions, and even frank nephritic syndrome.7
Following a study by Ward and co-workers that L-cystine dimethyl ester (CDME) and L-cystine methyl ester (CME), can bind to L-cystine crystal surfaces, acting as molecular imposters to slow down the crystallization of L-cystine,8 we recently reported the discovery of two novel L-cystine diamides that have been shown to be more effective than CDME as tailored L-cystine crystal growth inhibitors and with significantly better stability and in vivo activity profile.9 Herein, we present the design, synthesis, and evaluation of a small focused library of L-cystine diamides for the initial structure-activity relationship study that led to the discovery of the two potent inhibitors of L-cystine crystallization.
Design principle
CDME (1) is a dimethyl ester. Esters are known to be unstable in vivo and susceptible to esterase-mediated hydrolysis. For this reason, esters are commonly used prodrug forms and are readily converted in vivo to their precursor carboxylic acids.10, 11 Upon hydrolysis, CDME would be converted to L-cystine, the amino acid that is already in high concentration and causes stone formation in cystinuria patients. These concerns about CDME (1) prompted us to design the more stable amide derivatives of L-cystine.
As shown in Figure 1, a series of amide modification on the two α-carboxylates of L-cystine were designed to derive the various diamides (2), while a small methyl group was introduced to obtain a series of N,N′-dimethyl L-cystine diamide derivatives 3 to explore the effect of substitution at the α-amino groups of L-cystine on their ability to inhibit L-cystine crystallization. A total of 16 L-cystine derivatives were designed.
Figure 1.

Design of L-cystine diamides (2 and 3).
Chemical synthesis
As shown in Scheme 1, L-cystine diamides (2a–i) were readily synthesized through the amidation of Boc-protected L-cystine 6 using activated OSu or OBt ester and subsequent deprotection of Boc using 50% TFA in CH2Cl2 or 4 equiv. of 4 N HCl in dioxane, as we reported previously.9 Amidation using activated esters was found to give better reaction yields and fewer side products. The overall yields of the three step sequence ranged from 10% to 50%.
Scheme 1.

Synthesis of L-cystine diamides (2a–i) and N,N′-dimethyl L-cystine diamides (3b–i) from Boc-L-cystine (6) or L-thiazolidine-4-carboxylic acid (4).
Reaction conditions: i) Na, NH3 liquid; ii) air, FeCl3, pH 9; iii) (Boc)2O; iv) HOAt, EDC, DIEA; v) HNRR′; vi) 4 N HCl/dioxane
Several conditions were initially explored to directly methylate L-cystine but failed to obtain N,N′-dimethyl L-cystine. Later, we first obtained N-methyl L-cysteine through Na/NH3 reduction of L-thiazolidine-4-carboxylic acid (4) and then air oxidized the N-methyl cysteine in the presence of catalytic iron (III) chloride to afford the desired N,N′-dimethyl L-cystine 5.12, 13 Protection of the secondary amine with Boc anhydride afforded 7, which was activated through its activated ester and formed amides with a series of amines. The target N,N′-dimethyl L-cystine diamides 3b–i were obtained in 10% to 30% overall yield upon final Boc deprotection.
Effects of L-cystine diamides on the apparent aqueous solubility of L-cystine
In order to determine the effects of L-cystine diamides synthesized on the apparent aqueous solubility of L-cystine, a supersaturated solution of L-cystine was prepared in Millipore water according to the literature method.8 Then, 1 mM and 200 μM solutions of each L-cystine diamide synthesized were added to a supersaturated solution of L-cystine in water (1:100) to give supersaturated solutions of L-cystine containing 10 μM and 2 μM of a L-cystine diamide, respectively. The mixtures were then allowed to incubate at 25 °C for 72 h; the apparent solubility of L-cystine in the presence of 10 μM and 2 μM of each compound was then determined using the fluorescence assay we developed.9 CDME was used as a positive control and water was used as a blank control.
As shown in Figure 2, all L-cystine diamides 2a–i have equal or better activity than CDME at increasing the apparent aqueous solubility of L-cystine and thus inhibiting L-cystine crystallization while the corresponding methylated analogs, N,N′-dimethyl L-cystine diamides (3b–i), have little or no effect on the apparent aqueous solubility of L-cystine as compared to water control. The failure of N,N′-dimethyl L-cystine diamides 3b–i to inhibit crystallization of L-cystine could be due to the fact that the N-methyl substituent adversely affected intermolecular interaction (charge-charge and hydrogen bonding) between the methylated ammonium ions (–NH2(Me)+) of N,N′-dimethyl L-cystine diamides 3b–i and the carboxylates (–COO−) of L-cystine as discussed later in our modeling studies. The methylated L-cystine diamides 3b–i cannot bind to the cystine crystal surfaces and, thus, cannot inhibit the crystallization of L-cystine.
Figure 2.

Effect of L-cystine diamides (2 and 3) on the aqueous solubility of L-cystine in comparison to CDME (1) and water controls.
As illustrated in Figure 2, five of the non-methylated L-cystine diamides, namely L-cystine dicyclopropylamide (CDCPA, LH704, 2d), L-cystine bispiperidine (CDPIP, LH706, 2f), L-cystine bismorpholide (CDMOR, LH707, 2g), L-cystine bis(N′-methylpiperazide) (CDNMP, LH708, 2h), and L-cystine diethanolamide (CDEOA, LH709, 2i), were shown to have better activity than the positive control compound CDME and were selected for further dose-response characterization. In order to rank the test compounds, we determined the dose-response curves of the five L-cystine diamides in comparison to CDME and calculated EC2x, the effective concentration required to double the apparent solubility of L-cystine in water. As shown in Figure 3 and Table 1, all five L-cystine diamides were shown to have better EC2x than CDME with L-cystine bismorpholide (CDMOR, LH707, 2g) and L-cystine bis(N′-methylpiperazide) (CDNMP, LH708, 2h) being the most potent. CDMOR (2g) has an EC2x about 7-fold lower than CDME (0.86 vs 6.37 μM) while CDNMP (2h) is 24-fold more potent than CDME (EC2x of 0.26 vs 6.37 μM) at increasing the aqueous solubility of L-cystine. The upper plateau for 2g is lower than those for 2d, 2h, 2i, and CDME, but this can be attributed to a lower L-cystine concentration in the initial supersaturated solution used to test 2g, as we explained previously.9
Figure 3.

Effect of CDME and select L-cystine diamides 2d, 2g, 2h and 2i on the aqueous concentration of L-cystine. The error bars represent the standard deviation calculated from the triplicate measurements.
Table 1.
Effects of L-cystine diamides on the aqueous solubility of L-cystine in comparison to CDME
| Compound | Structure | Effect on L-cystine aqueous solubility | EBinding (kcal/mol)c | |
|---|---|---|---|---|
|
| ||||
| EC2xa (μM) | Ratiob | |||
| CDME |
|
6.37 | 1.0 | −316 |
| CDAA (LH701, 2a) |
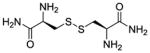
|
+d | +d | −287 |
| CDMA (LH702, 2b) |
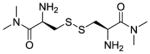
|
+d | +d | −296 |
| CDEA (LH703, 2c) |
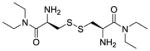
|
+d | +d | −288 |
| CDCPA (LH704, 2d) |
|
3.53 | 1.8 | −292 |
| CDPYR (LH705, 2e) |
|
+d | +d | −302 |
| CDPIP (LH706, 2f) |
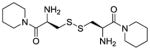
|
1.59 | 4.0 | −307 |
| CDMOR (LH707, 2g) |
|
0.86 | 7.4 | −324 |
| CDNMP (LH708, 2h) |
|
0.26 | 24.5 | −429 |
| CDEOA (LH709, 2i) |
|
2.02 | 3.2 | −307 |
| Me2-CDMA (LH710, 3b) |

|
− e | − e | −264 |
| Me2-CDEA (LH711, 3c) |
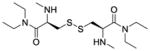
|
− e | − e | −235 |
| Me2-CDCPA (LH712, 3d) |
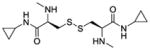
|
− e | − e | −260 |
| Me2-CDPYR (LH713, 3e) |

|
− e | − e | −247 |
| Me2-CDPIP (LH714, 3f) |

|
− e | − e | −234 |
| Me2-CDMOR (LH715, 3g) |
|
− e | − e | −267 |
| Me2-CDEOA (LH716, 3i) |
|
− e | − e | −273 |
EC2x refers to the concentration required to double the aqueous solubility of L-cystine.
Ratio refers to the improvement in potency over the control CDME.
The binding energies in kcal/mol for the binding of test compound to cystine crystal surface {100} were computed using the COMPASS forcefield in BIOVIA’s Materials Studio software after Monte Carlo searches of the configurational space for possible adsorption configurat ion in the presence of explicit water molecules (see experimental methods for details). It corresponds to the total non-bond energy (sum of van der Waals and electrostatic) released upon adsorption of the test molecule. H-bond interaction energy, when present, is included implicitly in the non-bond energy.
The effect on the aqueous solubility of l-cystine was similar to that of the control CDME and the expected EC2x would be similar to that of CDME, but was not measured.
There was no effect observed on the aqueous solubility of L-cystine at either 2 or 10 μM
Molecular modeling
The crystal morphology calculation and various adsorption/docking experiments were performed using BIOVIA’s Materials Studio software suite. The {100} set of faces had been identified to be the fast-growing faces. Binding energies of the L-cystine diamides onto the {100} surface of L-cystine crystal are listed in Table 1. The Bravais-Friedel Donnay-Harker (BFDH) calculated platelet morphology (data not shown) agrees well with the experimentally observed morphology.8, 14 The BFDH approach does not consider solvent effect; the fact that the computed morphology (with no solvent present) agrees with the experimentally observed in the presence of solvent (water) points to a strong inter-molecular interaction between the L-cystine molecules, which makes the L-cystine-water molecular interaction obscured in the L-cystine crystal morphology development. This is different from cases where intermolecular interactions inside the crystal is relatively weak compared to surface-solvent interactions.
As identified in the BFDH result and elsewhere,8 the {100} set of symmetry-related faces are fast-growing, giving rise to large relative surface area of the basal surfaces of {001}. Therefore, in order to inhibit growth, attachment of the feeding L-cystine molecules onto the fast growing {100} surfaces will need to be inhibited, as demonstrated experimentally in our study and in the previous CDME study.8
Various crystal growth mechanisms had been proposed, including formation of islands, terrace, attachment to steps, and kinks.15 Fundamental to these mechanisms is molecular binding to the surface/interface in study. Therefore, in part to establish a protocol for screening potential inhibitors, the present modeling effort focused on calculating binding energy of candidate inhibitors onto the fast-growing {100} surface of L-cystine crystal. Binding energies reported in Tables 1 contains results from modeling in the presence of solvent (water). Results obtained in the presence of water differ only slightly from that obtained in the absence of solvents. This is primarily due to the fact that, in adsorbed configuration, there is very little solvent-accessible space for the solvent to penetrate and assert its solvent dielectric shielding effect. We want to emphasize here that there are many other important roles a solvent can play, such as forming hydration shells around depositing molecules, and formation of electrical double layers covering the growing surfaces, all being important factors governing crystal growth/dissolution, but in the meantime are beyond the scope of the present work.
From this point on when referring to binding energies, they were all calculated in the presence of explicit water molecules. It corresponds to the total non-bond energy (sum of van der Waals and electrostatic including H-bond interaction energy, when present) released upon adsorption of the test molecule. The binding energy of an L-cystine molecule onto the {100} surface of L-cystine crystal is −85.8 kcal/mol per molecule (not shown in table 1). This value is substantially greater in magnitude than the energy released, −58.2 kcal/mol, for an L-cystine molecule coming off from a layer of L-cystine molecules on the {100} surface. This elevated binding energy can be explained by the fact that a single L-cystine molecule has greater degree of freedom in adjusting its configuration to maximize binding onto the surface than when packed into individual layers onto the {100} surfaces.
All the L-cystine derivatives considered have binding energies greater in magnitude than the L-cystine reference. The L-cystine diamides 2a–i and CDME, have surface binding energies between −287 ~ −429 kcal/mol. In particular, the binding energy of 2h is 43% greater in magnitude than CDME, which may explain the much greater inhibition efficiency observed experimentally. It is worth pointing out that, unlike other cystine derivatives, 2h has a large proportion (42%) of binding energy coming from the two N-methylpiperzine groups. Binding energies of dimethylated L-cystine diamides 3b–i are considerably lower than those of their corresponding unmethylated counterparts, averaging 47.6±15.9 kcal/mol lower. This is consistent with our experimental data showing that all dimethylated L-cystine diamides 3b–i had no effect on the apparent aqueous solubility of L-cystine. The reduced binding energies of the dimethylated L-cystine diamides can be explained by the steric hindrance of the methyl groups. Figure 4 shows a comparison of structural configurations of 2f and 3f, respectively, when the respective molecules are adsorbed onto the {100} surface of L-cystine. In the L-cystine crystal itself,16 the NH•••O hydrogen bonds, formed between the ammonium and carboxylate moieties, plays an important role in binding the L-cystine molecules together. The same hydrogen bonds are formed between the ammonium moiety from the L-cystine derivatives and the carboxylate moiety in the L-cystine on the crystal surface. In the inset showing hydrogen bonding details for the unmethylated 2f adsorption onto the {100} surface of L-cystine (Fig. 4A), a total of three hydrogen bonds (indicated by blue dashed lines) were formed between the 2f molecule and three different L-cystine molecules; in the methylated analog (3f) (Fig. 4B), the ammonium moieties were shielded by the methyl groups, and could only form one hydrogen bond with the carboxylate moiety in the substrate. Such difference could explain the 18% drop in adsorption energy onto the substrate upon methylation of 2f to 3f (Table 1).
Figure 4.

Structure configurations of CDPIP (LH706, 2f) and Me2-CDPIP (LH714, 3f), respectively, adsorbed onto the {100} surface of L-cystine crystal (in ball-n-stick representation). CDPIP and Me2-CDPIP molecules are in CPK representation at 70% of vdW radii; solvent (H2O) molecules are in line representation. The methyl groups in Me2-CDPIP are circled in green. Insets are made to show hydrogen bonding details (circled in yellow), with the CDPIP and Me2-CDPIP shown in line representation). Red: oxygen; grey: carbon; white: hydrogen; blue: nitrogen; yellow: sulfur.
In summary, we designed and synthesized a series of L-cystine diamides with or without Nα-methylation and evaluated their inhibitory activity of L-cystine crystallization. L-Cystine diamides 2a–i without Nα-methylation were found to be potent inhibitors of L-cystine crystallization while Nα-methylation of L-cystine diamides resulted in derivatives 3b–i without any inhibitory activity of L-cystine crystallization. Computational modeling indicates that Nα-methylation leads to a significant decrease in binding energy of the L-cystine diamides onto the fast-growing {100} surface of L-cystine crystal. Among the L-cystine diamides 2a–i without Nα-methylation, L-cystine bismorpholide (CDMOR, LH707, 2g) and L-cystine bis(N′-methylpiperazide) (CDNMP, LH708, 2h) are the most potent inhibitors of L-cystine crystallization. They are 7–24 times more potent than CMDE in inhibiting the crystallization of L-cystine. As we reported previously,9 both 2g and 2h are more stable than CDME and 2h has been shown to effectively inhibited L-cystine stone formation in vivo in a genetic mouse model of cystinuria.
Acknowledgments
We gratefully acknowledge the financial support of grant DK112782 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andreassen KH, Pedersen KV, Osther SS, Jung HU, Lildal SK, Osther PJS. Urolithiasis. 2016;44:65. doi: 10.1007/s00240-015-0841-x. [DOI] [PubMed] [Google Scholar]
- 2.Pereira DJ, Schoolwerth AC, Pais VM. Clin Nephrol. 2015;83:138. doi: 10.5414/cn108514. [DOI] [PubMed] [Google Scholar]
- 3.Saravakos P, Kokkinou V, Giannatos E. Urology. 2014;83:693. doi: 10.1016/j.urology.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 4.Strologo LD, Pras E, Pontesilli C, Beccia E, Ricci-Barbini V, de Sanctis L, Ponzone A, Gallucci M, Bisceglia L, Zelante L, Jimenez-Vidal M, Font M, Zorzano A, Rousaud F, Nunes V, Gasparini P, Palacín M, Rizzoni G. J Am Soc Nephrol. 2002;13:2547. doi: 10.1097/01.asn.0000029586.17680.e5. [DOI] [PubMed] [Google Scholar]
- 5.Moe OW. Lancet. 2006;367:333. doi: 10.1016/S0140-6736(06)68071-9. [DOI] [PubMed] [Google Scholar]
- 6.Mattoo A, Goldfarb DS. Semin Nephrol. 2008;28:181. doi: 10.1016/j.semnephrol.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Becker G. Nephrology. 2007;12:S4. doi: 10.1111/j.1440-1797.2006.00722.x. [DOI] [PubMed] [Google Scholar]
- 8.Rimer JD, An Z, Zhu Z, Lee MH, Goldfarb DS, Wesson JA, Ward MD. Science (New York, NY) 2010;330:337. doi: 10.1126/science.1191968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu L, Yang Y, Aloysius H, Albanyan H, Yang M, Liang J-J, Yu A-t, Shtukenberg A, Poloni LN, Kholodovych V, Ward MD, Sahota A. J Med Chem. 2016;59:7293. doi: 10.1021/acs.jmedchem.6b00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Nat Rev Drug Discovery. 2008;7:255. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 11.Hu L. In: Drug Delivery: Principles and Applications. Wang B, Siahaan TJ, Soltero RA, editors. Wiley & Sons; New York: 2005. p. 125. [Google Scholar]
- 12.Casey JP, Martin RB. J Am Chem Soc. 1972;94:6141. doi: 10.1021/ja00772a036. [DOI] [PubMed] [Google Scholar]
- 13.Menger FM, Caran KL. J Am Chem Soc. 2000;122:11679. [Google Scholar]
- 14.Poloni LN, Zhu Z, Garcia-Vázquez N, Yu AC, Connors DM, Hu L, Sahota A, Ward MD, Shtukenberg AG. Cryst Growth Des. 2017;17:2767. doi: 10.1021/acs.cgd.7b00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Yoreo JJ, Vekilov PG. Rev Mineral Geochem. 2003;54:57. [Google Scholar]
- 16.Moggach SA, Allan DR, Parsons S, Sawyer L, Warren JE. J Synchrotron Rad. 2005;12:598. doi: 10.1107/S0909049505019850. [DOI] [PubMed] [Google Scholar]