

Abstract
NUT midline carcinoma is an aggressive tumor that occurs mainly in the head and neck and, less frequently, the mediastinum and lung. Following identification of an index case of a NUTM1 fusion positive undifferentiated soft tissue tumor, we interrogated additional cases of primary undifferentiated soft tissue and visceral tumors for NUTM1 abnormalities. Targeted next-generation sequencing (NGS) was performed on RNA extracted from formalin-fixed paraffin-embedded tissue, and results validated by fluorescence in situ hybridization (FISH) using custom bacterial artificial chromosome (BAC) probes. Six patients were identified: mean age of 42 years [range: 3–71]; equal gender distribution; and, tumors involved the extremity soft tissues (N=2), kidney (N=2), stomach and brain. On systemic work-up at presentation all patients lacked a distant primary tumor. Morphologically, the tumors were heterogeneous, with undifferentiated round-epithelioid-rhabdoid cells arranged in solid sheets, nests and cords. Mitotic activity was generally brisk. Four cases expressed pancytokeratin, but in only two cases was this diffuse. NGS demonstrated the following fusions: BRD4-NUTM1 (three cases), BRD3-NUTM1, MXD1-NUTM1, and BCORL1-NUTM1. Independent testing by FISH confirmed the presence of NUTM1 and partner gene rearrangement. This study establishes that NUT-associated tumors transgress the midline and account for a subset of primitive neoplasms occurring in soft tissue and viscera. Tumors harboring NUTM1 gene fusions are presumably under-recognized, and the extent to which they account for undifferentiated mesenchymal, neuroendocrine, and/or epithelial neoplasms is unclear. Moreover, the relationship, if any, between NUT-associated tumors in soft tissue and/or viscera, and conventional NUT carcinoma, remains to be elucidated.
Keywords: NUT midline carcinoma, NUTM1, BCORL1, BRD3, BRD4, MXD1, fusions
INTRODUCTION
NUT midline carcinoma (also known as NUT carcinoma) is a clinically aggressive neoplasm that typically occurs in the head and neck(1) or mediastinum;(2, 3) although more recently, there have been reports of primary lung,(4-6) and renal involvement.(7, 8) Considered rare, the actual incidence of NUT midline carcinoma is unknown, and presumably significantly underestimated.(9) While initial reports suggested a pediatric predisposition, increased recognition of this entity has led to broader characterization amongst adults. There is no obvious sex predisposition.(10) Over 80% of patients die within the first year of diagnosis of NUT carcinoma;(9) locoregional and distant metastases are common and, aside from complete surgical resection, there is currently limited therapeutic benefit from chemo-radiotherapy.(9)
NUT carcinoma is considered a genetically-defined tumor, encompassing any malignant epithelial tumor neoplasm with rearrangement of NUTM1 (NUT [nuclear protein in testis] midline carcinoma family member 1(11)).(1, 9) Despite uncertainty with respect to its underlying histogenesis, NUT carcinoma has been classified as a form of poorly differentiated carcinoma – possibly a subtype of squamous cell carcinoma(12) – based on the frequent presence of squamous differentiation.(1) The most common NUTM1 fusion partner is BRD4, which occurs in roughly 70% of cases;(13) the remaining cases are reported to pair with BRD3(13) or NSD3 (also known as WHSC1L1),(14) or with an, as of yet, undefined fusion partner(s), which is referred to as NUT-variant carcinoma. There does not presently appear to be a relationship between patient outcome and NUTM1 fusion partner.(9, 10)
Next-generation sequencing (NGS) is gradually becoming routinely applied in the diagnostic pathology laboratory. Following the incidental discovery of an undifferentiated soft tissue tumor bearing a NUTM1 gene fusion by NGS testing, we proceeded to examine additional undifferentiated soft tissue and visceral tumors for NUTM1 rearrangement.
MATERIALS AND METHODS
Case Selection
The index case in this study had undergone routine diagnostic RNA Sequencing (RNASeq), which revealed NUTM1 rearrangement. As a result, retrospective archival searches were performed for other undifferentiated soft tissue and visceral tumors by the two senior authors (BCD/CRA; 2007-2017). Cases in which the soft tissues or viscera was purported to represent the site of primary disease were pulled for re-review. In view of the commonality of nuclear isomorphism amongst most translocation-associated tumors, only those cases that lacked significant pleomorphism were selected for RNASeq. This study received institutional Research Ethics Board approval.
Immunohistochemistry
The paraffin-embedded tissue blocks selected in all cases were confirmed to have adequate viable tumor, and all were properly formalin-fixed. Staining was performed for keratins (pancytokeratin, high- and low-molecular weight keratins), claudin-4, p63, S100, glial fibrillary acid protein, chromogranin, synaptophysin, actin, desmin CD34 and NUT using routine techniques (Supplementary Table 1). Appropriate on-slide positive controls were used throughout. Tumor immunoreactivity was scored based on the percentage of positive cells (0, no staining; 1+, <5%; 2+, 5% to 25%; 3+, 26% to 50%; 4+, 51% to 75%; and 5+, 76% to 100%).
RNA Sequencing
Briefly, for each case scrolls (3–4 at 10 microns) were cut from formalin-fixed paraffin-embedded tissue into Eppendorf tubes. RNA was extracted using the ExpressArt FFPE Clear RNA Ready kit (Amsbio, Cambridge, MA). RNA-seq libraries were prepared using 20-100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA), an enrichment-based assay that targets 507 known fusion-associated genes. Each sample was sequenced with 76 base-pair paired-end reads on an Illumina MiSeq at 8 samples per flow cell (~3 million reads per sample). The results were analyzed using the STAR aligner and Manta fusion caller as well as the JAFFA fusion caller utilizing BOWTIE2 aligner.(15, 16)
The mRNA expression levels of NUTM1, and the respective fusion partners, were evaluated and compared to those of other samples analyzed on the same targeted RNA sequencing platform.
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) for NUTM1 was performed on each case, followed by confirmation of each of the various fusion partners, including: BCORL1, BRD3, BRD4 and MXD1. Testing was performed as previously outlined in detail.(17) Briefly, custom bacterial artificial chromosome (BAC) clone probes were designed to flank the target genes, based on the UCSC genome browser (http://genome.ucsc.edu), and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA; http://bacpac.chori.org) (Supplementary Table 2). DNA from each BAC was isolated, then labeled with fluorochromes by nick translation. Slides were prepared using formalin-fixed paraffin-embedded tissue cut at 4 microns. The slides were deparaffinized, pretreated, and then hybridized with the denatured probes. Following an overnight incubation, the slides were rinsed, stained with 4′,6-diamidino-2-phenylindole (DAPI), mounted, and examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany).
RESULTS
Six cases were identified from institutional pathology archives; one of the cases (Patient 6) has previously been reported (Table 1).(8) The mean patient age was 42 years (range: 3-71 years). There were an equal number of females and males. Two tumors occurred in the extremity soft tissues and two in kidney, with individual cases involving brain (parietal lobe) and stomach wall (Table 1).
TABLE 1.
Clinical Attributes of Six Cases of NUT-Associated Tumor of Soft Tissue and Viscera.
| Case | Age (y) | Sex | Site | Treatment | Clinical Course | Status (mo) |
|---|---|---|---|---|---|---|
| 1* | 61 | M | Thigh, proximal, L | Biopsy | LN metastases | DOD 3 |
| 2 | 45 | M | Upper arm, L | Surgery, chemo, rads | LN, lung, soft tissue metastases | DOD 48 |
| 3 | 39 | F | Stomach wall | Surgery, chemo | Peritoneal carcinomatosis, LN, liver, spleen metastases | AWD 108 |
| 4 | 3 | M | Brain, parietal, L | Surgery, chemo | N/A | DOD 12 |
| 5 | 71 | F | Kidney, L | Biopsy | Lung metastases | DOD 2 |
| 6 | 36 | F | Kidney, R | Nephrectomy | Lung metastases | DOD 6 |
Abbreviations: ANED (alive no evidence of disease); AWD (alive with disease); DOD (dead of disease); L (left); LN (lymph node); mo (months); N/A (not available); R (right); y (years).
Index case
Grossly, where details were available, the tumors were gray-tan-white with hemorrhagic foci; they ranged from firm-friable, with one case described as ‘fleshy’. The margins tended to be infiltrative; the two soft tissue tumors were deep-seated and invaded surrounding skeletal muscle. The visceral lesions infiltrated into the brain and renal parenchyma; the gastric tumor was centered in the muscularis propria and extended to serosa. Microscopically, the tumors had a primitive appearance, with the cells arranged in solid sheets, nests and cords. One case contained an admixture of small round and epithelioid cells (Case 1) (Figure 1); two cases were largely comprised of epithelioid cells (Cases 2, 6) (Figure 2); one case was predominantly rhabdoid-polygonal (Case 3) (Figure 3); and, two cases predominantly contained small round cells (Cases 4, 5) (Figure 3). It should be emphasized that there was frequently considerable intra-tumoral variability, with many cases exhibiting morphologies ranging from round-rhabdoid-epithelioid to spindle shaped. The nuclei were round-ovoid with relative isomorphism, and frequent macronucleoli and irregular nuclear contours/clefts. Mitotic activity was generally brisk. Several of the cases contained interspersed multinucleated tumor giant cells (Figure 3). Where present, the intervening stroma ranged from collagenous to focally myxoid. Many of the cases contained sheets of loose/discohesive cells. No convincing squamous keratinization was identified in any of the cases. Four cases expressed cytokeratin, but this was only significant in two cases.
Figure 1.
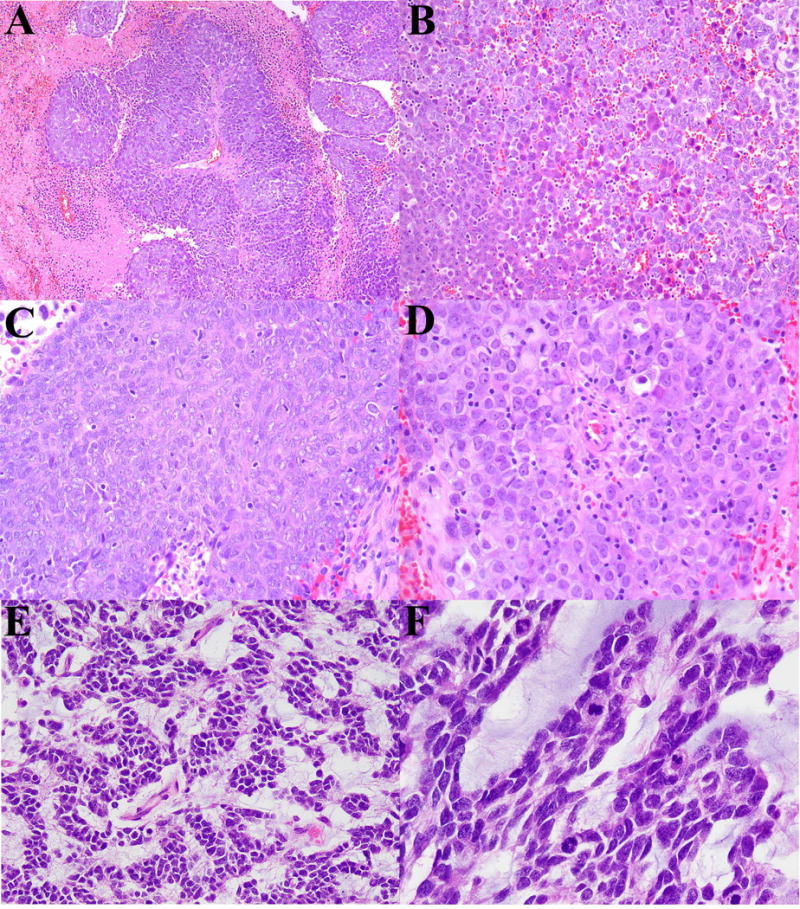
Patient 1: An intramuscular NUT-associated tumor. (A) Sheets of predominantly large epithelioid cells radiating from a vessel with an abrupt transitioning into areas of necrosis. (B) Juxtaposition of epithelioid-polygonal cells with differing cytoplasmic and nuclear characteristics. (C) Area of ovoid cells with syncytial pattern. (D) Sheets of epithelioid-rhabdoid cells with prominent nucleioli. Patient 4: A parietal lobe NUT-associated tumor. (E) Epithelioid-polygonal cells with a reticular-alveolar pattern and prominent myxoid stroma. (F) Higher magnification highlighting the nuclear molding, speckled chromatin and conspicuous mitotic activity.
Figure 2.
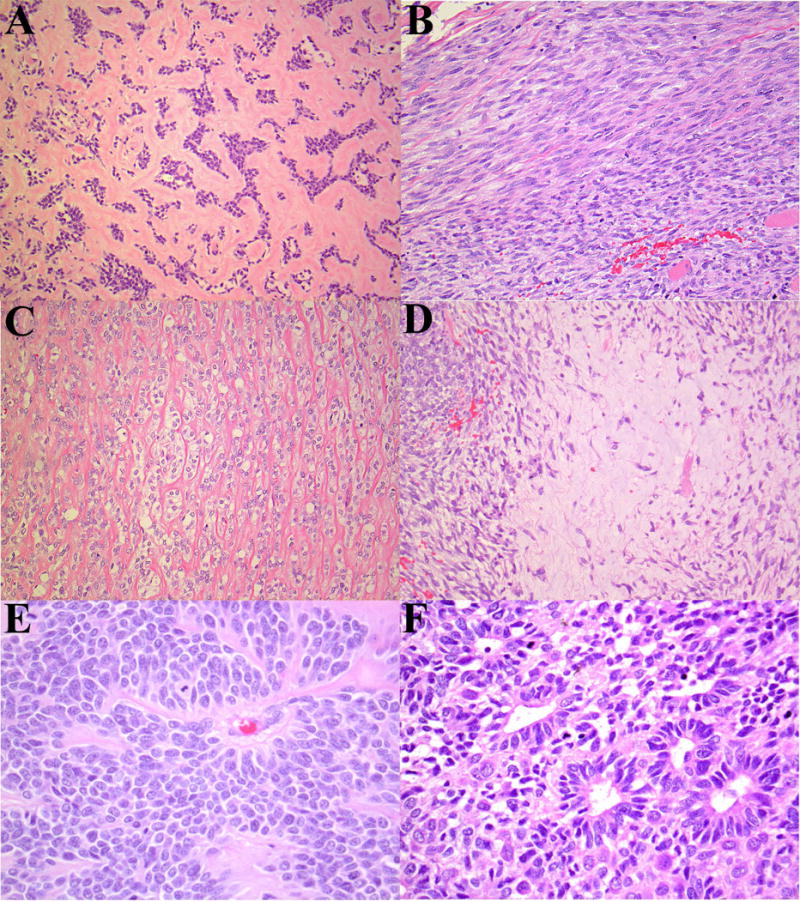
Patient 2: An intramuscular NUT-associated tumor with a markedly varied morphology. (A) Nests and cords of polygonal cells set within hyaline stroma, resembling myoepithelial carcinoma. (B) Spindle cells with a herringbone pattern. (C) Cords of epithelioid cells separated by wiry collagen, vaguely resembling sclerosing epithelioid fibrosarcoma. A soft tissue metastasis that occurred two years later, which retained many of the initial features; however, there was overall greater cellularity, atypia and mitotic activity: (D) Spindle-epithelioid cells with areas of chondromyxoid stroma. (E) Cellular nests of polygonal cells radiating around delicate blood vessels. The nuclei are large with delicately speckled chromatin. (F) Area of rosette formation.
Figure 3.
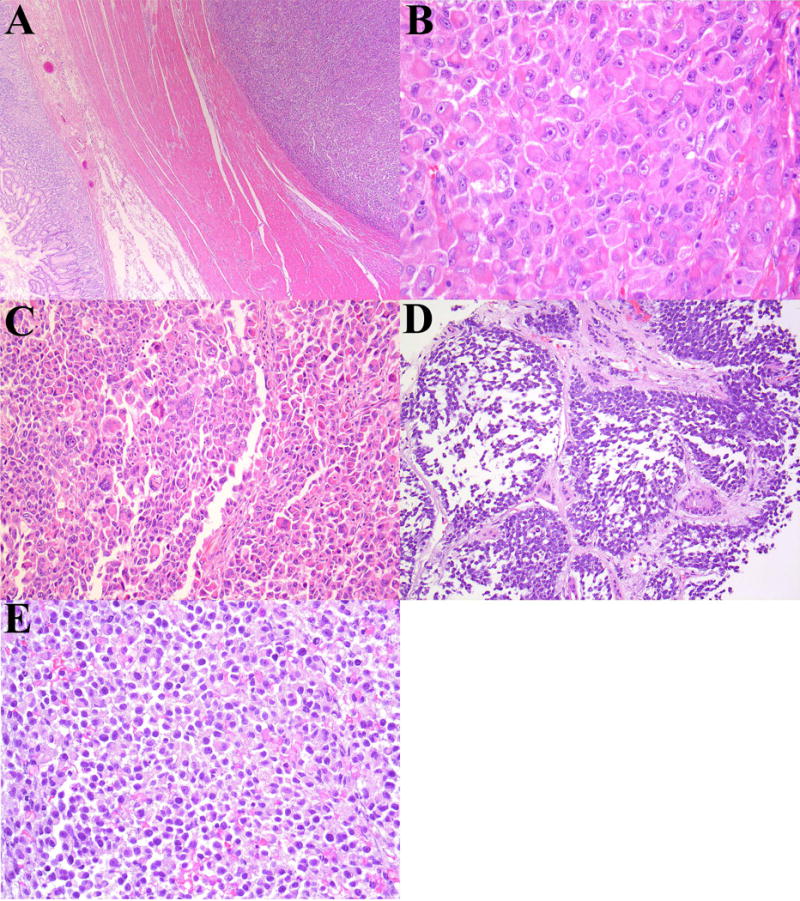
Patient 3: Gastric NUT-associated tumor. (A) The tumor was centered in the muscularis propria and extended to serosa. (B) Sheets and nests of polygonal-rhabdoid cells with dense eosinophilic cytoplasm. (C) Scattered nuclear atypia and multinucleated giant cells were present in several tumors. Patient 5: Renal NUT-associated tumor. (D) Infiltration of renal parenchyma by round-rhabdoid cells. (E) Sheets of discohesive cells with areas of degeneration. This tumor was predominantly necrotic. (F) Immunohistochemisty for claudin-4 in Patient 6; slightly more than half of this case was immunopositive (note entrapped tubules).
The most common NUTM1 fusion partner was BRD4 (N=3), with individual cases containing BCORL1, BRD3 and MXD1 (Table 2). Independent testing by FISH confirmed the presence of NUTM1 and partner gene rearrangement. Four cases demonstrated NUT immunoreactivity; immunopositivity was present in tumors with BRD4 and BRD3 gene fusions, but not in the two cases with the novel MXD1 and BCORL1 gene partners (Table 2). In one of the tumors, Case 2, immunohistochemical studies were repeated on freshly cut sections from two different surgical resections and both were negative for NUT staining. The remaining immunohistochemical findings are detailed in Table 2.
TABLE 2.
Immunohistochemical and Molecular Findings in Six Cases of NUT-Associated Tumor of Soft Tissue and Viscera.
| Pt | panCK | HMWK | LMWK | C-4 | p63 | S100 | GFAP | Syna | Chro | Actin | Desmin | CD34 | NUT | Molecular |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5+ | 5+ | 5+ | 1+ | 2+ | − | 5+ | 2+(w) | − | − | − | − | 5+ | BRD3-NUTM1 |
| 2 | − | − | − | − | − | − | − | − | − | 1+(w) | − | − | − | BCORL1-NUTM1 |
| 3 | 2+(w) | − | 2+(w) | − | − | − | 3+(w) | − | − | − | − | − | − | MXD1-NUTM1 |
| 4 | − | − | − | − | − | N/A | 2+ | 1+ | − | N/A | N/A | N/A | 5+ | BRD4-NUTM1 |
| 5 | 1+(w) | − | 1+(w) | N/A | − | − | 1+(w) | − | − | − | − | 5+ | BRD4-NUTM1 | |
| 6 | 5+* | 2+ | N/A | 4+ | 1+ | N/A | N/A | − | − | N/A | − | N/A | 3+ (w) | BRD4-NUTM1 |
Abbreviations: C-4 (Claudin-4); Chro (chromogranin); HMWK (high molecular weight keratin); LMWK (low molecular weight keratin); w (weak); N/A (not assessed); panCK (pancytokeratin); Pt (Patient number); Syna (synaptophysin); − (negative)
diffuse strong immunoreactivity for CK7.(8)
RNAseq demonstrated the presence of significant mRNA NUTM1 expression in all four cases tested, regardless of the fusion partner (Figure 5). Corresponding increases in mRNA expression among the fusion genes was observed for BCORL1 and MXD1, but not BRD3 or BRD4 (data not shown). The NUTM1 breakpoints were located at exon 5 (Case 1; NCBI Reference Sequence: NM_001284292.1) and exon 3 (Cases 2-3, 5-6; NM_001284292.1); the BRD3 breakpoint involved exon 9 (Case 1; NM_007371.3); BCORL1 exon 1 (Case 2; NM_021946.4); MXD1 exon 5 (Case 3; NM_002357.3); and, BRD4 exon 11 (Cases 5-6; NM_058243.2). These results imply that an identical NUTM1 breakpoint is present in BRD4, BCORL1 and MXD1; therefore, it appears that the specific breakpoint is unlikely to be a factor in the variable NUT protein expression identified by immunohistochemistry.
Figure 5.
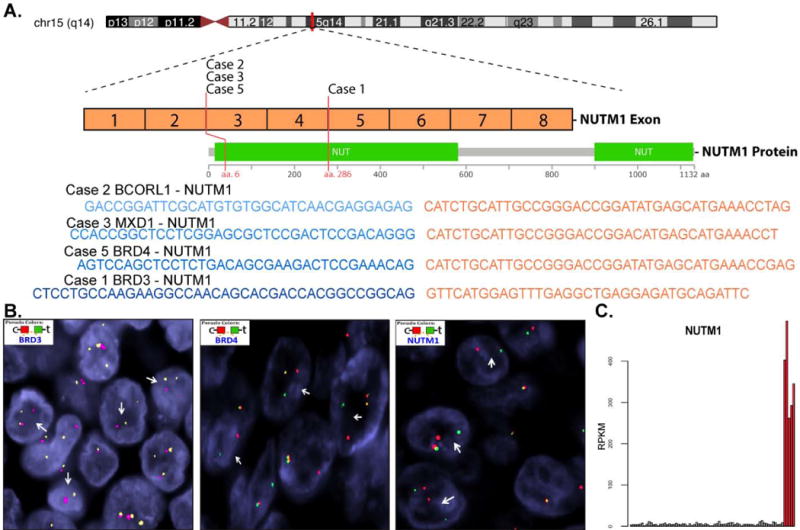
(A) Illustration of the NUT gene, and corresponding protein, highlighting the location of the gene breakpoint locations and fusion gene sequences. (B) Representative photomicrographs of fluorescence in situ hybridization break-apart probes for BRD3, BRD4 and NUTM1. (C) Bar chart illustrating mRNA expression profiles of NUTM1 (Cases 1–5).
On systemic work-up at initial presentation, none of the patients had evidence of a distant primary tumour. However, all patients subsequently developed lymph node and/or lung metastases. Patient 4, with a locally recurrent brain tumor, did not reveal evidence of extraneural disease on autopsy. At last follow-up, all except one patient had succumbed to disease, with four patients dying within the first year of diagnosis. The only patient still alive after 108 months, despite peritoneal carcinomatosis and lymph node metastases, had a MXD1-NUTM1 fusion (Table 1).
DISCUSSION
NUT midline carcinoma is a relatively recently recognized entity,(3, 18-21) and considered among the most aggressive of human solid malignancies.(22) Tumors tend to be distributed along the midline axis with a predilection for the head and neck, and mediastinum and lung. In addition, there are rare reports of primary bladder,(23) breast,(23) endometrium,(23) kidney(7, 8) and orbit(23) involvement. There is a propensity for early metastases to lymph nodes and lung, and tumors are almost inevitably fatal. Following discovery of an index case of an undifferentiated soft tissue tumor containing NUTM1 rearrangement, we sought to investigate the incidence of NUTM1-related fusions among undifferentiated tumors in the soft tissue and viscera. In addition to identifying primary NUTM1-rearanged undifferentiated tumors in the soft tissue extremities, kidney, stomach and brain, we report two novel NUTM1 fusion partners.
Our study cohort was characterized by a markedly heterogeneous phenotype, which lacked defining morphologic and/or immunohistochemical attributes. As is typical of most tumors characterized by reciprocal translocations, none manifested diffuse pleomorphism. Common architectural patterns included sheets, nests and cords of undifferentiated cells; the tumors were infiltrative, lacking ‘pushing’ borders. The cells varied from small to large; epithelioid, round, rhabdoid, and ovoid shapes were common. Necrosis was frequently present. In contrast to NUT midline carcinoma, which typically express cytokeratin expression,(23, 24) only a minority of our cases showed significant keratin positivity; none of the tumors in this series contained overt squamous differentiation in the form of keratinization. In addition, only a single case showed significant expression of claudin-4, a tight junction-associated protein expressed in most epithelial cells.(25) NUT midline carcinoma has been reported to express CD34, CD99 and synaptophysin. In our series, in addition to occasional synaptophysin expression, we identified the presence of glial fibrillary acid protein expression in several cases.
Notably, two of the cases in our series were negative for NUT by immunohistochemical testing. Each case was confirmed to contain abundant viable tumor that had been properly formalin-fixed and paraffin-embedded. In a previous series of NUT midline carcinoma NUT immunohistochemical testing, using a similar clone, was found to have a sensitivity of 87%, with four cases reported to be falsely negativity.(26) In that study two of the cases were, on re-review, found to be weakly positive, with one possibly attributable to post-mortem degradation; both of the negative cases were NUT-variant carcinoma, leading the authors to suggest variant fusion proteins may be expressed at lower levels than the BRD3 and BRD4 fusion products. Interestingly, the two negative cases in our series corresponded to the two novel NUTM1 fusion partners (BCORL1-NUTM1 and MXD1-NUTM1). By illustrating the presence of high levels of mRNA expression in all cases examined (Figure 4), our findings raise the possibility that, in a subset of cases, a post-transcriptional mechanism may account for the absence of NUT staining by immunohistochemistry.
Figure 4.
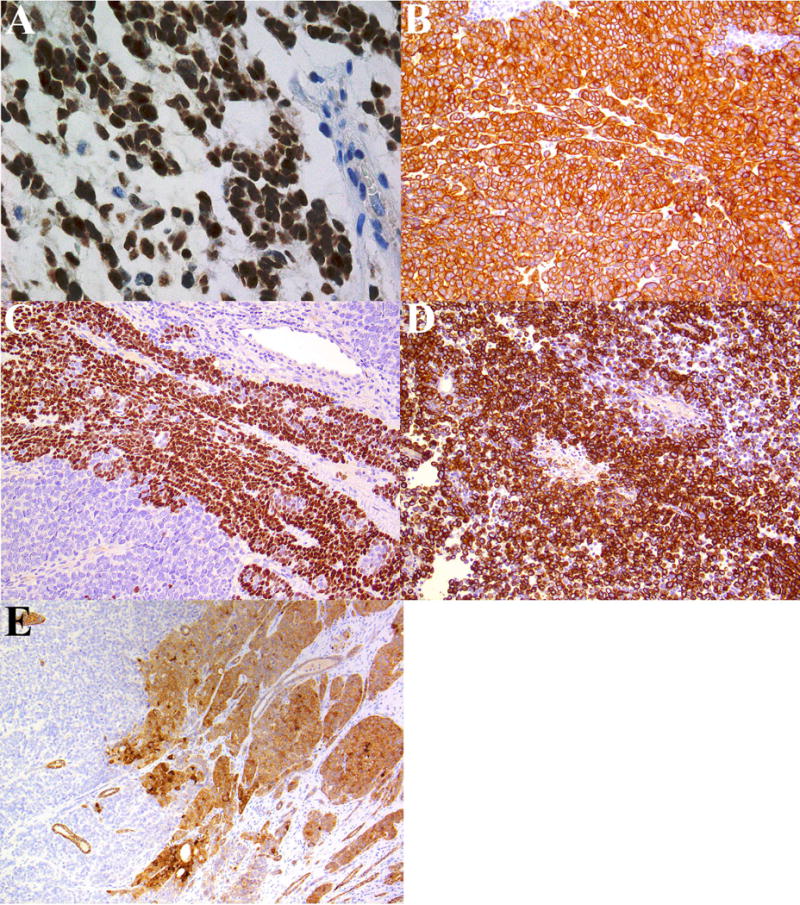
Patient 1: Immunohistochemistry highlighting (A) diffuse expression of NUT, (B) pancytokeratin, (C) patchy p63 and (D) glial fibrillary acid protein.
The sole pediatric patient in our series, presenting as a left parietal lobe tumor in a 3 year-old male, deserves further discussion. Due to its primitive morphology, this tumor was initially classified generically as a high-grade neuroepithelial neoplasm with embryonal features/PNET. This diagnosis was supported by focal GFAP and synaptophysin positivity. Given the combination of embryonal-appearing small cell components, alveolar pattern with myxoid growth and evidence of NUT expression we considered whether this unusual lesion might be related to the neoplasms recently described as CNS Ewing Family Tumors (EFT) with CIC-NUTM1 fusions.(27) On methylation profiling, however, this neoplasm did not cluster with tumors of the CNS EFT-CIC group or with any other embryonal or other neuroepithelial tumors included in the array algorithm (data not shown).
Based on frequent keratin expression it has been concluded NUT midline carcinoma represent a carcinoma.(23) The presence – often with abrupt transition – of squamous keratinization has further led to classification of NUT midline carcinoma as a subtype of squamous cell carcinoma.(28) Mesenchymal differentiation is reported to rarely occur.(29). Admittedly, keratin expression alone does not establish a diagnosis of carcinoma and it is a relatively consistent feature of several sarcomas (e.g., epithelioid sarcoma, desmoplastic small round cell tumor and synovial sarcoma). Undifferentiated small round cell tumors such as Ewing sarcoma, and those with BCOR rearrangement have also been reported to show keratin, epithelial membrane antigen and p63 expression.(30-33) The relationship between NUT-associated tumors of soft tissue, or viscera, and NUT midline carcinoma remains unclear. Some of the cases in the current series (Patient 1 and 5) bore morphologic and immunohistochemical overlap with reports of NUT midline carcinoma in the literature. This raises the possibility that at least some of the cases in this series fall under the rubric of NUT midline carcinoma. However, anatomic distribution notwithstanding, the morphology of two of the tumors is distinct from the histologic spectrum heretofore described in NUT midline carcinoma. For example, one case (Patient 2) had areas resembling myoepithelial carcinoma, sclerosing epithelioid fibrosarcoma and fibrosarcoma; this same patient also had PNET-like features, in addition to well-formed rosettes. The immunophenotype of the tumors in this series also appears to somewhat differ from the literature on NUT midline carcinoma. For example, in contrast to NUT midline carcinoma, where keratin positivity is frequently diffuse, only two cases in this series appeared to have significant keratin expression. Expression of p63 is likewise common in NUT midline carcinoma, but only observed in two cases in this series. Our cases possessed NUTM1 breakpoints similar to those previously reported;(18, 34) nevertheless, given differences in anatomic distribution, morphology and immunophenotype – and uncertainty regarding underlying histogenesis – we feel it prudent to classify the neoplasms in this series as a NUT-associated tumor, rather than NUT midline carcinoma.
The incidence of NUT-associated tumors is unknown, and it is presumed that cases in the soft tissue and viscera have heretofore gone unrecognized. In a series of unclassified malignancies and poorly differentiated carcinomas of the mediastinum, 3.5% were found to represent NUT midline carcinoma.(35) Another series examining poorly differentiated carcinomas in children and young adults demonstrated an incidence of 7%.(23) It is unclear if this range is similar for tumors occurring in the soft tissue and viscera. In addition to BRD4 and BRD3, we identified two novel NUTM1 fusion partners: BCORL1 and MXD1. Whether either of these genes accounts for any of the unknown gene partners in NUT-variant carcinoma remains to be determined. The protein encoded by BCORL1 (BCL6 corepressor-like 1) is a transcriptional corepressor homologue of BCOR; it is capable of interacting with Class II histone deacetylases and CtBP to mediate transcriptional repression.(36) Translocations involving BCORL1 have been previously reported in hepatocellular carcinoma,(37) ossifying fibromyxoid tumor(38) and endometrial stromal sarcoma.(39) The protein encoded by MXD1 (MAX dimerization protein 1) is a transcriptional repressor and it is a member of the MYC/MAX/MAD transcription family, with roles in apoptosis, differentiation and proliferation.(40, 41) The MYC/MAX/MAD network is important in the development of numerous malignancies, including, for example, neural crest tumors such as subsets of pheochromocytoma and neuroblastoma.(42) To our knowledge mutations, including translocations, involving MXD1 have not been reported to date.
In summary, we report six cases of primary undifferentiated tumors occurring in the soft tissue and viscera (brain, kidney, stomach wall) associated with NUTM1 rearrangement, and two novel NUTM1 fusion partners (BCORL1 and MXD1). Despite some overlap with NUT midline carcinoma, the tumors in this series differed in several regards, including: anatomic distribution, morphology and immunophenotype; of particular relevance is the fact that definitive evidence of epithelial differentiation could not be established among many of our cases. As a result, we have tentatively labelled these malignant neoplasms a NUT-associated tumor. It is conceivable this may represent an overarching category that also includes NUT midline carcinoma.
Supplementary Material
Acknowledgments
The authors are grateful to Illumina (San Diego, CA) for generously providing test kits; Ms. Evangeline Agro and Ms. Sharon Crafter for facilitating RNAseq testing; Mr. Anthony Griffin for assistance with clinical follow-up; and, Dr. Abha Gupta for her thoughtful review of an early draft of the manuscript.
Supported in part by: P50 CA140146-01 (CRA); P30-CA008748 (CRA); Kristen Ann Carr Foundation (CRA); Cycle for Survival (CRA)
Footnotes
Disclosures: The author(s) have no conflicts of interest to disclose.
References
- 1.French CA, Bishop JA, Lewis JS, et al. NUT carcinoma. In: El-Naggar AK, Chan JKC, Grandis JR, et al., editors. WHO Classification of Head and Neck Tumours. Lyon: International Agency for Research on Cancer; 2017. pp. 20–21. [Google Scholar]
- 2.French CA. Demystified molecular pathology of NUT midline carcinomas. J Clin Pathol. 2010;63:492–496. doi: 10.1136/jcp.2007.052902. [DOI] [PubMed] [Google Scholar]
- 3.Kubonishi I, Takehara N, Iwata J, et al. Novel t(15;19)(q15;p13) chromosome abnormality in a thymic carcinoma. Cancer Res. 1991;51:3327–3328. [PubMed] [Google Scholar]
- 4.Tanaka M, Kato K, Gomi K, et al. NUT midline carcinoma: report of 2 cases suggestive of pulmonary origin. Am J Surg Pathol. 2012;36:381–388. doi: 10.1097/PAS.0b013e31824230a8. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki S, Kurabe N, Ohnishi I, et al. NSD3-NUT-expressing midline carcinoma of the lung: first characterization of primary cancer tissue. Pathol Res Pract. 2015;211:404–408. doi: 10.1016/j.prp.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 6.Sholl LM, Nishino M, Pokharel S, et al. Primary Pulmonary NUT Midline Carcinoma: Clinical, Radiographic, and Pathologic Characterizations. J Thorac Oncol. 2015;10:951–959. doi: 10.1097/JTO.0000000000000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishop JA, French CA, Ali SZ. Cytopathologic features of NUT midline carcinoma: A series of 26 specimens from 13 patients. Cancer. 2016;124:901–908. doi: 10.1002/cncy.21761. [DOI] [PubMed] [Google Scholar]
- 8.Sirohi D, Garg K, Simko JP, et al. Renal NUT carcinoma: A case report. Histopathology. 2017 doi: 10.1111/his.13368. [DOI] [PubMed] [Google Scholar]
- 9.Chau NG, Hurwitz S, Mitchell CM, et al. Intensive treatment and survival outcomes in NUT midline carcinoma of the head and neck. Cancer. 2016;122:3632–3640. doi: 10.1002/cncr.30242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer DE, Mitchell CM, Strait KM, et al. Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin Cancer Res. 2012;18:5773–5779. doi: 10.1158/1078-0432.CCR-12-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.HGNC Database, HUGO Gene Nomenclature Committee (HGNC), EMBL Outstation - Hinxton, European Bioinformatics Institute, Wellcome Trust Genome Campus, Wellcome Trust Genome Campus, Cambridgeshire, CB10 1SD, UK www.genenames.org. Available at: http://www.genenames.org/cgi-bin/gene_symbol_report?hgnc_id=HGNC:29919. Accessed Sept- 30, 2017
- 12.French C. NUT midline carcinoma. Nat Rev Cancer. 2014;14:149–150. doi: 10.1038/nrc3659. [DOI] [PubMed] [Google Scholar]
- 13.French CA, Ramirez CL, Kolmakova J, et al. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27:2237–2242. doi: 10.1038/sj.onc.1210852. [DOI] [PubMed] [Google Scholar]
- 14.French CA, Rahman S, Walsh EM, et al. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discov. 2014;4:928–941. doi: 10.1158/2159-8290.CD-14-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Tsai WH, Ding Y, et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016;44:e47. doi: 10.1093/nar/gkv1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Schulz-Trieglaff O, Shaw R, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 17.Kao YC, Sung YS, Zhang L, et al. EWSR1 Fusions With CREB Family Transcription Factors Define a Novel Myxoid Mesenchymal Tumor With Predilection for Intracranial Location. Am J Surg Pathol. 2017;41:482–490. doi: 10.1097/PAS.0000000000000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.French CA, Miyoshi I, Kubonishi I, et al. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 2003;63:304–307. [PubMed] [Google Scholar]
- 19.French CA, Miyoshi I, Aster JC, et al. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19) Am J Pathol. 2001;159:1987–1992. doi: 10.1016/S0002-9440(10)63049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson LDR, Franchi A. New tumor entities in the 4th edition of the World Health Organization classification of head and neck tumors: Nasal cavity, paranasal sinuses and skull base. Virchows Arch. 2017 doi: 10.1007/s00428-017-2116-0. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21.Kees UR, Mulcahy MT, Willoughby ML. Intrathoracic carcinoma in an 11-year-old girl showing a translocation t(15;19) Am J Pediatr Hematol Oncol. 1991;13:459–464. doi: 10.1097/00043426-199124000-00011. [DOI] [PubMed] [Google Scholar]
- 22.Alekseyenko AA, Walsh EM, Wang X, et al. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015;29:1507–1523. doi: 10.1101/gad.267583.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.French CA, Kutok JL, Faquin WC, et al. Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol. 2004;22:4135–4139. doi: 10.1200/JCO.2004.02.107. [DOI] [PubMed] [Google Scholar]
- 24.Lemelle L, Pierron G, Freneaux P, et al. NUT carcinoma in children and adults: A multicenter retrospective study. Pediatr Blood Cancer. 2017 doi: 10.1002/pbc.26693. [DOI] [PubMed] [Google Scholar]
- 25.Schaefer IM, Agaimy A, Fletcher CD, et al. Claudin-4 expression distinguishes SWI/SNF complex-deficient undifferentiated carcinomas from sarcomas. Mod Pathol. 2017;30:539–548. doi: 10.1038/modpathol.2016.230. [DOI] [PubMed] [Google Scholar]
- 26.Haack H, Johnson LA, Fry CJ, et al. Diagnosis of NUT midline carcinoma using a NUT-specific monoclonal antibody. Am J Surg Pathol. 2009;33:984–991. doi: 10.1097/PAS.0b013e318198d666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sturm D, Orr BA, Toprak UH, et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell. 2016;164:1060–1072. doi: 10.1016/j.cell.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.French CA. NUT midline carcinoma. Cancer Genet Cytogenet. 2010;203:16–20. doi: 10.1016/j.cancergencyto.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.den Bakker MA, Beverloo BH, van den Heuvel-Eibrink MM, et al. NUT midline carcinoma of the parotid gland with mesenchymal differentiation. Am J Surg Pathol. 2009;33:1253–1258. doi: 10.1097/PAS.0b013e3181abe120. [DOI] [PubMed] [Google Scholar]
- 30.Moll R, Lee I, Gould VE, et al. Immunocytochemical analysis of Ewing's tumors. Patterns of expression of intermediate filaments and desmosomal proteins indicate cell type heterogeneity and pluripotential differentiation. Am J Pathol. 1987;127:288–304. [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada Y, Kuda M, Kohashi K, et al. Histological and immunohistochemical characteristics of undifferentiated small round cell sarcomas associated with CIC-DUX4 and BCOR-CCNB3 fusion genes. Virchows Arch. 2017;470:373–380. doi: 10.1007/s00428-017-2072-8. [DOI] [PubMed] [Google Scholar]
- 32.Li WS, Liao IC, Wen MC, et al. BCOR-CCNB3-positive soft tissue sarcoma with round-cell and spindle-cell histology: a series of four cases highlighting the pitfall of mimicking poorly differentiated synovial sarcoma. Histopathology. 2016;69:792–801. doi: 10.1111/his.13001. [DOI] [PubMed] [Google Scholar]
- 33.Specht K, Zhang L, Sung YS, et al. Novel BCOR-MAML3 and ZC3H7B-BCOR Gene Fusions in Undifferentiated Small Blue Round Cell Sarcomas. Am J Surg Pathol. 2016;40:433–442. doi: 10.1097/PAS.0000000000000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson-Wicking K, Francis RW, Stirnweiss A, et al. Novel BRD4-NUT fusion isoforms increase the pathogenic complexity in NUT midline carcinoma. Oncogene. 2013;32:4664–4674. doi: 10.1038/onc.2012.487. [DOI] [PubMed] [Google Scholar]
- 35.Evans AG, French CA, Cameron MJ, et al. Pathologic characteristics of NUT midline carcinoma arising in the mediastinum. Am J Surg Pathol. 2012;36:1222–1227. doi: 10.1097/PAS.0b013e318258f03b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pagan JK, Arnold J, Hanchard KJ, et al. A novel corepressor, BCoR-L1, represses transcription through an interaction with CtBP. J Biol Chem. 2007;282:15248–15257. doi: 10.1074/jbc.M700246200. [DOI] [PubMed] [Google Scholar]
- 37.Totoki Y, Tatsuno K, Yamamoto S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–469. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- 38.Kao YC, Sung YS, Zhang L, et al. Expanding the molecular signature of ossifying fibromyxoid tumors with two novel gene fusions: CREBBP-BCORL1 and KDM2A-WWTR1. Genes Chromosomes Cancer. 2017;56:42–50. doi: 10.1002/gcc.22400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allen AJ, Ali SM, Gowen K, et al. A recurrent endometrial stromal sarcoma harbors the novel fusion JAZF1-BCORL1. Gynecol Oncol Rep. 2017;20:51–53. doi: 10.1016/j.gore.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amati B, Land H. Myc-Max-Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Curr Opin Genet Dev. 1994;4:102–108. doi: 10.1016/0959-437x(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 41.Ayer DE, Kretzner L, Eisenman RN. Mad: a heterodimeric partner for Max that antagonizes Myc transcriptional activity. Cell. 1993;72:211–222. doi: 10.1016/0092-8674(93)90661-9. [DOI] [PubMed] [Google Scholar]
- 42.Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–667. doi: 10.1038/ng.861. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.