

Abstract
B lymphocytes migrate among varied micro-environmental niches during diversification, selection, and conversion to memory or antibody-secreting plasma cells. Aspects of the nutrient milieu differ within these lymphoid micro-environments and can influence signaling molecules such as the mechanistic Target of Rapamycin (mTOR). However, much remains to be elucidated as to the B cell-intrinsic functions of nutrient-sensing signal transducers that modulate B cell differentiation or antibody affinity. We now show that the amino acid-sensing mTOR complex 1 (mTORC1) is vital for induction of Bcl6 – a key transcriptional regulator of the germinal center fate – in activated B lymphocytes. Accordingly, disruption of mTORC1 after B cell development and activation led to reduced populations of antigen-specific memory B cells as well as plasma cells and germinal center B cells. In addition, induction of the germ line transcript that guides AID in selection of IgG1 heavy chain region during class switching required mTORC1. Expression of the somatic mutator AID was reduced by lack of mTORC1 in B cells, whereas point mutation frequencies in Ag-specific germinal center-phenotype B cells were only halved. These effects culminated in a B cell-intrinsic defect that impacted an anti-viral antibody response and drastically impaired generation of high-affinity IgG1. Collectively, these data establish that mTORC1 governs critical B cell-intrinsic mechanisms essential for establishment of GC differentiation and effective antibody production.
Introduction
BCR-initiated signals in mature lymphocytes are vital for antigen (Ag)-activated B cell proliferation and clonal expansion (1, 2). After several days and divisions, these signals can also yield functionally important class switch recombination as well as somatic hypermutation coupled to the iterative selection of B cells with higher affinity antigen (Ag)-specific BCR in germinal center reactions (3–6). Humoral immunity then stems from the differentiation of B lymphocytes into antibody-secreting plasmablasts and plasma cells (1, 4, 7). Activation-induced cytosine deaminase, AID, which is most highly induced in germinal centers, is essential for both classes of genome revision crucial in humoral immunity, i.e., antibody class switch recombination and the point mutations that diversify and heighten affinity in the memory and antibody repertoires (3, 4, 8–10). B cell activation and co-stimulation in the germinal centers and extrafollicular milieux occur in the context of a variegated interstitial environment (11–13). How sensors of environmental cues influence B lymphocyte-intrinsic mechanisms of differentiation and function that determine antibody response properties through somatic mutation is incompletely understood.
Ag-specific BCR-derived signals (2, 14) are modulated by CD19 along with a menu of additional B cell surface receptors (viz., CD40; IL-4R and IL-21R; TLR4 and 7) whose engagement and activity are crucial for the acquisition of a germinal center B (GCB) cell program (15–19). In the germinal center reaction, activated B cells receive cognate help from T cells that specialize into a TFH phenotype (3, 4). This reaction can involve iterative rounds of proliferation and AID-dependent somatic mutation in the dark zone, followed by restimulation and selection in a TFH–enriched light zone (5, 20–22). Diversification of the BCR on lymphoblasts by somatic mutation is one but not the only factor promoting high-affinity Ab (8, 23). The GC ultimately generates Ag-specific memory cells, plasmablasts, and long-lived plasma cells that constitutively secrete Ab (3, 4). The B lineage also generates plasma cells and some memory through extra-follicular responses – either TLR-driven or aided by T cell help (24–30). The Bmem population that arises from extra-follicular processes is dominated by unswitched IgM+ cells (28, 30). Along each of these cellular pathways, B cells encounter and depend on different arrays and densities of receptor ligands, suggesting that differences in signal transduction determine the relative efficiency of each outcome.
Many receptors that regulate B lineage fates increase activity of phosphatidyl-inositol 3-kinase (PI3K), a set of lipid kinases essential for development, survival, and functions of B cells (31). Phosphatidyl- inositol 3, 4, 5-triphosphate (PIP3) generated from PIP2 by this enzyme recruits pleckstrin homology domain-containing proteins and thereby activates downstream pathways (31). Activity of the mammalian mechanistic target of rapamycin, the serine-threonine kinase mTOR, is stimulated by PI3K (32, 33). mTORC1 activity also is regulated by sensing levels of nutrients such as glucose or certain amino acids (34). Thus, mTORC1 function is of particular interest as it integrates information about metabolite levels with conventional signal transduction by surface receptors. Rapamycin bound to an endogenous cellular protein acutely but incompletely inhibits the multi-protein mTOR complex 1 (mTORC1) by an allosteric mechanism dependent on the structural component Raptor (35). Of note, however, rapamycin cripples a second complex - mTORC2 - in lymphoid lineage and dendritic cells (36–39). Apart from PI3K, the activating inputs of the two respective mTOR complexes differ, as do their functional targets (35). Previous work has indicated that disruption of mTORC2 in B cells by genetic approaches cripples B cell development, survival, and antibody responses (32, 40). Accordingly, genetic approaches are essential for more fully understanding the molecular mechanisms related to mTOR.
Early work in this area showed that rapamycin blocked human B cell proliferation and Ab production in vitro (41). This finding may correlate with in vitro data indicating that mTORC1 inactivation attenuated switching to IgG1 for anti-CD40-activated B cells (42). Experiments in vitro and in mice, including deletion of mTOR with CD19-Cre, indicate that mTOR promotes Ab production and marginally increases high-affinity Ab (43–45), albeit with differences among the results. Intriguingly, systemic rapamycin administration during a primary response to immunization with influenza A virus dramatically decreased GC and GC B cell frequencies yet led to better heterosubtypic protection in cross-strain challenges four weeks after immunization (46). This cross-protection required CD4+ T cells as well as B cells, and the anti-influenza antibody specificities generated in rapamycin-treated mice appeared to differ from those of controls (46). With regard to mTOR activity, mTORC1 and 2 each is inhibited after hours of rapamycin treatment and each has crucial functions in CD4+ T cells that include follicular help to Ab production (37, 38, 47–49), and cell-intrinsic functions in dendritic cells as well (39). These studies of rapamycin and of mTOR – each of which impairs T cell differentiation into functional TFH - raise crucial unanswered questions as to which functions of mTORC1 are intrinsic to B lineage cells in vivo. Notable among these are: (i) what molecular defects of mTORC1-deficient B cells could explain a germinal center impact, (ii) whether lower Aicda gene expression is accompanied by defects of somatic hypermutation, (iii) is affinity maturation altered, and (iv) to what extent the mTORC1 function within B cells influences their capacity to generate memory.
To investigate B cell-intrinsic actions of mTORC1, we used multiple Cre-driven approaches to cause loss-of-function after establishment of normal B cell populations. In vivo and in vitro analyses with these systems revealed requirements for mTORC1 in promoting progression to germinal center and memory stages of the B lineage. Depletion of the core mTORC1 protein Raptor not only reduced AID expression but also limited the induction of germ line transcripts (GLT) that direct antibody class switching, leading to a B cell-intrinsic block in switched Ab in models of immunization or virus infection. These effects were associated with loss of BCL6 induction, weakened B cell support of a TFH population, and reductions in somatic point mutations. Strikingly, we found that quantitative decreases in high-affinity Ab deriving from mTORC1-deficient B cells were more profound than effects on somatic hypermutation. Collectively, the findings indicate that presence of mTORC1, a sensor of nutrient supply, critically impacts the qualities of the primary humoral response.
Materials and Methods
Reagents
Monoclonal antibodies (purified, biotinylated, or fluorophore-conjugated) were from BDPharmingen (San Jose, CA) or Tonbo Biosciences (San Diego, CA) unless otherwise specified. Recombinant B-cell Activating factor (BAFF) was purchased from Adipogen (San Diego, CA); IL-4, Cell Trace Violet, BrdU and CFSE (CFDA-SE) for proliferation assays, and TRIzol reagent for nucleic acid isolation were from ThermoFisher (Carlsbad, CA); LPS, tamoxifen, and 4-hydroxy-tamoxifen from Sigma-Aldrich (St. Louis, MO); NP-BSA and NP-PSA (porcine serum albumin) for ELISA and ELISpot, NP-KLH for immunizations and NP-O-succinimide (NP-Osu for NP-APC conjugation) from Biosearch Technologies (Novato, CA). SRBC were obtained from Remel (San Diego CA) via Thermo-Fisher and used within two weeks. Fluorescent APC protein Prozyme (Hayward, CA) was used in conjugation reactions with NP-Osu. Rapamycin and oligomycin were from EMD Millipore (Billerica, MA) and CellTak for Seahorse assays from Corning (Corning, NY).
Mice and B cell transfer models
All mice - C57BL/6J, B6-μMT, CD45.1 congenic, Ig CH allotype-disparate (IgHa), B6-Rptorf/f Cre [huCD20-CreERT2 (50); Rosa26-CreERT2 (37); Cγ1-Cre (51)] - were housed in ventilated micro-isolators under specified pathogen-free conditions in a Vanderbilt University mouse facility. A balanced mix of males and females was used starting at 6-8 wk of age and following approved mouse protocols, generally taking littermates that were housed together except that some CreERT2 Rptorf/f were bred separately from the CreERT2 Rptor+/+ mice used to control for effects of inducible Cre. In vivo analyses have found this wild-type control yields results no different from Creneg Rptorf/f controls. Other than huCD20-CreERT2 (50) and Rosa26-CreERT2 (37), breeding stock for each line was from JAX (Baa Habuh, ME). For influenza infection experiments, transfer recipient mice were moved to an ABSL-2 facility under separate protocol (J.V.W.) during periods of potential infectious nature and handled only by personnel with appropriate training and approval of Institutional Biosafety. Healthy donor and recipient mice of appropriate genotype were selected randomly for experiments without size or gender preference. For adoptive transfer experiments to measure primary immune responses donor (Rptor+/+, Rosa26-CreERT2+; Rptorf/f, Rosa26-CreERT2+) lymph node and spleen cell suspensions were depleted of T cells using biotinylated anti-Thy1.2 Ab and streptavidin-conjugated microbeads in the BD iMag system. Recipient mice then received intravenous (i.v.) transfers of 5–10×106 cells in sterile saline.
Infections and immunizations, ELISA and ELISpot
For influenza and other adoptive transfer experiments, Rosa26- CreERT2 mice of all three genotypes at the Rptor locus (Rptorf/f, Rptorf/+ and Rptor+/+) mice received a sequence of three tamoxifen injections every other day as described (32), i.e., 3 mg per injection, dissolved in safflower oil, and were harvested one week later. To measure the donor-specific humoral response stemming from transferred B cells in immune competent recipients, purified donor B cells were transferred intravenously (i.v.) into B6-IgHa allotype mice followed by immunization with NP20-ovalbumin as described (32). Alternatively, donor-derived GC-phenotype and NP-specific GC-phenotype B cells were enumerated after i.v. transfers of B cells purified from tamoxifen-injected mice into B6-CD45.1 recipients followed by immunization with NP-KLH [NP16-Keyhole Limpet Hemocyanin (KLH; 100 μg i.p.) precipitated in alum adjuvant [Imject, Thermo Fisher Scientific; (Carlsbad, CA)]. To analyze the characteristics of a serological response to influenza virus immunization and later infectious challenge, purified B cells from donor mice were transferred intravenously (i.v.) into μMT recipients that were then immunized by intra-peritoneal (i.p.) injection of the A/PR8/34 (H1N1) vaccine strain of influenza virus. After interval sera were collected from mice by phlebotomy (1 and 3 wk after immunization), mice were then challenged by intranasal administration of influenza A/X-31 (H3N2). Mice were monitored (with daily weights and clinical assessment to allow for humane endpoint termination if indicated) for 7 d post infection and then harvested (organs and terminal bleed for sera). For other immunization experiments, huCD20-CreERT2 mice (Rptorf/f, and Rptor+/+ controls) received serial tamoxifen injections as described for adoptive transfers. Seven days after the third such injection, mice were immunized with NP16-KLH as described above for the adoptive transfers into B6-CD45.1 recipients.
Measurement of humoral responses
ELISA were performed as described (11, 32). Sera from influenza-challenged mice were assayed using total viral lysate prepared from stocks of each strain (A/PR8/34; A/ X-31) or purified recombinant strain-specific H1 and H3 HA proteins (SinoBiological; Beijing). Relative levels of all- and high-affinity anti-NP antibodies were measured by ELISA capturing the hapten-binding Ab on plate-immobilized NP20-BSA or NP2-PSA, respectively; anti-KLH Ab were measured by ELISA with plate-bound KLH. Allotype- or isotype-specific antibodies were then detected using HRP-conjugated or biotinylated antibodies (Southern Biotechnology Associates, Birmingham AL). Antibody-secreting cells analyzed by ELISpot were quantitated using the ImmunoSpot Analyzer (Cellular Technology, Shaker Heights OH). Antigen specific germinal center and memory B cell populations were analyzed by flow cytometry using APC conjugated NP and an FL-3 dump channel consisting of 7-AAD (Thermo-Fisher, previously Invitrogen) and PerCP-Cy5.5 conjugated IgD (Biolegend, San Diego CA), F4/80, Gr1, CD11b, and CD11c (Tonbo Bio).
Immunoblots
Relative protein or phosphopeptide abundance in whole cell extracts was determined by immunoblotting as described (32, 37). In brief, denatured, reduced proteins separated by SDS-PAGE with a stacking – resolving gel system were transferred to polyvinylidene difluoride membranes by electrophoresis. For improved transfer of Raptor, gels first were incubated (5 min at 22 C) in Trypsin-EDTA (Gibco-BRL Life Technologies division of Thermo-Fisher). Membranes were probed with primary antisera at supplier-recommended dilutions, after blocking by pre-incubation with 5% milk, and rinsed with 0.1% Tween in Tris-buffered saline. After rinsing, membranes were incubated with goat anti-rabbit-specific - or for actin quantitation, donkey anti-goat - secondary anti-IgG Ab conjugated to Alexa680 nm fluorophore (Invitrogen Molecular Probes of Thermo-Fisher), rinsed, and bands visualized and quantified by scanning with an Odyssey Imaging system (Li-Cor, Lincoln NE). For measurement of phosphorylation of S6, anti-phosphopeptide antisera (Cell Signaling Technologies, Danvers MA) directed to the S235/S236 phosphorylated epitopes (a target of both p70 S6K and p90 RSK) were used. Other primary Ab for immunoblotting were from the same supplier [anti-S6 (5G10), anti-phospho S6K(T389), anti-S6K] or, for anti-cyclophilin B (Thermo-Fisher).
B cell cultures and assays of metabolism
Splenocytes were enriched for B cells by Thy1.1 or Thy1.2 depletion (dependent on mouse strain used) using biotinylated anti-Thy1.1 or -Thy1.2 mAb and the BD iMag system. For class switch cultures cells were cultured at a density of 0.5×106 cells/mL in 2 mL cultures on 12-well plates with BAFF; BAFF and LPS; or BAFF, LPS and IL-4. Unless otherwise specified cells were cultured in Iscove’s Modified Dulbecco’s Medium supplemented with 10% FBS. Cultures to measure effect of 2-deoxyglucose (2-DG) received agent (1.5 mM) on day 0 only. Cells were cultured 2-5 d, followed by flow cytometry and ELISA measurements of relative antibody concentrations in culture supernatants. Cultures for measurements of class switch, AID-GFP expression, and proliferation were carried out as described above. For proliferation assays cells were labeled after purification with either CFSE or Cell Trace Violet (as indicated) and then cultured 4 or 5 days (respectively) in the presence of BAFF, or BAFF, LPS and IL4. Radiotracer measurements of glycolysis were conducted as described (37).
Flow cytometry
Flow cytometric phenotyping of in vivo immune responses were conducted 5-10 days after booster immunization as indicated. For detection of NP-specific germinal center responses, spleen or lymph node cells (2×106) were stained with GL7, anti-CD95, -CD38, -B220, -IgM, NP-APC and a dump cocktail containing monoclonal antibodies for IgD, 7-AAD, CD11b, CD11c, F4/80, and Gr1. The flow cytometric gating strategy for analysis of NP-binding, Bmem, and GC B cell populations is shown in Supplemental Figure 1F. For intracellular staining of BCL6 and IRF4, cultured cells were stained for viability (7-AAD) and B220, followed by fixation and permeabilization (4 C for 30 min) using commercial transcription factor buffer (Tonbo), washing with permeabilization buffer (Tonbo), and then staining (1 hr at 4C) with anti-BCL6-APC (eBioscience) (5 μL/106 cells) or anti-IRF4 (2 μL/106 cells) (ThermoFisher). Cells were then washed and IRF4-stained cells were incubated (20 minutes at 4 C) with Alexa647 chicken anti-rabbit IgG second Ab, and washed with permeabilization buffer. For splenocytes recovered after immunization, samples were depleted of Thy1.2 as in the preceding section, stained (107 cells/sample) with IgD, CD19, GL7, and CD95 as well as 7AAD and B220, and then processed for direct and indirect immune fluorescent staining of BCL6 and IRF4 as for the cultured cells. All samples were then resuspended in wash buffer (2% FBS in PBS) followed by flow cytometry. For these and other flow cytometric analyses, fluorescence emission data on cell suspensions were collected on BD LSR or FACSCanto flow cytometers driven by BD FACS Diva software, then processed using Flow-Jo software (FlowJo LLC, formerly TreeStar, Ashland OR).
Seahorse Assays
Purified B cells (conditionally Raptor-depleted and wild-type) from lymphoid organs were plated (2×106 cells/ml in 12-well clusters) and then cultured (2 d) with BAFF alone, or LPS, BAFF, and IL-4. Viable cells recovered from these cultures were seeded at 150,000 cells per well on manufacturer-supplied 96-well plates after pre-coating wells with CellTak (Corning) at 2.5 μg/mL because optimization pilots showed this configuration to yield comparable adhesion along with more vigorous B lymphoblasts as compared to 22.4 μg/mL recommended by Seahorse while poly-D-lysine did not yield suitable adhesion. Measurements of extracellular pH were then made across time on an XFe96 metabolic flux analyzer (Agilent Technologies, Santa Clara CA) and converted to calculated or estimated rates of oxygen consumption, extracellular acidification, glycolysis, glycolytic flux, and glycolytic capacity using Microsoft Excel (Mac OS X) with formulae from Seahorse protocols.
Somatic Hypermutation Analyses
Wild Type (Rosa26- Cre ERT2+, Rptor+/+) or Rptor knockout (Rosa26-CreER T2+, Rptor f/f ->Δ/Δ) B cells were transferred into μMT recipients (5×106 cells per recipient). The mice were immunized with NP-KLH in alum as above, and harvested ten days later. Viable (Ghost dye-excluding cells in the viable lymphoid FSC × SSC gate) NP-specific germinal center B cells as well as naïve (GL7− IgD+) B cells were flow sorted and lysed for cellular DNA. [This viable cell gate is devoid of early apoptotic cells (i.e., annexin-V+ or cleaved caspase-3+ events).] Nested PCR was performed as previously described (43) and gel purified products were subcloned into TA vector [Promega (Madison, WI)] for sequencing. Sequences were aligned and scored for mutations using MEGA software.
Quantitation of mRNA by qPCR
mRNA was isolated from flow sorted germinal center phenotype B cells (dumpneg B220+ GL7+), memory B cells (dumpneg B220+ GL7neg), and naïve (dumpneg B220+ IgD+) B cells using TRIZOL reagent and then purified following the standard manufacturer’s protocol. B cell cultures grown to measure mRNA were harvested into TRIZOL, from which RNA was purified using the manufacturer’s protocol. RNA concentrations were measured using a NanoDrop spectrophotometer and then used to synthesize cDNA using the Promega AMV Reverse Transcriptase kit. All mRNA quantifications were normalized to HPRT. Primer pair sequences are tabulated and further details freely available upon request.
Statistical methods and tests of “significance”
The primary analyses were conducted on pooled data points from independent samples and replicates (minimum two, generally three, biologically and temporally independent replicate experiments for all data, with multiple independent samples in the case of two biological replicates), using an unpaired two-tailed Student’s t test with post-test validation of its suitability. Two-way ANOVA with Bonferroni correction for multiple comparisons was used for statistical analysis across ELISA titration curves to compare wild-type to each mutant sample set. When this determination indicated rejection of the null hypothesis (i.e., p<0.05), two-tailed comparisons of wild-type versus mutant were performed at single dilution values as above (typically, Students t-test). Data are displayed as mean (± SEM), i.e., ‘center values’ were ‘means’ as ‘average’. Results were considered statistically significant when the p value for the null hypothesis of a comparison was <0.05. Most p values are shown in the Figure panels, with some in Legends or Results text. All resulted from statistical tests of null hypotheses with post-hoc testing to assure appropriateness. Since the extent or direction of difference between samples was unknown, and regulations mandate reducing the number of animals used to the lowest feasible level, no statistical methods were used to determine pre-specified sample sizes and mice of both genders were selected in balanced numbers without bias. The experiments were not randomized and the investigators were not blinded during the experiments.
Tabulated details
A tabulation of names, sources, and stock numbers for Ab reagents and other such details are freely available on request.
RESULTS
Raptor is essential for a class-switched but not IgM response to hapten
We first used transfer models with a chemically activated Cre and conditional loss-of-function for Raptor to investigate how mTORC1 affects the capacity for B cells to yield antibody responses. When Raptor-deficient B cells were transferred into immune competent allotype-disparate recipients followed by immunization (Fig. 1A), the switched IgG1 anti-hapten response derived from donor (IgHb) B cells was completely eliminated, whereas Ag-specific IgMb concentrations were the same as those derived from WT control B cells (Fig. 1B). Internal control on immunization quality was provided by analyses of the recipient (a allotype) antibodies, which were no lower in recipients of Raptor-null B cells than controls. Similar data were obtained by measuring donor-allotype Ab secreting cells (ASCs) (Fig. 1C). Control analyses performed with B cells purified after tamoxifen injections showed that the chemically-induced Rosa26-Cre-mediated deletion was efficient. Little residual Raptor protein was detected, and phosphorylation of a protein downstream from mTORC1 was reduced substantially (Fig. 1D). Because the donor-derived B cells are a small minority of Ag-specific and activated B cells in these experiments, these data indicate that a switched response can have a strong B cell-autonomous dependence on mTORC1. Parallel work indicated that homozygous Rptor depletion led to a failure to elicit sustainable IgM (44). However, our results show that B cells can yield a normal primary IgM response despite essentially complete loss of mTORC1 function after Raptor depletion.
Figure 1.
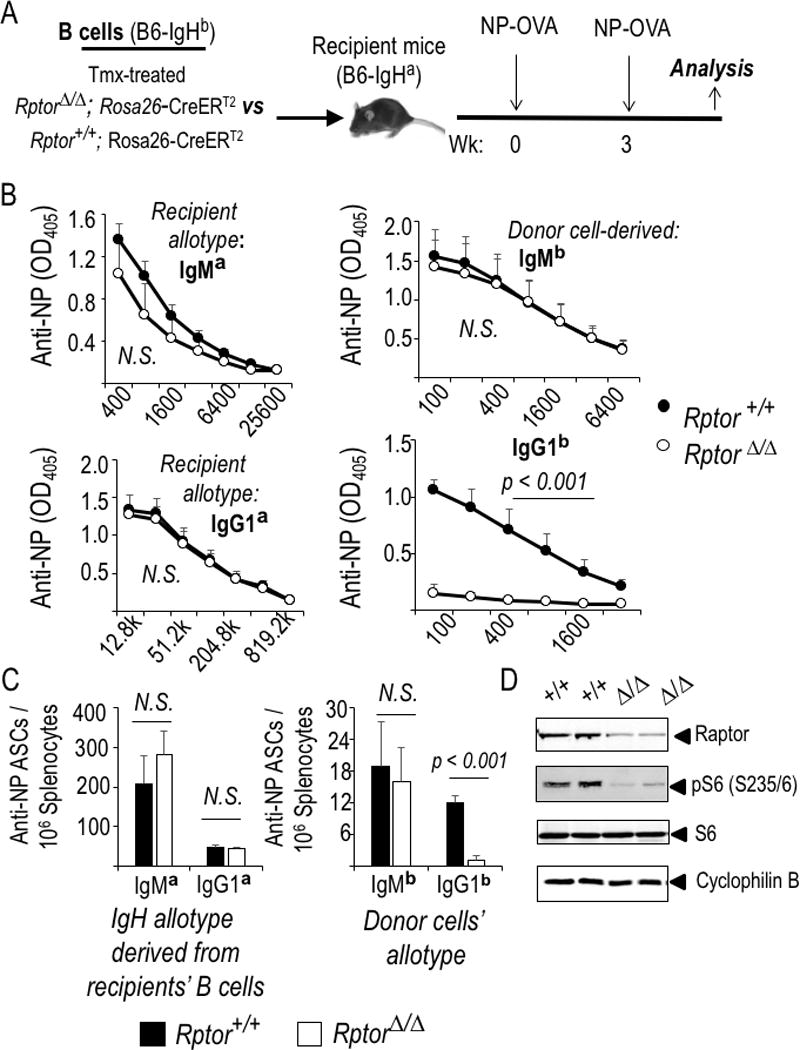
B cell-autonomous mTORC1 regulates the class switch and production of antibodies in response to NP-KLH immunization. (A) Equal numbers of B cells from tamoxifen-treated WT (Rptor +/+) or Raptor-deficient (Rptor fl/fl) CreERT2+ mice were transferred into CD45.1 (Fig. 2F, G) or IgHa-[a allotype]-disparate recipients that were then immunized following the schedule in the schematic. (B, C) Relative antibody concentrations (B), and prevalence of splenocytes that secrete Ag-specific Ab of the indicated isotype after harvesting immunized IgHa mice that received IgHb B cells (WT or Raptor-depleted, closed and open symbols, respectively) (C). (B) The mean (±SEM) data from three independent replicate experiments of Ig allotype disparate ELISA’s for all-affinity IgM/ G1a recipient antibody and IgM/G1b donor antibody responses. (C) Mean (±SEM) data from allotype-specific measurements of ASC by ELISPOT in the same three independent experiments (nine mice for each genotype of donor B cells). (D) Efficient mTORC1 inactivation via deletion of the conditional Rptor allele. Shown are immunoblots performed using the indicated Ab and extracts of B cells purified from mice of the indicated genotypes (one experiment representative of 2-3 replicates with biologically independent samples).
Reduced BCL6 expression and Ag-specific germinal center cells
Prior work with mTOR, rapamycin, and Rptor loss of function has indicated that mTORC1 promotes germinal center reactions (43–49) but the cellular or molecular mechanisms for this impairment are not known. We tested how disruption of mTORC1 in B cells affected proliferation, the populations of B cells, and their expression of key transcriptional mediators of the GC B cell program after immunization. To do so, we used both in situ Rptor gene disruption in transgenic models that allow B lineage-restricted tamoxifen activation of Cre-mediated Rptor gene inactivation (32, 52) (Fig. 2) and adoptive transfers into immunocompetent, allotype-disparate mice. In situ deletion in B cells driven by chemical activation of Cre using the huCD20-CreERT2 transgene yielded evidence of a reduction in the number of B cells recovered 4-5 wk after the onset of Raptor depletion (Fig. 2A, B; Supplemental Data Fig. 1A–C). Within the population of B cells, the frequency of GL7+ Fas+ (GCB-phenotype) cells was reproducibly reduced - albeit only to a modest extent (flow plots; Fig. 2C) - after immunization of tamoxifen-injected huCD20-CreERT2 mice that were Rptor f/f, leading to a reduction to ~2/3 control numbers (Fig. 2C). However, the frequency of NP-binding cells among B lymphocytes was more substantially affected by loss of Raptor than was the overall population (Fig. 2D). Cumulatively, the numbers of Ag-specific GC-phenotype B cells were reduced to about 0.4x those of control mice when naïve and activated B lineage cells were deficient for mTORC1 (Fig. 2D). Activation of the B lineage-restricted huCD20-driven CreERT2 led to sustained though not complete loss of Raptor in the B cell population present after immunization (Fig. 2E). Comparison of the data from longer-term loss-of-function (Fig. 2E) with the more immediate B cell isolation (Fig. 1D) suggests that the selective outgrowth of B cells in which Rptor deletion failed was at most modest. These results were complemented by independent experiments in which mTORC1-depleted B cells competed against a majority of normal B cells (Fig. 2F, G; Supplemental Fig. 1D, E). In this competitive setting, almost no NP-binding GL7+ B cells were observed after transfers of B cells from wild-type or Raptor-depleted B cells (CD45.2) into CD45.1 recipients followed by immunization.
Figure 2.
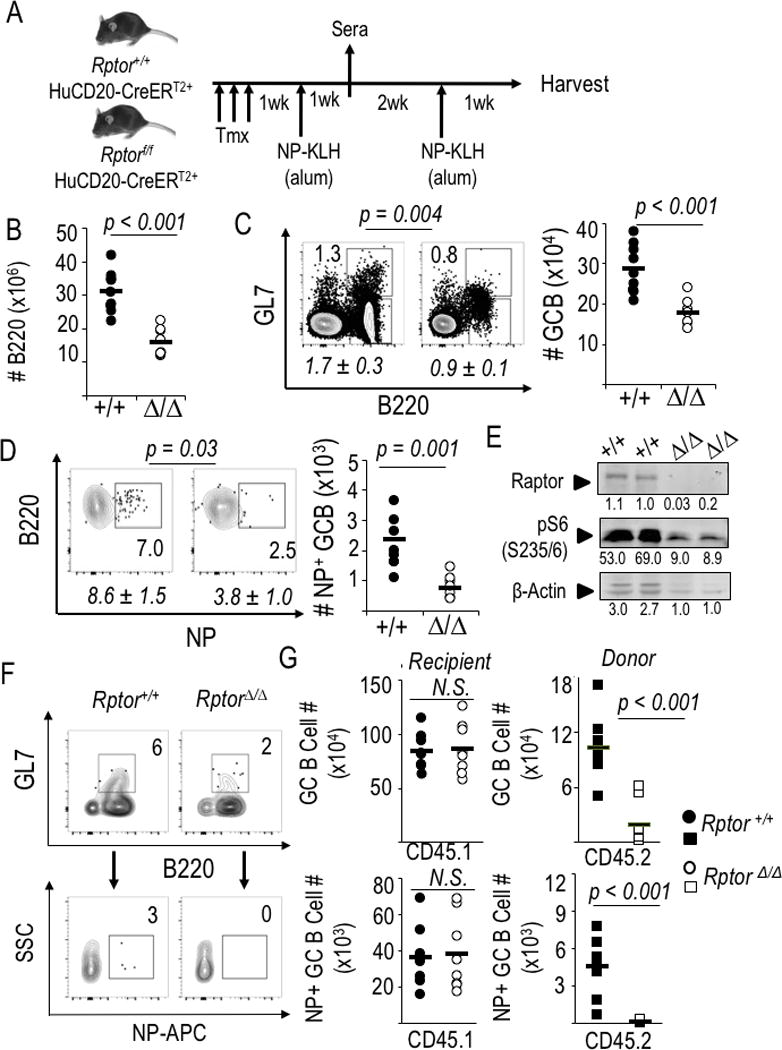
mTORC1 promotes production of class-switched antibodies by B cell-intrinsic mechanisms regulating Ag-specific GC B cells. (A) Schematic of experimental design used for panels B-E. (B-D) Splenocytes harvested 1 wk after the booster immunization were counted and analyzed by flow cytometry. (B) B cell counts in spleens at harvest after immunization and boost of huCD20-CreERT2 mice (Rptor +/+ or Δ/Δ). Each point, calculated from the enumeration of splenocytes and the fraction of B220+ cells [Supplemental Fig. 1A], represents an individual mouse [nine mice of each genotype, distributed evenly across four independent replicate experiments] and the horizontal bars represent means. (C, D) Preferential impact of intrinsic mTORC1 on Ag-specific germinal center B cells. (C) One representative result (with inset numbers representing percentages in the indicated GC-phenotype gate) as well as quantitation of the mean (±SEM) frequencies (italicized) of such B cells in the nine independent mice of each genotype in the four biological replicate experiments as in (B). (D) NP-binding B cells in the gate for viable, IgDneg, myeloid lineageneg cells that were GL7+ (germinal center phenotype), with a representative result from one experiment shown along with mean (±SEM) quantitation as in (C). (E) Recovered cells remained Raptor-depleted. CD19+ B cells were microbead-purified from spleens of tamoxifen-injected, immunized mice (huCD20-CreERT2+ Rptor+/+, and huCD20- CreERT2+ Rptor fl/fl), activated with LPS, cultured (2 d) and then analyzed by immunoblotting to measure Raptor levels and phosphorylation of S6 [anti-pS6(S235/236] as well as actin, shown as fluorescence units quantitated on the Odyssey IR imager. (F, G) Purified WT and Raptor-depleted B cells (as in Figure 1A) were transferred into CD45.1 recipients that were then immunized with NP-KLH, boosted 3 weeks later, and harvested one week after the boost. (F) Representative flow plots from within the viable cell, donor (CD45.2+) gate, with inset numbers representing the percentage germinal center-phenotype (dumpneg GL7+ B220+) B cells from spleens of CD45.1 recipient mice (upper panel pair) and the antigen-specific (NP-APC+) events in the GL7+ population (lower panel pair), which are then quantified in (G) with circles representing recipient mouse B cell phenotyping (CD45.1+) and squares representing the donor (CD45.2+) population (8 mice per donor cell genotype, in balanced distributions among three independent replicate experiments). Representative flow data for donor CD45.2 staining specificity are in Supplemental Fig. 1E.
Notwithstanding reduced proliferation of mTORC1-deficient B cells in vitro [(49), and data herein], these findings indicated that there is an intrinsic impairment of the differentiation program for GCB cells, beyond loss of surface IgD and induction of the GL7 epitope. To investigate this issue, we purified GL7+ IgD− CD38int viable (7-AADneg events in the lymphoid FSC × SSC gate) B cells by flow sorting after immunization of the tamoxifen-injected, huCD20-CreERT2+ mice. Quantitation of mRNA in these purified mTORC1-depleted cells revealed substantial or dramatic reductions in Irf4 and Bcl6 gene expression, respectively, in the viable GC-phenotype B cells (Fig. 3A). Consistent with this, Aicda mRNA also was lower (Fig. 3A). The GC makes major contributions to B cell memory (3, 4, 25, 29). Of note, Irf4 and Aicda mRNA levels also were lower in Raptor-depleted memory-phenotype B cells (Fig. 3B). In the development of follicular helper characteristics among CD4 T cells, mTORC1 improves translational efficiency of Bcl6 but not Irf4 mRNA (52). Consistent with this report, BCL6 protein was far lower in the Rptor-inactivated GCB subsets by intracellular staining after immunization as compared to WT controls (Fig. 3C). Analyses of T cells demonstrated links among transporters, uptake of leucine (via LAT1) and glutamine (via ASCT2), and differentiation (53, 54). When we tested if mTORC1 promotes gene expression for Slc7a5 and Slc1a5 (encoding LAT1 and ASCT2, respectively) in germinal center B cells, we found striking decreases in transporter induction (Fig. 3D). Consistent with this finding, mTORC1-deficient B lymphoblasts in vitro expressed less of these transporters (Supplemental Fig. 2A). Induction of the CD98 heavy chain – which pairs with LAT1 and ASCT2 along with other transport proteins, was also reduced (Fig. 3E). Together, these findings indicated that the capacity to express genes crucial for GC B cell growth and function is impaired in Raptor-depleted B cells. Rapamycin treatment during and after immunization almost completely eliminated mouse germinal centers (46), but rapamycin impacts both mTORC2 and mTORC1 and impacts all cells. Moreover, Tfh cells depend on both mTORC1 and mTORC2. GCs also were absent after disruption of mTORC2 via Rictor gene inactivation (32) or in mutant mice whose mTOR is functionally hypomorphic (43, 45). Therefore, we tested how B cell-intrinsic mTORC1 deficiency impacts GC anatomy. Immunofluorescent microscopy of spleens harvested after immunization of tamoxifen-injected huCD20-CreERT2+ mice (i.e., WT or Rptor Δ/Δ B cells) showed that an IgD-negative zone was organized within IgD+ IgM+ follicles surrounded by CD35+ cores (Supplemental Fig. 2B). These structures were present in essentially normal numbers [means (±SEM) of 43 (±5.1) for WT and 38 (±5.7) for Rptor Δ/Δ, p > 0.1], whereas germinal center structures with GL7+ centers depended on mTORC1 [mean values were 40 (±5.0) for WT versus 10 (±2.4) when B cells were Rptor Δ/Δ, p < 0.01] (Supplemental Fig. 2B). These anatomic data indicate that GC were reduced, but suggest that initial elements of a GC might form, possibly from a reduced number of B cells in which some mTORC1 remained. Notwithstanding, across the population, mTORC1 mediated at least a 10-fold increase of BCL6 in GL7+ B cells. When Raptor is depleted at the onset of B cell activation, the yield of Ag-specific memory-phenotype B cells also was reduced (Fig. 3F) to an extent that exceeds any effect on B cell numbers overall.
Figure 3.
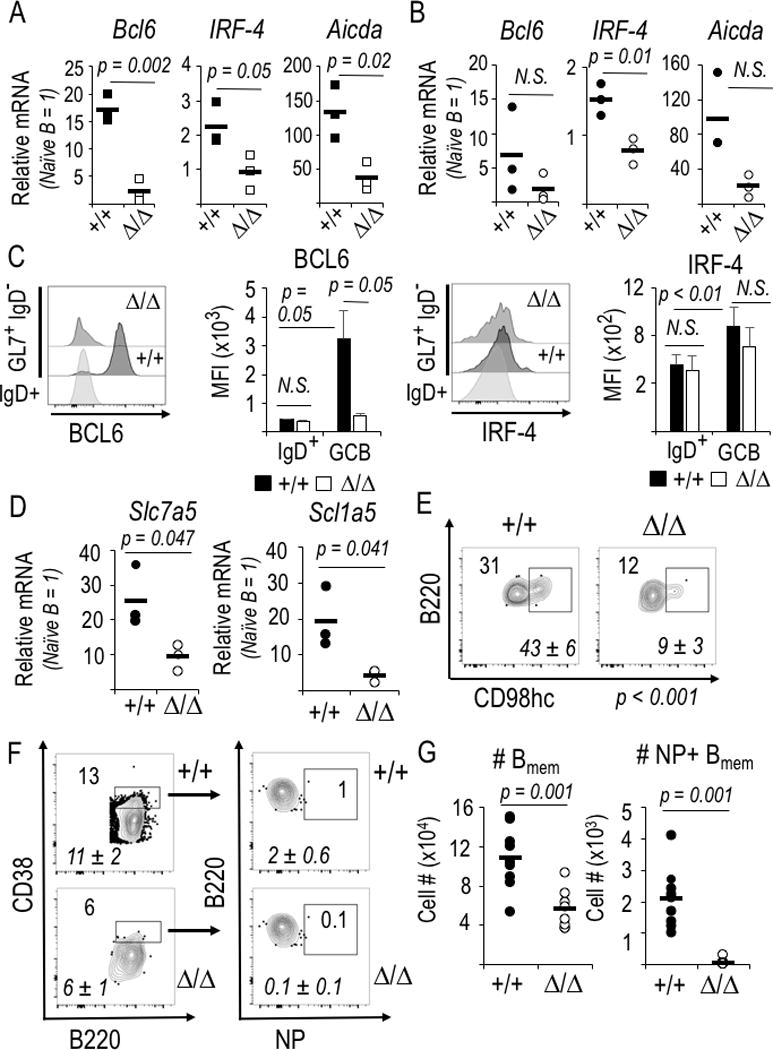
Bcl6 and Irf4 gene expression along with neutral amino acid transporters require mTORC1 in the GC-phenotype B cell. (A, B) Relative mRNA levels were quantified by qRT2-PCR using naïve, GC- and memory-phenotype B cells (huCD20-CreERT2+, either Rptor +/+ or Δ/Δ) purified from the splenocyte suspensions by flow sorting B cells according to IgD, GL7, CD38, and viability indices (FSC, SSC, and 7-AAD dye). Relative mRNA is normalized to flow-purified IgD+ GL7neg naïve B cells, whose relative mRNA level is defined as 1 for each amplification. Shown are mean (±SEM) measurements of independent samples in two biological replicate experiments, each with three each of WT (CreERT2+, Rptor+/+) and KO (CreERT2+, Rptorf/f). (A) Reduced mRNA encoding AID (Aicda) as well as BCL6 (Bcl6) and IRF4 (Irf4), transcription factors essential for GC B cell differentiation, in Raptor-depleted GL7+ B cells. (B) Reduced mRNA encoding IRF4 in Raptor-depleted memory-phenotype (IgD− GL7− CD38hi) B cells. (C) Induction of BCL6 in IgD− GL7+ B cells required mTORC1. Mice (huCD20-CreERT2+ Rptor +/+, and huCD20-CreERT2+ Rptor fl/fl)) were injected with tamoxifen and initially immunized with NP-KLH in alum as in Fig. 2A, boosted with the same amount of NP-KLH in alum one week later, harvested one week thereafter, processed as described in the Methods, followed by collection of 2 × 106 viable, dumpneg events on the flow cytometer. Shown to the left are representative flow profiles for intracellular staining of BCL6 in the GL7+ CD95+ gate or IgD+ control gate, as indicated, with B cells that were WT (+/+) or Raptor-depleted (Δ/Δ), flanked by a bar graph with the mean (±SEM) MFI for BCL6 in the replicate samples (two independent experiments, each with two mice of each genotype). To the right are the analogous data for intracellular IRF4 in splenic B cells of the same immunized mice. (D) mTORC1 dependence of large, neutral amino acid transporter gene expression in GC B cells. (E) Reduced cell surface CD98hc on Raptor-depleted GC-phenotype B cells. Representative and aggregate [italicized mean (±SEM)] flow cytometry results for IgD+ GL7neg B cells of the immunized mice. P<0.01 for the reduced CD98hc on mTORC1-depleted B cells relative to controls. (F, G) Formation of Ag-specific memory B cells requires mTORC1. (F) Representative flow phenotyping results for spleens from immunized and boosted huCD20-CreERT2 mice analyzed in Fig. 2A-E. Inset and italicized (mean ± SEM) numbers are as in Fig. 2C. (G) Absolute numbers of memory-phenotype and NP-binding memory B cells in the spleens of huCD20-CreERT2 mice, derived from phenotyping in (F).
Regulation of Iγ1 germ line transcript by mTORC1 and glycolysis in B cells
There were major defects of the class-switched anti-viral Ab response in rapamycin-treated mice, and anti-HA IgM increased due to rapamycin in this model (46), and reduced AID levels have been implicated as a mechanism for these findings (43). In contrast, Ag-specific IgM production failed within days after hapten-carrier immunization (44). We found in vitro switching to IgG1 was 3-fold higher when B cells expressed Raptor (Fig 4A). In addition to an effect on Aicda mRNA, however, the induction of Iγ1 germ line transcription that is essential for directing recombination to the switch region for IgG1 (55) was drastically reduced (Fig. 4B). Induction of Aicda gene expression and functional class switching require time and cell divisions (6). Even when flow cytometric gating restricted measurements to B lymphoblasts that had divided several times, the fractions of mTORC1-deficient B cells with switched Ab class (Supplemental Fig. 2C) and their levels of AID expression marked by fluorescence of an AID-GFP translational fusion were reduced (Fig. 4C; Supplemental Fig. 2D). Of note, AID protein expression was maintained among B cells that divided several times (Fig. 4C) though the frequency of cells that become GFP-positive was reduced. Irf4 and Bcl6 mRNA levels were substantially lower in Raptor-deficient B cells after activation (Fig. 4D; Supplemental Fig. 2E), as observed in the freshly sorted GC B cells after immunization (Fig. 3A). These defects culminated in a reduction in accumulation of class-switched Ab in vitro (Supplemental Fig. 2F) proportionally similar to the impaired generation of IgG1+ B cells. Thus, although there still is no in vitro system that accurately recapitulates the germinal center reaction, data in this model were congruent with those we obtained from in vivo GC-phenotype activated B cells. We conclude that mTORC1 promotes the induction of transcription factors essential for GC B cells, as well as gene targets crucial for Ig class switching.
Figure 4.
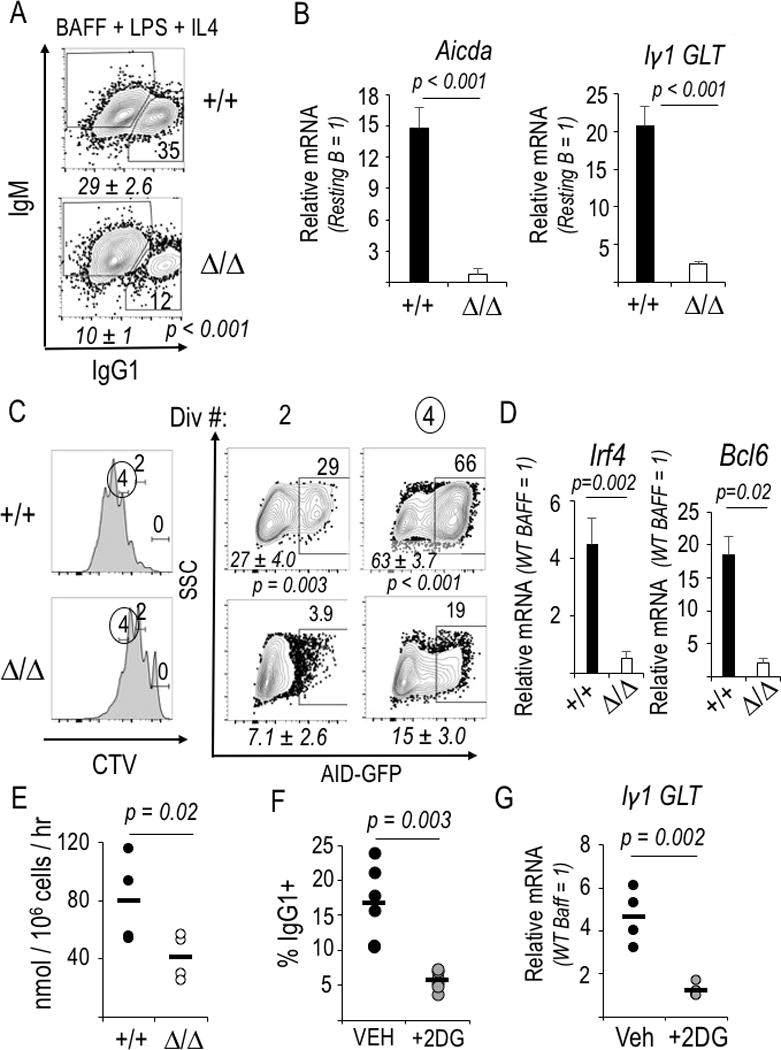
Glycolysis promotes IgCH germ line Iγ1 transcription and CSR. (A) Flow cytometric measurements of Ab class switching to IgG1 after WT and Raptor-depleted B cells were activated (LPS) and cultured with BAFF and IL-4. Shown are the flow plots of samples in one representative experiment. Italicized mean (±SEM) values in the three independent replicates [4 (WT) and 5 (Rptor Δ/Δ) samples] and inset numbers denote the fractions of IgG1+ cells in the viable B220+ gate for the particular experiment. (B) Decreased Aicda and germ line Iγ1 transcripts, measured by qRT2-PCR in mTORC1-deficient and control B cells 2 d after activation and culture as in (A). (n=3 independent replicate experiments). (C) Decreased AID in Raptor-depleted B lymphoblasts. WT and Raptor-deleted B cells encoding a AID-GFP BAC transgene were stained with CellTrace Violet before activation and culture as in (A), followed by flow cytometric analysis. Left panels: A representative division history (dye partitioning); Right: frequencies of GFP+ B cells in the gates for two (2) and four (4) divisions of one representative experiment, with italicized mean (±SEM) values for the samples in four independent replicate experiments. (D) Bcl6 and Irf4 mRNA levels in the samples of (C), measured by qRT2-PCR. (E) Reduced glycolysis in mTORC1-deficient B lymphoblasts. Purified B cells, activated and grown as in Fig. 4A, were assayed for flux of 3H from tritiated glucose into water via glycolytic generation of pyruvate. Mean (±SEM) results from four different samples, distributed equally in two biologically independent experiments. (F) As in Fig. 4A except 2-deoxyglucose (2DG) was added as indicated, with data from six independent experiments graphed as individual values (black or grey circles) along with each mean (horizontal line). (G) Glycolytic flux supports induction of Iγ1 germline transcripts (GLT). Results of qRT2-PCR using RNA of B cells cultured in BAFF alone (resting B cell control) or with IL-4 after LPS activation; each sample was normalized to the BAFF control culture. Shown are individual values from four independent experiments and mean results, displayed as in (F).
We then explored if there is a connection between mTOR regulation of cellular metabolism and the observed effects of Raptor depletion on class switching. Glycolysis in T cells is promoted by mTORC1 (47, 48). Flux of radiolabel from glucose into water via glycolytic generation of pyruvate was halved when equal numbers of WT and Rptor Δ/Δ B cells that had been activated and grown in vitro were assayed (Fig. 4E). These data were supported by an analogous decrease in extracellular acidification rates (Supplemental Fig. 3A). To decrease glycolytic flux experimentally, in a manner independent of the known impact of such a reduction on Tfh cells, we tested the effect of 2 deoxyglucose (2DG) on switching by purified B cells and found that it substantially reduced the frequency of IgG1+ IL-4-treated B lymphoblasts (Fig. 4F) without undermining viability (Supplemental Fig. 3B). Inhibition of glycolytic flux prevented induction of the Iγ1 germ line transcript (Fig. 4G), a proliferation-independent event. The frequency of B cells that became AID-GFP+ also decreased (Supplemental Fig. 3C), as did the activation-induced increases in BCL6 and IRF4 (Supplemental Fig. 3D), though the magnitudes of these effects were less striking than those stemming from lack of mTORC1 or the impact of glycolysis inhibition on Iγ1. We conclude that mTORC1 defects reduce glycolysis, and interference with glycolytic flux as a primary intervention attenuates Ab class switching via a failure to induce germ line transcription. However, additional mechanisms that regulate levels of AID likely collaborate in the overall reduction.
B cell-intrinsic Raptor function promotes SHM and high-affinity Ab
How B cell-intrinsic mTORC1 influences somatic hypermutation rates or affinity maturation in physiological conditions is unclear. Using the tamoxifen-triggered huCD20-CreERT2 system to effect Raptor depletion in B cells prior to immunization led to lower concentrations of Ab, particularly of switched isotype (Fig. 5A). Strikingly, the impact on high-affinity Ab (stably bound to low-valency NP in the ELISA) was as much as 20-fold greater than that on the all-affinity Ab (Fig. 5A), so that the NP2/NP20 ratio averaged 0.05x control. This result contrasts with a prior finding that B lineage-specific inactivation of the gene encoding the kinase mTOR resulted in a modest reduction of higher-affinity anti-NP Ab relative to all anti-NP, i.e., to about 0.9x control levels (43). The impact of mTORC1 also was evident in measurements of antibody-secreting cell (ASC) frequencies (Fig. 5B). The decreases in high-affinity Ab, together with the reduced AID, prompted us to explore underlying mechanism(s). Affinity maturation involves both the rate of imposing mutations and a mechanistically distinct process of selecting for coding sequence changes that affect BCR affinity for Ag. Accordingly, we quantified somatic mutations in non-coding (and hence non-selected) DNA of the immunoglobulin heavy chains of NP-binding B cells. This analysis revealed less frequent non-coding somatic mutations in NP-binding Raptor-depleted B cells (Fig. 5C). These data show that the reduction in mTORC1 limited both AID and the rate of mutation. However, the quantified decrease in mutation rate (~ 0.5x WT rate) was an order of magnitude less than the loss of high-affinity IgG1 anti-NP (~ 0.05x WT concentrations). These differences suggest that selective pressure on the B cells is higher while the mutation rate is lower, so that the combined alterations lead to an antibody response of dramatically lower affinity. Survival and affinity maturation depend on competition for a limiting pool of Tfh cells, and the capacity to sustain a Tfh phenotype in CD4 T cells requires sustained interactions with GC B cells (4, 5). Thus, it was striking that the B lineage defects after huCD20-CreERT2-driven Rptor gene disruption caused a profound decrease in activated CXCR5+ CD4+ T cells (Fig. 5D). These data are consistent with a model in which mTORC1-deficient B cells provide insufficient support for stabilization of a Tfh program among cognate CD4+ T cells with which they interact.
Figure 5.
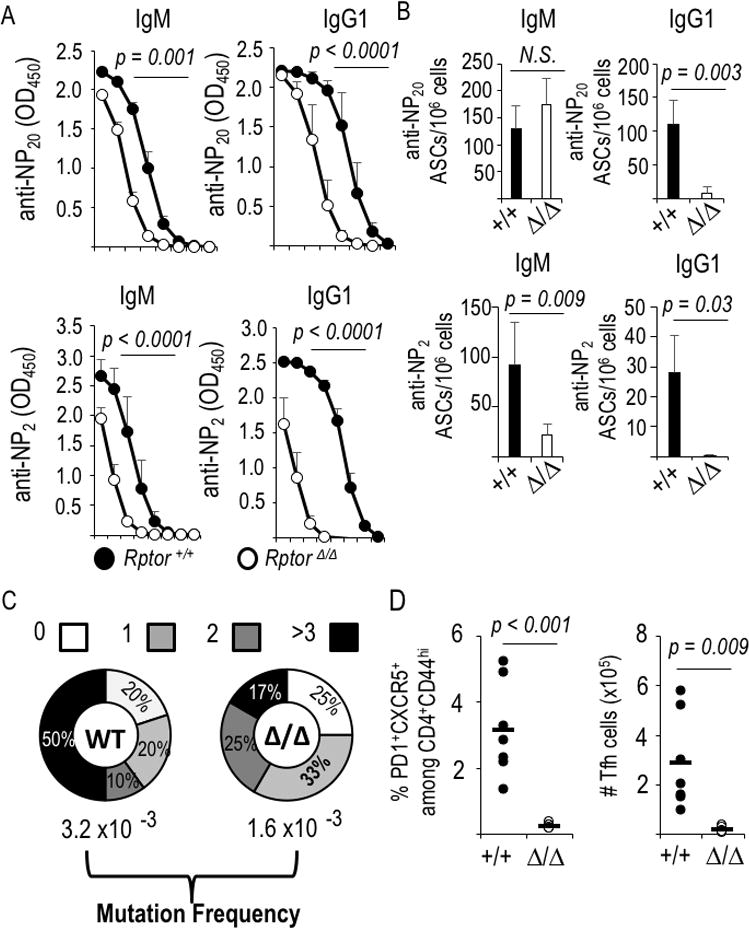
B cell-intrinsic mTORC1 promotes somatic mutation of Ig VH DNA and dramatically enhances affinity maturation. (A) All-affinity (anti-NP20) and high-affinity (anti-NP2) IgM and IgG1 in sera of immunized and boosted, tamoxifen-injected huCD20-CreERT2 mice that were either Rptor+/+ or Rptorf/f (as in Fig. 2A). Shown are the mean (±SEM) values across serial 4-fold dilutions for the independent mice (n=8, Cre+ WT; n=7 Cre+ Rptor Δ/Δ), in three separate experiments. (Initial dilutions: NP20, 1:400; NP2, 1:100). (B) Mean (±SEM) frequencies of ASCs quantified as all-affinity (anti-NP20) and high-affinity (anti-NP2) IgM- and IgG1-secreting cells in spleens of the immunized mice of Fig. 2 and Fig. 5a. (C) B cells from tamoxifen-treated CreERT2+ mice (WT and Rptor Δ/Δ) were transferred into μMT recipients that were immunized with NP-KLH. After flow purification of viable NP-binding IgDneg GL7+ B cells, rates of somatic mutation in a non-coding interval of VH186-2 of genomic DNA were scored [as in (43)]. (D) Raptor-deficient B cells fail to support the Tfh population. Splenocytes from immunized and boosted mice (as in Fig. 2A-E and Fig. 5A, B) were analyzed by flow cytometry to determine the fraction and number of Tfh (CXCR5+ PD-1+) cells among activated (CD44+) CD4+ T cells. Graphs display the values measured for each individual mouse, along with means (horizontal bars).
Because results in these and earlier studies might in part be due to limitations of the model systems, we used variant approaches. First, a hapten facilitates detection and semi-quantitation of affinity maturation in the overall Ab population, but the nature of the antigen potentially could influence results. We found here that mTORC1 depletion had a similar effect on concentrations of class-switched anti-KLH and anti-NP Ab after immunizations of the huCD20-CreERT2 mice: IgG1 anti-KLH was ~30-fold higher in sera of controls than in the mice whose B cells were Raptor-deficient, whereas IgM only decreased to ~ 0.5x control levels (Fig. 6A). Second, a potential concern with any work in which the signaling pathway is eliminated first is that the loss-of-function might yield an abnormal population of cells prior to immunization, or that results might be due to a defect arising at the time of initial B cell activation. Moreover, tamoxifen has strong effects on humoral immunity (56, 57). Thus, it was possible that either this strong nuclear hormone receptor ligand or the transfer model would somehow not be representative of physiological requirements. However, with Cγ1-Cre mice that were Rptor +/+ or f/f, all-affinity IgG1 was ~1/64th the control concentrations and no high-affinity anti-NP IgG1 was detectable whereas the IgM anti-NP response was normal (Fig. 6B). As expected, B cell numbers were normal (Fig. 6C) since only a low fraction of the B cell population would be activated by immunization and turn on transcription of the Cγ1-Cre allele. Nonetheless, the frequencies of GC-phenotype and of NP-binding B cells among them were reproducibly reduced (Fig. 6D). The prevalence of Tfh-phenotype T cells also was lower after Cγ1-Cre-driven Raptor depletion in vivo (Fig. 6E), consistent with both the results of experiments with huCD20-CreERT2 and the characteristics of each means of deleting the targeted Rptor gene segment. Because the Cγ1-Cre transgene acts only after activation and proliferation of B cells, these data indicate that (i) B cell-intrinsic mTORC1 is required even after initial B cell activation and (ii) the results are not a function of either the adoptive transfer system or the immune effects of tamoxifen.
Figure 6.
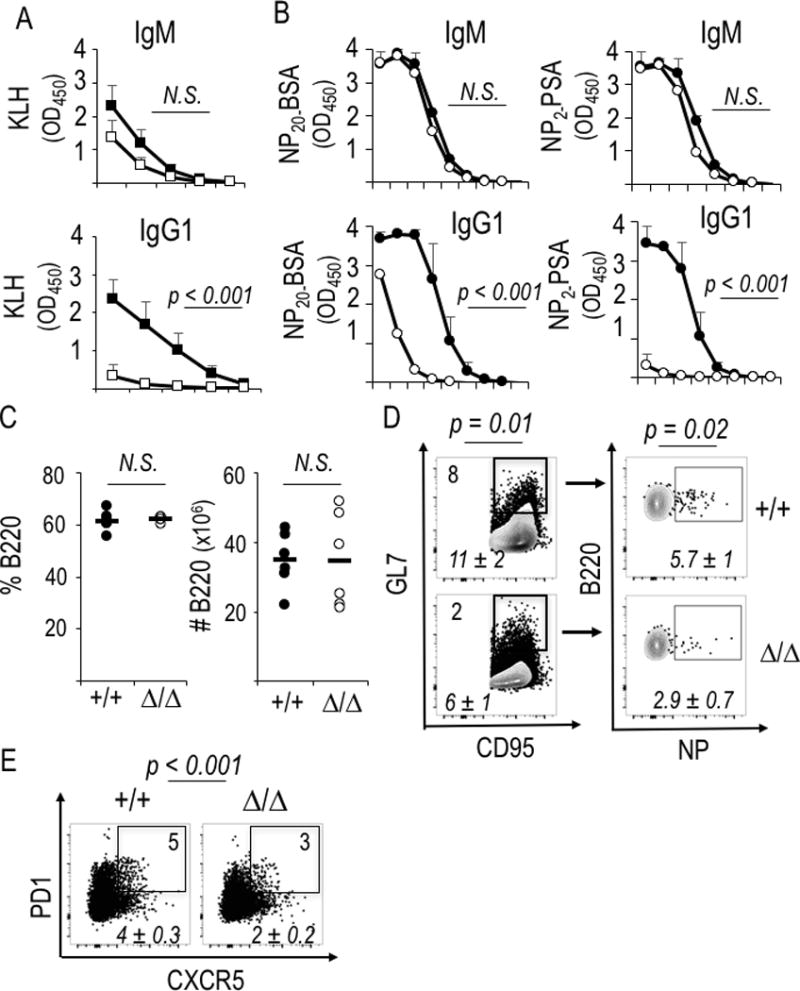
Raptor depletion causes reduced switching and affinity maturation in hapten- or tamoxifen-independent systems. (A) Relative concentrations of anti-KLH Ab were measured by ELISA using sera from the eight mice immunized with NP-KLH for analyses of Fig. 5A. Shown are mean (±SEM) ELISA results across serial four-fold dilutions (1:50 to 1:12,800). After screening statistical significance by ANOVA, the null hypothesis was tested pairwise and achieve the indicated P values for IgG1 in samples at the dilutions 1:80, 1:200, and 1:800. (B-E) Cγ1-Cre knock-in mice (Rptor +/+ or f/f, six of each genotype distributed equally in two independent experiments) were immunized as in Fig. 2A. Shown are serological analyses (B) as well as B cell numbers (C), flow staining to measure the frequencies of B cells of GC phenotype (D), or (E) Tfh cells as in Fig. 5D. Shown in (B) are ELISA determinations of circulating anti-NP Ab (all-affinity, left panels; high affinity, panels to right) across serial 10-fold dilutions (1:10 to 1:108), with each point representing mean (±SEM) data for the individual samples (six WT, six mutant). After screening the overall curves by ANOVA, statistical significance was tested pairwise and yielded the indicated P values for samples at the dilutions 1:10 through 1:104. (D, E) Shown are flow data from a representative mouse of each genotype in one experiment, with inset numbers and italicized means (±SEM) as in Fig. 2C.
B cell-intrinsic mTORC1 inhibits heterosubtypic Ab generation after influenza immunization
To evaluate B cell-intrinsic function(s) of mTORC1 in immune memory using an immunization model incapable of generating new B cells, we used chemical activation of Cre-mediated Rptor gene deletion followed by transfers of the resultant B cells into B cell-deficient mice (Fig. 7A). Recipient mice were then immunized by inoculation with one strain of influenza (A/PR8/34; H1N1) at a non-permissive site, followed 6 wk later by intranasal inoculation with a recombinant influenza (FLU) strain of heterologous surface type (A/ X-31; H3N2). Based on reports of rapamycin-resistant mTORC1 (39), we tested heterozygous (Δ/+) B cells as well as homozygous loss-of-function. Measurements of the antibodies were performed both with virus lysates and with recombinant strain-specific hemagglutinin (HA) proteins, which are surface-displayed and a major target of protective antibodies (58). The post-challenge response as measured with virus lysates demonstrated that Raptor depletion caused modest increases in IgM anti-influenza concentrations after infection to elicit Ab responses at a memory phase after immunization (Fig. 7B). Consistent with systemic rapamycin effects, major reductions in the anti-viral IgG1 and IgG2c were observed in mice whose B cells were mTORC1-deficient (Fig. 7B). Type-specific antibody responses were measured at three stages – (i) the primary response (3 wk), (ii) a pre-challenge estimate of more durable Ab concentrations, and (iii) post-challenge at a time point (1 wk post inoculation) chosen to bias the measurement of class-switched Ab toward those derived from recall responses of memory B cells. The primary IgM response to the immunizing H1 strain (PR8) was robust and not reduced by mTORC1 inactivation (Fig. 7C, upper row panels). Ab directed against the X-31 HA (H3) were virtually undetectable in the primary response derived from WT B cells (Fig. 7C, lower row panels). However Raptor-depleted B cells yielded a low but reproducible and statistically significant increase in IgM that bound HA of the X-31 challenge strain (Fig. 7D). Complete loss of Raptor eliminated the primary IgG1 and IgG2c responses against the immunogen subtype, H1. Strikingly, Rptor haplo-insufficiency reduced but did not eliminate Ag-specific IgG1 while IgG2c anti-H1 was almost completely eliminated. After a period for formation of memory, inoculation with a different influenza subtype at a site permissive for replication rapidly stimulated anti-HA responses in which the IgM anti-H1 levels derived from Raptor-depleted B cells were higher than for the WT controls (Fig. 7C, left panel of upper row). While little IgG1 or IgG2c anti-H1 Ab were detected (Fig 7C), anti-H3 reactivities were elicited by intranasal X-31 inoculation. Switched and IgM responses were robust for controls, whereas Rptor Δ/Δ B cells yielded only IgM production (Fig. 7C, lower row of panels). However, the secondary virus challenge elicited substantial anti-H3 IgG1 from haplo-insufficient Rptor Δ/+ B cells akin to the primary anti-H1 Ab response (Fig. 7C, middle panels). The strength of haplo-insufficiency in recall humoral immunity was intriguing in that at most a modest effect was noted if Raptor was present during a primary immunization and only then was one allele inactivated (44). The straightforward basis for such findings would be that the integrity of mTORC1 is more critical during the B cell activation and the establishment of memory than once a memory B cell population is established. However, extensive direct comparisons will be needed to test this model. Notwithstanding, we conclude that graded reductions in B cell-intrinsic mTORC1 progressively eliminated the Ig class switch while reprogramming the repertoire of B cell responses. Consequently, the primary response yielded a modest amount of heterosubtypic IgM Ab against the HA of the X-31 challenge strain (H3) and an increase in the IgM anti-H3 upon specific challenge.
Figure 7.
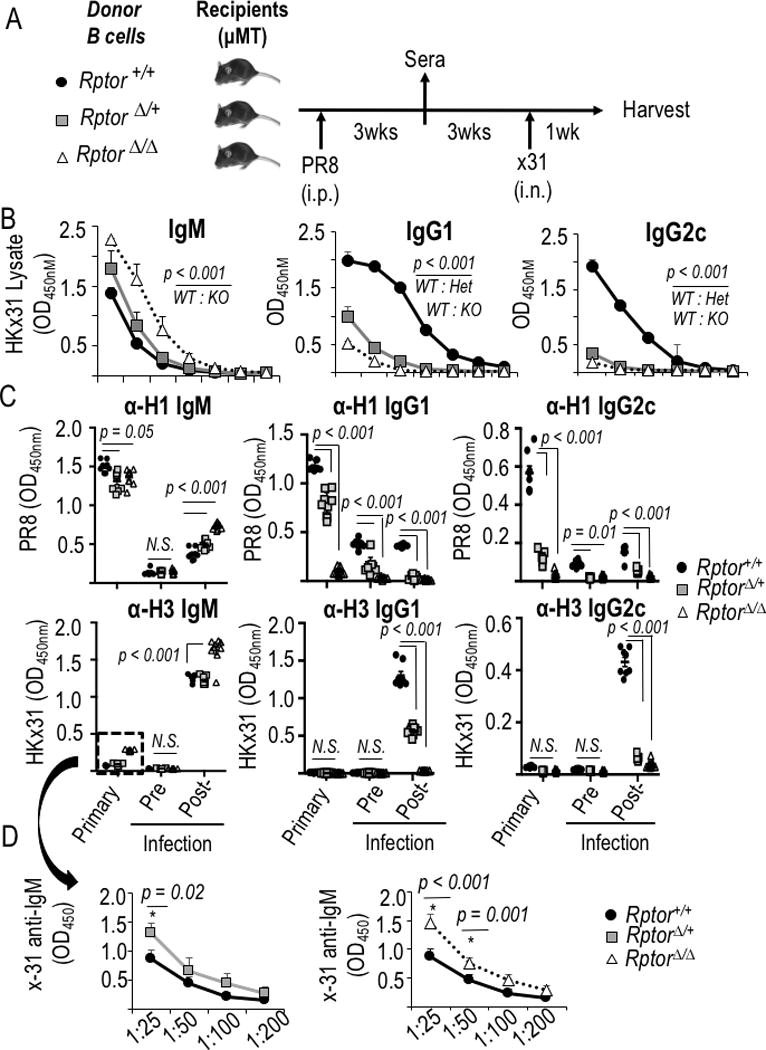
mTORC1 level programs the spectrum of anti-influenza Ab in primary and recall responses. (A) Schema of adoptive transfer, immunization, and respiratory challenge experiments to test B cell-intrinsic function of mTORC1 in humoral responses to influenza virus. Donor B cells were harvested from Rosa26-CreERT2 mice with wild-type or flox alleles of Rptor after tamoxifen injections as in Fig. 1A. (B) Mean (±SEM) ELISA results on sera 1 wk after PR8-immunized mice were challenged with intranasal X-31 strain influenza virus, using lysates of X-31 virions to capture anti-FLU Ab in sera from mice whose B cells were WT, haplo-insufficient for Rptor, or homozygous for the deleted Rptor allele. Isotype-specific ELISA results after serial two-fold dilutions starting at 1:50 serum dilution. For sera of WT versus Rptor-depleted B cell recipients, p≤0.001 by ANOVA calculated for each of the three isotypes as specified in the Methods, followed by pairwise comparisons at single dilutions. (C) Mean (±SEM) results of ELISA performed with recombinant HA proteins to capture anti-H1 (PR8) and anti-H3 (X-31) Ab of the indicated isotypes, using sera (1:100 dilutions) from the mice in (B) after identification of serum dilutions in the linear range of the assay. Data capture the sera from individual recipient mice for each genotype of donor B cells [8 Rptor +/+, 9 Rptor Δ/+, and 9 Rptor Δ/Δ, each distributed evenly among three independent experiments]. (D) Titrations for cross-reactive IgM anti-H3 in the primary response to immunization with A/PR8, separating panels comparing Δ/+ (left) and Δ / Δ (right) to the same WT controls for clarity. Significance testing was performed using two-way ANOVA with Bonferroni correction for multiple comparisons, followed by testing the null hypothesis for pairwise comparisons at 1:25 dilutions.
Using this non-lethal challenge inoculum, we also explored how much protection against illness resulted from the cross-reactive (X-31, or H3-binding) IgM response to immunization with A/PR8 influenza. At most, subtle differences were observed in the lung pathology, amount of weight loss, and the number of infiltrating cells recovered from lungs one week after intranasal challenge (Supplemental Fig. 4A-C). Modest differences suggestive of some protective effect were not statistically significant, and the protection was at most very modest. Activated T cells were in the inflamed lungs in similar numbers (Supplemental Fig. 4D-G). Consistent with evidence in the protein immunization systems (Fig. 1–6), however, lack of mTORC1 reduced the populations of GC and memory-phenotype B cells in spleens of the μMT recipients (Supplemental Fig. 4H, I). We conclude that a B cell-intrinsic defect of mTORC1 liberated production of heterospecific anti-HA Ab, but that the resultant humoral memory was insufficient for any substantial protective effect.
DISCUSSION
Elucidation of extrinsic and cell-intrinsic regulators of antibody generation and qualities is central to the potential to manipulate humoral responses after vaccination, during and after an infection, or in disease due to autoantibodies. Previous work has provided evidence that mTOR, mTORC1 and mTORC2 promote humoral immunity. Several recent papers underscored T cell-intrinsic functions of mTORC1 impact T follicular helper phenotype and functions (47–49). Other findings suggest that both mTORC2 (32) and mTORC1 exert cell-intrinsic functions in B lineage ontogeny and function (42–44, 59). However, crucial unanswered questions remain about the mechanisms for these findings, and the interplay between mTORC1 and metabolism in the B cell contribution to antibody regulation. We have shown here that following depletion of Raptor, an essential component of mTORC1, in B cells, inclusion of activated B cells in GC is drastically reduced when the analyses involve physiological frequencies of Ag-specific cells in the naïve repertoire and immunization. Furthermore, mTORC1 was crucial for normal induction of the GC-determining transcription factor BCL6. AID expression was increased when this signaling pathway was active, accompanied by increased rates of somatic mutations. Thus, one critical function of mTORC1 in B cells is to support acquisition of the full functionality of lymphocytes whose activation sufficed for loss of IgD and gain of GL7 expression. Ultimately, perturbations of mTORC1 that were restricted to B cells sufficed to alter the nature of antiviral Ab specificities. Moreover, our studies connect mTORC1 effects on class switching to increased glycolysis in B cells. Thus, in a setting independent from Tfh contributions, mTORC1 promoted glycolytic flux of TLR-activated B cells. Strikingly, reduced glycolytic flux limited the capacity to undergo Ab class switching, express AID, and induce a germ line transcript crucial for Ig heavy chain switch region targeting by AID. Collectively, these findings are consistent with a capacity of the metabolic programming - and perhaps micro-environments deficient in nutrients - to impact the qualities of humoral immunity.
Our data reveal that at physiological precursor frequencies and with antigen-carrier immunization akin to vaccination, mTORC1 in B cells strongly promotes affinity maturation and the primary humoral response as well as recall Ab production. The underlying frequency of somatic mutation was halved among the reduced pool of NP-binding GC-phenotype B cells when Raptor was depleted. While absolutes rarely apply in immunity, the somatic mutations of antibodies are a fundamental property of the germinal center reaction (3, 4, 60). Accordingly, it is intriguing to consider how this reduction may relate to the dynamics of germinal center reactions. CXCR4-dependent iterative shuttling of B cells between the GC dark and light zones is a prominent feature of these dynamics (3, 4, 20–22), and although Aicda mRNA levels are on average at most 2-fold different between DZ and LZ B cells (61, 62) it is thought that mutations are imposed in the proliferating B cells of the DZ. Inactivation of CXCR4 gene expression in B cells disrupted shuttling and trapped B cells in the dark zone. Quantitation of SHM in Cxcr4-deficient GC-phenotype B cells after influenza infection, i.e., in the absence of iterative LZ/DZ shuttling, about a 50% reduction (22). This effect size matches what we have determined for mTORC1-deficient B cells. Intriguingly, work in parallel to our studies found that rapamycin inhibited the movement of transferred B1-8 B cells from light to the dark zones of GC in mice immunized prior to the transfer (63), but the effects of this intervention on mTORC2, somatic mutation, and affinity of the resulting Ab are not clear. It is tempting to relate the elegant findings in this transfer system to the results of B cell-restricted Raptor depletion. Among caveats, however, one is that with a physiological Ag-specific B cell repertoire prior to immunization, depletion of B cells’ mTORC1 via Cγ1-Cre expression undermined B cell support for the Tfh population. Moreover, Raptor-deficient B cells yielded a drastically reduced number of Ag-specific GC phenotype B cells, and GC-like structures with few B cells. Thus, SHM was reduced in Raptor- and CXCR4-deficient B cell to a quantitatively similar degree. However, understanding the relative contributions of shuttling versus impaired GC functionality due to mTORC1-deficiency of Ag-specific B cells under physiological conditions awaits further studies. Our data indicate that mTORC1 integrity promotes higher concentrations of high-affinity Ab to a greater extent than the effect on somatic hypermutation. This disparity points to more stringent selection, which appears to differ from the finding that a ubiquitously-expressed hypomorphic mTOR led to a very modest affinity reduction quantitatively similar to a decrease in JH4 SHM to about 0.6 × WT frequency (43).
Our data from immunization experiments in vivo confirm and extend earlier evidence that loss of Raptor decreased switching to IgG1 in vitro (42). However, earlier work had indicated that hyperactivity of PI3K suppressed CSR as well as Aicda gene expression (64). PI3K actions in immunity in vivo may mostly be attributable to mTORC1 and mTORC2 since disease resulting from constitutive PI3K activity operates primarily along rapamycin-sensitive pathways (33). Thus, increased PI3K had, on the surface, the same effect as a block of mTORC1 activity. Although mTORC2-depleted B cells appeared to switch normally in vitro (32) some of this apparent paradox might be due to mTORC2 or other signaling resulting from sustained increases in PIP3. For instance, a suppressive role of mTORC2 could fit with observed increases in switched Ab after using catalytic site inhibitors of mTOR (42). Closer to mTORC1, inactivation of Tsc1 via Cd19-Cre prior to full B cell maturation caused sustained increases in mTORC1 activity and developmental defects of B cells along with a mild defect of CSR and Ab responses (65). More recently, although the effect on developmental progression was not reported, Tsc1 inactivation via mb1-Cre also increased mTORC1 activity (as expected) and undermined affinity maturation, while a separate gain-of-function mutant in RRaga confined B cells to the DZ but did not alter VH somatic mutation rates (63). Subject to a caveat from the likely abnormalities of B cell development, these findings suggest that excess mTORC1 undermines CSR and affinity selection but not SHM per se. All together, then, sustained increases in mTORC1 may reduce rates of somatic mutation but our data indicate that decreases in mTORC1 activity undermine SHM in a physiological repertoire in a primary response to immunization.
Ultimately, an important aspect of dissecting these mechanisms is to assess how they affect immunization, infection, or auto-immune processes. Rapamycin in vivo can limit auto-Ab production in mouse models, as can a combination of 2DG and metformin (44, 66, 67). These rapamycin effects involve decreased plasma cell generation and function (42, 44). In addition 2DG dramatically reduces the efficiency of plasma cell differentiation (Li J, Raybuck AL, and Boothby M, unpublished observations). In this regard, 2DG reduced IgG1 in media after anti-IgM-activated B cells were provided help from mixtures of follicular T cells (Tfh and Tfr) ex vivo (68). Our findings suggest that this includes a B cell-specific effect involving switching. One of the more striking findings has been that rapamycin treatment during a primary immunization led to dramatic cross-protection against lethal challenge by an HA-disparate influenza virus notwithstanding the virtual elimination of germinal centers (46). CD4+ cells as well as B cells needed to be present for this protective effect, DC function also is rapamycin-sensitive (39), and recent findings have highlighted a requirement for mTORC1 in TFH cell development and function (47–49). Evidence herein, generated with genetic approaches, shows that the block to forming class-switched antibodies against HA generated by loss of mTORC1 is B cell-intrinsic. Moreover, although the use of transfers into B cell deficient recipients means that there would be an element of homeostatic proliferation, the results provide support for a licensing of the emergence of IgM clones specific for epitopes on an HA-subtype differing from the immunogen and definitively map this to the B cell. Indeed, our evidence indicates that even a partial restriction of mTORC1 activity restricted to B cells sufficed to increase cross-protective anti-HA IgM. However, the durability of the humoral memory and extent of protection against disease are not clear, and probably need to invoke rapamycin’s effects on mTORC2 as well as mTORC1 in follicular T cells and others. Indeed, IgM memory B cells persist for extended periods yet the cross-reactive anti-HA antibody concentrations had declined within a month, so it may be that a substantial fraction of the response was extra-follicular or derived from shorter-lived plasma cells (26). In any event, CD4 T cells were required for the protective effect on survival upon influenza challenge in the rapamycin-treated immunization model, as were B cells (46). Our work indicates that a haplo-insufficient B cell-autonomous, mTORC1-specific mechanism suffices to reduce switching and liberate the emergence of heterosubtypic anti-HA IgM. However, these Ab did not improve the clinical outcome. These findings suggest that the protective effect required rapamycin action on the CD4 T cells, and perhaps also DC, during and after immunization.
Supplementary Material
Acknowledgments
We gratefully acknowledge assistance from T. Laufer in expediting receipt of crucial genetic stock and in sharing protocols for preparation of NP-derivatized fluorophore proteins, and R. Bonami for helpful guidance on SHM analysis approaches.
This work was supported by NIH grants (M.R.B., AI113292, HL106812; J.V.W. AI085062) for the experiments, and additional support via NIH T32 HL069765 (C.L.W.), Korean Ministry of Science NRF-2014M3A9D5A01073841 (K.W.L.), NIH Shared Instrumentation Grant 1S10OD018015 as well as scholarships via the Cancer Center Support Grant (CA068485) and Diabetes Research Center (DK0205930) to help defray costs of Vanderbilt Cores.
Abbreviations used
- IRF
interferon response factor
- AID
activation-induced deaminase
- GC
germinal center
- mTOR
mammalian (or, mechanistic) target of rapamycin
- MZ
marginal zone
- TFH
T follicular helper
- Bmem
memory B cell; phosphatidyl- inositol 3, 4, 5-triphosphate (PIP3)
- SHM
somatic hypermutation
- KLH
Keyhole Limpet Hemocyanin
- BAFF
B-cell Activating factor
- FSC
forward-scattered (light) channel
- SSC
side-scattered (light) channel
- 2-DG
2-deoxyglucose
- ASC
antibody-secreting cell
- GLT
germ line transcript
- HA
hemagglutinin
Footnotes
No conflicting interests
Disclosures The authors declare no competing financial interests.
References
- 1.Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol. 2010;11:681–688. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- 2.Kurosaki T, Shinohara H, Baba Y. B cell signaling and fate decisions. Annu Rev Immunol. 2010;28:21–55. doi: 10.1146/annurev.immunol.021908.132541. [DOI] [PubMed] [Google Scholar]
- 3.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–445. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 4.Shlomchik M, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. 2012;247:52–63. doi: 10.1111/j.1600-065X.2012.01124.x. [DOI] [PubMed] [Google Scholar]
- 5.Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature. 2014;509:637–640. doi: 10.1038/nature13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rush JS, Liu M, Odegard V, Unniraman S, Schatz DG. Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc Natl Acad Sci USA. 2012;102:13242–13247. doi: 10.1073/pnas.0502779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarlinton D, Radbruch A, Hiepe F, Dörner T. Plasma cell differentiation and survival. Curr Opin Immunol. 2008;20:162–169. doi: 10.1016/j.coi.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Muramatsu M, Nagaoka H, Shinkura R, Begum NA, Honjo T. Discovery of activation-induced cytidine deaminase, the engraver of antibody memory. Adv Immunol. 2007;94:1–36. doi: 10.1016/S0065-2776(06)94001-2. [DOI] [PubMed] [Google Scholar]
- 9.Neuberger MS. Antibody diversification by somatic mutation: from Burnet onwards. Immunol Cell Biol. 2008;86:124–132. doi: 10.1038/sj.icb.7100160. [DOI] [PubMed] [Google Scholar]
- 10.Conley ME, Dobbs A, Farmer D, Kilic S, Paris K, Grigoriadou S, Coustan-Smith E, Howard V, Campana D. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227. doi: 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- 11.Cho SH, Raybuck A, Stengel K, Wei M, Beck T, Volanakis E, Thomas JW, Hiebert S, Haase V, Boothby MR. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. 2016;537:234–238. doi: 10.1038/nature19334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abbott RK, Thayer M, Labuda J, Silva M, Philbrook P, Cain DW, Kojima H, Hatfield S, Sethumadhavan S, Ohta A, Reinherz EL, Kelsoe G, Sitkovsky M. Germinal Center Hypoxia Potentiates Immunoglobulin Class Switch Recombination. J Immunol. 2016;197:4014–4020. doi: 10.4049/jimmunol.1601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jellusova J, Cato M, Apgar JR, Ramezani-Rad P, Leung C-R, Chen C, Richardson A, Conner E, Benschop RJ, Woodgett JR, Rickert RC. Gsk3 is a metabolic checkpoint regulator in B cells. Nat Immunol. 2017;18:303–312. doi: 10.1038/ni.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pierce SK, Liu W. The tipping points in the initiation of B cell signaling: how small changes make big differences. Nat Rev Immunol. 2010;10:767–777. doi: 10.1038/nri2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foy TM, Laman J, Ledbetter J, Aruffo A, Claassen E, Noelle RJ. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J Exp Med. 1994;180:157–163. doi: 10.1084/jem.180.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banchereau J, Bazan F, Blanchard D, Brière F, Galizzi JP, van Kooten C, Liu YJ, Rousset F, Saeland S. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 17.Ozaki K, Spolsk R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC, 3rd, Liu C, Schwartzberg PL, Leonard WJ. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 18.Lau CM, Broughton C, Tabor A, Akira S, Flavell RA, Mamula M, Christensen S, Shlomchik MJ, Viglianti G, Rifkin I, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pone EJ, Zan H, Zhang J, Al-Qahtani A, Xu Z, Casali P. Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: relevance to microbial antibody responses. Crit Rev Immunol. 2010;30:1–29. doi: 10.1615/critrevimmunol.v30.i1.10. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science. 2007;315:528–531. doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- 21.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bannard O, Horton R, Allen CD, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity. 2013;39:912–924. doi: 10.1016/j.immuni.2013.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, Kelsoe G. Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity. 2016;44:542–552. doi: 10.1016/j.immuni.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gourley TS, Wherry EJ, Masopust D, Ahmed R. Generation and maintenance of immunological memory. Semin Immunol. 2004;16:323–333. doi: 10.1016/j.smim.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 25.Good-Jacobson KL, Tarlinton DM. Multiple routes to B-cell memory. Int Immunol. 2012;24:403–408. doi: 10.1093/intimm/dxs050. [DOI] [PubMed] [Google Scholar]
- 26.Oracki SA, Walker JA, Hibbs M, Corcoran L, Tarlinton DM. Plasma cell development and survival. Immunol Rev. 2010;237:140–159. doi: 10.1111/j.1600-065X.2010.00940.x. [DOI] [PubMed] [Google Scholar]
- 27.Pape KA, Taylor JJ, Maul R, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bortnick A, Chernova I, Quinn WJ, 3rd, Mugnier M, Cancro MP, Allman D. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J Immunol. 2012;188:5389–5396. doi: 10.4049/jimmunol.1102808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takemori T, Kaji T, Takahashi Y, Shimoda M, Rajewsky K. Generation of memory B cells inside and outside germinal centers. Eur J Immunol. 2014;44:1258–1264. doi: 10.1002/eji.201343716. [DOI] [PubMed] [Google Scholar]
- 30.Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, Reynaud CA, Weil JC. Multiple layers of B cell memory with different effector functions. Nat Immunol. 2009;10:1292–1299. doi: 10.1038/ni.1814. [DOI] [PubMed] [Google Scholar]
- 31.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3:317–33. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 32.Lee K, Heffington L, Jellusova J, Nam KT, Raybuck AL, Cho SH, Thomas JW, Rickert RC, Boothby M. Requirement for Rictor in homeostasis and function of mature B lymphoid cells. Blood. 2013;122:2369–2379. doi: 10.1182/blood-2013-01-477505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lucas CL, Kuehn H, Zhao F, Niemela J, Deenick E, Palendira U, Avery D, Moens L, Cannons J, Biancalana MM, Stoddard J, Ouyang W, Frucht DM, Rao VK, Atkinson TP, Agharahimi A, Hussey AA, Folio LR, Olivier KN, Fleisher TA, Pittaluga S, Holland SM, Cohen JI, Oliveira JB, Tangye SG, Schwartzberg PL, Lenardo MJ, Uzel G. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15:88–97. doi: 10.1038/ni.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sengupta S, Peterson TT, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarbassov DD, Ali SM, Sengupta S, Sheen J, Hsu P, Bagley A, Markhard A, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 37.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–753. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delgoffe GM, Pollizzi K, Waickman A, Heikamp E, Meyers D, Horton M, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosborough BR, Raïch-Regué D, Matta B, Lee K, Gan B, DePinho RA, Hackstein H, Boothby M, Turnquist HR, Thomson AW. Murine dendritic cell rapamycin-resistant and rictor-independent mTOR controls IL-10, B7-H1, and regulatory T-cell induction. Blood. 2013;121:3619–3630. doi: 10.1182/blood-2012-08-448290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Hu T, Hua C, Gu J, Zhang L, Hao S, Liang H, Wang X, Wang W, Xu J, Liu H, Liu B, Cheng T, Yuan W. Rictor is required for early B cell development in bone marrow. PLoS One. 2014;9:e103970. doi: 10.1371/journal.pone.0103970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aagaard-Tillery KM, Jelinek D. Inhibition of human B lymphocyte cell cycle progression and differentiation by rapamycin. Cell Immunol. 1994;156:493–507. doi: 10.1006/cimm.1994.1193. [DOI] [PubMed] [Google Scholar]
- 42.Limon JJ, So L, Jellbauer S, Chiu H, Corado J, Sykes S, Raffatell M, Fruman DA. mTOR kinase inhibitors promote antibody class switching via mTORC2 inhibition. Proc Natl Acad Sci USA. 2014;111:E5076–5085. doi: 10.1073/pnas.1407104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang S, Pruitt M, Tran D, Du Bois W, Zhang K, Patel R, Hoover S, Simpson R, Simmons J, Gary J, Snapper C, Casellas R, Mock B. B cell-specific deficiencies in mTOR limit humoral immune responses. J Immunol. 2013;191:1692–1703. doi: 10.4049/jimmunol.1201767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones DD, Gaudette B, Wilmore J, Chernova I, Bortnick A, Weiss B, Allman D. mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest. 2016;126:4250–4261. doi: 10.1172/JCI86504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang S, Readinger J, DuBois W, Janka-Junttila M, Robinson R, Pruitt M, Bliskovsky V, Wu J, Sakakibara K, Patel J, Parent C, Tessarollo L, Schwartzberg PL, Mock B. Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood. 2011;117:1228–1238. doi: 10.1182/blood-2010-05-287821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keating R, Hertz T, Wehenkel M, Harris T, Edwards B, McClaren J, Brown S, Surman S, Wilson Z, Bradley P, Hurwitz J, Chi H, Doherty PC, Thomas P, McGargill M. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat Immunol. 2013;14:1266–1276. doi: 10.1038/ni.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ray JP, Staron M, Shyer J, Ho PC, Marshall H, Gray S, Laidlaw B, Araki K, Ahmed R, Kaech S, Craft J. The Interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity. 2015;43:690–702. doi: 10.1016/j.immuni.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown S, Cloer C, Kishton R, Gao X, Youngblood B, Do M, Li M, Locasale J, Rathmell J, Chi H. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity. 2016;45:540–554. doi: 10.1016/j.immuni.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Lin X, Pan Y, Wang J, Chen P, Huang H, Xue H-H, Gao J, Zhong X-P. Critical roles of mTOR complex 1 and 2 for T follicular helper cell differentiation and germinal center responses. eLife. 2016;5:e17936. doi: 10.7554/eLife.17936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science. 2012;336:1178–1181. doi: 10.1126/science.1213368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casola S, Cattoretti G, Uytterspro N, Koralov S, Seaga J, Hao Z, Waisman A, Egert A, Ghitza D, Rajewsky K. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci USA. 2006;103:7396–7401. doi: 10.1073/pnas.0602353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi W, Gupta S, Ricker E, Manni M, Jessberger R, Chinenov Y, Molina H, Pernis AB. The mTORC1-4E-BP-eIF4E axis controls de novo Bcl6 protein synthesis in T cells and systemic autoimmunity. Nat Commun. 2017;8:254. doi: 10.1038/s41467-017-00348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14:500–508. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X, Sun SC. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40:692–705. doi: 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matthews AJ, Zheng S, DiMenna LJ, Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol. 2014;122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dayan M, Zinger H, Kalush F, Mor G, Amir-Zaltzman Y, Kohen F, Sthoeger Z, Mozes E. The beneficial effects of treatment with tamoxifen and anti-oestradiol antibody on experimental systemic lupus erythematosus are associated with cytokine modulations. Immunology. 1997;90:101–108. doi: 10.1046/j.1365-2567.1997.00122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Babina M, Kirn F, Hoser D, Ernst D, Rohde W, Zuberbier T, Worm M. Tamoxifen counteracts the allergic immune response and improves allergen-induced dermatitis in mice. Clin Exp Allergy. 2010;40:1256–1265. doi: 10.1111/j.1365-2222.2010.03472.x. [DOI] [PubMed] [Google Scholar]
- 58.Couch RB. Seasonal inactivated influenza virus vaccines. Vaccine. 2008;26(Suppl 4):D5–9. doi: 10.1016/j.vaccine.2008.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iwata TN, Ramírez J, Tsang M, Park H, Margineantu D, Hockenbery D, Iritani BM. Conditional Disruption of Raptor Reveals an Essential Role for mTORC1 in B Cell Development, Survival, and Metabolism. J Immunol. 2016;197:2250–2260. doi: 10.4049/jimmunol.1600492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Toyama H, Okada S, Hatan M, Takahashi Y, Takeda N, Ichii H, Takemori T, Kuroda Y, Tokuhisa T. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity. 2002;17:329–339. doi: 10.1016/s1074-7613(02)00387-4. [DOI] [PubMed] [Google Scholar]
- 61.Victora GD, Dominguez-Sola D, Holmes A, Deroubaix S, Dalla-Favera R, Nussenzweig M. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphoma. Blood. 2012;120:2240–2248. doi: 10.1182/blood-2012-03-415380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado D, Li S, Imami K, Selbach M, Virgilio M Di, Bullinger J, Rajewsky K. PI3 Kinase and FOXO1 Transcription Factor Activity Differentially Control B Cells in the Germinal Center Light and Dark Zones. Immunity. 2015;43:1075–1086. doi: 10.1016/j.immuni.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 63.Ersching J, Efeyan A, Mesin L, Jacobsen J, Pasqual G, Grabiner B, Dominguez-Sola D, Sabatini DM, Victora GD. Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity. 2017;46:1045–1058. doi: 10.1016/j.immuni.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Omori SA, Cato M, Anzelon-Mills A, Puri K, Shapiro-Shelef M, Calame K, Rickert RC. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity. 2006;25:545–557. doi: 10.1016/j.immuni.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 65.Ci X, Kuraoka M, Wang H, Carico Z, Hopper K, Shin J, Deng X, Qiu Y, Unniraman S, Kelsoe G, Zhong XP. TSC1 Promotes B Cell Maturation but Is Dispensable for Germinal Center Formation. PLoS One. 2015;10:e0127527. doi: 10.1371/journal.pone.0127527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu T, Qin X, Kurepa Z, Kumar KR, Liu K, Kanta H, Zhou XJ, Satterthwaite AB, Davis LS, Mohan C. Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus. J Clin Invest. 2007;117:2186–2196. doi: 10.1172/JCI30398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7:274ra18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sage PT, Ron-Harel N, Juneja VR, Sen DR, Maleri S, Sungnak W, Kuchroo VK, Haining WN, Chevrier N, Haigis M, Sharpe AH. Suppression by TFR cells leads to durable and selective inhibition of B cell effector function. Nat Immunol. 2016;17:1436–1446. doi: 10.1038/ni.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.