

Abstract
The transition of effector T cells or memory precursors into distinct long-lived memory T cell subsets is not well understood. While many molecules made by antigen presenting cells (APCs) can contribute to clonal expansion and effector cell differentiation, it is not clear if clonal contraction and memory development is passive or active. Using respiratory virus infection, we found that CD8 T cells that cannot express the TNF family molecule LIGHT are unimpaired in their initial response and clonally expand to form effector cell pools. Thereafter, LIGHT-deficient CD8 T cells undergo strikingly enhanced clonal contraction with resultant compromised accumulation of both circulating and tissue resident memory cells. LIGHT expression at the peak of the effector response regulates the balance of several pro- and anti-apoptotic genes, including Akt, and has a preferential impact on the development of the peripheral memory population. These results underscore the importance of LIGHT activity in programming memory CD8 T cell development, and suggest that CD8 effector T cells can dictate their own fate into becoming memory cells by expressing LIGHT.
Introduction
Memory CD8+ T cells are critical in limiting the successful establishment of secondary infections (1, 2). It is now known that remarkable diversity can exist within the memory T cell pool, which was initially divided only into CD62L+CCR7+ central memory cells (TCM) and CD62L−CCR7− effector memory cells (TEM) (3). This led to the hypothesis that TEM cells that patrol non-lymphoid tissue and rapidly produce effector cytokines act as a first line of defense, whereas TCM cells, because of their ability to proliferate extensively, act as a second line of defense by generating secondary effector cells to bolster the ongoing response (4). However, subsequent studies have found that greater heterogeneity exists even within the TCM and TEM populations (5–7), and the markers CXCR3, CD27 and CD43 have been proposed to refine the functional characteristics of memory subsets (8–10). Adding to this growing complexity, resident memory T cells (TRM) can become committed to permanent retention within the tissue to which they localize (11), and they have been suggested to enhance the recruitment of circulating memory T cells to the site of infection (12). More recently, CX3CR1 has also been used to divide effector CD8+ T cells and their memory counterparts into three distinct subsets (CX3CR1neg, CX3CR1int, CX3CR1hi) (13, 14), with CX3CR1int cells (peripheral memory cells) considered to be responsible for patrolling non-lymphoid tissue and to possess the highest potential for self-renewal and differentiation (13).
Altogether, the effectiveness of protective immunity against pathogens provided by memory T cells likely depends on the extent of their generation, localization, and survival (7, 15). Following the resolution of acute infection, only a small fraction of activated (effector and memory-precursor) CD8+ T cells present at the peak of the immune response survive as long-lived memory cells. This suggests it is crucial to understand the precise molecular signals that regulate the transition of effector CD8+ T cells or activated memory-precursor CD8+ T cells, into the diverse memory subsets that can accumulate and persist at high frequencies.
A number of co-stimulatory and co-inhibitory molecules in the immunoglobulin (Ig) superfamily (e.g., CD28, PD-1) or the TNF receptor superfamily (e.g., OX40 and CD27) have been described to control effector T cell accumulation and differentiation, along with cytokines such as IL-12 and type 1 interferons (IFN-I); and thus indirectly contribute to memory development, since often the size of the memory pool is a corresponding fraction of the effector pool (16, 17). Furthermore, it is well established that the cytokines IL-15 and IL-7 provide essential survival signals to already differentiated memory cells (18). However, the factors that dictate the conversion of effector cells or memory-precursor cells into long-lived memory populations are not well appreciated. Some have hypothesized that clonal contraction and memory development is a passive and random process brought about by withdrawal of the antigenic stimulus and is pre-programmed within the first few days of infection or antigen encounter (12, 17, 19). Additionally, the contribution of pro- versus anti-apoptotic molecules, signals through death receptors (e.g., Fas, TNFR1 or TRAILR) and autophagy to clonal contraction and hence the number and type of subsequent memory cells generated, has been debated with no clear consensus (18, 20). Thus, it is not clear whether there are individual molecular interactions that are major contributors to the formation, divergence, or persistence of distinct memory subpopulations, at or after the peak of the effector response.
Here, we show that the TNF superfamily molecule LIGHT (Lymphotoxin-like, exhibits inducible expression, competes with HSV glycoprotein D for HVEM, a receptor expressed by T lymphocytes) is one protein that may impart essential signals for memory CD8+ T cell development. LIGHT (also known as TNFSF14) is an inducible molecule that can be expressed on the surface of activated/effector CD8+ and CD4+ T cells, as well as other immune cells such as dendritic cells (DCs), monocytes and neutrophils (21, 22). It has two receptors, the Herpes Virus Entry Mediator (HVEM) and Lymphotoxin β-Receptor (LTβR) that are found on a wide variety of hematopoietic and non-hematopoietic cells (23). LIGHT is pro-inflammatory as shown by studies of knockout animals that get less severe disease in models of intestinal inflammation, graft versus host disease (GVHD), transplantation and asthma (24–27). LIGHT has also been shown to be important for resistance against parasitic infection of mice such as Leishmania and Chlamydia (28–30). But the exact source, role, and target of LIGHT activity in many of these studies has not been well defined and is likely to be diverse.
In this report, we focused on the role of LIGHT, expressed specifically by CD8+ T cells, in controlling memory development in response to respiratory virus infection. We show that CD8+ T cell-derived LIGHT does not control the expansion of effector cells but rather, acts at and after the peak of the primary response to profoundly influence the development of resident and circulating memory CD8+ T cells of all subtypes, and in particular CD27hiCD43hiCX3CR1int cells. These studies suggest that CD8+ T cells by expressing LIGHT, can dictate their own fate into becoming memory cells, and imply that LIGHT might be a valuable target for promoting long-lived CD8+ T cell immunity.
Materials and Methods
Mice
Eight to twelve-week-old female C57BL/6 (CD45.2) and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1/Ly5.1) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). HVEM−/− mice, and wild type and LIGHT−/− OT-I transgenic mice were bred and maintained at the University of Florida animal facility. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Florida (OLAW Assurance # A3377-01).
Viruses and infections
Recombinant vaccinia virus Western Reserve strain (VacV-WR) and virus expressing full-length ovalbumin (rVacV-WR-OVA) were grown in HeLa cells and subsequently tittered on VeroE6 cells as described previously (47). Recombinant influenza virus expressing SIINFEKL peptide (PR8-OTI) was a generated as previously described (48). Mice were infected with 2 × 104 PFU of VacV-WR-OVA via the intra-nasal route in a volume of 10μl. For challenge experiments, PR8-SIINFEKL was administered at a normally lethal dose of 2 × 107 PFU in 40μl volume via the intra-nasal route.
CD8+ T cell adoptive transfer
For adoptive transfer experiments, 5 × 104 naive WT or HVEM−/− OT-I CD8 T cells were purified from spleens with MACS technology (Miltenyi Biotec) and transferred into WT C57BL/6 mice via the intra-venous route as described previously (47). Enriched cells were 99% specific CD8+TCR-transgenic T cells. One day later, mice were infected with rVacV-WR-OVA as above. OT-I expansion and effector formation were detected by flow staining of the transgenic TCR-α and TCR-β chains after gating on CD8+ T cells. In some cases, CD8 T cells were labeled with CFSE (Molecular Probes) by incubating 107 purified cells/mL with 5 μM CFSE for 10 minutes at 37°C. Cells were then washed three times in HBSS containing 2.5% FCS.
Flow cytometry
Preparation of cells, extracellular/intracellular staining, data acquisition, and data analysis were performed as described previously(10, 33, 49). To label all cells in compartments contiguous with the vasculature, animals were injected (i.v) with anti-mouse CD45.2-APC eFlour (clone104, eBioscience), which was allowed to circulate for three minutes prior to sacrifice. Following sacrifice, single cell preparations of lung tissue were stained with antibodies against CD8, CD44, Vα2, Vβ5, CD69 and CD103α antibodies. Lung DC and macrophage stains included antibodies for CD11c, CD11b, MHC-II, CD103 and Siglec-F. Other innate cells such as NK cells, inflammatory monocytes and neutrophils present in the lungs and mediastinal lymph nodes (MLN) were identified by staining with CD3, CD11b, NKp46, NK1.1, Ly6C and Ly6G. The MLN DC staining panel included B220, CD3, CD11b, CD11c, MHC-II, CD8 and CD4 (50).
Immunofluorescence studies
At various time-points post infection with VacV, spleens or lungs were harvested and immediately snap frozen in OCT on dry ice and procedure was performed as previously described (10, 47). The OCT embedded 5 μm thick cryo-sections of spleen were cut using a Microm HM 505E cryostat and prepared on super frost glass slides for immunofluorescence microscopy. Each slide of cryo-sections was washed with 1 ml phosphate buffered saline (PBS) and non- specific binding was blocked by incubating sections with 2% horse serum for 30 minutes at 4°C. After blocking, the sections were washed with cold PBS and incubated overnight in the dark at 4°C with fluorophore conjugated antibodies as indicated in the figure legends. Sections were washed three times with PBS, mounted with cytoseal and covered with a glass coverslip. The stained sections were observed and analyzed at wavelength 488 nm for FITC (green), 543 nm for PE (red) and 647 for APC (purple) labeling, using an EVOS fl, (Advanced Microscopy Group inverted immunofluorescence microscope) and images were captured by 4×, 10×, 20× and 40× objectives, keeping all the conditions of the microscope and settings of the software identical for all treatments and controls.
Gene array
Lungs from WT OT-I and LIGHT−/− OT-I recipient mice were harvested and CD27hiCD43hi effector CD8 T cell subset was sort purified by fluorescence activated cell sorting (FACS). Total lung RNA from this subset was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions (10, 49, 51). Total RNA was subsequently treated with DNase I (Qiagen) and further purified using an RNeasy Mini Kit (Qiagen). A total of 1 mg high-quality total RNA (RNA integrity number = 0.7) was then reverse transcribed using the First Strand Synthesis Kit (Qiagen) and subsequently loaded onto mouse apoptotic RT2 profiler array according to the manufacturer’s instructions (Qiagen). Qiagen’s online Web analysis tool was used to produce comparative heat maps, and fold change was calculated by determining the ratio of mRNA levels to control values using the D threshold cycle (Ct) method (22DDCt). All data were normalized to an average of five housekeeping genes, Gusb, Hprt, Hsp90ab1, Gapdh, and Actb. PCR conditions used: hold for 10 min at 95°C, followed by 45 cycles of 15 s at 95°C and 60 s at 60°C.
Statistical analysis
Tests were performed using GraphPad Prism 5.0 software (GraphPad, San Diego, CA). Statistics were done using two-tailed, unpaired Student’s t test with 95% confidence intervals unless otherwise indicated. Unless otherwise indicated, data represent the mean ± SEM; with *p < 0.05 and **p < 0.01, considered statistically significant.
Results
LIGHT expressed by CD8 T cells regulates clonal contraction and memory generation
We investigated the role of LIGHT expressed by CD8 T cells during an antiviral response using T cells lacking this molecule. A small number of wild-type (WT) and LIGHT-deficient naïve OT-I CD8+CD44lo T cells expressing Vα2Vβ5 and specific for H-2Kb/OVA257-264 were adoptively transferred into naïve WT C57BL/6 mice. Recipient mice were infected intra-nasally the following day with recombinant vaccinia virus (VacV) Western Reserve expressing the full-length ovalbumin protein (rVacV-WR-OVA). CD8+ T cells were then analyzed in the lungs and spleen on days 6 and 8, day 15, and day 40 post-infection, representing the acute expansion phase, the contraction phase, and the maintenance/memory phases of the primary response, respectively. Based on accumulation of the transferred cells, and production of IFNγ, the generation of the primary effector pool was found to be normal on day 6 even when the transferred CD8 T cells did not express LIGHT. A moderate decrease in numbers was seen by day 8 (Fig. 1A-D). However, this defect became more pronounced during the contraction phase of the response with ~60% of LIGHT−/− CD8+ T cells by day 15, followed by a greater loss of LIGHT−/− CD8 T cells (~90%) by day 40, the time when memory generation to this virus is complete in the lungs (Fig. 1A-D). Similar results were also observed in the spleen (Supplementary Fig. 1). To control for the potential that adoptive transfer of OT-I CD8+ T cells could alter the endogenous response to viral infection, we monitored the immunodominant CD8+ T cell population specific for VacV (B8R) and observed the development of normal primary and memory responses in the lungs and spleen (Supplementary Fig. 2). These data suggested that LIGHT expression by antigen-specific CD8+ T cells is critical for generating both effector memory (lung) and central memory (spleen) cells.
Figure 1. LIGHT expressed by CD8 T cells regulates formation of memory cells.
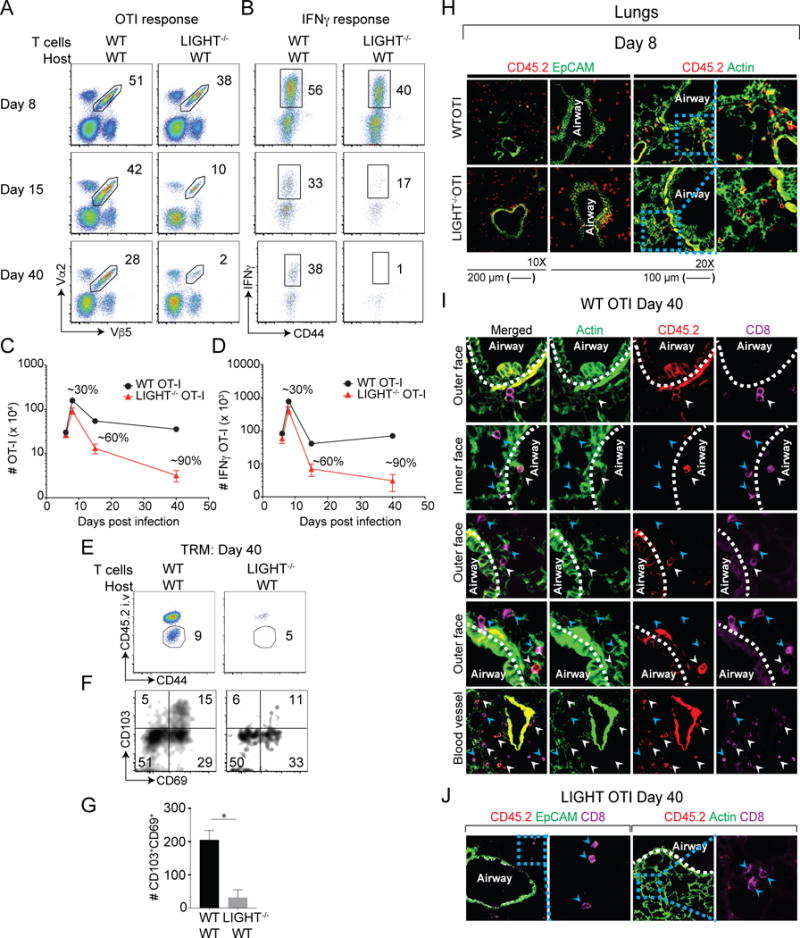
Equal numbers (5 × 104) of WT and LIGHT−/− naïve (CD44lo) OT-I (Vα2+Vβ5+) transgenic CD8 T cells were adoptively transferred into BL/6 mice and infected with rVacV-WR-OVA (2 × 104 PFU i.n) the following day. (A-D) Lungs were harvested at days 6, 8, 15 and 40 post-infection and stained for (A) CD8, CD44, Vα2, and Vβ5 and frequencies of OT-I CD8 T cells determined. (C) OT-I cells from lungs of recipients were re-stimulated in vitro with OVA peptide and stained intra-cellularly with IFN-γ. Absolute numbers of (C) OT-I cells and (D) IFNγ+ OT-I cells from lungs were quantified post-infection. (E-G) At day 40 post-infection, recipient mice were injected with anti-CD45.2 antibody intravenously, three minutes before euthanizing. (H-J) Equal numbers (5 × 105) of WT and LIGHT−/− naïve (CD44lo) OT-I (Vα2+Vβ5+) transgenic CD45.2+CD8 T cells were adoptively transferred into CD45.1+SJL mice and infected with rVacV-WR-OVA (2 × 104 PFU i.n) the following day. IF analysis of frozen (H) lung sections from recipient mice stained for CD45.2 (red) and CD8 (magenta) at day 8 post-infection. Similar analysis for was done at day 40 post-infection in (I & J) the lungs. ActinGreen or EpCAM (green) were used to visualize lung morphology. Bars, 200 μm & 100 μm respectively. Similar results were obtained in two independent experiments. Representative plots of CD45.2 and CD44 were pre-gated on CD8+OT-I (Vα2+Vβ5+) cells. The gated CD45.2 negative cells were analyzed for the expression of CD103 and CD69 using flow cytometry and total numbers of cells calculated in lungs. *, P<0.05; and results are the mean ± SEM (n = 3 mice/group). Similar results were obtained in four independent experiments.
In order to determine whether LIGHT affects the tissue resident memory (TRM) CD8+ T cell subset, we intravenously injected WT and LIGHT−/− CD8+ T cell recipients with fluorophore conjugated anti-CD45.2 antibody prior to sacrifice to distinguish between circulating memory cells (CD45.2+; vascular) from lung resident memory cells (CD45.2−; parenchyma, airway). We observed reduced frequencies of both subsets including lower numbers of CD103+CD69+ TRM cells in recipients that received LIGHT−/− CD8+ T cells (Fig. 1E-G). However, the proportion of TRM subsets was normal in these fractions (Fig. 1F), implying that LIGHT does not control TRM differentiation but regulates the generation or persistence of TRM cells. Overall, our data suggest that CD8+ T cell-derived LIGHT plays a minor role in effector CD8+ T cell expansion but is critical for the generation of large numbers of circulating and tissue-resident memory CD8+ T cells and its primary activity is displayed from the peak of the effector response throughout the phase when CD8+ T cells contract in numbers.
Immuno-histological analyses showed that LIGHT−/− CD8+ T cells localized normally within the lung tissue, similar to WT CD8+ T cells at the peak of the response (day 8). Both WT and LIGHT−/− CD8 T cells were found near the parenchyma as well as in contact with the airway epithelium in the lung tissue (Fig. 1H). Also, no significant change was observed in the localization of WT or LIGHT−/− CD8+ T cells in the spleen. LIGHT−/− CD8+ T cells were observed both in the red pulp as well as the white pulp and bridging channel, similar to WT CD8+ T cells (Supplementary Fig. 3A). At day 40, WT CD8+ T cells were found at multiple sites within the lung tissue, such as close to the airway epithelium, in the airways, in the parenchyma away from the epithelium, as well as near the blood vessels (Fig. 1I). In striking contrast, very few LIGHT−/− CD8+ T cells (CD45.2+) were detected at day 40, while endogenous LIGHT-sufficient CD8 T cells (purple only) were detected in these lung samples (Fig. 1J). Similar results were also observed in the spleen where the transferred WT CD8+ T cells were detected in the marginal zone, the red pulp and the PALS area, but LIGHT−/− CD8+ T cells were very few in number (Supplementary Fig. 3B). These data validated the flow cytometry results. Thus, LIGHT deficiency does not impact the tissue specific localization of CD8+ T cells but seems critical for their long-term tissue persistence.
Intact early activation, proliferation, differentiation and antigen independent homeostatic turnover in the absence of LIGHT
As there was a small defect observed in the accumulation of LIGHT-deficient CD8+ T cells in the lung tissue at day 8 post-infection, we asked if this was due to a defect in early activation, early proliferation or differentiation. We observed no change in the expression of either CD25 or CD69 when LIGHT was not expressed by CD8+ T cells, both in the lungs and the draining lymph nodes at day 6 post-infection (Fig. 2A). There were also no differences in the early (day 6) and later (day 8) proliferation with all T cells being Ki67 positive at these time points (Fig. 2B). We then examined the expression of IFNγ, TNF and CD107α in response to SIINFEKL peptide stimulation to determine whether LIGHT expression imparts changes in T cell poly-functionality (Fig. 2C). Again, no major differences in effector function were observed in the absence of LIGHT except for a small defect in IFNγ production by LIGHT−/− CD8+ T cells. Similarly, at day 8, the LIGHT deficiency did not significantly alter the expression of the transcription factors T-bet, Eomes, Blimp-1 or pSTAT5, which are known to play critical roles in CD8+ T cell differentiation (Fig. 2D). Moreover, WT and LIGHT−/− CD8+ T cells upregulated their expression of the lung trafficking molecule CXCR3 and downregulated their expression of CD62L and CD127 to the same extent (Fig. 2E). Overall, this shows that LIGHT does not play a major role in regulating early activation, proliferation or differentiation of effector CD8+ T cells responding to virus-expressed antigen.
Figure 2. Intact early activation, proliferation, poly-functionality and differentiation of CD8 T cells in the absence of LIGHT.
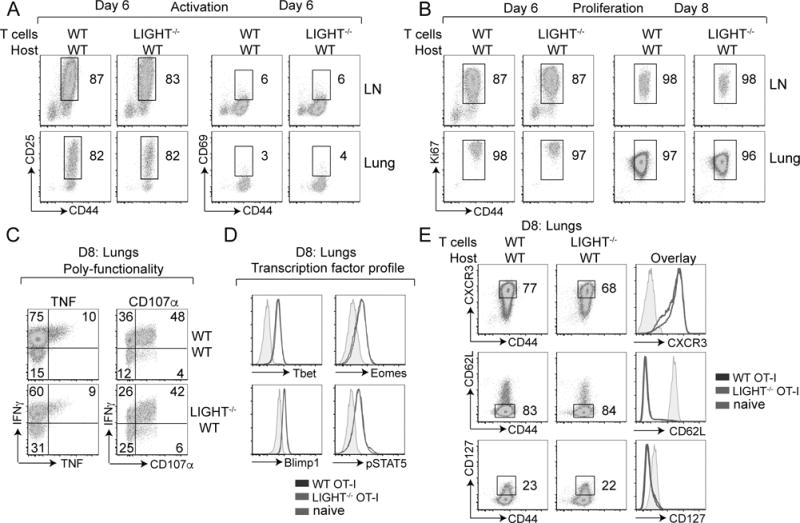
Mice that received WT or LIGHT−/− CD8 T cells in Fig 1 were analyzed on day 6 and 8 post-infection. (A-E) Lung cells were stained extracellularly with antibodies to CD8, Vα2, Vβ5, CD44, CD25, CD69, CXCR3, CD62L, IL7Rα (CD127); (B) intranuclearly with Ki67; (C) intracellularly with antibodies to TNF, IFNγ following re-stimulation with SIINFEKL peptide during which CD107α was added; and (D) intra-nuclearly with antibodies to t-bet, eomes, blimp1, pStat5, without peptide re-stimulation.
Impact of LIGHT deficiency on effector and memory CD8 T cell subsets
We next asked whether deficiency of LIGHT expression might affect the types of CD8+ T cell subsets generated in response to infection (31). Using CD27 and CD43 expression, memory CD8 T cells can be segregated into four subsets (7). The CD27hi subset of memory cells is known to proliferate to a greater extent compared to CD27lo cells when re-activated (8–10). On the other hand, CD27lo cells were shown to localize at the pathogen entry site within the secondary lymphoid organs and hence, were suggested to confer better protection against blood borne pathogens (9). In contrast, we have recently shown that CD27hi cells provide optimal protection against lethal respiratory VacV infection due to their localization near airway epithelium as well as their flexibility in differentiation (10). During the peak of the primary CD8+ T cell response, we found that both WT OT-I CD8+ T cells and LIGHT−/− OT-I CD8+ T cells were equivalently able to differentiate into CD27hi and CD27lo effector cells in the lungs (Fig. 3A). However, by day 40 post-infection, the percentages of the CD43hi subsets (particularly CD27hiCD43hi cells) were reduced in the absence of LIGHT (Fig. 3A). Although all LIGHT−/− CD8+ memory T cell subsets were reduced by ~10-15 fold at this time-point compared to their WT counterparts, there was greater than a 30-fold reduction in the CD27hiCD43hi subset (Fig. 3B). Similar results were found when analyzing the spleen (Supplementary Fig. 4). Thus, although LIGHT expression by CD8+ T cells is required for the generation of all memory subsets, it has a greater impact on the development of the CD27hiCD43hi memory subset which has been previously been implicated in providing optimal protection against lethal respiratory virus infection (10).
Figure 3. Differential impact of a LIGHT deficiency on memory CD8 T cell subsets.
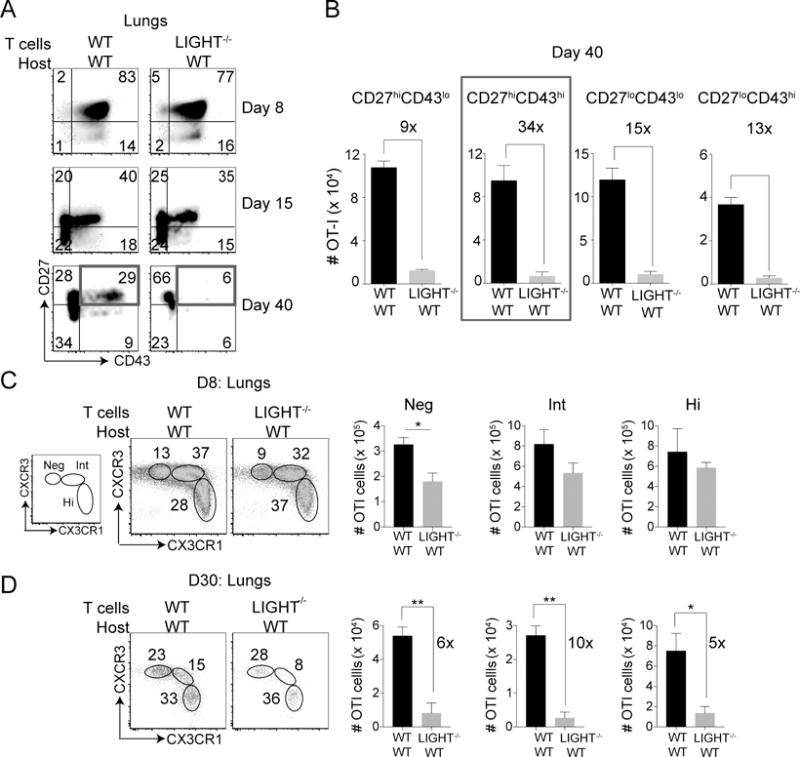
(A) Mice that received WT or LIGHT−/− CD8 T cells as in Fig 1 were analyzed on day 8, 15 and 40 post-infection. Lung cells were stained with extracellular antibodies for CD8, Vα2, Vβ5, CD44, CD27, CD43. (B) Absolute cell numbers of CD27 vs. CD43 subsets at day 40 post-infection were quantified. Similar results were obtained in three independent experiments and results are the mean ± SEM (n = 3 mice/group). (C-D) Lungs of WT or LIGHT−/− CD8 T cell recipient mice were harvested at (C) day 8 and (D) day 30 post-infection and stained for CD8, Vα2, Vβ5, CD44, CXCR3 and CX3CR1 and corresponding cell numbers of subsets were quantified. Single experiment with 4 mice in a group and results are the mean ± SEM. Statistical significance was performed using student t test with * >0.05 and ** >0.005.
Effector CD8+ T cells have been characterized based on differential expression of the fractalkine receptor CX3CR1 (13, 14). CX3CR1hi cells are the most terminally differentiated whereas CX3CR1neg cells the least differentiated. Effector cells with intermediate expression of CX3CR1 have distinct surveillance properties in non-lymphoid tissue exhibiting the highest potential for self-renewal and differentiation (13). At the peak of the effector response, we found that CD8+ T cells regardless of being WT or LIGHT-deficient, formed CXCR3+CX3CR1neg, CXCR3+CX3CR1int and CXCR3−CX3CR1hi populations, but there was a significant reduction in the CXCR3+CX3CR1neg population in the absence of LIGHT (Fig. 3C). Similar results were also observed when CXCR3 was replaced by CD27 (Supplementary Fig. 4C). These data show that the small defect in accumulation of CD8+ T cells in the lungs at the peak of the effector response (Fig. 1A-D) was mainly confined to reduced numbers of the less differentiated CX3CR1neg cells. The CD27hiCD43hi cells, which were most depleted at late time points, both in the lungs and the spleen of LIGHT−/− CD8+ T cells, consisted of both CX3CR1int and CX3CR1neg cells, but not CX3CR1hi cells (Fig. 3A, 3B and Supplementary Fig. 4). Interestingly, by day 30 post-infection, there was a much greater reduction (10-fold) in the CX3CR1int cell population compared to the CX3CRneg and CX3CR1hi subsets (5 to 6-fold) in LIGHT−/− CD8+ T cells compared to their WT counterparts (Fig. 3D). Overall, these data show that, initially (at the peak of the CD8 T cell response), LIGHT plays an important role in the accumulation of CXCR3+CX3CR1neg effector CD8+ T cells in the lungs, and later (following contraction), LIGHT is important for all CD8+ memory T cell subsets.
Intact homeostatic proliferation in absence of LIGHT
Although we found no difference in proliferation of LIGHT-deficient effector CD8+ T cells at day 8, we observed a strong reduction in the proportion of highly proliferating LIGHT−/− CD8+ T cells at day 15, as assessed by the intensity of Ki67 staining (Fig. 4A, top). There was also a significant reduction in the total numbers of both Ki67hi and Ki67lo LIGHT−/− CD8+ T cells at this time-point (Fig. 4A, bottom). To test whether the LIGHT deficiency led to impaired homeostatic turnover in an antigen-independent manner, we adoptively transferred T cells into RAG-deficient mice, which have surplus endogenous IL-7 and IL-15 that enable cells to undergo proliferation without the need for an antigenic stimulus. Based on CFSE dilution, WT and LIGHT-deficient OT-I CD8+ T cells underwent similar homeostatic proliferation in the absence of antigen over the initial 4 days (Fig. 4B). Moreover, the accumulation of the transferred CD8+ T cells and the percentages of proliferating cells (based on a high intensity of Ki67 staining) were similar in the RAG−/− hosts at all time-points tested over 40 days (Fig. 4C & 4D). Therefore, enhanced contraction of LIGHT−/− CD8+ T cells (Fig. 1A-D) was not due to their impaired ability to undergo antigen-independent homeostatic proliferation. Rather, it was likely due to a greater propensity to undergo apoptosis during the contraction phase of the response and hence, related to a defect in their capacity to survive, during and following the peak of the primary effector response to viral infection.
Figure 4. Intact homeostatic proliferation in the absence of LIGHT.
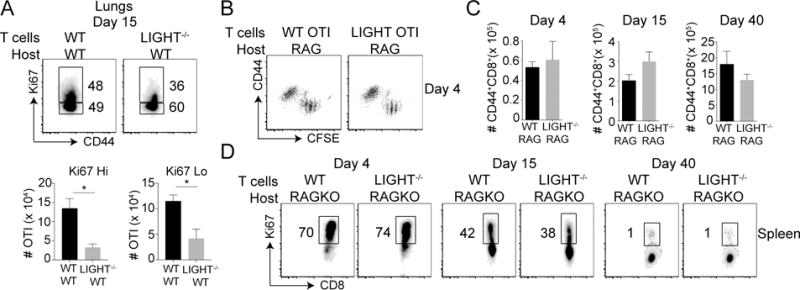
(A) Lung cells from mice that received WT or LIGHT−/− CD8 T cells were analyzed on day 15 post-infection with VacV as in Fig. 1. After staining cells for CD8, Vα2, Vβ5, and CD44, cells were stained intra-nuclearly with Ki67 (B-D) Equal numbers (5 × 105) of WT and LIGHT−/− naïve (CD44lo) OT-I (Vα2+Vβ5+) transgenic CD8 T cells were CFSE labeled and adoptively transferred into RAG-deficient mice without infection. Spleens were harvested from recipient mice at day 4, 15 and 40 post-infection and cells were stained extracellularly for CD8, Vα2, Vβ5, CD44, and intranuclearly with Ki67. *, P<0.05. Similar results were obtained in two independent experiments and results are the mean ± SEM (n = 3 mice/group).
LIGHT regulates expression of pro and anti-apoptotic factors
We then asked whether LIGHT-deficiency affects the balance of pro- versus anti-apoptotic molecules in the virus-responding CD8+ T cells. Since the most severe defect was in the CD27hiCD43hi memory subset, we isolated cells based on these markers at day 8 (the peak of the effector response), a time-point when the initial loss was observed in LIGHT−/− CD8+ T cells (Fig. 5A). Comparing fold-change between LIGHT−/− CD8+ T subset and WT CD8+ T cell subset, we found that the expression of the majority of genes associated with apoptosis were not changed more than two-fold (Fig. 5B). However, three genes that promote apoptosis were highly upregulated in LIGHT-deficient subset, namely Caspase-2, Bid and Bad. Correspondingly, four genes that enhance survival were downregulated including Akt1, Atf5, Igfr1 and Nol3 (Fig. 5B). In particular, Caspase-2 was upregulated ~60-fold while Akt1 gene expression was reduced ~30-fold (Fig. 5C). Thus, LIGHT expression regulates the balance of pro- and anti-apoptotic genes in CD8+ T cells and thereby, provides an essential survival signal allowing a proportion of effector CD8+ T cells to transition into long-lived memory cells.
Figure 5. LIGHT regulates the survival of effector CD8 T cells.
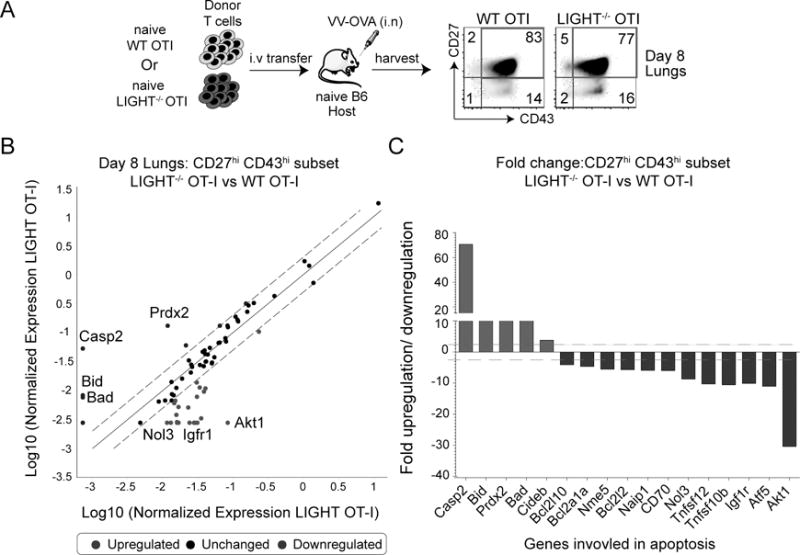
(A) Lungs from mice that received WT or LIGHT−/− CD8 T cells were harvested at day 8 post-infection with VacV and stained for CD8, Vα2, Vβ5, CD44, CD27 and CD43. The CD27hiCD43hi subset was sort purified and total mRNA was isolated. (B) Transcript levels of apoptotic genes were measured using affymetrix mouse apoptotic gene arrays and presented as a scatter plot with dotted lines representing 2-fold differences between the two groups. (C) Bars represent fold change in transcript levels of the CD27hiCD43hi subset between LIGHT-deficient CD8 T cells and WT CD8 T cells. Inset: Lung cells from WT and LIGHT−/− OT-I recipient mice were stained intra-nuclearly with pAKT.
HVEM as a ligand for CD8 T cell-expressed LIGHT
We next asked which LIGHT receptor (HVEM or LTβR) was required to generate memory cells. Mice that lack LTβR are devoid of lymph nodes and have other abnormalities associated with LTβR expression on stromal/tissue structural cells (32), hence we focused on HVEM. WT OT-I CD8+ T cells were adoptively transferred into either naïve WT or HVEM−/− recipient mice and their response compared to LIGHT−/− OT-I CD8+ T cells transferred into WT recipients. Strikingly, HVEM−/− host receiving WT CD8+ T cells exhibited a severe defect in the percentages of memory CD8+ T cells, identical to the defect observed in WT recipients that received LIGHT−/− CD8+ T cells (Fig. 6A). This defect was also reflected in the total memory CD8+ T cell numbers, both in the lungs and the spleen (Fig. 6B). This suggests that HVEM on endogenous cells plays an important role as a ligand for LIGHT expressed by CD8+ T cells. However, whether along with HVEM, LTβR is also involved in imparting signal to LIGHT on CD8+ T cells cannot be completely excluded. Nevertheless, HVEM seems to play a non-redundant role as a ligand because even though LIGHT-LTβR interactions are presumably intact in HVEM−/− mice, memory CD8 T cell development in the lungs is severely compromised.
Figure 6. Endogenous HVEM acts as a ligand for LIGHT made by CD8 T cells.
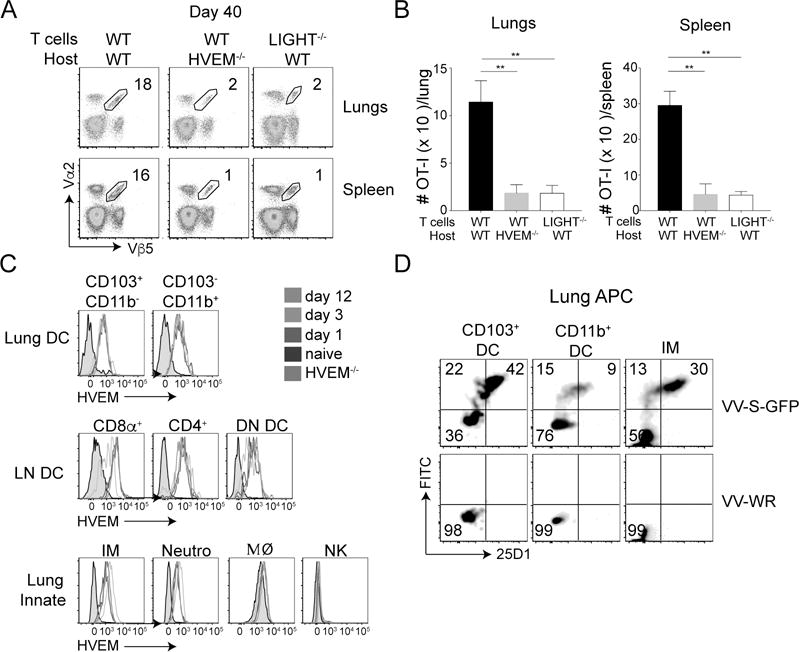
(A & B) Equal numbers (5 × 104) of WT CD8 T cells were adoptively transferred into naive WT and HVEM−/− mice, and compared to LIGHT−/− CD8 T cells transferred into WT mice. All recipient mice were infected with rVacV-WR-OVA (2 × 104 PFU i.n) the following day. At day 40 post-infection, lung and spleen cells were stained for CD8, Vα2, Vβ5, and CD44 and analyzed by flow cytometry. *, P<0.05, **, P<0.01. Similar results were obtained in two independent experiments and results are the mean ± SEM (n = 3 mice/group). (C) Lungs and mediastinal lymph nodes from WT CD8 T cell recipient mice were harvested on day 0 (naïve), 1, 3 and 12 post-infection. Dendritic cells and various innate cells were stained for HVEM. Grey histogram represents cells from HVEM−/− mice. (D) Lung cells were incubated in vitro with either recombinant VV-S-OVA-GFP or VV-WR at MOI of 1. Six-hours post-incubation, cells were stained with antibodies specific for lung DC subsets and inflammatory monocytes and 25D1 that recognizes S-OVA-MHC-I complexes.
We then assessed the expression of HVEM in the lung and draining lymph nodes during the response to intranasal VacV infection. We found that HVEM was highly expressed on both CD103+ and CD11b+ DCs (CD11c+ MHCII+) within the lungs and all three DC subsets (CD8α+DC, CD4+DC and CD8α−CD4− double negative [DN] DC) within the mediastinal lymph nodes (Fig. 6C). HVEM was also highly expressed on inflammatory monocytes (Ly6ChiCD11b+), and neutrophils (Ly6GhiCD11b+), but not on macrophages (SiglecFhiCD11b−) or NK cells (NKp46+CD11bintNK1.1+) (Fig. 6C). This suggests that multiple cell types in the lung or lymph nodes have the potential to engage CD8+ T cell expressing LIGHT via HVEM. However, as we did not observe a role for LIGHT in homeostatic antigen-independent responses (Fig. 4), we hypothesized that HVEM expression would be most relevant on APCs.
To understand the potential for lung APCs to present virus-derived antigenic peptides, mouse lung APCs were infected in vitro with recombinant VacV expressing green fluorescent protein (GFP) and the SIINFEKL peptide of ovalbumin (VacV-S-GFP). GFP fluorescence and staining with the 25D1 antibody to detect SIINFEKL peptide-MHC complexes showed that CD103+ DCs and inflammatory monocytes were most susceptible to infection and capable of presenting the antigenic peptide (Fig. 6D). Overall, these results suggest that HVEM expressed on endogenous innate immune cells, most likely CD103+ DCs or inflammatory monocytes, provides a trans-activating signal when it engages LIGHT on CD8+ T cells to directly or indirectly alters the balance of pro- versus anti-apoptotic factors and to allow for the survival and development of long-lived memory cells.
LIGHT is important for the generation of memory CD8 T cells in response to respiratory influenza virus infection
Finally, we asked if the requirement for LIGHT expressed by CD8+ T cells was only restricted to VacV infection or if LIGHT was essential for memory to other respiratory viruses. Experiments were carried as described above with one exception: recipient mice were infected with a recombinant influenza virus PR8 expressing the ovalbumin peptide SIINFEKL, genetically unrelated positive sense RNA virus. Transfer of LIGHT−/− OT-I CD8+ T cells into WT recipients again showed a severe defect in memory cell development at day 40 in the lungs, compared to their WT counterparts (Fig. 7A). A control for viral reactivity also showed that endogenous CD8+ T cell memory to viral nucleoprotein (NP) was unaffected in the recipients of LIGHT−/− CD8+ T cells (Fig. 7A). Thus, LIGHT expressed by CD8+ T cells plays a crucial role in generating CD8+ T cell memory to two genetically distinct respiratory viruses.
Figure 7. LIGHT controls memory and recall responses to influenza virus infection.
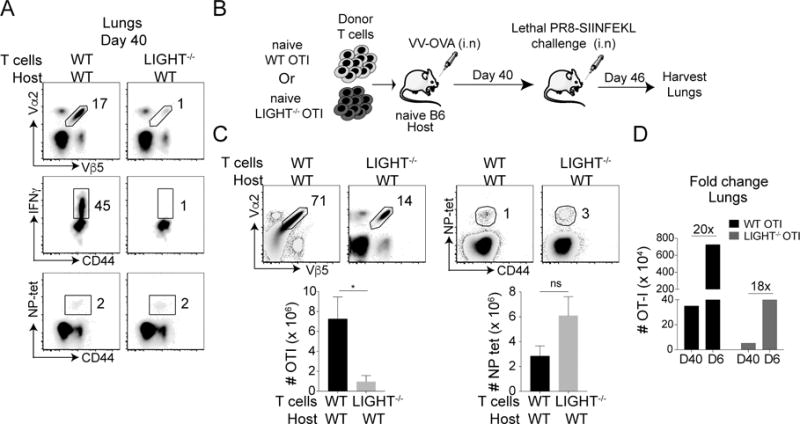
(A) Equal numbers (5 × 104) of WT and LIGHT−/− naïve (CD44lo) OT-I (Vα2+Vβ5+) transgenic CD8 T cells were adoptively transferred into WT mice and infected with PR8-SIINFEKL (i.n) the following day. Lungs were harvested at day 40 post-infection and stained for CD8, CD44, Vα2, Vβ5 and NP-tetramer (top and bottom). OT-I cells from lungs of recipients were re-stimulated in vitro with OVA peptide and stained intra-cellularly with IFN-γ (middle). (B & C) WT & LIGHT−/− CD8 T cell recipient mice that were previously infected with rVacV-WR-OVA (i.n) were re-infected with PR8-SIINFEKL (i.n) after 40 days from primary infection. Lungs were harvested at day 6 and stained for CD8, CD44, Vα2, Vβ5 and NP-tetramer. (D) Numbers and fold change in the cell numbers between WT CD8 T cells and LIGHT−/− CD8 T cells from day 40 post-primary infection to day 6 post re-infection was quantified. *, P<0.05. Similar results were obtained in two independent experiments and results are the mean ± SEM (n = 3 mice/group).
To further expand on this, we assessed the response to secondary influenza virus challenge. Recipients of WT or LIGHT−/− OT-I CD8+ T cells previously infected with rVacV-OVA were re-infected with PR8-SIINFEKL after 40 days (Fig. 7B). At day 6 post-PR8 infection, there was a dramatic defect in the accumulation of LIGHT−/− CD8 T cells in the lungs compared to WT T cells (Fig. 7C). As a control for virus reactivity no change was observed in the response of endogenous LIGHT-sufficient NP-reactive cells (Fig. 7C). Also, during this time period, no gross difference was observed in the fold expansion of LIGHT−/− memory CD8+ T cells compared to WT memory CD8+ T cells (Fig. 7D), even though the total numbers of transferred CD8 T cells that accumulated in both recipients at day 46 were different. This shows the defect in the secondary response was due to the low memory CD8+ T cell precursor frequency at the time of virus re-challenge. Collectively, our data indicate that, LIGHT does not participate in the expansion phase of the primary or secondary responses to respiratory viruses but plays a critical role in determining the frequency and type of memory CD8+ T cells that are generated and survive the phase of clonal contraction.
Discussion
The development of pulmonary memory CD8+ T cells is a vital component of immunity to respiratory viruses (8, 10, 33–36). Therefore, understanding the molecular signals necessary for the generation of long-lived memory CD8+ T cells in the lungs is of potential importance to the design of effective immunization strategies against respiratory viruses. Herein, our work identified a crucial role for LIGHT, a TNF family molecule expressed by activated CD8+ T cells, in regulating the development of memory CD8+ T cell subsets in the lungs. LIGHT−/− CD8+ T cells exhibited no gross defects in their activation, proliferation, functionality or trafficking to the lungs in response to respiratory virus infection, however, very low number of LIGHT−/− CD8+ T cells survived after the peak of the effector response resulting in a severely compromised memory compartment. Although LIGHT regulated all memory subsets, it had a particularly strong impact on the accumulation of cells with the phenotype CD27hiCD43hiCX3CR1int, which comprise the recently designated peripheral memory pool (13). Importantly, this phenomenon was not restricted to a single virus infection and we showed the crucial role of LIGHT with two genetically distinct viruses, suggesting that LIGHT activity might be a common feature for development of strong cellular immunity to many respiratory viral infections.
Over the years, a number of TNF family molecules have been shown to participate in responses to viruses, and the principal model emerging from these studies proposes that the molecules act together (concurrent use) and in a temporal manner (kinetic use) to drive and sustain clonal expansion in both primary and secondary T cell responses allowing for the development of large populations of effector CD8+ and CD4+ T cells (37). In this regard, previous work has highlighted roles of several TNF family interactions such as CD27/CD70, OX40/OX40L, 4-1BB/4-1BBL, and GITR/GITRL during systemic infection with LCMV, mCMV, or VacV, and intranasal infection with influenza virus (15, 16, 38). The general consensus is that these molecules act predominantly during the early phase of the antiviral T cell response, as effector cells begin to accumulate in the draining lymph nodes and infected tissue, since the lack of these molecules often causes a significant defect in accumulation of high numbers of T cells at the peak of the primary or secondary response. In many infection models, this early defect has also been accompanied by a parallel effect on the size of the subsequent memory cell pool. However, none of the molecules studied to date had an effect strictly during the later phase of the response (i.e., clonal contraction and memory development). In that sense, our data highlights that LIGHT clearly has a distinct and non-redundant function compared to other TNF family proteins because LIGHT deficiency had a negligible effect on early effector cell differentiation and accumulation over the first 6 days of either a primary or secondary response. These findings further substantiate the use of kinetic model of temporal activity for TNF family molecules. Importantly, they demonstrate the essential pro-survival or anti-apoptotic effect of the LIGHT-HVEM interaction on differentiated effector CD8 T cells as they mature and transition into long-lived memory cells.
Previously, no gross changes in either T cell or B cell responses were reported when LIGHT-deficient hosts were intravenously infected with vesicular stomatitis virus (VSV) or intraperitoneally infected with influenza virus (32, 39), in contrast to the notable phenotype observed in our study. This discrepancy could be explained by the use of CD8+ T cell specific LIGHT-deficiency in our system rather than the use of host completely deficient in LIGHT. LIGHT is expressed by several immune cells including T cells, DCs, monocytes and neutrophils, and though the mechanism is not well understood, increasing evidence suggests that LIGHT also has anti-inflammatory properties in addition to its pro-inflammatory activities (40, 41). Moreover, since LIGHT can interact with two receptors, multiple cell-cell interactions might be abrogated in whole LIGHT-deficient mice complicating the interpretation of the prior virus studies or otherwise obscuring a phenotype.
Another important observation was with respect to the impact of LIGHT on the balance of alternate memory subsets. The role of TNFR molecules in the development of memory T cell subsets has not been studied in great detail. Increasing evidence suggests that specific subsets within the memory population might have superior protective characteristic against certain pathogens emphasizing the need to understand the molecular signals that regulate the development of these subsets (9, 10). CD27hiCD43hi T cells are known to localize close to pathogen entry sites in the lungs and have better proliferative capacity than CD27lo T cells; hence, loss of these cells is expected to strongly compromise protective immunity against respiratory virus infection (10). More recently, Gerlach et al., described a memory subset expressing intermediate levels of CX3CR1 called ‘peripheral memory cells’ that predominantly surveyed the peripheral tissue at steady state (13). These cells are also largely CD27hiCD43hi and therefore, may be critical in orchestrating recall responses against pathogens that infect mucosal sites such as the lung. Intriguingly, LIGHT deficiency caused a significant reduction in the CD27hiCD43hiCX3CR1int peripheral memory cell compartment, further supporting the importance of LIGHT in controlling the development of memory subsets important for fighting pulmonary infections. In recent years, another subset of non-circulating memory cells called tissue resident memory cells (TRM), have been demonstrated to permanently reside within tissues including the lung, and provide immunity against respiratory virus challenge such as influenza (35, 36). Rather than being cytolytic, this subset seems to mediate its effect by enhancing local innate immunity as well as rapidly recruiting memory cells from circulation. LIGHT expression on CD8+ T cells also greatly contributed to the accumulation of optimal numbers of lung tissue resident memory cells (TRM), further substantiating its overall importance. Recently it was suggested that T cells generated around the peak of the effector response seed lung TRM cells (42). Hence it is likely that the defect in CD8 T cell numbers in the lungs at day 8 post-infection contributes to the TRM defect. In line with this we observed that at day 8 post-infection the defect in lung CD8+ T cell numbers were mainly due to specific reduction in CXCR3+CX3CR1neg population, which is known to give rise to TRM cells (13). These findings raise the question of whether other TNF family interactions also play similar or distinct roles in memory subset development. A recent report demonstrated a role for 4-1BBL in the generation of TRM cells in the context of intranasal influenza virus infection, suggesting other molecules, in addition to LIGHT, can also be relevant (43).
A focus for future studies is how the LIGHT-HVEM interaction controls memory development. CD8+ T cells transferred into HVEM-deficient hosts recapitulated the defect observed when LIGHT-deficient T cells were transferred into intact hosts, suggesting that LIGHT expressed on the surface of the effector CD8 T cell interacts with HVEM expressed on endogenous cell. The most logical cells would be APCs that are infected with the virus following respiratory infection, such as CD103+ DC or inflammatory monocytes, as we implied. However, since HVEM is widely expressed on many different innate immune cells, future studies with conditional deletion of HVEM in specific cell types will be required to understand the essential cell type that provides HVEM signal. Furthermore, at present, it is not clear whether HVEM is functional and transmits a signal when it engages LIGHT. This again is the most logical hypothesis as HVEM can signal through several pro-inflammatory pathways, including NF-κB, and has the potential to induce maturation, differentiation, survival, and inflammatory activity in the cell that expresses it, including a DCs or monocytes. An alternative hypothesis is that LIGHT itself is functional on CD8+ T cells. Similar to other TNF family proteins, LIGHT has only a short cytoplasmic domain; but nevertheless, it has the potential to transmit intracellular signals (21, 23). Little is known whether signaling through LIGHT can affect T cell responses in vivo, but in vitro studies have shown that engagement of LIGHT expressed on the surface of T cells can induce proliferation (44, 45). Experiments with macrophages have substantiated the idea that LIGHT can signal because its engagement on these cells was found to induce some inflammatory molecules and enhance activation of several pathways including those involving ERK, PI3-kinase, Akt, and NF-κB (46). In our studies, when transferring LIGHT−/− CD8+ T cells into WT hosts, the endogenous VacV and influenza-specific CD8+ T cell responses were normal despite the profound defect in transferred LIGHT−/− CD8+ T cell responses. Although difficult to interpret, because endogenous LIGHT-sufficient T cells were responding in these hosts, the results might imply that HVEM signaling in APCs or other cells were intact, leading to the conclusion that the defect in T cell memory was specifically due to the lack of signals through LIGHT into the responding T cells.
In summary, our findings provide new insights into the role of LIGHT expressed specifically by CD8+ T cells in the generation of memory CD8 T cell subsets. Because the factors controlling the development of the mucosal memory CD8+ T cell compartment are not fully understood, our novel results suggest that targeting LIGHT and its binding partners are worth considering when formulating the design of vaccines to generate strong immune memory.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health Tetramer Core Facility (contract HHSN272201300006C) for provision of MHC/peptide tetramer containing the B8R peptide (20–27; TSYKFESV)/H-2Kb, conjugated to APC. We also thank Dr. Amanda Posgai for her assistance with technical editing.
This study was supported by NIH grants AI77079 and AI087734 to S.S.-A, and AI67341 to MC. P.D. was supported through The American Association of Immunologists Careers in Immunology Fellowship Program. V.T. was supported by NIH grant T32 AR007603-15.
References
- 1.Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol. 2009;9:153–161. doi: 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- 2.Lauvau G, Boutet M, Williams TM, Chin SS, Chorro L. Memory CD8(+) T Cells: Innate-Like Sensors and Orchestrators of Protection. Trends Immunol. 2016;37:375–385. doi: 10.1016/j.it.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 4.Lanzavecchia A, Sallusto F. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. 2000;290:92–97. doi: 10.1126/science.290.5489.92. [DOI] [PubMed] [Google Scholar]
- 5.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 6.Bachmann MF, Wolint P, Schwarz K, Oxenius A. Recall proliferation potential of memory CD8+ T cells and antiviral protection. J Immunol. 2005;175:4677–4685. doi: 10.4049/jimmunol.175.7.4677. [DOI] [PubMed] [Google Scholar]
- 7.Hikono H, Kohlmeier JE, Ely KH, Scott I, Roberts AD, Blackman MA, Woodland DL. T-cell memory and recall responses to respiratory virus infections. Immunol Rev. 2006;211:119–132. doi: 10.1111/j.0105-2896.2006.00385.x. [DOI] [PubMed] [Google Scholar]
- 8.Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL. Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J Exp Med. 2007;204:1625–1636. doi: 10.1084/jem.20070322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE. Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity. 2013;38:1250–1260. doi: 10.1016/j.immuni.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abboud G, Desai P, Dastmalchi F, Stanfield J, Tahiliani V, Hutchinson TE, Salek-Ardakani S. Tissue-specific programming of memory CD8 T cell subsets impacts protection against lethal respiratory virus infection. J Exp Med. 2016;213:2897–2911. doi: 10.1084/jem.20160167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 12.Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–161. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- 13.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, von Andrian UH. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity. 2016;45:1270–1284. doi: 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bottcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Hochst B, Eickhoff S, Rieckmann JC, Russo C, Bauer T, Flecken T, Giesen D, Engel D, Jung S, Busch DH, Protzer U, Thimme R, Mann M, Kurts C, Schultze JL, Kastenmuller W, Knolle PA. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat Commun. 2015;6:8306. doi: 10.1038/ncomms9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goulding J, Tahiliani V, Salek-Ardakani S. OX40:OX40L axis: emerging targets for improving poxvirus-based CD8(+) T-cell vaccines against respiratory viruses. Immunol Rev. 2011;244:149–168. doi: 10.1111/j.1600-065X.2011.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salek-Ardakani S, Croft M. Tumor necrosis factor receptor/tumor necrosis factor family members in antiviral CD8 T-cell immunity. J Interferon Cytokine Res. 2010;30:205–218. doi: 10.1089/jir.2010.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Leeuwen EM, Sprent J, Surh CD. Generation and maintenance of memory CD4(+) T Cells. Curr Opin Immunol. 2009;21:167–172. doi: 10.1016/j.coi.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed R, Bevan MJ, Reiner SL, Fearon DT. The precursors of memory: models and controversies. Nat Rev Immunol. 2009;9:662–668. doi: 10.1038/nri2619. [DOI] [PubMed] [Google Scholar]
- 20.Williams MA, Holmes BJ, Sun JC, Bevan MJ. Developing and maintaining protective CD8+ memory T cells. Immunol Rev. 2006;211:146–153. doi: 10.1111/j.0105-2896.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 21.Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, Spear PG, Ware CF. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- 22.Morel Y, Schiano de Colella JM, Harrop J, Deen KC, Holmes SD, Wattam TA, Khandekar SS, Truneh A, Sweet RW, Gastaut JA, Olive D, Costello RT. Reciprocal expression of the TNF family receptor herpes virus entry mediator and its ligand LIGHT on activated T cells: LIGHT down-regulates its own receptor. J Immunol. 2000;165:4397–4404. doi: 10.4049/jimmunol.165.8.4397. [DOI] [PubMed] [Google Scholar]
- 23.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Anders RA, Wang Y, Turner JR, Abraham C, Pfeffer K, Fu YX. The critical role of LIGHT in promoting intestinal inflammation and Crohn’s disease. J Immunol. 2005;174:8173–8182. doi: 10.4049/jimmunol.174.12.8173. [DOI] [PubMed] [Google Scholar]
- 25.del Rio ML, Schneider P, Fernandez-Renedo C, Perez-Simon JA, Rodriguez-Barbosa JI. LIGHT/HVEM/LTbetaR interaction as a target for the modulation of the allogeneic immune response in transplantation. Am J Transplant. 2013;13:541–551. doi: 10.1111/ajt.12089. [DOI] [PubMed] [Google Scholar]
- 26.Brown GR, Lane GW, Whittington BJ. Disparate role of LIGHT in organ-specific donor T cells activation and effector molecules in MHC class II disparate GVHD. J Clin Immunol. 2010;30:178–184. doi: 10.1007/s10875-009-9337-1. [DOI] [PubMed] [Google Scholar]
- 27.Doherty TA, Soroosh P, Khorram N, Fukuyama S, Rosenthal P, Cho JY, Norris PS, Choi H, Scheu S, Pfeffer K, Zuraw BL, Ware CF, Broide DH, Croft M. The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nat Med. 2011;17:596–603. doi: 10.1038/nm.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai H, Chen S, Xu S, Sun Y, Bai Q, Lu C, Chen Y, Fu X, Xu G, Chen L. Deficiency of LIGHT signaling pathway exacerbates Chlamydia psittaci respiratory tract infection in mice. Microb Pathog. 2016;100:250–256. doi: 10.1016/j.micpath.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Xu G, Liu D, Okwor I, Wang Y, Korner H, Kung SK, Fu YX, Uzonna JE. LIGHT Is critical for IL-12 production by dendritic cells, optimal CD4+ Th1 cell response, and resistance to Leishmania major. J Immunol. 2007;179:6901–6909. doi: 10.4049/jimmunol.179.10.6901. [DOI] [PubMed] [Google Scholar]
- 30.Stanley AC, de Labastida Rivera F, Haque A, Sheel M, Zhou Y, Amante FH, Bunn PT, Randall LM, Pfeffer K, Scheu S, Hickey MJ, Saunders BM, Ware C, Hill GR, Tamada K, Kaye PM, Engwerda CR. Critical roles for LIGHT and its receptors in generating T cell-mediated immunity during Leishmania donovani infection. PLoS Pathog. 2011;7:e1002279. doi: 10.1371/journal.ppat.1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buchholz VR, Graf P, Busch DH. The origin of diversity: studying the evolution of multi-faceted CD8+ T cell responses. Cell Mol Life Sci. 2012;69:1585–1595. doi: 10.1007/s00018-012-0967-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheu S, Alferink J, Potzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. 2002;195:1613–1624. doi: 10.1084/jem.20020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salek-Ardakani S, Moutaftsi M, Sette A, Croft M. Targeting OX40 promotes lung-resident memory CD8 T cell populations that protect against respiratory poxvirus infection. J Virol. 2011;85:9051–9059. doi: 10.1128/JVI.00619-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salek-Ardakani S, Flynn R, Arens R, Yagita H, Smith GL, Borst J, Schoenberger SP, Croft M. The TNFR family members OX40 and CD27 link viral virulence to protective T cell vaccines in mice. J Clin Invest. 2011;121:296–307. doi: 10.1172/JCI42056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slütter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, Harty JT. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Science Immunology. 2017;2:eaag2031. doi: 10.1126/sciimmunol.aag2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. 2016;16:79–89. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- 37.Croft M. The TNF family in T cell differentiation and function–unanswered questions and future directions. Semin Immunol. 2014;26:183–190. doi: 10.1016/j.smim.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wortzman ME, Clouthier DL, McPherson AJ, Lin GH, Watts TH. The contextual role of TNFR family members in CD8(+) T-cell control of viral infections. Immunol Rev. 2013;255:125–148. doi: 10.1111/imr.12086. [DOI] [PubMed] [Google Scholar]
- 39.Sedgmen BJ, Dawicki W, Gommerman JL, Pfeffer K, Watts TH. LIGHT is dispensable for CD4+ and CD8+ T cell and antibody responses to influenza A virus in mice. Int Immunol. 2006;18:797–806. doi: 10.1093/intimm/dxl016. [DOI] [PubMed] [Google Scholar]
- 40.Mana P, Linares D, Silva DG, Fordham S, Scheu S, Pfeffer K, Staykova M, Bertram EM. LIGHT (TNFSF14/CD258) is a decisive factor for recovery from experimental autoimmune encephalomyelitis. J Immunol. 2013;191:154–163. doi: 10.4049/jimmunol.1203016. [DOI] [PubMed] [Google Scholar]
- 41.Krause P, Zahner SP, Kim G, Shaikh RB, Steinberg MW, Kronenberg M. The tumor necrosis factor family member TNFSF14 (LIGHT) is required for resolution of intestinal inflammation in mice. Gastroenterology. 2014;146:1752–1762 e1754. doi: 10.1053/j.gastro.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, Fujisawa M, Chikaishi T, Komeda J, Itoh J, Umemura M, Kyusai A, Tomura M, Nakayama T, Woodland DL, Kohlmeier JE, Miyazawa M. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med. 2016;213:3057–3073. doi: 10.1084/jem.20160938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou AC, Wagar LE, Wortzman ME, Watts TH. Intrinsic 4-1BB signals are indispensable for the establishment of an influenza-specific tissue-resident memory CD8 T-cell population in the lung. Mucosal Immunol. 2017 doi: 10.1038/mi.2016.124. [DOI] [PubMed] [Google Scholar]
- 44.Shi G, Luo H, Wan X, Salcedo TW, Zhang J, Wu J. Mouse T cells receive costimulatory signals from LIGHT, a TNF family member. Blood. 2002;100:3279–3286. doi: 10.1182/blood-2002-05-1404. [DOI] [PubMed] [Google Scholar]
- 45.Wan X, Zhang J, Luo H, Shi G, Kapnik E, Kim S, Kanakaraj P, Wu J. A TNF family member LIGHT transduces costimulatory signals into human T cells. J Immunol. 2002;169:6813–6821. doi: 10.4049/jimmunol.169.12.6813. [DOI] [PubMed] [Google Scholar]
- 46.Lim SG, Suk K, Lee WH. Reverse signaling from LIGHT promotes pro-inflammatory responses in the human monocytic leukemia cell line, THP-1. Cell Immunol. 2013;285:10–17. doi: 10.1016/j.cellimm.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 47.Flynn R, Hutchinson T, Murphy KM, Ware CF, Croft M, Salek-Ardakani S. CD8 T cell memory to a viral pathogen requires trans cosignaling between HVEM and BTLA. PLoS One. 2013;8:e77991. doi: 10.1371/journal.pone.0077991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenkins MR, Webby R, Doherty PC, Turner SJ. Addition of a prominent epitope affects influenza A virus-specific CD8+ T cell immunodominance hierarchies when antigen is limiting. J Immunol. 2006;177:2917–2925. doi: 10.4049/jimmunol.177.5.2917. [DOI] [PubMed] [Google Scholar]
- 49.Goulding J, Bogue R, Tahiliani V, Croft M, Salek-Ardakani S. CD8 T cells are essential for recovery from a respiratory vaccinia virus infection. J Immunol. 2012;189:2432–2440. doi: 10.4049/jimmunol.1200799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abboud G, Tahiliani V, Desai P, Varkoly K, Driver J, Hutchinson TE, Salek-Ardakani S. Natural Killer Cells and Innate Interferon Gamma Participate in the Host Defense against Respiratory Vaccinia Virus Infection. J Virol. 2015;90:129–141. doi: 10.1128/JVI.01894-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goulding J, Abboud G, Tahiliani V, Desai P, Hutchinson TE, Salek-Ardakani S. CD8 T cells use IFN-gamma to protect against the lethal effects of a respiratory poxvirus infection. J Immunol. 2014;192:5415–5425. doi: 10.4049/jimmunol.1400256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.