

Summary
Objective
Gastric inhibitory polypeptide plays a role in glucose and lipid metabolism and is associated with obesity and insulin resistance. The objective of this study is to confirm the anti‐obesity effects of the gastric inhibitory polypeptide receptor antagonist, SKL‐14959, on diet‐induced obesity mice.
Method
Diet‐induced obesity mice at 20 weeks of age were administered with or without SKL‐14959 for 96 d. Body weight and food intake were monitored throughout the experiment. Mice were sacrificed, and physiological and biochemical markers were measured, and then histochemical and gene expression analyses were also performed. In further studies, mice were orally gavaged with [14C]‐oleic acid to investigate the excursion of digested lipids.
Results
SKL‐14959 significantly suppressed weight gain without affecting food intake, decreased triacylglycerol contents in the liver and the muscle and the intensity stained with oil‐red in the liver. It also improved plasma glutamic pyruvic transaminase and 3‐hydroxybutyrate levels in addition to notably down‐regulated relative gene expression of srebf1 and dgat1 in the liver despite not altering in the adipose tissue. Furthermore, SKL‐14959 showed remarkable inhibition of lipid uptake in the adipose tissue after the oil challenge.
Conclusion
SKL‐14959 inhibited lipids uptake and improved lipids metabolism, results in suppression of body‐weight gain.
Keywords: anti‐obesity, fatty liver, GIP, GIP receptor antagonist
Introduction
Obesity has been a major health problem in developed countries and is a growing issue in the developing world. The world‐wide epidemic of obesity is about to increase the risk of the incidence of diabetes mellitus, insulin resistance, fatty liver, hyperlipidaemia, hypertension, cardiovascular diseases and some forms of cancer 1, 2. Several anti‐obesity agents such as orlistat, sibutramine and rimonabant have been approved for clinical uses in many countries including in the United States or Europe. However, these drugs with the exception of orlistat were withdrawn from the market due to the serious adverse effects of cardiac dysfunction or psychiatric behaviour 3. Recently, lorcaserin and exendine, a selective serotonin 2C (5HT2c) receptor agonist and Qsymia, a combination of phetermine and topiramate, and liraglutide were approved by the US Food and Drug Administration 4. Thus, an ideal anti‐obesity agent without adverse effects has yet to be developed.
Gastric inhibitory polypeptide or glucose‐dependent insulinotropic polypeptide (GIP) is an incretin hormone, which is released from duodenal K‐cell in a glucose‐dependent manner 5, 6. The physiological function of GIP plays a key role in promoting fatty acid incorporation and synthesis 7, 8 and enhancing of lipoprotein lipase (LPL) activity in explant adipose tissues or 3 T3‐L1 cell 9, 10. Increased postprandial GIP levels have been associated with obesity in both human 11, 12, 13, 14, 15 and mice 16. Furthermore, GIP infusion increased the rate of chylomicron clearance in dogs 17, and both endogenous and exogenous GIP were shown to lower plasma triacylglycerol (TG) levels in rats in response to intraduodenal fat infusion 18.
Gastric inhibitory polypeptide receptor knock‐out mice fed high‐fat diets showed suppression of body weight gain compared with wild mice fed the same high‐fat diets 19. In addition, Nasteska et al. 20 reported that chronic reduction of GIP improved obesity and insulin resistance in transgenic mice fed a high‐fat diet. Moreover, administration of GIP receptor antagonist, (Pro3) GIP, prevented the development of obesity and insulin resistance in diet‐induced obesity (DIO) mice and ob/ob mice 21, 22. However, this compound has also the effect of a partial or full agonist against GIP receptor 23. Nakamura et al. 24 reported that a candidate, SKL‐14959 is a novel low molecular chemical compound and GIP specific receptor antagonist, had a strong binding affinity for the GIP receptor and inhibited GIP‐stimulated cAMP production. Additionally, SKL‐14959 suppressed insulin secretion in oral glucose tolerance test and postponed TG clearance from the circulation in oil tolerance test. In the point of clinical applications, Roux‐en‐Y gastric bypass surgery, which includes the removal of GIP‐secreting K cells from the upper small intestine, results in body weight loss and improved insulin resistance 25. Thus, it seems that GIP has a bifacial characterization, at least, it is apparent from the contribution to obesity, insulin resistance and diabetes mellitus.
The objective of the present study is to confirm the hypothesis that inhibition of lipid uptake by GIP receptor antagonist, SKL‐14959 on DIO mice model, can indicate an anti‐obesity effect.
Material and methods
SKL‐14959 was synthesized in our laboratory. [14C]‐oleic acid was purchased from Daiichi Pure Chemicals Co., Ltd. (Tokyo, Japan). All other chemicals used were of molecular biology grade.
Animal study
Diets‐induced obesity model
All experiments were approved by the Committee on Animal Care of Sanwa Kagaku Kenkyusho, SKK. Institute of Cancer Research (ICR) male mice obtained from Clea Japan (Osaka, Japan) at 10 weeks of age were fed laboratory standard powder chow, CRF‐1 (Oriental Yeast Co., Ltd., Tokyo, Japan). Mice were allowed ad libitum access to food and water and were maintained in a plastic cage at a constant temperature of 23 ± 2°C under a 12‐h light–dark cycle (lights on from 7:00 am to 7:00 pm). At 16 weeks of age, mice were divided into two groups of low‐fat diet (LFD) fed (Oriental Yeast Co., Ltd. Tokyo, Japan; fat 5%, protein 16.8%, carbohydrate 60.9%; 358.9 kcal 100 g−1) and high‐fat diet (HFD) fed (Oriental Yeast Co., Ltd., Tokyo, Japan; beef tallow 40%, corn starch 10%, sugar 9%, vitamin mixture 1%, mineral mixture 4% and casein 36%; 563.4 kcal 100 g−1) 26 and acclimated individually in plastic cages.
Drug evaluation
At 20 weeks of age, the HFD group was divided into two groups, HFD control and treatment with SKL‐14959, the body mass and food intake were matched, and the study was performed to evaluate anti‐obesity for 95–96 d. The beginning of administration was defined as day1. The treatment group was given SKL‐14959 by 0.133% mixed diets during the test. Body mass and food intake were measured until the end of the study. Anatomy was split into 2 d to match the condition of mealtimes in a day. Mice were anaesthetized using ether and mouse lengths, from nose‐to‐anus, and were measured using callipers before the mice were sacrificed and dissected. Body mass index (BMI) was calculated as body weight (g) divided by the length (cm) squared. Blood samples were withdrawn from inferior vena cava and centrifuged to separate plasma and then kept at – 80°C until each was assayed. Liver, heart, kidney, spleen, lung, adipose tissues, mesenteric, epididymal, perirenal and retroperitoneal adipose pads and soleus, gastrocnemius and vastus lateralis muscles were removed, and their wet weight measured. For histological analyses, sections of the liver and adipose tissue were either immediately stored in liquid nitrogen or fixed in 10% neutral‐buffered formalin. The remaining sections of the liver and adipose tissue were immediately transferred to – 80°C and stored until TG and gene expression analysis were performed.
Lipid distribution in single [14C]‐oleic acid administration
ICR male mice at 20 weeks of age were divided into two groups on the basis of body weight in the morning after 16‐h fast and then orally administered with or without SKL‐14959 (100 mg kg−1 body weight) prepared with 0.5% sodium carboxymethylcellulose solution 15 min prior to oral administration of [14C]‐oleic acid (18.5 MBq g−1). The mice were immediately released into an occlusive metabolic cage Metabolica from Yamagen Co., Ltd. (Tokyo, Japan) after the challenge and then euthanized under ether anaesthesia at 60 and 120 min. Blood samples were collected from the inferior vena cava, and epididymal adipose tissue, soleus muscles and liver were removed and weighed. Sections of all samples including blood were oxidized using a combustion system (ASC‐113, Aloka), and the radioactivity in the absorbent (Carbo‐Sorb E, PerkinElmer), which absorbed yields of 14C‐carbon dioxide, was measured using a liquid scintillation counter (LS6000TA, Beckman). Those data were expressed as a concentration of each whole tissue.
Triacylglycerol content
Triglyceride analysis was measured with the modified procedure previously reported 27. In brief, the liver and the muscle samples were homogenized in chloroform‐methanol mixture (2:1 v / v) and then extracted with the same reagent and phosphate buffer saline. Organic layers were dried up under the stream of nitrogen and reconstructed in isopropanol‐TritonX‐100 (9:1). The TG content was determined using L‐type Wako TG‐H (Wako Pure Chemicals, Osaka, Japan).
Histological analysis
The small piece of the epididymal adipose tissue fixed with 10% neutral‐buffered formalin was embedded in paraffin. These paraffin blocks were sectioned at a thickness of 2 μm and stained with eosin and haematoxylin. To examine the size of adipocytes, the number of cells was individually counted three areas scanned under the 15 power 20 magnification and then expressed as the mean ± standard error (i.e. a larger number means smaller size) 28. The small piece of the liver fixed with 10% neutral‐buffered formalin was dipped into 10% to 20% sucrose solution in 0.1 M phosphate buffer at 4°C all day and then frozen in liquid nitrogen with freezing medium (compound Tissue‐Tek). From the blocks, 10 μm‐thick sections were prepared and then stained with Oil red O. To examine the degree of steatosis, images were captured using a microscope (Olympus BX‐50, Tokyo, Japan) under the 10 power 10 magnification.
β‐Hydroxyacyl‐CoA dehydrogenase assay
β‐Hydroxyacyl‐CoA dehydrogenase (β‐HAD) assay was measured with the procedure previously reported 29. Liver and muscle segments were weighed and immediately homogenized in 20 volumes of ice‐cold 20 mM phosphate buffer (pH 7.4) including in 5.4 M glycerol, 5 mM 2‐mercaptoethanol, 0.5 mM ethylenediaminetetraacetic acid and 0.02% bovine serum albumin. The homogenate was centrifuged 600 g for 10 min before the supernatant was used for enzyme assay. All homogenate operations were performed at 0–4°C. The reaction mixture contained 95 mM pyrophosphate buffer (pH 7.3) including in 0.2 mM nicotinamide adenine dinucleotide, 4.8 mM acetoacetyl‐CoA as a substrate and its supernatant. The activity was determined for 5 min at 25°C by monitoring the rate of disappearance of nicotinamide adenine dinucleotide by measuring the absorbance at 339 nm. One unit of the activity was defined as the conversion of 1.0 μ mol−1 of substrate to product per minute and was expressed as U g−1 tissue.
RNA extraction and real‐time polymerase chain reaction
Total RNA was isolated from the frozen liver and the adipose tissues using the RNeasy Mini Kit (QIAGEN, Valencia, CA) following the manufacturer's instruction. In brief, approximately 100 mg organs were homogenized in 1 mL of Trizol reagent (Invitrogen, Carlsbad, CA) using zirconia beads and were purified total RNA. Two micrograms of total RNA was reverse‐transcripted to complementary DNA using the High Capacity cDNA Reverse Transcription Kit (Applied biosystems, Foster City, CA). Polymerase chain reactions were performed using the LightCycler480 (Roche, USA). Polymerase chain reactions for each target cDNA were carried out using commercially supplied ready‐to use primer and probe sets by Applied Biosystems (TaqMan gene expression assays): sterol regulatory element binding transcription factor 1, Srebf1 (Mm00550338_m1), diacylglycerol O‐acyltransferase 1, Dgat1 (Mm00515643_m1), fatty acid synthase, Fasn (Mm01253292_m1), acetyl‐coenzyme A carboxylase alpha, Acaca (Mm01304257_m1), peroxisome proliferator activated receptor alpha, Ppara (Mm00440939_m1), uncoupling protein 2, Ucp2 (Mm00627598_m1), hormone sensitive lipase, Lipe (Mm00495359_m1) and TATA box binding protein, TBP (Mm0111222) as housekeeping gene. Relative gene expression was analysed by the ⊿⊿CT method 30 and normalized with internal control, TBP. We validated TBP as housekeeping gene by confirming its expression level was not significantly different between each group. LFD was defined as 1.0 and expressed mean ± standard deviation as fold change.
Blood analysis
Plasma glutamic oxaloacetic transaminase (GOT), glutamic pyruvic transaminase (GPT), non‐esterified free acid (NEFA), TG, total cholesterol, low‐density lipoprotein (LDL), high‐density lipoprotein (HDL) and glucose levels (Glu) were measured with L‐type Wako GOT‐J2, L‐type Wako GPT‐J2, NEFA‐HA Test Wako, L‐type Wako TG‐H, L‐type CHO H, L‐Type LDL‐C, L‐Type HDL‐C and L‐Type Glucose 2 (Wako Pure Chemical Industries, Osaka, Japan), respectively. 3‐Hydroxybutyrate was determined by Ketone test liquid (Sanwa Kagaku Kenkyusho, Nagoya, Japan) using auto analyser (7170, Hitachi Ltd., Tokyo, Japan). Plasma GIP, insulin, and adiponectin levels were measured using enzyme‐linked immunosorbent assay kits (LINCO RESEARCH, INC., Missouri, U.S.A., Shibayagi, Gumma, Japan, and Otsuka Pharmaceutical Ltd., Tokushima, Japan, respectively).
Statistical analysis
Data were expressed as means ± standard error. Two‐way analysis of variance followed by Dunnett's test were performed in the body weight, body weight gain and food intake, and others were performed by unpaired t‐test for comparison of the LFD and the HFD group or of the HFD treated with or without SKL‐14959 group and less than 0.05 of p value was statistically significant.
Results
Effects of SKL‐14959 treatment on body weight gain and food intake and tissue mass
Two‐way repeated measures analysis of variance (mouse groups across time) was used to analyse body weight, weight gain, food intake and tissue mass. Intake calories in the LFD, HFD control and SKL‐14959‐treated group through the test were 1638 ± 6.6, 1877 ± 10 and 1845 ± 9.5 kcal, respectively. The body mass gain in the HFD control group was significantly increased compared with the LFD group, whereas the food intake was significantly decreased. The body weight gain in the SKL‐14959‐treated group was significantly suppressed compared with the HFD control group without the impact of food intake (p < 0.001), and the suppression percent of body mass was 7.0% (Figure 1A–1C). BMI was not significantly decreased (Figure 1E).
Figure 1.
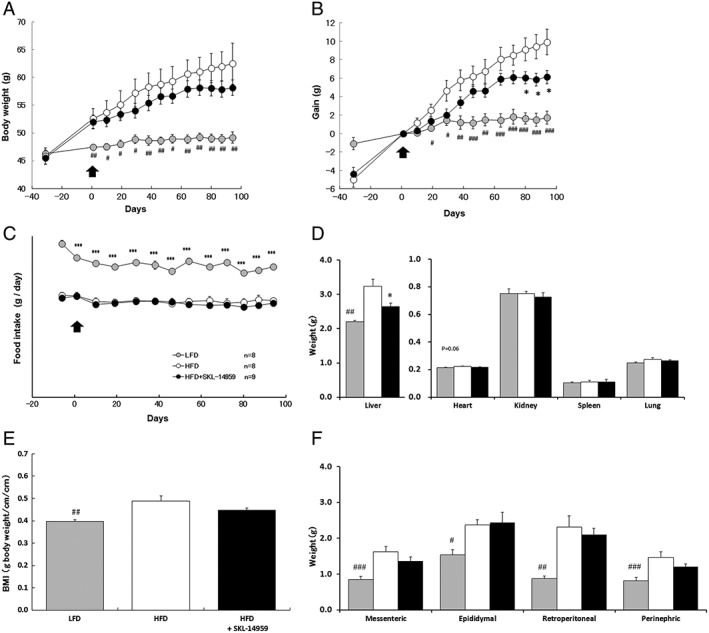
Effects of SKL‐14959 on body weight (A), body weight gain (B), daily food intake (C), organ weights (D), body mass index (BMI) (E) and adipose tissue weights (F). Grey, open and closed circle or bar express LFD, HFD and HFD with SKL‐14959 group, respectively. Arrow is the date of beginning administration, Day1. Data are means ± standard error (eight to nine mice per group). # p < 0.05, ## p < 0.01, ### p < 0.001 vs. LFD group, *p < 0.05 vs. HFD control group (t‐test).
Weights of the each adipose pad, mesenteric, epididymal, retroperitoneal, and perirenal, the liver and BMI in the HFD control group were significantly increased compared with the LFD group (p < 0.01). The SKL‐14959‐treated group was significantly decreased in the liver weight (p < 0.05) and was not significantly decrease BMI and the adipose tissues compared with the HFD control group (Figure 1D and 1F). The heart, kidney, spleen and lung weight did not show important differences between all groups (Figure 1D).
Biochemical markers in plasma
Plasma GPT activity and total cholesterol, LDL, HDL, GIP and insulin concentrations in the HFD control group were significantly elevated compared with the LFD group, whereas the SKL‐14959‐treated group tended have lower plasma levels of GOT and GPT activity (p = 0.08 and p < 0.05, respectively), 3‐hydroxybutyrate (p = 0.09) compared with the HFD control group. Plasma NEFA, TG, glucose and adiponectin concentrations were not significantly different between all groups (Table 1).
Table 1.
Plasma parameters treated with SKL‐14959 for 95–96 days on diet‐induced obesity in mice.
| Marker | LFD | HFD | HFD + SKL‐14959 | |
|---|---|---|---|---|
| GOT | (IU/L) | 46 ± 9.0 | 60 ± 6.6 | 47 ± 2.9 |
| GPT | (IU/L) | 27 ± 3.2 | 48 ± 7.2* | 27 ± 2.8*** |
| NEFA | (μEq/L) | 1.80 ± 0.1 | 1.76 ± 0.2 | 1.98 ± 0.2 |
| TG | (mg/dL) | 100 ± 11 | 108 ± 17 | 122 ± 19 |
| T‐Cho | (mg/dL) | 112 ± 3.9 | 209 ± 5.4** | 190 ± 7.8 |
| LDL | (mg/dL) | 11.6 ± 0.9 | 38 ± 7.1** | 28 ± 3.2 |
| HDL | (mg/dL) | 88.5 ± 3.4 | 165 ± 12** | 153 ± 6.1 |
| Glucose | (mg/dL) | 213 ± 9.7 | 253 ± 11 | 232 ± 9.3 |
| 3‐hydroxybutyrate | (mg/dL) | 132 ± 9.7 | 193 ± 31 | 135 ± 14 |
| GIP | (pg/mL) | 28.7 ± 5.5 | 75.2 ± 14.4** | 87 ± 10 |
| Insulin | (ng/mL) | 3.4 ± 1.3 | 9.0 ± 2.2* | 9.6 ± 2.3 |
| Adiponectin | (μg/mL) | 15.7 ± 1.3 | 14.2 ± 1.0 | 13.7 ± 1.1 |
Data are means ± standard error (eight to nine mice per group).
p < 0.05.
p < 0.01 vs. low‐fat diet (LFD) group.
p < 0.05 vs. HFD control group (t‐test).
GIP, gastric inhibitory polypeptide; GOT, glutamic oxaloacetic transaminase; GPT, glutamic pyruvic transaminase; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; NEFA, non‐esterified free acid; T‐Cho, total cholesterol; TG, triacylglycerol.
Triacylglycerol content and β‐hydroxyacyl‐CoA dehydrogenase activity in the liver and the muscles
Hepatic and muscle lipid accumulation in the HFD control group were increased compared with the LFD group. TG contents in the SKL‐14959‐treated group showed significant decrease in the liver (p < 0.05) and showed a tendency to decrease in the muscles contrasted with the HFD control (Figure 2A–2C). Hepatic and muscle β‐HAD activities in the HFD control group were increased compared with the LFD group, but there was no significant difference between the results for the SKL‐14959‐treated group and the HFD control group (Figure 2D–2F).
Figure 2.
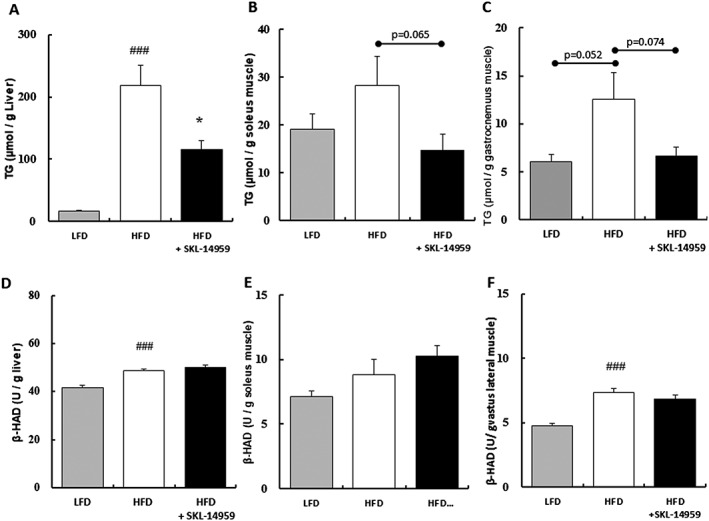
Effects of SKL‐14959 on triacylglycerol (TG) contents in the liver (A), soleus muscle (B), and gastronectin muscle (C), and β‐hydroxyacyl‐CoA dehydrogenase (β‐HAD) activity in the liver (D), soleus muscle (E) and vastus lateralis muscle (F). Data are means ± standard error (eight to nine mice per group). ### p < 0.001 vs. LFD group, *p < 0.05 vs. HFD control group (t‐test).
Histological analysis of the liver and the adipocytes
In histochemical analysis in the liver, the HFD control group showed significant higher intensity by oil‐red staining compared with the LFD group (p < 0.001), whereas the SKL‐14959‐treated group showed a significant decrease in the intensity compared with the HFD control group (p < 0.05, Figure 3A–3D). In the adipose tissues, the size of the epididymal adipocytes in the HFD control group was significantly larger than the LFD group (p < 0.001), but the sizes of the adipocytes were not significantly different between the SKL‐14959‐treated group and the HFD control group (Figure 3E–3H).
Figure 3.
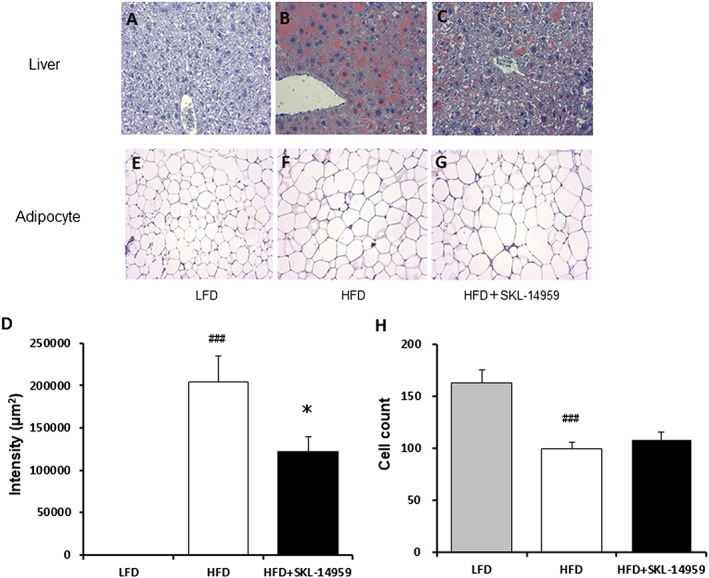
Histological analysis of SKL‐14959 treatment on lipid droplets in the liver (A–D) and on the size in the epididymal adipocytes (E–H) represented mice in each group (eight to nine mice per group). Data are means ± standard error (eight to nine mice per group). ### p < 0.001 vs. LFD group, *p < 0.05 vs. HFD control group (t‐test).
Relative gene expression using real‐time polymerase chain reaction
To investigate gene expression levels regulating lipogenesis and lipolysis in the liver and the adipose tissue, we performed gene analysis. In the liver, the HFD control group showed significant enhancement of relative gene expression of SREBP‐1 (Srebf1) and PPAR‐alpha (Ppara) compared with the LFD group (p < 0.05), whereas the SKL‐14959‐treated group showed significant down‐regulation of SREBP‐1 (Serbf1, p < 0.05), but not PPAR‐alpha (Ppara), compared with the HFD control group (Figure 4A). Moreover, the SKL‐14959‐treated group also indicated significant down‐regulation of the relative gene expression of DGAT‐1 (Dgat1, p < 0.001) in the final step of TG production, but the relative gene expression of FAS (Fasn) and ACC1 (Acaca) did not changed (Figure 4A). On the other hand, the relative gene expression in the adipose tissue did not significantly alter relative to the HFD control group (Figure 4B).
Figure 4.
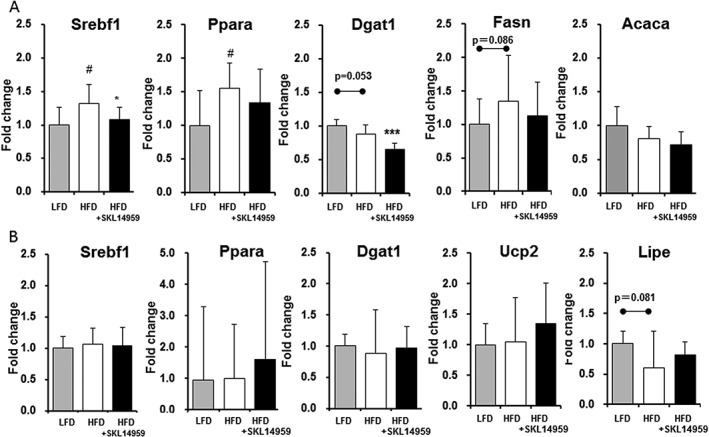
The relative gene expression of SREBP1 (Srebf1), PPAR‐alpha (Ppara), DGAT‐1 (Dgat1), FAS (Fasn) and ACC1 (Acaca) in the liver (A) and SREBP1 (Srebf1), PPAR‐alpha (Ppara), DGAT‐1 (Dgat1), UCP2 (Ucp2) and HSL (Lipe) in the adipose tissue (B) with regard to SKL‐14959 treatment. Values are expressed as means ± standard deviation (eight to nine mice per group). # p < 0.05 vs. LFD group (t‐test), *p < 0.05 vs. HFD control group (t‐test).
Lipid distribution after a single administration of [14C]‐oleic acid
To investigate the excursion of ingested lipids on SKL‐14959, [14C]‐oleic acid was acutely administered. SKL‐14959 significantly decreased the radioactivity in the adipose tissues at 120 min in contrast with control (p < 0.05), whereas it did not evidence a significant difference in the liver and the muscle (Figure 5).
Figure 5.
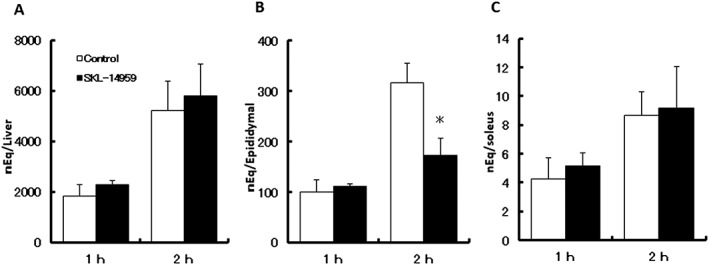
Effects of SKL‐14959 on the radioactivity in liver (A), epididymal adipose tissue (B) and soleus muscle (C) after oral [14C]‐oleic acid gavage. The 0.5% sodium carboxymethyl cellulose solution with or without SKL‐14959 (100 mg / kg body weight) was orally administered 15 min prior to the oil challenge. Values are expressed as means ± standard error (four to six mice per group). *p < 0.05 versus control group (t‐test).
Discussion
Weight loss have resulted from either suppression of energy intake or increase of the energy expenditure or both. From the viewpoint of energy consumption, the SKL‐14959‐treated group did not significantly enhance β‐HAD activity in the liver and the muscle (Figure 2D–2F) nor significantly up‐regulate the relative gene expression of PPAR‐alpha (ppara) in the liver and the adipose tissue, UCP‐2 (ucp2) and hormone sensitive lipase (Lipe) in the adipose tissue compared with the HFD control group (Figure 4A and 4B). It might be because GLP‐1 made up for the effect 31; alternatively, raised β‐HAD activity in the HFD control group compared with the LFD group was already reached maximal effectiveness, so that further combustible effects of SKL‐14959 were not observed 32. These results suggested that SKL‐14959 might not have direct action or have only a subtle effect of lipolysis.
From the viewpoint of energy intake, the processing of lipids anabolism using exogenous lipids, [14‐C]‐oleic acid, was evaluated, and the radioactivity increased time dependently until 6 h after gavage in the preliminary study (data not shown), suggesting that evaluated points in the present study could reflect the processing anabolic state. The SKL‐14959‐treated group indicated suppression of the radio activity in the adipose tissues but not in the liver and the muscle, contrast to the vehicle group 2 h after oil challenge (Figure 5). According to gene analysis of lipogenesis in the liver and the adipose tissue, the SKL‐14959‐treated group showed significant down‐regulation relative gene expression of SREBP1 (srebf1) and DGAT1 (dgat1) in the liver (Figure 4A) compared with the HFD control group but did not vary in the adipose tissue (Figure 4B). Moreover, lipid contents in the liver and muscle of the SKL‐14959‐treated group dropped to 45% and that level of the HFD control group, respectively (Figure 2). However, there is no report that GIP receptor expresses in liver and muscles in rodents 33, 34. Thus, that suggested that the suppression of body weight gain and lipid accumulation might be not direct, but indirect effect of GIP receptor signalling especially could be secondary to reduced insulin release 29. We also had reported that SKL‐14959 trended to decrease LPL activity in post‐heparin plasma 25. This enzyme is secreted by adipose and muscles, which is anchored to the surface of endothelial cell and hepatic sinusoids. Plasma LPL binding to extracellular proteoglycan has associated with lipid intake and the control of lipid metabolism in remote organs. NEFA is a triglyceride degradation product by lipase, which is incorporated into cell thought the transporter. Thus, these results suggested that SKL‐14959 inhibited the incorporation of fatty acid into the adipocytes, the liver and the muscle by suppressing LPL activity, and glucose utilization is likely to promote in the liver and the muscle by energy depletion.
Actually, the SKL‐14959‐treated group trended to decline the plasma ketone bodies level compared with the HFD control group (Table 1). Ketone bodies in blood are biomarker of energy source instead of diminished glucose utilities in diabetic hyperglycaemia 35, 36, which are high levels in individuals with diabetic hyperglycaemia 36 or in obese control relative to lean in DIO rat 37. Thus, this result suggested that SKL‐14959 might improve the glucose metabolism in peripheral tissues, resulted in decrease lipid deposition. Therefore, SKL‐14959 might indicate prominent effects in the liver and the muscle, not adipocyte, and might have the suppressive effect of ectopic lipid deposition in systemic body. It is possible that SKL‐14959 might also have the anti‐diabetic effects.
In histological analysis, the SKL‐14959‐treated group did not affect the size of adipocytes compared with the HFD control group (Figure 3). In the present study, this phenotype is consisted with the result that plasma concentrations of adiponectin secreted from the adipocytes in inverse proportion to its size. Okuno et al. reported 38 that troglitazone increased the number of small adipocytes in obese rat. These results suggested that SKL‐14959 did not possess the potency of adipocyte proliferation such as peroxide‐per gamma agonists.
There are some limitations in the present study. The evaluation period was short. To elucidate the effect of SKL‐14959 in the weight loss, glucose tolerance and insulin sensitivity, it would have done the long‐term study. Another one, the effect of neither normal nor low fat diet was evaluated. Furthermore, it remains undetermined which agent is superior to another because SKL‐14959 was not directly compared with sibutramine or lipase inhibitor in weight loss in the same study.
In the point of clinical implications, because the target of SKL‐14959 is not central anorexiants, it may be low risk of psychiatric behaviour. Additionally, this compound is an oral agent, and patients can avoid all the trouble from getting injection.
In conclusion, SKL‐14959, the GIP receptor antagonist, suppressed weight gain and lipids deposit in the liver and the muscle regardless of food intake and indirect action of lipolysis. Additionally, SKL‐14959 tended to improve diabetes status by raising glucose availability in peripheral tissue and acutely inhibited lipids intake into the adipocyte. Thus, SKL‐14959 has more effect on the inhibition of the lipid anabolism for calorie intake than on the enhancement of the lipid catabolism for energy expenditure that results in the long‐lasting reduction of adiposity and suppression of weight gain. Therefore, it is possible that SKL‐14959 may be a desirable new candidate for obesity and diabetic mellitus.
Conflict of interest statement
The authors declared no conflicts of interest.
Funding
This study is not receiving any funding at all. All authors have been employed at pharmaceutical company, Sanwa Kagaku Kenkyusho, Co.,LTD.
Author Contributions
H. Noda and Y. Tsubamoto conceived the study. H. Tanimoto, M. Okamoto and Y. Mizuno performed experiments. M. Takeuchi operated the gene analysis. We greatly appreciate the excellent technical assistance by Chika Ohtani and Mai Nampei. M. Okamoto reviewed the manuscript.
Nakamura, T. , Tanimoto, H. , Mizuno, Y. , Okamoto, M. , Takeuchi, M. , Tsubamoto, Y. , and Noda, H. (2018) Gastric inhibitory polypeptide receptor antagonist, SKL‐14959, suppressed body weight gain on diet‐induced obesity mice. Obesity Science & Practice, 4: 194–203. doi: 10.1002/osp4.164.
References
- 1. Paul Z, Alberti KGMM, Jonathan S. Global and societal implications of the diabetes epidemic. Nature 2001; 13: 782–787. [DOI] [PubMed] [Google Scholar]
- 2. Jeffrey MF. Obesity: causes and control of excess body fat. Nature 2009; 459: 340–342. [DOI] [PubMed] [Google Scholar]
- 3. Ashish KK, Neha D. Drug treatment of obesity: current status and future prospects. Eur J Intern Med 2015; 26: 89–94. [DOI] [PubMed] [Google Scholar]
- 4. Vidya N, Linda PD. Obesity: current and potential pharmacotherapeutics and targets. Pharmacol Ther 2017; 170: 116–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 1973; 37: 826–828. [DOI] [PubMed] [Google Scholar]
- 6. Pederson RA, Schubert HE, Brown JC. Gastric inhibitory polypeptide. Its physiologic release and insulinotropic action in the dog. Diabetes 1975; 24: 1050–1056. [DOI] [PubMed] [Google Scholar]
- 7. Beck B, Max JP. Hypersensitivity of adipose tissue to gastric inhibitory polypeptide action in the obese zucker rat. Cell Molec O Biologico 1987; 33: 555–562. [PubMed] [Google Scholar]
- 8. Oben J, Morgan L, Fletcher J, Marks V. Effect of the entero‐pancreatic hormones, gastric inhibitory polypeptide and glucagon‐like polypeptide‐1(7‐36) amide, on fatty acid synthesis in explants of rat adipose tissue. J Endocrinol 1991; 130: 267–272. [DOI] [PubMed] [Google Scholar]
- 9. Eckel RH, Fujimoto WY, Brnzell JD. Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocyte. Diabetes 1979; 28: 1141–1142. [DOI] [PubMed] [Google Scholar]
- 10. Knapper J, Puddicombe S, Morgan L, Fletcher J. Investigations into the actions of glucose‐dependent insulinotropic polypeptide and glucagon‐like peptide(1‐36)amide on Lipoprotein lipase activity in explants of rat adipose tissue. Nutrition 1995; 125: 183–188. [DOI] [PubMed] [Google Scholar]
- 11. Creutzfeldt W, Ebert R, Willims B, Frerichs H, Brown JC. Gastric inhibitory polypeptide (GIP) and insulin in obesity: increased response to stimulation and defective feedback control of serum levels. Diabetologia 1978; 14: 15–24. [DOI] [PubMed] [Google Scholar]
- 12. Ebert R, Frerichs H, Creutzfeldt W. Impaired feedback control of fat induced gastric inhibitory polypeptide (GIP) secretion by insulin obesity and glucose intolerance. Eur J Clin Investig 1979; 9: 129–135. [DOI] [PubMed] [Google Scholar]
- 13. Elahi D, Andersen DK, Muller DC, Tobin JD, Brown JC, Andres R. The enteric enhancement of glucose‐stimutated insulin release: the role of GIP in aging, obesity, and non‐insulin‐dependent diabetes mellitus. Diabetes 1984; 33: 950–957. [DOI] [PubMed] [Google Scholar]
- 14. Salera M, Giacomoni P, Pironi L, Cornia G, Capelli M, Marini A. Gastric inhibitory polypeptide release after oral glucose: relationship to glucose intolerance, diabetes mellitus and obesity. J Clin Endocrinol Metab 1982; 55: 329–336. [DOI] [PubMed] [Google Scholar]
- 15. Vilsvoll T, Krarup T, Sonne J. Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J Clin Endocrinol Metab 2003; 88: 2706–2713. [DOI] [PubMed] [Google Scholar]
- 16. Flatt PR, Cj B, Kwasowski P, Swanston‐Flatt SK, Marks V. Abnormalities of GIP in spontaneous syndromes of obesity and diabetes in mice. Diabetes 1983; 32: 433–435. [DOI] [PubMed] [Google Scholar]
- 17. Wasada T, McCorlkle K, Harris V, Kawai K. Effect of gastric inhibitory polypeptide on plasma levels of chylomicron triglycerides in dog. J Clin Invest 1981; 68: 1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ebert R, Nauck M, Creutzfeldt W. Effect of exogenous or endogenous gastric inhibitory polypeptide (GIP) on plasma triglyceride responses in rats. Horm Metab Res 1991; 23: 517–521. [DOI] [PubMed] [Google Scholar]
- 19. Miyawaki K, Yamada Y, Ban N, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002; 8: 738–742. [DOI] [PubMed] [Google Scholar]
- 20. Nasteska D, Harada N, Suzuki K, et al. Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high‐fat diet conditions. Diabets 2014; 63: 2332–2343. [DOI] [PubMed] [Google Scholar]
- 21. Gault VA, McClean PL, Cassidy RS, Irwin N, Flatt PR. Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high‐fat and cafeteria diets. Diabetologia 2007; 50: 1752–1762. [DOI] [PubMed] [Google Scholar]
- 22. Irwin N, McClean PL, O'Haete FPM, Gault VA, Harriott P, Flatt PR. Early administration of the glucose‐dependent insulinotropic polypeptide receptor antagonist (Pro3) GIP prevents the development of diabetes and related metabolic abnormalities associated with genetically inherited obesity in ob/ob mice. Diabetologia 2007; 50: 1532–1540. [DOI] [PubMed] [Google Scholar]
- 23. Brian F, Muller TD, Christoffer C, Perez‐Tilve D, DiMarchi RD, Tschop MH. Reappraisal of GIP pharmacology for metabolic diseases. Trends Mol Med 2016; 22: 359–376. [DOI] [PubMed] [Google Scholar]
- 24. Nakamura T, Tanimoto H, Mizuno Y, Tsubamoto Y, Noda H. Biological and functional characteristics of a novel low‐molecular‐weight antagonist of glucose‐dependent insulinotropic polypeptide receptor, SKL‐14959, in vitro and in vivo. Diabetes Metabol Obes 2012; 14: 511–517. [DOI] [PubMed] [Google Scholar]
- 25. Irwin N, Flatt PR. Evidence for beneficial effects of compromised gastric inhibitory polypeptide action in obesity‐related diabetes and possible therapeutic implications. Diabetologia 2009; 52: 1724–1731. [DOI] [PubMed] [Google Scholar]
- 26. Han L‐H, Takaku T, Li J, Kimura Y, Okuda H. Anti‐obesity action of oolong teas. Int J Obes 1999; 23: 98–105. [DOI] [PubMed] [Google Scholar]
- 27. Folch BJ, Lees M, Stanley GHS. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 1957; 226: 497–509. [PubMed] [Google Scholar]
- 28. Kato H, Ohue M, Kato K, et al. Mechanism of amelioration of insulin resistance by β3‐adrenoceptor agonist AJ‐9677 in the KK‐Ay/Ta diabetic obese mouse model. Diabetes 2001; 50: 113–122. [DOI] [PubMed] [Google Scholar]
- 29. Zhou H, Yamada Y, Tsukiyama K, et al. Gastric inhibitory polypeptide modulated adiposity and fat oxidation under diminished insulin action. Biochem Biophys Res Commun 2005; 335: 937–942. [DOI] [PubMed] [Google Scholar]
- 30. Kenneth JL, Thomas DS. Analysis of relative gene expression data using real‐time quantitative PCR and the 2‐d⊿⊿CT method. Methods 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 31. Nathalie P, Lynn FC, Buchan AMJ, et al. Glucose‐dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. Am J Physiol Endocrinol Metab 2003; 284: E931–E939. [DOI] [PubMed] [Google Scholar]
- 32. Negel T, Clinton RB, Susan MB, et al. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid‐induced insulin resistance in rodents. Diabetes 2007; 56: 2085–2092. [DOI] [PubMed] [Google Scholar]
- 33. G‐CYip R, Michael OB, Timothy JK, Michael W. Functional GIP receptors are present on adipocyte. Endocrynology 1998; 139: 4004–4007. [DOI] [PubMed] [Google Scholar]
- 34. Ted BU, Eva M, Donald CB, Michael JB, Tom IB. Gastric inhibitory polypeptide receptor, a member of the seretin‐vasoactive intestinal peptide receptor family, is wodely distribution in peripheral organs and the brain. Endocrinology 1993; 133: 2861–2870. [DOI] [PubMed] [Google Scholar]
- 35. Felts PW, Crofford OB, Park CR. Effect of infused ketone bodies on glucose utilization in the dog. J Clin Invest 1964; 43: 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuvaraj M, Jagadish V, Henna C, et al. Association of ketone body levels with hyperglycemia and type 2 diabetes in 9398 Finnish Men. Diabetes 2013; 62: 3618–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haung BW, Chiang MT, Yao HT, Chiang W. The effect of high‐fat and high fructose diets on glucose torelance and plasma lipid and leptin levels in rats. Diabetes Metab Obesity 2004; 6: 120–126. [DOI] [PubMed] [Google Scholar]
- 38. Okuno A, Tamemoto H, Tobe K, et al. Toroglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese zucker rats. J Clin Invest 1998; 101: 1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]