

Abstract
Continuous activation of hypoxia-inducible factor (HIF) is important for progression of renal cell carcinoma (RCC) and acquired resistance to anti-angiogenic multi-kinase and mTOR inhibitors. Recently, HIF2α antagonists PT2385 and PT2399 were developed and are being evaluated in a Phase I clinical trial for advanced or metastatic clear cell RCC (ccRCC). However, resistance to HIF2α antagonists would be expected to develop. In this study, we identified signals activated by HIF2α deficiency as candidate mediators of resistance to the multi-kinase inhibitor sunitinib. We established sunitinib-resistant tumor cells in vivo and created HIF2α-deficient variants of these cells using CRISPR/Cas9 technology. Mechanistic investigations revealed that a regulator of the serine biosynthesis pathway, phosphoglycerate dehydrogenase (PHGDH), was upregulated commonly in HIF2α-deficient tumor cells along with the serine biosynthesis pathway itself. Accordingly, treatment with a PHGDH inhibitor reduced the growth of HIF2α-deficient tumor cells in vivo and in vitro by inducing apoptosis. Our findings identify the serine biosynthesis pathway as a source of candidate therapeutic targets to eradicate advanced or metastatic ccRCC resistant to HIF2α antagonists.
Keywords: CRISPR/Cas9, HIF2α, serine biosynthesis, PHGDH, renal cell carcinoma
INTRODUCTION
In the last decade, molecularly targeted therapeutics, such as anti-angiogenic multityrosine kinase inhibitors (TKIs) or mammalian target of rapamycin (mTOR) inhibitors have been widely used for patients with metastatic or recurrent renal cell carcinoma (RCC) (1). However, these types of therapies are not expected to have curative effects because they extend progression-free survival only slightly due to acquisition of resistance to these drugs (2). Therefore, it is necessary to elucidate and overcome the molecular mechanisms of resistance to these drugs.
We previously reported that glucose transporter protein type 1 (GLUT1) and hexokinase-2 (HK2) (3,4), which function as key molecules in the glycolytic pathway, are upregulated in clear cell RCC (ccRCC) through suppression of microRNAs targeting GLUT1 or HK2. Similar to many other cancers, RCC is dependent on aerobic glycolysis for ATP production, a phenomenon known as the Warburg effect (5–7); therefore, glucose metabolism to lactate is increased in the presence of sufficient oxygen. However, dysregulation of metabolic pathways occurs when some genes are mutated, allowing cells to avoid the undesirable environment caused by chemotherapy or radiation (8). Accordingly, developing a deeper understanding of the mechanisms through which ccRCC acquires drug resistance will facilitate the development of novel treatment options for advanced ccRCC.
Hypoxia-inducible factor (HIF) is known to accelerate glycolysis, and continuous HIF activation was thought to be critical for RCC progression and acquired resistance to TKIs and mTOR inhibitors (9). Although HIF2α is thought to be undruggable, HIF2α antagonists (PT2399 and PT2385) are currently being developed (10,11). These HIF2α antagonists have been shown to have excellent inhibitory effects against tumours in vivo compared with sunitinib, a TKI and used as a standard first-line therapy for metastatic ccRCC (1). In addition, PT2399 has also been shown to lead to improved outcomes with regard to progression-free survival in patients with advanced or metastatic ccRCC who had prior TKIs (11). Although HIF2α antagonists have shown promising potency, ccRCCs requiring long treatment periods with HIF2α antagonists acquired resistance through HIF mutations that prevent PT2399 from entering the cavity or preserve HIF2 dimers due to structural changes (11). Therefore, improvement of current HIF2α antagonists may be necessary to overcome the effects of structural mutations and achieve better outcomes.
Accordingly, we established sunitinib-resistant 786-o (SU-R-786-o) ccRCC cells and knocked out HIF2α in this cell line (HIF2α-KO-SU-R-786-o cells) in order to elucidate the survival mechanism of HIF2α-deficient ccRCC cells. To identify key molecules within the cells which could still proliferate, we performed quantitative proteomics (12) and RNA sequencing. From this analysis, we identified phosphoglycerate dehydrogenase (PHGDH), a key enzyme involved in serine biosynthesis that functions to convert the glycolytic intermediate 3-phosphoglycerate (3-PG) to 3-phosphohydroxypyruvate (3-PHP). siRNA and two new PHGDH inhibitors (NCT-503 and CBR-5884) were used in HIF2α-KO-SU-R-786-o cells (13,14) in vitro or in vivo analyses. Additionally, metabolomics analysis was performed in HIF2α-KO-SU-R-786-o cells.
MATERIALS AND METHODS
Cell culture and establishment of SU-R-786-o cells
Human RCC cell lines 786-o, A498, ACHN, Caki1, and Caki2, were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). 786-o, A498, Caki1, and Caki2 cell lines were acquired in 2010, and ACHN was acquired in 2017. They were maintained in culture for no more than 30 continuous passages. ATCC authenticated cell lines with short tandem repeat profiling. All cell lines were tested and found negative for mycoplasma (e-Myco Mycoplasma PCR Detection Kit; iNtRON, Korea). These cell lines were incubated as previously described (15). Gavage feeding of sunitinib (40 or 25 mg/kg, five times a week; biorbyt, CA, USA) was performed in mice. The tumour sizes and weights were measured and calculated as previously described (15). The chemical structures of sunitinib is in Supplementary Figure 1A.
CRISPR-Cas9 mediated gene knockout and lentivirus-mediated gene expression
We used CRISPR-Cas9-mediated genome editing to achieve gene knockout with lentiCRISPR v2 (Addgene Plasmid #52961), which was a kind gift from Dr. Feng Zhang. The sgRNAs targeting HIF2α were designed as previously described (15). The sgRNA sequence, 5′-GCTGATTGCCAGTCGCATGA-3′ or 5′-CAAGGCCTCCATCATGCGAC-3′, which targeted the exon of HIF2α, was cloned into lentiCRISPR v2. For PHGDH overexpression in cells, we used pLJM5-WT PHGDH, which was a gift from Dr. David Sabatini (Addgene plasmid #83901) (14). We produce lentivirus as previously described (15).
Cell proliferation, apoptosis, and colony formation assays
CBR-5884 (Focus Biomolecules, Plymouth Meeting, PA, USA) and NCT-503 (Aobious, Gloucester, MA, USA) were used as PHGDH inhibitors (13,14). The chemical structures of these inhibitors are in Supplementary Figure 1B and 1C. Cell proliferation was determined with XTT assays (Roche Applied Science, Tokyo, Japan) according to the manufacturer’s instructions. Cell apoptosis assays were carried out by flow cytometry (CytoFLEX analyzer; Beckman Coulter, Brea, CA, USA) using a FITC Annexin V Apoptosis Detection Kit (BD Biosciences, Bedford, MA, USA) according to the manufacturer’s recommendations. As a positive control, we used 5 μg/mL cycloheximide (Sigma, St. Louis, MO, USA) in apoptosis assay. We performed colony formation assay as previously described (15).
In vivo tumour xenograft model
A mixture containing 100 μL HIF2α-KO-SU-R-786-o cells (2 × 106 cells) and 100 μL Matrigel Matrix (Corning, Bedford, MA, USA) was injected subcutaneously into the flanks of female nude mice (BALB/c nu/nu, 6- to 8-weeks-old). NCT-503 was prepared in a vehicle of 5% ethanol, 35% PEG 300 (Sigma), and 60% of an aqueous 30% hydroxypropyl-β-cyclodextrin (Sigma) solution and injected at 40 mg/kg intraperitoneally once daily, beginning the day after tumour injection. The dose was adjusted according to the weight of each mouse, and the volume of injection did not exceed 150 μL. All the animal experiments were approved by the animal care review board of Kagoshima University.
RNA extraction and quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was isolated using Isogen (Nippon Gene, Tokyo, Japan) according to the manufacturer’s protocol. We applied a SYBR-green quantitative PCR-based array approach as previously described (15), and the following primers were used: HIF2α, forward primer, 5′-CGGAGGTGTTCTATGAGCTGG-3′ and reverse primer, 5′-AGCTTGTGTGTTCGCAGGAA-3′; and GUSB, forward primer, 5′-CGTCCCACCTAGAATCTGCT-3′ and reverse primer, 5′-TTGCTCACAAAGGTCACAGG-3′.
RNA sequencing and quantitative proteomics analyses
RNA sequencing was performed by Eurofins Japan. mRNA profiles were generated by single-read deep sequencing using Illumina HiSeq 2500/2000. In vitro proteome-assisted multiple reaction monitoring for protein absolute quantification (iMPAQT) analysis was performed at Kyusyu University in Japan (12).
Metabolite analysis
Metabolome analysis was performed at Human Metabolome Technologies (HMT, Tsuruoka, Japan, http://humanmetabolome.com). Cellular metabolites were extracted according to the manufacturer’s protocol. Metabolome analysis was performed by capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS). Metabolite peaks were quantified and normalised according to protein concentrations.
Immunoassays
Immunoblotting was carried out as previously described (15) with diluted (1:1000) anti-PHGDH antibodies (HPA021241; Sigma), anti-HIF2α antibodies (ab51608; Abcam, Cambridge, MA, USA), anti-glyceraldehyde 3-phosphate dehydrogenase antibodies (GAPDH; MAB374, EMD Millipore, Billerica, MA), and anti-β-actin antibodies (bs-0061R; Bioss, Woburn, MA, USA). Immunohistochemistry were performed using an UltraVision Detection System (Thermo Scientific, Fremont, CA, USA) according to the manufacturer’s instructions. The primary rabbit monoclonal antibodies against Ki67 (ab92742; Abcam) were diluted 1:100. For immunofluorescence analyses, nuclei were stained with DAPI (1 μg/mL; Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA), and slides were mounted in Fluoromount (Diagnostic Biosystems, Pleasanton, CA, USA). Anti-PHGDH antibodies (HPA021241; Sigma) were used as the primary antibody at a dilution of 1:100, and binding was visualised using secondary antibodies conjugated to Alexa Fluor 488 (ab150077; Abcam).
Bioinformatics analysis
The Cancer Genome Atlas (TCGA) cohort database for 534 patients with ccRCC (KIRC) was used for analysis of clinical relevance (16–19). Gene set enrichment analysis (GSEA) was performed to identify enriched pathways using open source software v2.0 (www.broad.mit.edu).
Statistical analysis
Statistical analysis was carried out as previously described (15).
RESULTS
Establishment of sunitinib-resistant ccRCC cells
We injected 786-o ccRCC cells into mice subcutaneously and started sunitinib treatment after tumour formation to establish sunitinib-resistant ccRCC cells (Fig. 1A, left). After tumours acquired resistance to sunitinib, tumours were extracted and harvested. Tumours showing HIF2α expression were selected for subsequent experiments as SU-R-786-o cells (Fig. 1A, right). We confirmed that SU-R-786-o cells showed resistant to sunitinib compared with parental cells in cell proliferation and xenograft assays (Fig. 1B and 1C).
Figure 1. Establishment of sunitinib resistant ccRCC cells.
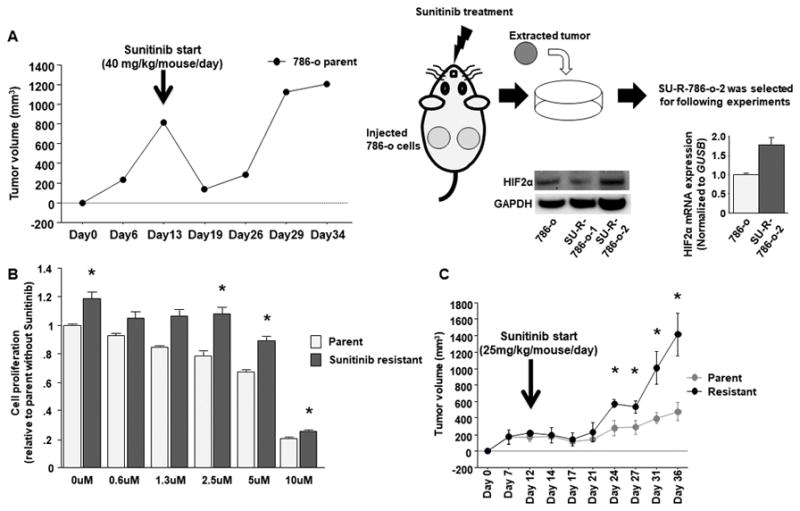
A, Time course of tumor volumes in nude mice after subcutaneous injection of parental 786-o cells which was used to acquire resistance to sunitinib out of 6 parts from 3 mice treated by sunitinib (40mg/kg/mouse/day) (left). Schema of the way to establish sunitinib resistant ccRCC cells (right). B, Cell proliferation assay between parental and SU-R-786-o cells with or without sunitinib (* P < 0.05). C, Time course of comparison between tumor volumes of parental 786-o and SU-R-786-o cells (n = 3 for each group) in nude mice after subcutaneous injection under sunitinib treatment (25mg/kg/mouse/day) (* P < 0.05). On Day 36, average volume of sunitinib resistant tumors became nearly 3 times as large as its parental cells (Parent vs Resistant: 480.9 cm3 vs 1412.2 cm3).
HIF2α knockout by CRISPR/Cas9 in SU-R-786-o cells
HIF upregulation was postulated to be associated with resistance to inhibitors of the vascular endothelial growth factor (VEGF) and mTOR pathways (9). Therefore, we next attempted to knock out HIF2α using the CRISPR/Cas9 system in SU-R-786-o cells, because 786-o parent cell depends more on HIF2α than on HIF1α (10). The expression of HIF2α in HIF2α-KO-SU-R-786-o cells was confirmed by immunoblotting analysis (Fig. 2A). Known HIF target genes were downregulated in HIF2α-KO-SU-R-786-o cells, and most upregulated genes in 786-o treated with a HIF2α antagonist in a previous paper (10) were actually upregulated in HIF2α-KO-SU-R-786-o cells, indicating that HIF2α-KO-SU-R-786-o cells mimicked treatment with the HIF2α antagonist (Fig. 2B). Surprisingly, HIF2α-KO-SU-R-786-o cells showed dramatic morphological changes from a spindle shape to a round cell shape, whereas no distinct changes were observed in SU-R-786-o cells transduced by empty vector (Fig. 2C). Cell proliferation was significantly decreased in HIF2α-KO-SU-R-786-o cells compared with that in control cells (Fig. 2D).
Figure 2. Establishment of HIF2α knock out SU-R-786-o cells.

A, Immunoblotting analysis showed that HIF2α was significantly depleted in sunitinib resistant 786-o cells. B, RSEM values of HIF targets genes from RNA-seq expression data between 786-o empty and HIF2α-KO-SU-R-786-o cells. C, Representative images of HIF2α-KO-SU-R-786-o cells. D, Cell proliferation assay between control and HIF2α-KO-SU-R-786-o (* P < 0.0001).
Identification of PHGDH in the serine biosynthesis pathway in HIF2α-KO-SU-R-786-o cells
RNA sequencing with 786-o parent, SU-R-786-o, empty-SU-R-786-o, and HIF2α-KO-SU-R-786-o cells showed 11 commonly upregulated genes in SU-R-786-o and HIF2α-KO-SU-R-786-o cells (Fig. 3A and 3B). Quantitative proteomics analyses with 786-o parent, SU-R-786-o, and HIF2α-KO-SU-R-786-o cells showed 141 commonly upregulated genes in SU-R-786-o and HIF2α-KO-SU-R-786-o cells (Fig. 3A and 3C). From the combined RNA sequencing and proteomics analysis, we identified PHGDH (Fig. 3A). The RNA sequencing and proteomics data were approved by the DNA Data Bank of Japan (DDBJ) with accession number DRA006007, and Japan Proteome Standard Repository/Database (jPOST) with accession number PXD007222, respectively. We also performed pathway analyses with the 141 upregulated genes identified in proteomics analysis; GSEA, GO-BP, and KEGG identified the serine biosynthesis pathway as a significantly enriched pathway (Fig. 3D and Suppl. Fig. 2).
Figure 3. RNA sequence and quantitative proteomics analyses identified key molecule and pathway for HIF2α-KO-SU-R-786-o cells.
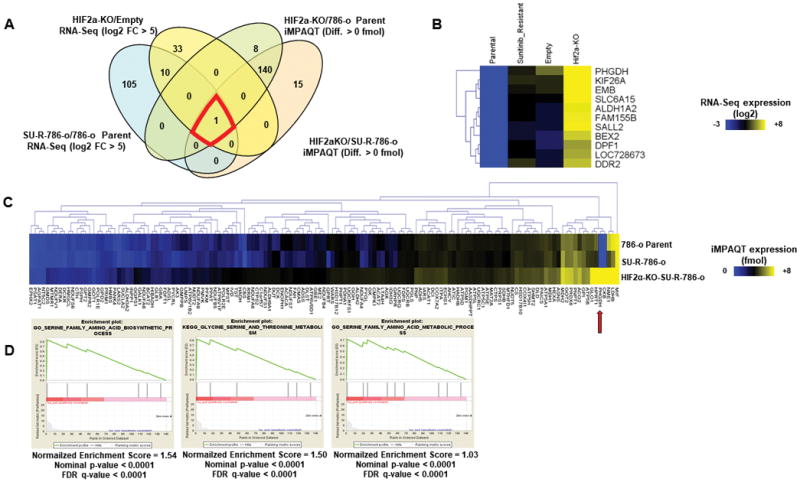
A, Venn diagram between RNA sequence and proteomics analyses indicated PHGDG as key molecule for HIF2α-KO-SU-R-786-o cells. B, Heat map of 11 common genes from RNA-seq. C, heat map of 141 genes from proteomics analysis. D, GSEA indicating serine biosynthesis and metabolism as significantly enriched pathways.
Effects of PHGDH inhibition in HIF2α-KO-SU-R-786-o cells
Immunoblotting analyses showed that PHGDH was dramatically elevated in HIF2α-KO-SU-R-786-o cells (Fig. 4A). Cell proliferation was inhibited by si-PHGDH transfection in HIF2α-KO-SU-R-786-o cells compared to empty cells (Fig. 4B). We also used two novel PHGDH inhibitors, CBR-5884 and NCT-503, for cell proliferation assays, and found that these compounds showed potent inhibitory effects in HIF2α-KO-SU-R-786-o cells (Fig. 4C and Suppl. Fig. 3A). In addition, CBR-5884 significantly induced apoptosis in HIF2α-KO-SU-R-786-o cells (Fig. 4D). We also found its apoptotic effect in ccRCC cell lines, such as A498, ACHN, and Caki1 cells (Suppl. Fig. 3B). We then performed xenograft assays with NCT-503 because CBR-5884 was unstable in mouse plasma, whereas NCT-503 had been used successfully in prior xenograft assays (13,14). We found that tumour growth was significantly suppressed in the mice treated with NCT-503 compared with that in vehicle-treated mice (Fig. 4E). Immunohistochemistry with Ki67 antibodies indicated that the administration of NCT-503 significantly reduced cell proliferation in HIF2α-KO-SU-R-786-o cells in vivo, relative to vehicles (Fig. 4F).
Figure 4. PHGDH inhibition by si-RNA and inhibitor.

A, Immunoblotting analysis showed that PHGDH expression was dramatically elevated in HIF2α-KO-SU-R-786-o cells. B, Cell proliferation assay by PHGDH si-RNA. C, Cell proliferation assay by PHGDH inhibitor (CBR-5884). (* P < 0.0167). D, Apoptosis assay by CBR-5884 (* P < 0.0167). E, Time course of tumor volumes in nude mice treated by vehicle or NCT-503 after subcutaneous injection of HIF2α-KO-SU-R-786-o cells (* P < 0.01) (n = 5 for each group). F, Ki67 positive cells were calculated from five independent tumor sections per group and expressed as the mean ± SD Immunohistochemical staining of in the harvested tumors (* P = 0.0209).
PHGDH overexpression in ccRCC cells
We overexpressed PHGDH in 786-o and A498 parental cells (Fig. 5A). As observed following HIF2α knockout in SU-R-786-o cells, 786-o and A498 cells that showed PHGDH strong overexpression exhibited a change in morphology from a spindle shape to a round cell shape (Fig. 5B). Using immunofluorescence analyses, 786-o cells that showed weak PHGDH overexpression did not show altered cell morphology and still maintained a spindle shape (Fig. 5C, left upper, white arrow, and right). However, 786-o and A498 cells showing strong expression of PHGDH exhibited a homogeneous change in shape to round cells (Fig. 5C). The number of colonies were significantly increased in PHGDH overexpressed 786-o or A498 cells compared to parental cells (Fig. 5D).
Figure 5. PHGDH over expression in 786-o and A498 parental cells.

A, Immunoblotting analysis showed that PHGDH expression was dramatically elevated in 786-o and A498 cells. B, Representative images of parental and PHGDH overexpressed 786-o or A498 cells. C, Immunofluorescence analysis; PHGDH overexpressed 786-o or A498 cells showed morphological changes from a spindle to a round cell shape. The graph showed the ratio between spindle and round cells in PHGDH overexpressed 786-o or A498 cells. D, Representative image of colony formation in parental and PHGDH overexpressed 786-o or A498 cells. The graph showed the ratio of number of colonies between parental and PHGDH overexpressed cells (*P < 0.05).
PHGDH gene amplification was correlated with poor overall survival in ccRCC
According to TCGA database, patients with PHGDH gene amplification (n = 24) had poor overall survival and disease-free survival in comparison to patients without amplification (n = 500; P = 0.0003 and 0.0329, respectively; Fig. 6A). A multivariate Cox proportional hazards model also showed that PHGDH gene amplification was an independent predictor of overall survival (Fig. 6B and Supplementary Table 1). We also analysed relationship between PHGDH copy number alteration and Von Hippel–Lindau (VHL) mutation, HIF1A, or HIF2A. There was no significant relationship between PHGDH copy number alteration and VHL mutation (P = 0.491), or HIF1A expression (P = 0.0649) (Suppl. Fig. 4A and 4B). However, there was a significant negative correlation between PHGDH copy number alteration and EPAS1 (HIF2A) expression (P = 0.0062) (Suppl. Fig. 4B).
Figure 6. Kaplan-Meier survival plots for PHGDH gene amplification groups in a TCGA cohort.
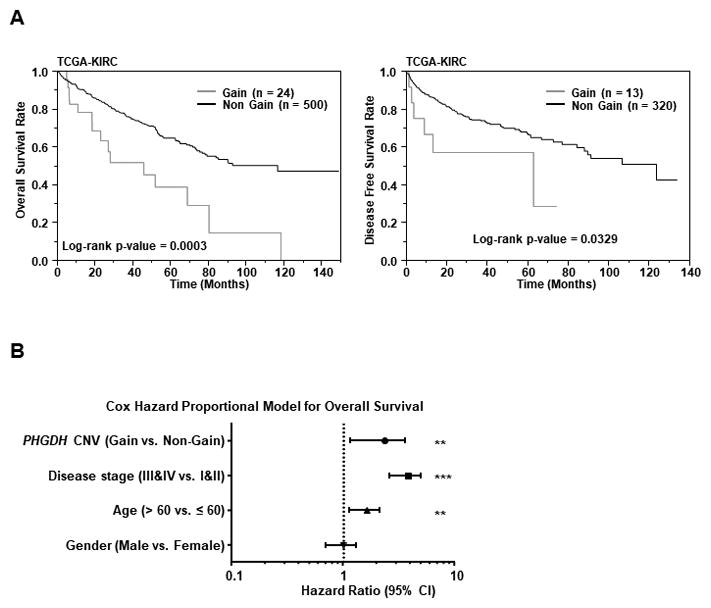
A, Overall survival (left) and Disease-free survival periods (right) were significantly reduced in the patients with PHGDH gene amplification in comparison to patients without amplification (P =0.0003 and P = 0.0329 respectively). B, Cox proportional analysis for the prediction of overall survival, and summary of the analysis.
Metabolite analysis in 786-o parent, SU-R-786-o, and HIF2α-KO-SU-R-786-o cells
As shown in the heat map (Fig. 7A), intracellular metabolites in 786-o parent, SU-R-786-o, and HIF2α-KO-SU-R-786-o cells were clearly altered among the 3 cell lines. Moreover, serine biosynthesis, including 3-phosphoserine (3P Ser), serine, and glycine, accumulated in HIF2α-KO-SU-R-786-o cells compared with those in 786-o parent and SU-R-786-o cells (Fig. 7B). Interestingly, the tricarboxylic acid (TCA) cycle was also activated in HIF2α-KO-SU-R-786-o and SU-R-786-o cells compared with that in 786-o parent cells (Suppl. Fig. 5 and Supplementary Table 2).
Figure 7. Serine biosynthesis pathway was up-regulates in HIF2α-KO-SU-R-786-o cells.

A, Heat map diagram of metabolites among 786-o parent cells, SU-R-786-o cells, and HIF2α-KO-SU-R-786-o cells. Precise information of all metabolites are listed in Supplementary Table 2. B, Intracellular concentration of key metabolites involved in glycolysis and serine biosynthesis pathway. Representative metabolites such as glucose 6-phosphate (G6P), 3-phosphoglycerate (3PG), 2-phosphoglycerate (2PG), pyruvate, lactate, 6-phosphogluconate (6PG), 3-Phosphoserine (3P Ser), Serine (Ser), Glycine (Gly), and Acetyl-CoA (AcCoA) are shown here. Others are listed in Supplementary Table 2.
DISCUSSION
In the absence of oxygen or the presence of a mutated VHL gene, HIF1α and HIF2α are stabilised and induce the expression of various transcriptional target genes, such as VEGF, PDGF, and TGF-α, to facilitate angiogenesis, migration, and proliferation through the development of an aggressive phenotype and resistance to chemotherapy and radiation therapy, thereby supporting the metabolic shift that underlies RCC tumourigenicity (8,20). While current targeted therapies, including sorafenib, sunitinib, bevacizumab, pazopanib, and axitinib, have targeted the downstream effects of HIF activity, the HIF complex was previously considered “undruggable” (21). In pVHL-deficient ccRCCs, not HIF1α but HIF2α displayed enhancing proliferation and resistance to replication stress through c-Myc activation (22). Recently, two studies showed that HIF2α could be targeted by novel inhibitors (10,11). Chen et al showed that the novel HIF2α inhibitor PT2399 suppressed tumor growth by 60% across patient-derived ccRCC xenograft (PDX) in 56% of PDX lines; the mechanism of action was shown to require interruption of heterodimer complex with HIF1β and prevention of gene transcription (11). In addition, they identified HIF mutations that prevented PT2399 from entering the cavity or preserved HIF-2 dimers due to structural changes, thereby contributing to the resistance mechanism. They also referred to the development of second-generation inhibitors and/or complementary approaches, including other potential drug-binding pocket inhibitors (23). Therefore, our results from HIF2α-knockout cells were different from their findings of resistance to PT2399 and may indicate the involvement of a more fundamental mechanism, e.g., PHGDH-based serine production.
Many studies have shown that the serine biosynthesis pathway plays a critical role in various cancers (13,14,24–29). PHGDH, the first enzyme in serine biosynthesis from glycolysis, is amplified or overexpressed in various types of cancers (28,29). In a ccRCC cohort from TCGA database, patients with PHGDH gene amplification had poor overall survival and disease-free survival in comparison to patients without amplification; therefore, PHGDH could be targeted not only for patients showing resistance to HIF2α antagonists but also for patients showing PHGDH gene amplification. TCGA database also showed no significant relationship of PHGDH copy number alteration with VHL mutation, or HIF1A expression but a significant negative correlation of it with EPAS1 (HIF2A) expression. These data may support our finding that expression of PHGDH was revived in HIF2α-knockout cells, on the other hand they might indicate that only PHGDH copy number alteration cannot explain the mechanism underlying RCC develop in the presence of wild type VHL. Recently, PHGDH inhibitors (NCT-503 and CBR-5884) were developed by different laboratories (13,14). Although NCT-503 has only been applied for in vivo experiments because of the instability of CBR-5884 in mouse plasma, both inhibitors showed cell inhibitory effects in this study. Therefore, clinical trials with these inhibitors or next-generation PHGDH inhibitors are needed to improve cancer treatment options in the near future. Moreover, because we did not investigate the mechanisms of serine involvement in this paper, further studies are also necessary to improve our understanding of the underlying pathways of cancer metabolism.
Cancer cells have a characteristic metabolic phenotype, termed aerobic glycolysis or the Warburg effect, whereby there is increased metabolism of glucose to lactate in the presence of sufficient oxygen (5,7). Additionally, metabolic pathways can be altered to avoid undesirable conditions thorough acquisition of mutations in ccRCCs (8). Notably, in our study, endogenous metabolites were clearly altered among 786-o parent, SU-R-786-o, and HIF2α-KO-SU-R-786-o cells. TCA cycle, which includes succinic acid, fumaric acid, and D-2-hydroxyglutarate for control of energy production, biosynthesis, and the redox state, was particularly activated in SU-R-786-o cells compared with 786-o parent cells. Additionally, metabolites shifted to serine and glycine biosynthesis in HIF2α-deficient SU-R-786-o cells. Because metabolite alterations among these cells are a response to escape from undesirable conditions and promote survival, drugs targeting each induced metabolism may be efficacious. Therefore, HIF inhibitors, which suppress glycolysis through GLUT-1, HK2, pyruvate kinase muscle 2 (PKM2) repression (30), may be useful in patients showing resistance to sunitinib, whereas PHGDH inhibitors, which suppress serine biosynthesis, may be useful in patients showing resistance to HIF2α antagonists.
In conclusion, we demonstrated that there was a diverse range of metabolites in ccRCC and that activation of serine biosynthesis could be targeted by inhibition of PHGDH.
Supplementary Material
Acknowledgments
Financial support
This study was supported by the KAKENHI (KIBAN-B) 16H05464 (M. Nakagawa) and 17H04332 (H. Enokida), KAKENHI (KIBAN-C) 16K11015 (H. Yoshino) and 17K11148 (T. Itesako), KAKENHI (WAKATE-B) 17K16799 (M. Yonemori), and the Department of Veterans Affairs VA Merit Review 101BX001123 (R. Dahiya), and NIH RO1CA199694 (R. Dahiya).
We thank Mutsumi Miyazaki for excellent laboratory assistance. This study was supported by the KAKENHI (KIBAN-B) 16H05464 (M. Nakagawa) and 17H04332 (H. Enokida), KAKENHI (KIBAN-C) 16K11015 (H. Yoshino) and 17K11148 (T. Itesako), KAKENHI (WAKATE-B) 17K16799 (M. Yonemori), and the Department of Veterans Affairs VA Merit Review 101BX001123 (R. Dahiya), and NIH RO1CA199694 (R. Dahiya).
Footnotes
DISCLOSURE STATEMENT
The authors declare no conflicts of interest.
References
- 1.Garcia-Donas J, Leandro-Garcia LJ, Gonzalez Del Alba A, Morente M, Alemany I, Esteban E, et al. Prospective study assessing hypoxia-related proteins as markers for the outcome of treatment with sunitinib in advanced clear-cell renal cell carcinoma. Ann Oncol. 2013;24:2409–14. doi: 10.1093/annonc/mdt219. [DOI] [PubMed] [Google Scholar]
- 2.Margulis V, Master VA, Cost NG, Leibovich BC, Joniau S, Kuczyk M, et al. International consultation on urologic diseases and the European Association of Urology international consultation on locally advanced renal cell carcinoma. European urology. 2011;60:673–83. doi: 10.1016/j.eururo.2011.06.042. [DOI] [PubMed] [Google Scholar]
- 3.Yamasaki T, Seki N, Yoshino H, Itesako T, Yamada Y, Tatarano S, et al. Tumor-suppressive microRNA-1291 directly regulates glucose transporter 1 in renal cell carcinoma. Cancer science. 2013;104:1411–9. doi: 10.1111/cas.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshino H, Enokida H, Itesako T, Kojima S, Kinoshita T, Tatarano S, et al. Tumor-suppressive microRNA-143/145 cluster targets hexokinase-2 in renal cell carcinoma. Cancer science. 2013;104:1567–74. doi: 10.1111/cas.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY) 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warburg O. On the origin of cancer cells. Science (New York, NY) 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 7.Gogvadze V, Zhivotovsky B, Orrenius S. The Warburg effect and mitochondrial stability in cancer cells. Molecular aspects of medicine. 2010;31:60–74. doi: 10.1016/j.mam.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature reviews Urology. 2010;7:277–85. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. The Lancet Oncology. 2009;10:992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
- 10.Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature. 2016;539:107–11. doi: 10.1038/nature19795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting Renal Cell Carcinoma with a HIF-2 antagonist. Nature. 2016;539:112–7. doi: 10.1038/nature19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsumoto M, Matsuzaki F, Oshikawa K, Goshima N, Mori M, Kawamura Y, et al. A large-scale targeted proteomics assay resource based on an in vitro human proteome. Nature methods. 2017;14:251–58. doi: 10.1038/nmeth.4116. [DOI] [PubMed] [Google Scholar]
- 13.Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E1585. doi: 10.1073/pnas.1521548113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452–8. doi: 10.1038/nchembio.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshino H, Yonemori M, Miyamoto K, Tatarano S, Kofuji S, Nohata N, et al. microRNA-210-3p depletion by CRISPR/Cas9 promoted tumorigenesis through revival of TWIST1 in renal cell carcinoma. Oncotarget. 2017;8:20881–94. doi: 10.18632/oncotarget.14930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nature reviews Cancer. 2002;2:673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 21.Koehler AN. A complex task? Direct modulation of transcription factors with small molecules. Current opinion in chemical biology. 2010;14:331–40. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer cell. 2008;14:435–46. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu D, Potluri N, Lu J, Kim Y, Rastinejad F. Structural integration in hypoxia-inducible factors. Nature. 2015;524:303–8. doi: 10.1038/nature14883. [DOI] [PubMed] [Google Scholar]
- 24.Ou Y, Wang SJ, Jiang L, Zheng B, Gu W. p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. The Journal of biological chemistry. 2015;290:457–66. doi: 10.1074/jbc.M114.616359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frezza C. Cancer metabolism: Addicted to serine. Nat Chem Biol. 2016;12:389–90. doi: 10.1038/nchembio.2086. [DOI] [PubMed] [Google Scholar]
- 26.Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer discovery. 2014;4:1406–17. doi: 10.1158/2159-8290.CD-14-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell reports. 2014;7:1248–58. doi: 10.1016/j.celrep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 28.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–50. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature genetics. 2011;43:869–74. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–44. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.