

Abstract
Previous studies demonstrated that selenium in the form of sodium selenate reduces neurofibrillary tangle formation in Alzheimer’s disease models. Hyperphosphorylation of tau, which leads to formation of neurofibrillary tangles in Alzheimer’s disease, is increased by endoplasmic reticulum (ER) stress. Selenoprotein S (SelS) is part of an ER membrane complex that removes misfolded proteins from the ER as a means to reduce ER stress. Selenate, as with other forms of selenium, will increase selenoprotein expression. We therefore proposed that increased SelS expression by selenate would contribute to the beneficial actions of selenate in Alzheimer’s disease. SelS expression increased with ER stress and decreased under conditions of elevated glucose concentrations in the SH-SY5Y neuronal cell line. Reducing expression of SelS with siRNA promoted cell death in response to ER stress. Selenate increased SelS expression, which significantly correlated with decreased tau phosphorylation. Restricting SelS expression during ER stress conditions increased tau phosphorylation, and also promoted aggregation of phosphorylated tau in neurites and soma. In human postmortem brain, SelS expression coincided with neurofibrillary tangles, but not with amyloid-β plaques. These results indicate that selenate can alter phosphorylation of tau by increasing expression of SelS in Alzheimer’s disease and potentially other neurodegenerative disorders.
Keywords: Alzheimer’s disease, endoplasmic reticulum stress, neurofibrillary tangle, selenium, selenoprotein, tau
INTRODUCTION
According to recent reports, selenium in the form of sodium selenate reduces neurofibrillary tangle formation [1, 2]. The studies suggest selenate acts as an agonist for protein phosphatase 2A (PP2A), which targets tau phosphorylation. However, selenate supplementation also upregulates expression of selenoproteins [3–5]. These proteins contain selenocysteine (Sec), the 21st amino acid [6]. Selenate is reduced and converted to selenophosphate, and utilized for Sec synthesis directly on the Sec tRNA [7]. Selenoproteins have important roles in oxidative and endoplasmic reticulum (ER) stress pathways that could potentially influence tau hyperphosphorylation [8, 9]. Selenium deficiency is associated with developmental and neurological disorders [9], highlighting the crucial antioxidant functions of selenium.
Selenoprotein S is a glucose-regulated ER-bound selenoprotein involved in inflammation and removal of misfolded proteins from the ER [8]. SelS is also regulated by inflammatory cytokines, and by ischemic conditions in brain astrocytes [10, 11]. Genetic analysis demonstrated that a promoter single-nucleotide polymorphism that decreases SelS expression in humans is correlated with higher serum levels of inflammatory cytokines, suggesting an important role for SelS in regulating inflammation [12]. SelS resides in the ER and links derlin-1, a shuttle protein that removes misfolded proteins from the ER to be ubiquitinated and transported by the p97 ATPase for proteasomal degradation [13]. Thus, impairment of SelS may play a role in disorders involving misfolded proteins, which include Alzheimer’s disease and other neurodegenerative disorders.
In this study, we examine the function of SelS in ER stress in neuronal cells, its regulation by selenate and role in tau phosphorylation, and expression in postmortem human Alzheimer’s disease brain. We report that SelS is associated with neurofibrillary tangles and regulates phosphorylation of tau in response to ER stress.
MATERIALS AND METHODS
SH-SY5Y culture conditions
SH-SY5Y cells were maintained in DMEM with 10% FBS. For experiments, cells were plated in multiwell plates and differentiated by exposure to Neurobasal media (Invitrogen) supplemented with B27 (Invitrogen) for 48 h. For selenate treatment, cells were grown in Roth-Schweitzer media [14] with 0, 0.1, 1.0, or 10 μM sodium selenate in place of sodium selenite.
ER stress
Cells were differentiated for 48 h in Neurobasal-A media (Invitrogen) supplemented with B27 (Invitrogen), and then treated for 20 h with thapsigargin (1 μM), brefeldin A (1 μM), or tunicamycin (100 nM). Thapsigargin induces stress by depleting the ER calcium stores [15]. Brefeldin A prevents anterograde transport of proteins from the ER to the Golgi [16]. Tunicamycin prevents the N-glycosylation of glycoproteins necessary for their transport out of the ER, inducing the misfolded protein response [17]. These organic compounds were solubilized in DMSO to make 10,000X stock concentrations. DMSO (0.01%) was used as a vehicle control for studies with these compounds. Following chemical induction of ER stress, cells were stained or harvested for mRNA or protein as described below.
Variation of glucose in culture media
SH-SY5Y cells were maintained in low (5 mM) glucose DMEM (Invitrogen) with 10% FBS (Invitrogen). Cells were then plated onto 35 mM dishes, and media was changed after 1 day to glucose-free Neurobasal-A media supplemented with varying levels of glucose (5 mM, 25 mM, or 50 mM). Mannitol was added at 45 mM, 20 mM, or 0 mM to maintain the same osmolarity at all glucose concentrations.
SelS siRNA
SH-SY5Y cells grown in DMEM with 5% FBS were transfected with SelS (5′-ACGGAAAUCGGACAGAAAGUU-3′) or negative control siRNA (Thermo Scientific) by introducing with Lipofectamine 2000 transfection reagent (Life Technologies) for 4 h. Media was then changed to Neurobasal media with B27 supplement, followed by an additional transfection after a 48-h differentiation. ER stress was induced 4 h after the second transfection. After 24 h, cell viability was assayed with an MTS assay kit (Promega) according to manufacturer’s instructions, or cells were harvested for protein or mRNA studies.
Stable transfection with P301L tau
SH-SY5Y cells were transfected with a pCDNA3.1 vector (Invitrogen) containing a Tau clone with the P301L mutation [18] using Lipofectamine (Invitrogen) per manufacturer’s instructions. DMEM with 10% FBS was supplemented with G418 (1 mg/ml) to select for transfected cells. Selected cells were then plated at low density on 96 well plates, and wells containing single cells identified. Following proliferation, colonies were removed with trypsin and replated in selection media. Cells expressing mutant Tau were maintained in media with 0.2 mg/ml G418 until changing to differentiation media (Neurobasal supplemented with B27 as described above) during experiments.
Western blot
Lysates were prepared from SH-SY5Y cells by sonication in RIPA buffer (Cell Signaling Technology) with addition of 1 mM PMSF, as per manufacturer’s instructions. Proteins in the lysates were separated via SDS-PAGE and transferred to PVDF membrane by electroblotting. The PVDF membranes were blocked in Odyssey blocking buffer (Licor Biosciences) for 1 h and incubated with either a rabbit polyclonal SelS antibody (1 : 1000, Sigma), mouse monoclonal tau antibody (1 : 1000, Millipore) or rabbit polyclonal phospho-tau antibodies for residues T212, S214, T236 (AT180 clone), and S262 (1 : 1000, Invitrogen). Co-incubation with a mouse monoclonal alpha-tubulin antibody (Novus Biologicals) was used to confirm even loading of protein. We used secondary antibodies conjugated with HRP, followed by visualization using an enhanced chemiluminescence kit (GE Biosciences), or secondary antibodies labeled with infrared fluorophores, followed by imaging with the Odyssey infrared system (Li-Cor Biosciences).
Quantitative PCR
RNA was extracted with Trizol (Invitrogen) per manufacturer’s instructions. cDNA was synthesized from RNA using a reverse transcriptase kit (ABI). qPCR reactions contained cDNA, oligonucleotide primers, and Platinum SYBR Green qPCR SuperMix (Invitrogen). qPCR reactions were run on a Roche LightCycler 480 II. The following primer sequences were used: SelS forward: AAA CGG AAA TCG GAC AGA AA; SelS reverse: CCT CCT TCA CCA GAC AAC G; Selenoprotein W (SelW) forward: GCC GTC CGA GTC GTT TAT TGT G; SelW reverse: GCT ACC ATC ACT TCA AAG AAC CCG; XBP1 forward: TTA CGA GAG AAA ACT CAT GGC C, XBP1 reverse; GGG TCC AAG TTG TCC AGA ATG C, 18S rRNA forward: GCA ATT ATT CCC CAT GAA CG, 18S rRNA reverse: GGG ACT TAA TCA ACG CAA GC, UBC forward: ATT TGG GTC GCG GTT CTT G, UBC reverse: TGC CTT GAC ATT CTC GAT GGT, HPRT forward: TGA CAC TGG CAA AAC AAT GCA, HPRT reverse: GGT CCT TTT CAC CAG CAA GCT.
PCR and DNA gel electrophoresis
For XBP1 splice variant investigations, reactions contained cDNA, XBP1 primers (forward: AGG CTC GAA TGA GTG AGC TGG AAC A; reverse: TCA TTC CCC TTG GCT TCC GCC T) and GoTaq Green Master Mix (Promega), and amplified with a Bio-Rad Thermal Cycler. The products were then run on a 3% agarose gel and imaged on a UV transilluminator.
Immunofluorescence
Cells plated onto coverslips were fixed in 4% paraformaldehyde (PFA) in PBS for 30 min and kept in PBS at 4°C until ready for staining. Cells were blocked with Chemiblocker (Millipore) diluted 1 : 2 in PBS with 0.1% Triton X-100 (Harleco) for 1 h. After washes with PBS and PBST, the cells were incubated in primary antibodies (anti-phospho-tau for residue 214, and mouse anti-tau) diluted 1 : 100 in Chemiblocker/PBST with 1% bovine serum albumin (BSA) for 1 h at room temperature. Secondary antibodies conjugated with Alexa 488 and Alexa 546 were diluted 1 : 250 and left on the cells for 30 min at room temperature. Following subsequent washes, the coverslips were mounted onto slides using Vectashield containing DAPI to label nuclei (Vector Laboratories).
Human brain histology
Paraffin-embedded brain tissue sections were obtained from the Honolulu-Asia Aging Study, an ongoing project that has monitored the health and lifestyle of Japanese-American men born between 1900 and 1919 and residing on Oahu, Hawaii. Summary information of subjects was published previously [19]. 10 μm sections of medial temporal cortex were deparaffinized, and ‘antigen unmasking’ was achieved by heating to 95°C at 15 psi in a pressure cooker in an alkaline/EDTA solution (Trilogy, Cell Marque), followed by 3 min in 90% formic acid. The tissue was blocked in PBS with 5% serum species-matched to secondary antibody, followed by avidin and biotin blocking steps. The tissue was then incubated at 4◦C overnight with an anti-SelS rabbit polyclonal antibody (1 : 100, Sigma) in 2.5% serum. After several washes, the tissue was incubated with a biotinylated anti-rabbit secondary antibody and ABC™ reagent (Vector Laboratories). HRP signals were developed with 3,3-diaminobenzidine hydrochloride (DAB, Vector Labs) and alkaline phosphatase detected with BCIP reactions. For double labeling with either tau or amyloid-β, tissues were then blocked in 5% normal horse serum, avidin, and biotin solutions and incubated overnight at 4°C with anti-amyloid-β 1–16 mouse monoclonal antibody (Chemicon).
Microscopy
Bright-field images of human brain tissue were obtained using a Zeiss Axioskop II Plus. Images of fluorescently labeled cells were taken on a Zeiss LSM Pascal laser-scanning or an Olympus IX81 DSU confocal microscope and analyzed with ImageJ software.
Statistical analysis
Differences between two groups were analyzed using a Student t-test. One-way ANOVA was performed with Tukey’s post hoc test to evaluate the differences between three or more groups, and two-way ANOVA with Tukey’s post hoc test was used for comparisons with more than one variable. A p value ≤ 0.05 was considered statistically significant.
RESULTS
SelS and ER stress in neuronal cells
SelS is involved in removal of misfolded proteins from the ER lumen and protects against ER stress [20]. As pathology of AD is marked by aggregates of misfolded amyloid-β and tau proteins, we hypothesized that SelS could function to remove amyloid-β and tau to prevent the buildup of amyloid plaques and neurofibrillary tangles. We first tested whether SelS is involved in protecting against ER stress in neurons, as it has been reported to do in other cell types [10, 11, 21]. We used three separate agents to induce ER stress via distinct mechanisms. In response to the ER stress induced by each agent, we consistently observed large increases in SelS mRNA (Fig. 1A, above) as well as SelS protein (Fig. 1A, below). To confirm induction of ER stress, we also looked at mRNA expression and splicing of the ER stress marker XBP1 [22]. Unconventional splicing of XBP1 to encode for an active transcription factor is induced following ER stress by the endonuclease inositol-requiring enzyme 1 (IRE1) [23]. Thapsigargin and tunicamycin significantly increased XBP1 mRNA levels (Fig. 1B, above). ER stress also led to splicing of XBP1 mRNA (Fig 1B, below). These findings confirm that SelS is regulated by ER stress in neural cells.
Fig. 1.
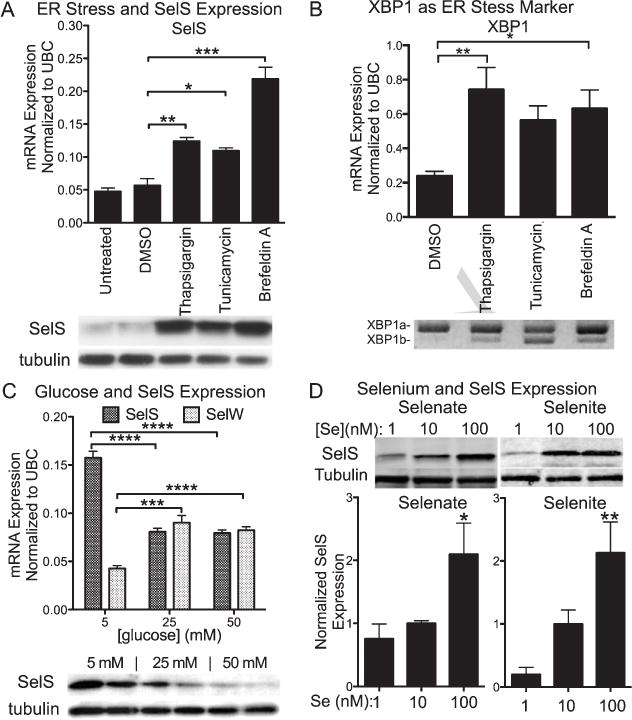
Regulation of expression of SelS in SH-SY5Y cells by ER stress and selenium. A) Above: SelS mRNA measured with qPCR is increased following ER stress induced by tunicamycin, brefeldin A, or thapsigargin. Below: SelS protein (21 kDa) measured by western blot is also increased. Tubulin was used as a loading control. B) Above: XBP1 mRNA expression also increases with ER stress induction. Below: XBP1 splicing occurs following treatment with thapsigargin, brefeldin A, or tunicamycin. C) Glucose increases expression of SelS in SH-SY5Y cells. Above: Quantitative reverse-transcriptase PCR measurements of SelS message levels in cells grown with different glucose levels. Expression of SelS decreases as glucose levels increase. In contrast, expression of another selenoprotein, SelW, is slightly elevated as glucose levels are increased. Below: SelS protein is decreased at higher glucose concentrations, shown by western blot. Alpha-tubulin is shown to demonstrate even loading. D) Sodium selenate (left) and selenite (right) both increase SelS expression. SH-SY5Y cells were grown in 0 Se media for 48 h, followed by supplementation with 1, 10, or 100 nm selenate or selenite for 3 days. Representative blots are shown above. Bar graphs show quantitation of SelS protein levels normalized to tubulin, relative to the 10 nM selenium samples. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Regulation of SelS expression by hyperglycemic conditions and selenium supplementation
Type 2 diabetes can increase the risk of Alzheimer’s disease. SelS was initially discovered as a glucose-regulated protein [24]. However, this has not been tested in brain cells. We found that hyperglycemic conditions (>10 mM glucose) for neuronal cells in culture suppressed SelS mRNA and protein levels. SelS expression was greatest at a glucose concentration near the normal range in brain (5 mM) [25] and decreased as glucose increased (Fig. 1C, above) [10, 11, 21]. Expression in 25 mM glucose was about 50% of the levels found at 5 mM glucose, when normalized to all three housekeeping genes. We also examined expression of SelS protein levels by western blot (Fig. 1C, below). As found with mRNA, SelS protein levels are decreased in 25 and 50 mM glucose conditions compared to 5 mM glucose, confirming an apparent downregulation of SelS in hyperglycemic conditions. Thus hyperglycemia with type II diabetes could suppress SelS expression and restrict its actions.
We investigated selenate and selenite supplementation on expression of SelS. As shown in Fig. 1D, increases in both selenate and selenite concentrations significantly increased SelS expression (Fig. 1D), This confirms that selenate can be utilized as a selenium source for selenoprotein synthesis in neuronal cells.
Selenoprotein S expression in human brain and SH-SY5Y neuronal cells
SelS has not previously been investigated in human brain. We first examined the location of SelS in postmortem human cortex using brain tissue from the Honolulu-Asia Aging Study. We observed SelS immunolabeling across cortical layers with slightly higher density in upper layers (Fig. 2A). We observed SelS immunoreactivity in small, branched cells as well as large pyramidal neurons in upper cortical layers. We also examined SelS immunoreactivity in SH-SY5Y neuronal cells (Fig. 2B). SelS was found in a ‘perinuclear’ pattern characteristic of ER-localized proteins. Immunoreactivity extended throughout the cell body surrounding the nucleus. SelS was also found in punctate labeling within cell processes.
Fig. 2.
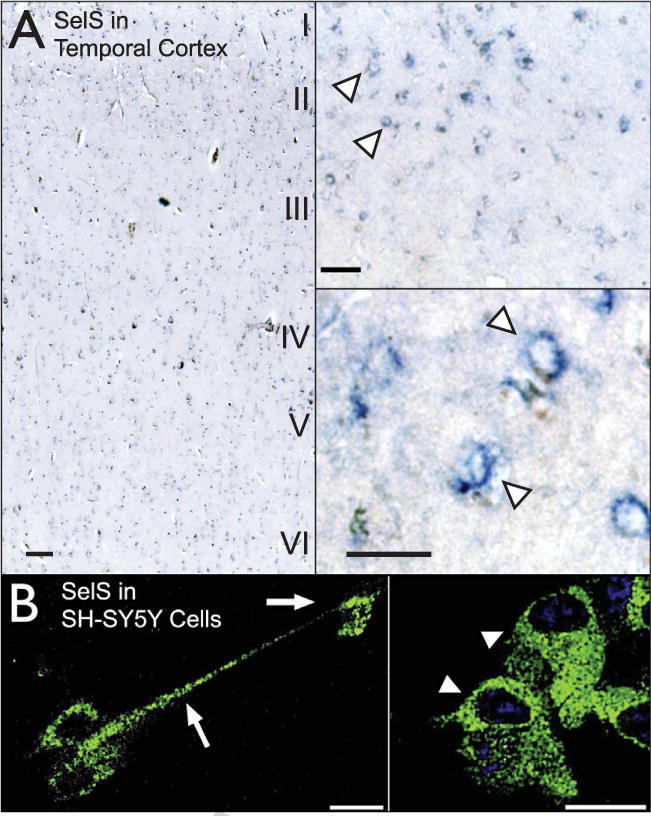
Selenoprotein S in human temporal cortex and SH-SY5Y cells. A) SelS (blue) distribution across cortical layers in the medial temporal cortex (left) and in individual cells. SelS was found in small cells in upper cortical layers as well as in larger pyramidal neurons. Expression was predominantly perinuclear (arrowhead), characteristic of proteins localized to the endoplasmic reticulum. Scale bars: 20 μm. B) SelS in SH-SY5Y neuroblastoma cells, with perinuclear labeling similar to that found in human cortex. SelS was also present in neurites (right, arrow). Scale bars: 20 μm.
Knockdown of SelS with siRNA
We investigated the role of SelS in neuroprotection by determining responses to oxidative stress following knockdown of SelS. We transfected SH-SY5Y cells with siRNA directed at both transcript variants. SelS expression was subtly but significantly reduced by over 25% (Fig. 3A). Knockdown of SelS decreased cell viability when compared to incubation with control siRNA (Fig 3B). Thapsigargin and brefeldin A further decreased cell viability compared to vehicle controls, with the combination of knockdown of SelS and induction of ER stress causing the greatest reduction. However, SelS siRNA with tunicamycin did not further decrease cell viability compared with tunicamycin and control siRNA, possibly indicating that tunicamycin occludes further reduction from SelS mRNA knockdown, and vice versa.
Fig. 3.
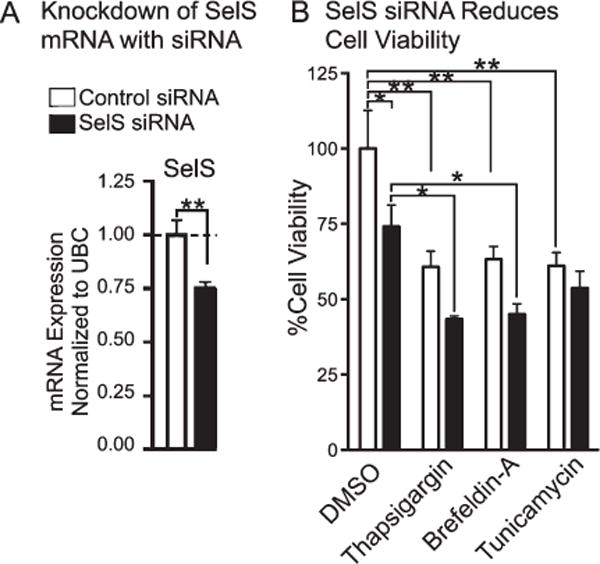
Cell viability of siRNA-transfected neuronal cells measured by MTS assay following challenge with various inducers of ER stress. A) Decrease in SelS mRNA from SelS siRNA. B) Suppression of SelS significantly decreased cell viability of cells for most treatments. ∗p < 0.05, ∗∗p < 0.01, Student’s t-test (left) or Tukey’s post hoc test (right), n = 4.
Selenate and tau phosphorylation
We examined if SelS might have a role in the selenate-induced reduction of tau phosphorylation. We examined tau phosphorylation in SH-SY5Y cells expressing the tau P301L mutation. Differentiated cells were treated for 1 h in OA before switching to Roth-Schweitzer media [14] with 0, 0.1, 1 or 10 μM sodium selenate. Tau phosphorylation was then assayed in cell lysates by western blots (Fig. 4A). Increasing the selenate concentration did not alter total tau levels relative to tubulin (Fig. 4B). There was a trend toward decreased phosphorylation at threonine 212 (pT212) with increasing selenate concentrations, but the changes were not significant (Fig. 4C). However, phosphorylation at serine 214 (pS214) (Fig. 4D) and threonine 236 (pT236, detected with the AT180 antibody) (Fig. 4E) were significantly reduced after normalizing to total tau. There was a significant negative correlation between the decreased pS214 and pT236 and increased SelS expression (Fig. 4F), indicating a potential role for SelS in reducing tau phosphorylation.
Fig. 4.
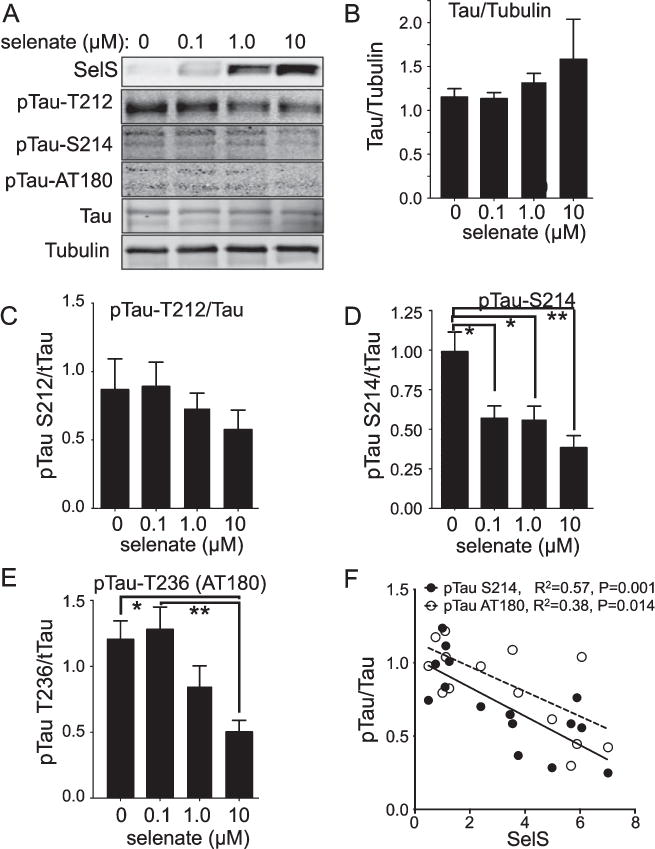
Selenate-induced reductions in tau phosphorylation correlate negatively with SelS expression. SH-SY5Y cells were grown in 0 Se media for 48 h. Following 1 h treatment with 80 nM okadaic acid, cells were grown in 0, 0.1, 1.0, or 10 M sodium selenate for 24 h. A) Western blots for tau phosphorylated at different sites, as well as total tau with tubulin as a loading control. B) Quantitation of tau relative to tubulin, showing no change. C-E) Quantitation of westerns for pTau T212 (C), S214 (D), and S236 (AT180, E). F) Negative correlation of pTau S214 and AT180 relative to SelS. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Tukey’s post hoc test, n = 4.
Tau phosphorylation regulated by SelS and ER stress
Recent studies have shown a connection between ER stress and formation of neurofibrillary tangles [26, 27]. We investigated whether loss of SelS expression would promote phosphorylation of tau under various conditions of ER stress. We used siRNA to knockdown expression of SelS in SH-SY5Y cells, and induced ER stress with tunicamycin to promote accumulation of unfolded proteins. SelS siRNA did not significantly reduce SelS protein relative to a negative control siRNA, shown in Fig. 5A and quantified in 5F. This is likely due to the limited knockdown of SelS mRNA (approximately 25%, as shown in Fig. 3A) combined with cell-to-cell variability. However, SelS siRNA did prevent an increase in SelS protein in response to tunicamycin, allowing examination of ER stress response without increased levels of SelS. SelS siRNA increased phosphorylation of tau at pT212 (Fig. 5C), but this increase was not further promoted by tunicamycin. However, combined tunicamycin treatment and knockdown of SelS together significantly increased phosphorylation of tau at the pS214 site (Fig. 5D). Phosphorylation at the tau serine 262 (pS262) site, as well as total tau, was not altered (Fig. 6E–F). Thus ER stress, when not accompanied by an increase in SelS, results in increased tau phosphorylation, suggesting that upregulation of SelS has a preventative role in neurofibrillary tangle formation.
Fig. 5.
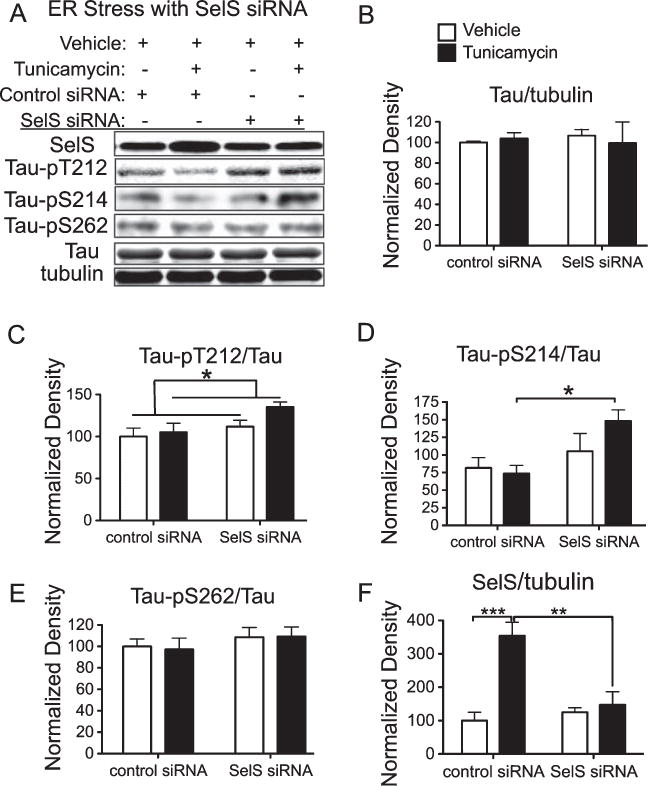
Knockdown of SelS along with ER stress promotes phosphorylation of tau. SelS was knocked down with siRNA in SH-SY5Y cells with and without induction of ER stress by tunicamycin. Phosphorylation of tau was detected with antibodies recognizing tau phosphorylated at specific sites. A) Representative blots with antibodies to SelS, Tau-pT212, pS214, and S262, total tau, and tubulin as a loading control. B) Following knockdown of SelS, the increase in protein expression induced by tunicamycin is significantly reduced. C) Tau-pT212 phosphorylation increases with tunicamycin treatment. D) Tau-pS214 phosphorylation is significantly increased only after SelS knockdown and treatment with tunicamycin. E) There is no significant change in Tau-pS262 phosphorylation. F) Total tau relative to tubulin does not change significantly with ER stress or SelS knockdown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Tukey’s post hoc test, n = 4.
Fig. 6.
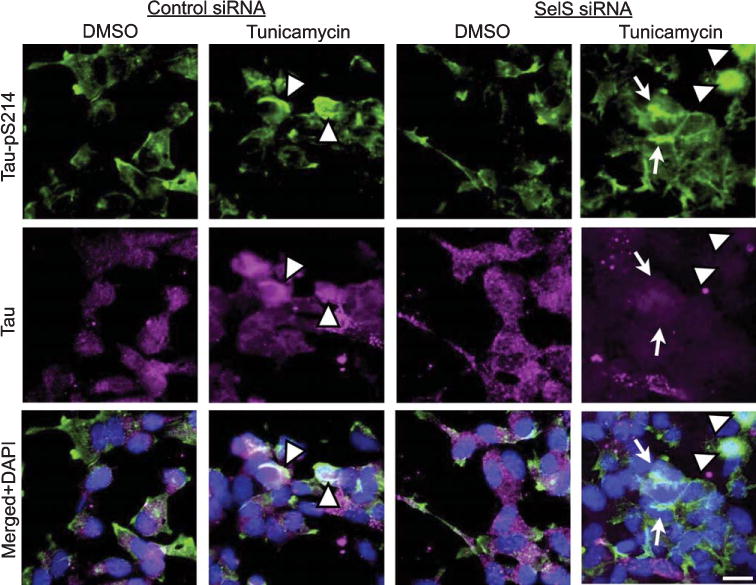
Tunicamycin and SelS siRNA together increase pTau aggregates in cell bodies and neurites. Immunolabeling for Tau-pS214 (green, top row), tau (magenta, second row), or combined (last row) with DAPI (blue). Tunicamycin induced aggregates of pTau in cell bodies (arrowheads), while tunicamycin and SelS siRNA combined also increased pTau aggregates in neuronal processes (arrows). Scale bar: 20 μm.
We also investigated morphological changes in cells following ER stress and SelS siRNA using immunocytochemistry. As shown in Fig. 6, tunicamycin induced aggregates of tau and tau pS214 immunoreactivity in cell bodies (arrowheads). Tunicamycin and SelS siRNA combined also increased pS214 immunoreactivity in neurites (arrows). The increased phosphorylated tau in soma and neurites is consistent with the promotion of neurofibrillary tangle formation.
Selenoprotein S expression in Alzheimer’s disease
We examined SelS in postmortem Alzheimer’s brain tissue in relation to Alzheimer’s disease markers, including amyloid-β plaques (deposits of the amyloid-β peptide) and neurofibrillary tangles. We compared SelS expression with tau, a primary component of neurofibrillary tangles and another hallmark of Alzheimer’s disease. We observed a strong correlation of the immunoreactivity for tau with SelS (Fig. 7A). SelS immunoreactivity was more pronounced in neurons with neurofibrillary tangles. To determine the spatial relationship between SelS and deposition of amyloid-β, tissue was immunostained with antibodies to both SelS and amyloid-β (Fig. 7B). We did not find a correlation between SelS and amyloid-β plaques. This is in contrast to our previous observations of selenoprotein P (Sepp1) immunoreactivity in relation to amyloid-β in postmortem Alzheimer’s brain tissue [6]. Thus SelS expression correlates with neurofibrillary tangles, but not amyloid-β plaques.
Fig. 7.
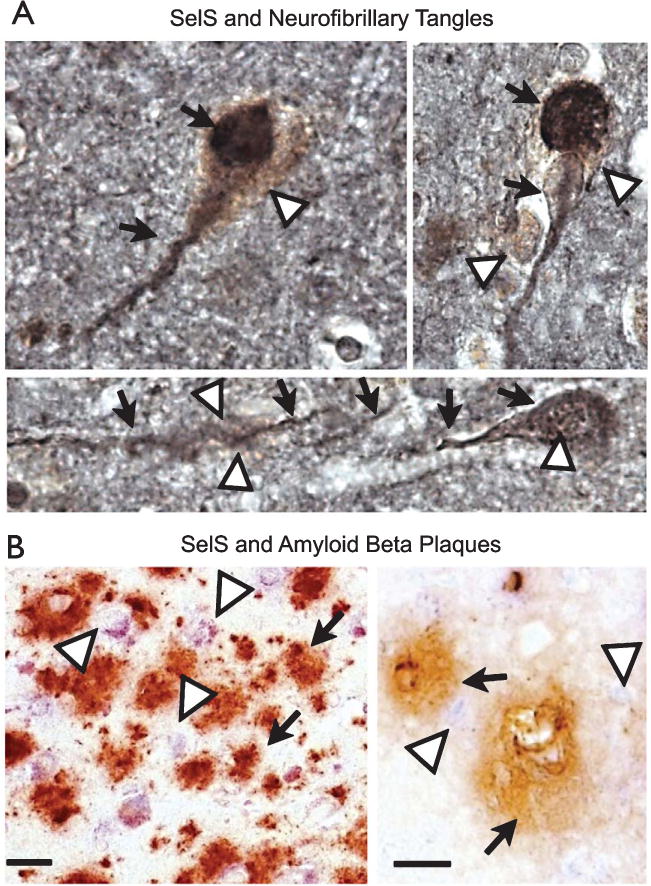
SelS expression is associated with neurofibrillary tangles in human postmortem temporal cortex. A) SelS expression (brown, white arrowheads) is found in cells expressing tangles, as shown by immunoreactivity to tau (black, indicated by black arrows). B) SelS (blue, shown by white arrowheads in top panel) did not appear to have any relationship to amyloid plaques (brown, shown by black arrows). Scale bars: 10 μm (left), 20 μm (right).
DISCUSSION
In these studies, we found that SelS increases in negative correlation with tau phosphorylation. The restriction of SelS expression during ER stress results in increased tau phosphorylation, and also decreases cell viability. In postmortem human brain, we found SelS in neurons containing neurofibrillary tangles. These studies suggest that increased SelS expression may contribute to a reduction in tau phosphorylation by decreasing ER stress.
The unfolded protein response (UPR) is triggered by accumulation of misfolded proteins in the ER [28]. SelS is induced by the UPR [24] and forms a complex with the derlin proteins to assist in retrotranslocation of misfolded proteins from the ER [13, 29]. SelS is upregulated in astrocytes by ischemia and brain injury to reduce apoptotic cell death [10, 30]. Recent studies indicate that ER stress accompanies Alzheimer’s disease pathology and may potentiate cell loss in the disease [31, 32]. Markers of ER stress are upregulated in cortical and hippocampal neurons [33], and may increase tau phosphorylation [26, 27, 34]. Neurofibrillary tangles form when tau, which is involved in organizing microtubule formation, becomes hyperphosphorylated and loses its association with tubulin. This causes neurons to lose internal structure and form dense, insoluble masses. Additionally, tangles could further promote ER stress [35].
Other forms of selenium can also decrease pathology in different Alzheimer’s disease models. Sodium selenite can inhibit tau phosphorylation and cell death in vitro [36]. Selenite can also prevent streptozotocin-induced cognitive decline and oxidative damage [37] and reduce secretase activity and amyloid-β production in vitro [38]. Selenomethionine can reduce tau phosphorylation, amyloid-β, and cognitive decline in Alzheimer’s disease mouse models [39, 40], and protects against oxidative stress from amyloid-β in vitro [41]. The glutathione peroxidase mimetic and organoselenium compound ebselen can reduce tau phosphorylation [42]. Other synthetic organoselenium compounds can improve cognitive impairments induced by streptozotocin and amyloid-β [43–46], and inhibit Cu-induced amyloid-β aggregates [47]. In contrast to supplementation studies, selenium deficiency increases amyloid-β deposition in an Alzheimer’s disease mouse model [48].
Both inorganic and organic forms of selenium can act on both tau phosphorylation and amyloid-β. Both inorganic and organic forms of selenium can also increase synthesis of selenoproteins [49], which may facilitate these actions. However, different forms vary in facilitation of selenoprotein production and toxicity. The toxicity of sodium selenate is much lower than sodium selenite [1, 2, 49], and its benefit for selenium supply compared to its toxicity is more favorable than many forms. These properties, along with the potential for direct action on PP2A, indicate selenate may be a preferable form of selenium supplementation for prevention and treatment of Alzheimer’s disease and related disorders.
In our studies, preventing upregulation of SelS in response to tunicamycin-induced ER stress through the use of siRNA resulted in increased phosphorylation of tau and accumulation of phosphorylated tau in neurite processes. These results suggest that SelS may serve as an important negative regulator of formation of neurofibrillary tangles. While upregulation of SelS in response to cellular stress would provide a compensatory action against tangle formation, this regulation may ultimately be overwhelmed over the course of the disease. Knockdown of SelS with siRNA also reduced cell viability (Fig. 3), despite the limited reduction of SelS mRNA and protein. This indicates an essential role for the protein in normal cell function. Changes in SelS expression and tau phosphorylation may be greater than depicted due to loss of cells most affected by siRNA.
Selenate supplementation may be a promising treatment for Alzheimer’s disease. Selenate decreases tau phosphorylation in models of neurofibrillary tangle formation, and may additionally decrease amyloid-β [50]. Selenate also reduces tau hyperphosphorylation and brain pathology in traumatic brain injury [51], and reduces seizures in rodent epilepsy models [52]. According to recent reports, selenium in the form of sodium selenate reduces neurofibrillary tangle formation [1, 2]. Additionally, selenate may reduce release of amyloid-β, the major component of amyloid plaques [50]. The studies suggest selenate acts as an agonist for protein phosphatase 2A (PP2A), which reduces tau phosphorylation [53]. Knockdown or deletion of PP2A prevents selenate-induced dephosphorylation of tau [1, 2]. This supports the argument made in previous studies that selenate acts directly as an agonist of PP2A. However, dietary selenate is usually converted to other forms of selenium [49]. Selenium supplements generally increase serum levels of organic selenium and not inorganic selenium [54], and selenium transport to the brain is mediated by selenoprotein P (Sepp1) [55]. Regardless, our study and others demonstrate that selenate supplementation upregulates expression of selenoproteins [49], which could mediate or contribute to dephosphorylation of tau.
The presence of prominent SelS labeling in neurons containing tangles from postmortem tissue from individuals with Alzheimer’s disease as compared to controls is consistent with these findings, namely that these neurons had undergone significant stress resulting in upregulation of SelS. However, as Alzheimer’s disease progresses, expression may be limited by availability of selenium, and the increased expression may be insufficient to prevent tangle formation. The correlation of SelS expression with tau and not amyloid-β can be expected from the ER localization of SelS, and suggests its actions are limited to intracellular and not extracellular actions.
Interestingly, SelS is downregulated under hyperglycemic conditions. This suggests that brain expression of SelS might be reduced in diabetics with hyperglycemia. Diabetes greatly increases the risk of developing Alzheimer’s disease, with abnormal insulin signaling and accumulation of misfolded proteins common to both disorders [56, 57]. Our findings suggest that hyperglycemia could promote neurofibrillary tangle formation by reducing SelS levels.
The results reported herein indicate an important role for SelS in tau phosphorylation, and suggest a role for this protein in studies of selenate and dephosphorylation of tau. However, regardless of whether SelS and selenoproteins have any direct action on tau phosphorylation, selenate treatment will nonetheless increase selenoprotein expression. Therefore, we need a better understanding of the consequences of this increased expression in order to utilize selenate as a treatment for Alzheimer’s disease and other disorders. Moreover, selenate may have multiple targets both in and outside of the central nervous system, and identification of specific proteins regulated by selenate could provide more specific targets for therapeutic intervention.
Acknowledgments
We thank Kristen Ewell for assistance with the Honolulu-Asia Aging Study tissue, Robert Nichols and Arjun Raman for helpful comments and suggestions, and Virginia Lee and colleagues for kindly providing the P301L tau clone. This work was supported by grants to FPB and PG from the RCMI Translational Research Network (RTRN, SGP14-161) and to FPB from the Hawaii Community Foundation (08PR-43031 and 11ADVC-49233), as well as NIH grant G12-MD007601 that supports the JABSOM imaging and histology cores.
Footnotes
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-1208r1).
References
- 1.Corcoran NM, Martin D, Hutter-Paier B, Windisch M, Nguyen T, Nheu L, Sundstrom LE, Costello AJ, Hovens CM. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J Clin Neurosci. 2010;17:1025–1033. doi: 10.1016/j.jocn.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 2.van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Gotz J, Ittner LM. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci U S A. 2010;107:13888–13893. doi: 10.1073/pnas.1009038107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosse AC, Pallauf J, Hommel B, Sturm M, Fischer S, Wolf NM, Mueller AS. Impact of selenite and selenate on differentially expressed genes in rat liver examined by microarray analysis. Biosci Rep. 2011;30:293–306. doi: 10.1042/BSR20090089. [DOI] [PubMed] [Google Scholar]
- 4.Hill KE, Xia Y, Akesson B, Boeglin ME, Burk RF. Selenoprotein P concentration in plasma is an index of selenium status in selenium-deficient and selenium-supplemented Chinese subjects. J Nutr. 1996;126:138–145. doi: 10.1093/jn/126.1.138. [DOI] [PubMed] [Google Scholar]
- 5.Persson-Moschos M, Alfthan G, Akesson B. Plasma selenoprotein P levels of healthy males in different selenium status after oral supplementation with different forms of selenium. Eur J Clin Nutr. 1998;52:363–367. doi: 10.1038/sj.ejcn.1600565. [DOI] [PubMed] [Google Scholar]
- 6.Bellinger FP, He QP, Bellinger MT, Lin Y, Raman AV, White LR, Berry MJ. Association of selenoprotein p with Alzheimer’s pathology in human cortex. J Alzheimers Dis. 2008;15:465–472. doi: 10.3233/jad-2008-15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bulteau AL, Chavatte L. Update on selenoprotein biosynthesis. Antioxid Redox Signal. 2015;23:775–794. doi: 10.1089/ars.2015.6391. [DOI] [PubMed] [Google Scholar]
- 8.Bellinger FP, Raman AV, Reeves MA, Berry MJ. Regulation and function of selenoproteins in human disease. Biochem J. 2009;422:11–22. doi: 10.1042/BJ20090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pillai R, Uyehara-Lock JH, Bellinger FP. Selenium and selenoprotein function in brain disorders. IUBMB Life. 2014;66:229–239. doi: 10.1002/iub.1262. [DOI] [PubMed] [Google Scholar]
- 10.Fradejas N, Pastor MD, Mora-Lee S, Tranque P, Calvo S. SEPS1 gene is activated during astrocyte ischemia and shows prominent antiapoptotic effects. J Mol Neurosci. 2008;35:259–265. doi: 10.1007/s12031-008-9069-3. [DOI] [PubMed] [Google Scholar]
- 11.Gao Y, Hannan NR, Wanyonyi S, Konstantopolous N, Pagnon J, Feng HC, Jowett JB, Kim KH, Walder K, Collier GR. Activation of the selenoprotein SEPS1 gene expression by pro-inflammatory cytokines in HepG2 cells. Cytokine. 2006;33:246–251. doi: 10.1016/j.cyto.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Curran JE, Jowett JB, Elliott KS, Gao Y, Gluschenko K, Wang J, Abel Azim DM, Cai G, Mahaney MC, Comuzzie AG, Dyer TD, Walder KR, Zimmet P, MacCluer JW, Collier GR, Kissebah AH, Blangero J. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37:1234–1241. doi: 10.1038/ng1655. [DOI] [PubMed] [Google Scholar]
- 13.Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. Inaugural Article: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A. 2005;102:14132–14138. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roth S, Zhang S, Chiu J, Wirth EK, Schweizer U. Development of a serum-free supplement for primary neuron culture reveals the interplay of selenium and vitamin E in neuronal survival. J Trace Elem Med Biol. 2010;24:130–137. doi: 10.1016/j.jtemb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Copanaki E, Schurmann T, Eckert A, Leuner K, Muller WE, Prehn JH, Kogel D. The amyloid precursor protein potentiates CHOP induction and cell death in response to ER Ca2+ depletion. Biochim Biophys Acta. 2007;1773:157–165. doi: 10.1016/j.bbamcr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Preston AM, Gurisik E, Bartley C, Laybutt DR, Biden TJ. Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia. 2009;52:2369–2373. doi: 10.1007/s00125-009-1506-5. [DOI] [PubMed] [Google Scholar]
- 17.Chan S-W, Egan PA. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005;19:1510–1512. doi: 10.1096/fj.04-3455fje. [DOI] [PubMed] [Google Scholar]
- 18.Nasreddine ZS, Loginov M, Clark LN, Lamarche J, Miller BL, Lamontagne A, Zhukareva V, Lee VM, Wilhelmsen KC, Geschwind DH. From genotype to phenotype: A clinical pathological, and biochemical investigation of frontotemporal dementia and parkinsonism (FTDP-17) caused by the P301L tau mutation. Ann Neurol. 1999;45:704–715. doi: 10.1002/1531-8249(199906)45:6<704::aid-ana4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 19.Rueli RH, Parubrub AC, Dewing AS, Hashimoto AC, Bellinger MT, Weeber EJ, Uyehara-Lock JH, White LR, Berry MJ, Bellinger FP. Increased selenoprotein P in choroid plexus and cerebrospinal fluid in Alzheimer’s disease brain. J Alzheimers Dis. 2015;44:379–383. doi: 10.3233/JAD-141755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shchedrina VA, Zhang Y, Labunskyy VM, Hatfield DL, Gladyshev VN. Structure-function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxid Redox Signal. 2010;12:839–849. doi: 10.1089/ars.2009.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim KH, Gao Y, Walder K, Collier GR, Skelton J, Kissebah AH. SEPS1 protects RAW264.7 cells from pharmacological ER stress agent-induced apoptosis. Biochem Biophys Res Commun. 2007;354:127–132. doi: 10.1016/j.bbrc.2006.12.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samali A, Fitzgerald U, Deegan S, Gupta S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int J Cell Biol. 2010;2010:830307. doi: 10.1155/2010/830307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 24.Gao Y, Feng HC, Walder K, Bolton K, Sunderland T, Bishara N, Quick M, Kantham L, Collier GR. Regulation of the selenoprotein SelS by glucose deprivation and endoplasmic reticulum stress - SelS is a novel glucose-regulated protein. FEBS Lett. 2004;563:185–190. doi: 10.1016/S0014-5793(04)00296-0. [DOI] [PubMed] [Google Scholar]
- 25.Kleman AM, Yuan JY, Aja S, Ronnett GV, Landree LE. Physiological glucose is critical for optimized neuronal viability and AMPK responsiveness in vitro. J Neurosci Methods. 2008;167:292–301. doi: 10.1016/j.jneumeth.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Resende R, Ferreiro E, Pereira C, Oliveira CR. ER stress is involved in Abeta-induced GSK-3beta activation and tau phosphorylation. J Neurosci Res. 2008;86:2091–2099. doi: 10.1002/jnr.21648. [DOI] [PubMed] [Google Scholar]
- 27.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kudo T, Katayama T, Imaizumi K, Yasuda Y, Yatera M, Okochi M, Tohyama M, Takeda M. The unfolded protein response is involved in the pathology of Alzheimer’s disease. Ann N Y Acad Sci. 2002;977:349–355. doi: 10.1111/j.1749-6632.2002.tb04837.x. [DOI] [PubMed] [Google Scholar]
- 29.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 30.Fradejas N, Serrano-Perez Mdel C, Tranque P, Calvo S. Selenoprotein S expression in reactive astrocytes following brain injury. Glia. 2011;59:959–972. doi: 10.1002/glia.21168. [DOI] [PubMed] [Google Scholar]
- 31.Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- 32.Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer’s disease: A novel neuronal trigger for inflammation and Alzheimer’s pathology. J Neuroinflammation. 2009;6:41. doi: 10.1186/1742-2094-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005;110:165–172. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- 34.Gotz J, David D, Hoerndli F, Ke YD, Schonrock N, Wiesner A, Fath T, Bokhari L, Lim YA, Deters N, Ittner LM. Functional genomics dissects pathomechanisms in tauopathies: Mitosis failure and unfolded protein response. Neurodegener Dis. 2008;5:179–181. doi: 10.1159/000113696. [DOI] [PubMed] [Google Scholar]
- 35.Abisambra JF, Jinwal UK, Blair LJ, O’Leary JC, 3rd, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, Cockman M, Suntharalingham A, Li P, Jin Y, Atkins CA, Dickey CA. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci. 2013;33:9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee YJ, Kim JE, Kwak MH, Go J, Yang SY, Kwon HS, Kim BC, Kim JM, Hwang DY. Selenium treatment significantly inhibits tumor necrosis factor-alpha-induced cell death and tau hyperphosphorylation in neuroblastoma cells. Mol Med Rep. 2014;10:1869–1874. doi: 10.3892/mmr.2014.2442. [DOI] [PubMed] [Google Scholar]
- 37.Ishrat T, Parveen K, Khan MM, Khuwaja G, Khan MB, Yousuf S, Ahmad A, Shrivastav P, Islam F. Selenium prevents cognitive decline and oxidative damage in rat model of streptozotocin-induced experimental dementia of Alzheimer’s disease. Brain Res. 2009;1281:117–127. doi: 10.1016/j.brainres.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Gwon AR, Park JS, Park JH, Baik SH, Jeong HY, Hyun DH, Park KW, Jo DG. Selenium attenuates A beta production and A beta-induced neuronal death. Neurosci Lett. 2010;469:391–395. doi: 10.1016/j.neulet.2009.12.035. [DOI] [PubMed] [Google Scholar]
- 39.Song G, Zhang Z, Wen L, Chen C, Shi Q, Zhang Y, Ni J, Liu Q. Selenomethionine ameliorates cognitive decline, reduces tau hyperphosphorylation, and reverses synaptic deficit in the triple transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2014;41:85–99. doi: 10.3233/JAD-131805. [DOI] [PubMed] [Google Scholar]
- 40.Lovell MA, Xiong S, Lyubartseva G, Markesbery WR. Organoselenium (Sel-Plex diet) decreases amyloid burden and RNA and DNA oxidative damage in APP/PS1 mice. Free Radic Biol Med. 2009;46:1527–1533. doi: 10.1016/j.freeradbiomed.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiong S, Markesbery WR, Shao C, Lovell MA. Seleno-L-methionine protects against beta-amyloid and iron/hydrogen peroxide-mediated neuron death. Antioxid Redox Signal. 2007;9:457–467. doi: 10.1089/ars.2006.1363. [DOI] [PubMed] [Google Scholar]
- 42.Xie L, Zheng W, Xin N, Xie JW, Wang T, Wang ZY. Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem Int. 2012;61:334–340. doi: 10.1016/j.neuint.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 43.Pinton S, Bruning CA, Sartori Oliveira CE, Prigol M, Nogueira CW. Therapeutic effect of organoselenium dietary supplementation in a sporadic dementia of Alzheimer’s type model in rats. J Nutr Biochem. 2013;24:311–317. doi: 10.1016/j.jnutbio.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 44.Pinton S, da Rocha JT, Gai BM, Prigol M, da Rosa LV, Nogueira CW. Neuroprotector effect of p,p′-methoxyl-diphenyl diselenide in a model of sporadic dementia of Alzheimer’s type in mice: Contribution of antioxidant mechanism. Cell Biochem Funct. 2011;29:235–243. doi: 10.1002/cbf.1741. [DOI] [PubMed] [Google Scholar]
- 45.Pinton S, da Rocha JT, Zeni G, Nogueira CW. Organoselenium improves memory decline in mice: Involvement of acetylcholinesterase activity. Neurosci Lett. 2010;472:56–60. doi: 10.1016/j.neulet.2010.01.057. [DOI] [PubMed] [Google Scholar]
- 46.Pinton S, Souza AC, Sari MH, Ramalho RM, Rodrigues CM, Nogueira CW. p,p′-Methoxyl-diphenyl diselenide protects against amyloid-beta induced cytotoxicity in vitro and improves memory deficits in vivo. Behav Brain Res. 2013;247:241–247. doi: 10.1016/j.bbr.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Wang Y, Li W, Mao F, Sun Y, Huang L, Li X. Design, synthesis, and evaluation of multitarget-directed selenium-containing clioquinol derivatives for the treatment of Alzheimer’s disease. ACS Chem Neurosci. 2014;5:952–962. doi: 10.1021/cn500119g. [DOI] [PubMed] [Google Scholar]
- 48.Haratake M, Yoshida S, Mandai M, Fuchigami T, Nakayama M. Elevated amyloid-beta plaque deposition in dietary selenium-deficient Tg2576 transgenic mice. Metallomics. 2013;5:479–483. doi: 10.1039/c3mt00035d. [DOI] [PubMed] [Google Scholar]
- 49.Hoefig CS, Renko K, Kohrle J, Birringer M, Schomburg L. Comparison of different selenocompounds with respect to nutritional value vs. toxicity using liver cells in culture. J Nutr Biochem. 2011;22:945–955. doi: 10.1016/j.jnutbio.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 50.Chen P, Wang L, Wang Y, Li S, Shen L, Liu Q, Ni J. Phosphoproteomic profiling of selenate-treated Alzheimer’s disease model cells. PLoS One. 2014;9:e113307. doi: 10.1371/journal.pone.0113307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shultz SR, Wright DK, Zheng P, Stuchbery R, Liu SJ, Sashindranath M, Medcalf RL, Johnston LA, Hovens CM, Jones NC, O’Brien TJ. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain. 2015;138:1297–1313. doi: 10.1093/brain/awv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones NC, Nguyen T, Corcoran NM, Velakoulis D, Chen T, Grundy R, O’Brien TJ, Hovens CM. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol Dis. 2012;45:897–901. doi: 10.1016/j.nbd.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 53.Sontag JM, Sontag E. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci. 2014;7:16. doi: 10.3389/fnmol.2014.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vinceti M, Grill P, Malagoli C, Filippini T, Storani S, Malavolti M, Michalke B. Selenium speciation in human serum and its implications for epidemiologic research: A cross-sectional study. J Trace Elem Med Biol. 2015;31:1–10. doi: 10.1016/j.jtemb.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 55.Burk RF, Hill KE. Selenoprotein P-expression, functions, and roles in mammals. Biochem Biophys Acta. 2009;1790:1441–1447. doi: 10.1016/j.bbagen.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cole AR, Astell A, Green C, Sutherland C. Molecular connexions between dementia and diabetes. Neurosci Biobehav Rev. 2007;31:1046–1063. doi: 10.1016/j.neubiorev.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 57.Li L, Holscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: A review. Brain Res Rev. 2007;56:384–402. doi: 10.1016/j.brainresrev.2007.09.001. [DOI] [PubMed] [Google Scholar]