

Abstract
The increasing popularity of biomimetic design principles in nanomedicine has led to therapeutic platforms with enhanced performance and biocompatibility. This includes the use of naturally derived cell membranes, which can bestow nanocarriers with cell-specific functionalities. Herein, we report on a strategy enabling efficient encapsulation of drugs via remote loading into membrane vesicles derived from red blood cells. This is accomplished by supplementing the membrane with additional cholesterol, stabilizing the nanostructure and facilitating the retention of a pH gradient. We demonstrate the loading of two model drugs: the chemotherapeutic doxorubicin and the antibiotic vancomycin. The therapeutic implications of these natural, remote-loaded nanoformulations are studied both in vitro and in vivo using animal disease models. Ultimately, this approach could be used to design new biomimetic nanoformulations with higher efficacy and improved safety profiles.
Keywords: biomimetic nanotechnology, remote loading, cell membrane, biocompatible, drug delivery
COMMUNICATION
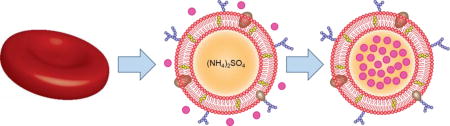
Remotely loaded RBC vesicles are fabricated by enriching the naturally derived cell membrane with additional cholesterol. Two model drugs, doxorubicin and vancomycin, are successfully loaded and show efficacy in animal models of disease. The reported approach may pave the way for biomimetic nanoformulations with high potency and safety
The field of nanomedicine has grown significantly over the past several decades.[1] Starting with the U.S. Food and Drug Administration’s approval of Doxil, a liposomal formulation of doxorubicin (Dox), numerous other nanoparticulate drugs have since been translated for clinical use.[2] Nanodelivery has distinct advantages compared with the administration of free drug, including improved blood residence, sustained release, and enhanced tumor targeting. All of these benefits ultimately lead to improved efficacy and patient compliance, better enabling clinicians to care for those with life-threatening diseases.
More recently, researchers have begun focusing on next-generation platforms that can build upon the initial successes of the drug delivery field. Along these lines, there has been a significant shift towards bioinspired or biomimetic design principles.[3] This is motivated by the fact that biological systems have been carefully cultivated over time by the process of evolution. Despite the significant advances made in bottom-up, synthetic nanofabrication, it is still incredibly difficult to replicate the complex functionalities found in nature. One such biomimetic strategy involves the combination of cell-derived membranes with artificial nanocarriers, leading to the generation of natural–synthetic hybrids with unique functionalities.[4] Benefits can include enhanced biocompatibility, lowered immunogenicity, heightened affinity for toxins, and the ability to target diseased tissues.[5] This approach has been proven highly generalizable and has utility across a wide range of different applications.
The remote loading of drugs into liposomal carriers represents one of the most highly used and effective means of nanoencapsulation.[6] In this work, we explored a strategy to adapt this approach for use with red blood cell (RBC) membrane-derived vesicles (Figure 1). It was demonstrated that membrane vesicles supplemented with exogenous cholesterol could more effectively retain a pH gradient, which was used to drive the successful loading of two model drugs: the chemotherapeutic Dox and the antibiotic vancomycin (Vanc). The Dox formulation (Dox-RBC) was used to treat a murine model of breast cancer, significantly controlling tumor growth, while the Vanc formulation (Vanc-RBC) was used to reduce bacterial load in a methicillin-resistant Staphylococcus aureus (MRSA) skin infection model.
Figure 1.

Remote loading into RBC vesicles. Natural cell membrane vesicles are fashioned from RBC ghosts enriched with cholesterol (yellow). Ammonium sulfate (orange) is used to generate a pH gradient, which facilitates accumulation of the drug (X) inside the cholesterol-enriched RBC vesicle.
To fabricate cholesterol-enriched RBC vesicles, membrane ghosts were derived from purified RBCs by hypotonic lysis. Free cholesterol was then incubated with the ghosts in 300 mM ammonium sulfate, followed by physical disruption to induce vesiculation. Finally, the outer phase was exchanged with phosphate buffered saline (PBS) to create a pH gradient. At increasing cholesterol input, the final nanovesicles exhibited a corresponding increase in size up to approximately 150 nm (Figure 2a). To characterize the membrane sidedness, which is important for retention of the membrane’s biological functions,[7] non-denaturing immunoblotting was employed (Figure 2b). When probed for either the intracellular or extracellular domains of the transmembrane protein CD47, ammonium sulfate-loaded vesicles had a predominantly right-side-out orientation, which was retained after transferring the vesicles to PBS. In contrast, vesicles made in water, which lacks the ions necessary to preserve orientation,[8] displayed equivalent amounts of both signals. Co-loading of the pH-sensitive dye LysoSensor Green was used to assess membrane integrity (Figure 2c). With increasing cholesterol, higher fluorescence was observed, indicating the retention of ammonium sulfate within the vesicles. This effect plateaued at a 5% input, and it was demonstrated that the enriched vesicles were also impermeable to calcein dye (Figure 2d). Fluorescence microscopy showed that the signal of dye loaded into the vesicles colocalized well with dye labeling the membrane, further confirming successful encapsulation and retention (Figure 2e). Over time, the enriched vesicles showed little change in size when incubated in serum-containing media (Figure 2f). Overall, the improved stability of the cholesterol-enriched vesicles likely stems from the ability of the molecule to positively regulate membrane fluidity.[9]
Figure 2.

Characterization of cholesterol-enriched RBC vesicles. a) Size of vesicles with varying cholesterol inputs (n = 3; mean ± SD). b) Dot blot intensities of various RBC vesicles probed with antibodies against the intracellular or extracellular domains of CD47 (n = 3; mean ± SD). c) Fluorescence spectrum of ammonium sulfate-loaded RBC vesicles when co-loaded with a pH-sensitive LysoSensor Green dye. d) Leakage of calcein from RBC vesicles (n = 3; mean ± SD). e) Confocal fluorescence imaging of ammonium sulfate-loaded RBC vesicles co-loaded with LysoSensor Green (red: DiD, green: LysoSensor Green; scale bar = 5 µm). f) Size of enriched RBC vesicles over time in serum-containing media (n = 3; mean ± SD).
We next sought to demonstrate remote loading into the RBC vesicles. Dox, which represents the most classical example of a remotely loaded drug,[6a] was first employed. After fabricating the vesicles at a 5% cholesterol input, loading yield was quantified using various Dox input concentrations (Figure 3a). With increasing inputs, it was observed that more Dox could be loaded, up to approximately 10%. This process was also dependent on both temperature (Figure 3b) and time (Figure 3c), with optimal loading occurring at 37 °C and saturating at 40 minutes of reaction time. Observation of Dox-RBC negatively stained with uranyl acetate by transmission electron microscopy revealed vesicular structures (Figure 3d). Notably, elongated crystals were visualized, consistent with reports on Dox-loaded synthetic liposomes.[10] Drug release was also evaluated over a period of 2 days at different pH values (Figure 3e). Regardless of pH, a burst release occurred in the first 12 hours, followed by more gradual, sustained release. Lowering the pH from a physiological value of 7.4 down to 5.0 facilitated drug release, likely due to the instability of RBC membrane under acidic conditions.
Figure 3.

Dox loading into RBC vesicles. a) Loading yield at different drug inputs (n = 3; mean ± SD). b) Loading yield at different temperatures (n = 3; mean ± SD). c) Loading yield over time (n = 3; mean ± SD). d) Transmission electron microscopy images of Dox-RBC after negative staining with uranyl acetate (scale bar = 100 nm). Inset depicts a single Dox-RBC particle (scale bar = 50 nm). e) Dox release at pH 5.0 or pH 7.4 (n = 3; mean ± SD).
Dox-RBC was incubated with murine 4T1 breast cancer cells for 24 hours to evaluate its potency in vitro. The nanoformulation showed better efficacy compared to free Dox (Figure 4a). This may be explained by the fact that nanoparticulate delivery enables cellular uptake via endocytic pathways,[11] possibly via a clathrin-mediated mechanism as reported previously for membrane-derived nanoparticles.[12] Enhanced uptake was confirmed by flow cytometric analysis (Figure 4b,c). When visualized by fluorescence microscopy, it was revealed that Dox-RBC facilitated retention of drug across the entirety of the cell; the free drug was only found in the nuclei, clearly delineated as large round objects during imaging (Figure 4d). This ability to facilitate uptake and enhance intracellular retention is a unique advantage of nanodelivery and could help to overcome drug-resistance by lessening drug efflux.[13]
Figure 4.

Antitumor activity of Dox-RBC. a) Viability of 4T1 breast cancer cells after incubation with free Dox or Dox-RBC for 24 hours (n = 4; mean ± SD). b) Representative flow cytometry histograms of control 4T1 cells or 4T1 cells incubated with free Dox or Dox-RBC. c) Mean fluorescence intensities from (b) (n = 3; mean ± SD). d) Confocal fluorescence imaging of cells incubated with either free Dox or Dox-RBC (red: Dox; scale bar = 10 µm). e) Growth kinetics of 4T1 tumors implanted orthotopically into BALB/c mice and treated with free Dox or Dox-RBC (n = 6; mean ± SEM). ** p < 0.01, *** p < 0.001 (compared to Dox-RBC at day 21); Student’s t-test. f) Survival of the mice in (e) over time (n = 6). * p < 0.05, ** p < 0.01 (compared to Dox-RBC); log-rank test. g) Enumeration of metastatic nodules present on the lungs after treatment (n = 3; mean ± SD). * p < 0.05, *** p < 0.001 (compared to Dox-RBC); Student’s t-test. h) Body weight of treated tumor-bearing mice over time (n = 6; mean ± SD). (i) Representative H&E-stained histological sections of cardiac tissue after treatment (scale bar = 250 µm). (j) Quantification of Dox fluorescence in tumors of mice administered with free Dox or Dox-RBC (n = 3; mean ± SEM).
Therapeutic efficacy was evaluated using an orthotopic 4T1 breast cancer model. Tumors were implanted in the mammary fat pads of female BALB/c mice. Once palpable, treatment was initiated using either Dox-RBC or free Dox. The nanoformulation had a marked effect, significantly slowing tumor growth compared to free Dox (Figure 4e). Median survival was also extended from 25.5 days for the untreated control group to 39 days for the Dox-RBC group (Figure 4f). Notably, 33% of the mice receiving nanoparticles had complete regressions and remained tumor-free. Dox-RBC was also better at preventing the formation of lung metastases, reducing the number of nodules after 21 days of treatment (Figure 4g). With regards to safety, mice subjected to a full course of Dox-RBC treatment did not experience any weight loss (Figure 4h). It should be noted that rapid tumor growth likely counteracted any weight loss for the free Dox group. The difference in safety could more readily be seen upon histopathological examination of the cardiac tissue (Figure 4i). Chronic myocardial toxicity with free Dox is a well-known occurrence in the clinic.[14] For this group, we observed that the myocardial fibers became thin and loose, with varying degrees of rupture. On the other hand, the hearts of mice treated with Dox-RBC did not show any significant alterations in fiber architecture. In all, it was shown that Dox-RBC could more effectively control tumor growth compared with free drug while also displaying evidence of increased safety. The improved performance of the nanoformulation could likely be attributed to its ability to preferentially deliver drug to the tumors (Figure 4j).
Following the antitumor studies, we explored if the remote loading strategy could be generalized to additional payloads. For this purpose, the antibiotic Vanc, another amine-containing small molecule drug, was employed. At increasing input concentrations, a loading yield of up to 40% was achieved (Figure 5a). The higher yield compared with Dox may be explained by the higher molecular weight of Vanc. Like Dox loading, the optimal temperature was 37 °C (Figure 5b), whereas saturation took longer and occurred at 1 hour (Figure 5c). Drug release kinetics followed a similar trend, with an initial burst observed in the first several hours and lower pH accelerating the process (Figure 5d). The absolute rate of release was slower when compared with Dox, and it took approximately 48 hours for all the drug to be released at pH 5.0.
Figure 5.

Vanc loading into RBC vesicles. a) Loading yield at different drug inputs (n = 4; mean ± SD). b) Loading yield at different temperatures (n = 4; mean ± SD). c) Loading yield over time (n = 3; mean ± SD). d) Release of Vanc over time at pH 5.0 or pH 7.4 (n = 3; mean ± SD).
Subsequently, we sought to test the nanoformulation against MRSA. A Förster resonance energy transfer (FRET) study was performed to demonstrate the interaction of the bacteria with the membrane vesicles (Figure 6a). As increasing amounts of bacteria were incubated with FRET pair-doped RBC vesicles, more donor signal was recovered, signaling that fusion was occurring. This effect could potentially be leveraged to facilitate delivery of loaded drugs to bacteria upon contact.[15] Vanc-RBC or free Vanc was then incubated with MRSA at increasing concentrations for 24 hours, and the results confirmed that the remotely loaded drug still retained its activity (Figure 6b). No effect was observed from RBC vesicles alone.
Figure 6.

Antibacterial activity of Vanc-RBC. a) Fluorescence recovery of donor dye after incubation of FRET pair-doped RBC vesicles with MRSA (n = 6; mean ± SD; linear regression). b) Bacteria count after incubation with free Vanc, RBC vesicles, or Vanc-RBC (n = 3; mean ± SD). c) Skin lesion size on mice challenged with MRSA and treated with free Vanc or Vanc-RBC (n = 6; mean ± SEM). *** p < 0.001 (compared to Vanc-RBC); Student’s t-test. d) Enumeration of bacteria in the skin tissue on day 5 (n = 6; geometric mean ± SD). *** p < 0.001, **** p < 0.0001 (compared to Vanc-RBC); Student’s t-test. e) Representative macroscopic images of bacterial challenge site on day 5. f) Representative H&E-stained histological sections of skin from the challenge site on day 5 (scale bar = 200 µm).
Therapeutic efficacy of the Vanc-RBC formulation was tested using a murine skin infection model. Following subcutaneous challenge with MRSA, mice were administered either free Vanc or Vanc-RBC. Over 5 days, the nanoformulation proved to be exceptionally potent, completely inhibiting the formation of skin lesions (Figure 6c,e). The free Vanc group experienced subtle lesion formation, which was a large improvement over the untreated controls. While the difference in physical effect between the treatment groups was slight, bacterial enumeration revealed that Vanc-RBC outperformed the free drug by 3 orders of magnitude (Figure 6d). Histological analysis showed that MRSA-induced lesions were marked by the loss of cilia and columnar morphology, characteristic of acanthosis and apoptosis (Figure 6f). These symptoms were decreased with free Vanc, although hyperkeratosis with aberrant hair shafts could still be observed. Samples from the Vanc-RBC group appeared normal. Ultimately, the enhanced performance of the nanoformulation could likely be attributed to its retention at the treatment site, preventing the Vanc from diffusing away.
In conclusion, we have demonstrated a method of remote loading drugs into naturally derived RBC vesicles. The process is greatly facilitated by supplementing the cell membrane with additional cholesterol, which facilitates the maintenance of a pH gradient. This concept was demonstrated using two small molecule model drugs, a chemotherapeutic and an antibiotic. In both cases, the nanoformulations were able to outperform the corresponding drugs in the free form using appropriate animal disease models. By employing a naturally derived nanocarrier, it is possible to leverage the benefits of nanoparticulate delivery, such as improved localization to diseased tissue, with fewer concerns regarding safety and immunogenicity. The work presented here can help to provide guiding principles for the development of next-generation, remote-loaded drug platforms.
Supplementary Material
Acknowledgments
This work is supported by the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under Grant Number HDTRA1-16-1-0013 and by the National Institutes of Health under Award R01CA200574.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.a) Wang AZ, Langer R, Farokhzad OC. Annu. Rev. Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]; b) Farokhzad OC, Langer R. ACS Nano. 2009;3:16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 2.a) Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Clin. Pharmacol. Ther. 2008;83:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]; b) Anselmo AC, Mitragotri S. AAPS J. 2015;17:1041–1054. doi: 10.1208/s12248-015-9780-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Nat. Rev. Drug Discov. 2011;10:521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]; b) Fang RH, Jiang Y, Fang JC, Zhang L. Biomaterials. 2017;128:69–83. doi: 10.1016/j.biomaterials.2017.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Proc. Natl. Acad. Sci. U.S.A. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y, O'Connor DE, Zhang L. Nano Lett. 2014;14:2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Luk BT, Fang RH, Hu CM, Copp JA, Thamphiwatana S, Dehaini D, Gao W, Zhang K, Li S, Zhang L. Theranostics. 2016;6:1004–1011. doi: 10.7150/thno.14471. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hu CM, Fang RH, Luk BT, Zhang L. Nat. Nanotechnol. 2013;8:933–938. doi: 10.1038/nnano.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hu CM, Fang RH, Copp J, Luk BT, Zhang L. Nat. Nanotechnol. 2013;8:336–340. doi: 10.1038/nnano.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Fritze A, Hens F, Kimpfler A, Schubert R, Peschka-Suss R. Biochim. Biophys. Acta. 2006;1758:1633–1640. doi: 10.1016/j.bbamem.2006.05.028. [DOI] [PubMed] [Google Scholar]; b) Haran G, Cohen R, Bar LK, Barenholz Y. Biochim. Biophys. Acta. 1993;1151:201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 7.Hu CM, Fang RH, Luk BT, Chen KN, Carpenter C, Gao W, Zhang K, Zhang L. Nanoscale. 2013;5:2664–2668. doi: 10.1039/c3nr00015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kant JA, Steck TL. Nat. New Biol. 1972;240:26–28. doi: 10.1038/newbio240026a0. [DOI] [PubMed] [Google Scholar]
- 9.de Meyer F, Smit B. Proc. Natl. Acad. Sci. USA. 2009;106:3654–3658. doi: 10.1073/pnas.0809959106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barenholz Y. J. Control. Release. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 11.a) Iversen TG, Skotland T, Sandvig K. Nano Today. 2011;6:176–185. [Google Scholar]; b) Sahay G, Alakhova DY, Kabanov AV. J. Control. Release. 2010;145:182–195. doi: 10.1016/j.jconrel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nguyen TDT, Pitchaimani A, Koirala MB, Muhammad F, Aryal S. RSC Adv. 2016;6:33003–33008. [Google Scholar]
- 12.Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z. Adv Mater. 2015;27:7043–7050. doi: 10.1002/adma.201503323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng X, Morgenstern R, Nystrom AM. Biomaterials. 2014;35:1227–1239. doi: 10.1016/j.biomaterials.2013.10.042. [DOI] [PubMed] [Google Scholar]
- 14.Chatterjee K, Zhang JQ, Honbo N, Karliner JS. Cardiology. 2010;115:155–162. doi: 10.1159/000265166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pornpattananangkul D, Olson S, Aryal S, Sartor M, Huang CM, Vecchio K, Zhang L. ACS Nano. 2010;4:1935–1942. doi: 10.1021/nn9018587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.