

Abstract
Devastating human neuromuscular disorders have been associated to defects in the ATP synthase. This enzyme is found in the inner mitochondrial membrane and catalyzes the last step in oxidative phosphorylation, which provides aerobic eukaryotes with ATP. With the advent of structures of complete ATP synthases, and the availability of genetically approachable systems such as the yeast Saccharomyces cerevisiae, we can begin to understand these molecular machines and their associated defects at the molecular level. In this review, we describe what is known about the clinical syndromes induced by 58 different mutations found in the mitochondrial genes encoding membrane subunits 8 and a of ATP synthase, and evaluate their functional consequences with respect to recently described cryo-EM structures.
Keywords: mitochondrial diseases, F1Fo ATP synthase structure, mitochondrial DNA (mtDNA), MT-ATP6, MT-ATP8
Introduction
Mitochondria support aerobic respiration and produce the bulk of cellular ATP by oxidative phosphorylation (OXPHOS) (Saraste, 1999). Electrons provided by the oxidation of fatty acids and carbohydrates are shuttled to oxygen along four respiratory chain (RC) complexes (I–IV) embedded in the inner mitochondrial membrane (IMM), producing water and releasing the energy necessary to pump protons from the mitochondrial matrix to the intermembrane space (IMS). This results in the formation of transmembrane electrochemical ion gradient across the IMM, also called the proton-motive force (pmf). The outer side of the IMM is positively charged (the p-side) while the inner side is negatively charged (the n-side). The pmf enables the F1Fo ATP synthase to produce ATP from ADP and inorganic phosphate (Boyer, 1997). The OXPHOS complexes contain ~90 structural proteins of which 13 are encoded by the mtDNA in humans.
More than 150 distinct genetic mitochondrial dysfunction syndromes characterized by a diminished OXPHOS capacity have been described (Tuppen et al., 2010; Hejzlarova et al., 2014; Chinnery, 2015; Ng and Turnbull, 2015; Stewart and Chinnery, 2015; Xu et al., 2015). These diseases affect at least 1 in 5,000 live human births (Skladal et al., 2003). Typical clinical traits include visual/hearing defects, encephalopathies, cardiomyopathies, myopathies, diabetes, liver, and renal dysfunctions (Table 1; Dimauro and Schon, 2003; Zeviani and Carelli, 2007; Vafai and Mootha, 2012). Many known cases result from alterations in mtDNA (~15%, e.g., NARP, MILS, LHON), which occur as a result of this DNA's high susceptibility to mutations because of the nearby ROS production and the poor effectiveness of the mitochondrial DNA repair system (Wallace, 2010). More than 600 different point mutations and innumerable large-scale rearrangements of mtDNA have been implicated in human diseases (Lott et al., 2013).
Table 1.
Diseases and syndromes caused by mutations in ATP8 and ATP6.
| Disease/Syndrome | Phenotypes |
|---|---|
| Apical hypertrophic cardiomyopathy (AHCM) and neuropathy | primary disease of the myocardium (the muscle of the heart) in which a portion of the myocardium is hypertrophied (thickened) without any obvious cause, creating functional impairment of the cardiac muscle; neuropathy is damage to or disease affecting nerves, which may impair sensation, movement, gland or organ function, or other aspects of health, depending on the type of nerve affected |
| Ataxia | genetic disorders characterized by slowly progressive incoordination of gait and is often associated with poor coordination of hands, speech, and eye movements, with full mental capacity |
| Autism | neurodevelopmental disorder, characterized by impaired social interaction, verbal and non-verbal communication, and restricted and repetitive behavior; noticeably affected by mitochondrial dysfunction which impairs energy metabolism |
| Charcot-Marie-Tooth syndrome (CMT) | hereditary disorders that damage the nerves in arms and legs (peripheral nerves); symptoms usually begin in feet and legs, but they may eventually affect hands and arms |
| Encephalopathy | abnormal brain function or brain structure, symptoms may be mental or physical dysfunctions, depending on what part of the brain is being affected |
| Epilepsy with Brain Pseudoatrophy | brain disorder manifesting by seizures, dementia, convulsions, loss of control on muscles, difficulties with talking |
| Episodic Weakness | muscular disorders (myopathies) that are only present after exercise or are exacerbated by exercise, skeletal muscle diseases, may be accompanied by neurological symptoms |
| Hereditary Spastic Paraplegia (HSP) | heterogeneous disorder characterized by lower extremity spasticity and weakness |
| Familial Bilateral Striatal Necrosis (FBSN) | acute neurological syndrome associated with radiological findings, respiratory illnesses presenting with an array of neurological findings, including axial ataxia, grimacing, mutism, head nodding, and high-pitched cry |
| Infantile cardiomyopathy | severe, eventually fatal, cardiac arrhythmias, characterized pathologically by cardiac hypertrophy and by a distinctive type of focal degeneration of the muscle cells, which lose their myofibrils, undergo marked mitochondrial hyperplasia, become rounded in shape and enlarged, and resemble histiocytes |
| Leber Hereditary Optic Neuropathy (LHON) | maternally inherited disease leading to acute bilateral blindness due to loss of the optic nerve and papillomacular bundle nerve fibers, predominantly in young men |
| Left Ventricular HyperTrabeculation syndrome (LVHT) (noncompaction) | myocardial abnormality of the apex, characterized by multiple, myocardial cotyledo-like protrusions and interwoven strings, all lined by endocardium; in three quarters of the cases associated with neuromuscular disorders |
| Maternally Inherited Diabetes and Deafness syndrome (MIDD) | form of diabetes that is often accompanied by hearing loss, especially of high tones, characterized by high blood sugar levels (hyperglycemia) resulting from a shortage of the hormone insulin |
| Maternally Inherited Leigh Syndrome (MILS) | early-onset progressive neurodegenerative disorder with a characteristic neuropathology consisting of focal, bilateral lesions in one or more areas of the central nervous system, manifesting with (encephalopathy), lactic acidosis, seizures, heart disease (cardiomyopathy), breathing (respiratory) abnormalities, and developmental delays |
| Mesial Temporal Lobe Epilepsies with Hippocampal Sclerosis (MTLE-HS) | chronic neurological condition characterized by recurrent, unprovoked epileptic seizures (epilepsy) which originate in the temporal lobe of the brain, its pathophysiological substrate is usually hippocampal sclerosis, the most common epileptogenic lesion encountered in patients with epilepsy |
| Metabolic Syndrome (MS) | disorder characterized by a group of metabolic abnormalities including hyperglycemia, hypertension, hyperlipaemia and central obesity, which are the risk factors of cardiovascular disease and diabetes |
| Motor Neuron Syndrome (MNS) | cognitive impairment, exercise intolerance, and progressive muscle weakness |
| Myopathy, Lactic Acidosis, and Sideroblastic Anemia (MLASA) | mitochondrial disorder specific to skeletal muscle and bone marrow with myopathy, lactic acidosis, and sideroblastic anemia with ringed sideroblasts |
| Neurogenic Ataxia Retinis Pigmentosa syndrome (NARP) | mitochondrial disease affecting nervous system manifesting with pain in the arms and legs (sensory neuropathy), muscle weakness, problems with balance and coordination (ataxia), vision loss caused by retinitis pigmentosa or changes in the light-sensitive tissue that lines the back of the eye |
| Periodic paralyzes | genetic diseases that lead to weakness or paralysis (rarely death) from common triggers such as cold, heat, high carbohydrate meals, not eating, stress or excitement, and physical activity of any kind |
| Schizophrenia | mental disorder, often characterized by abnormal social behavior and failure to recognize what is real |
| SpinoCerebellar Ataxia (SCA) | ataxia that is due to dysfunction of the cerebellum |
| Tetralogy of Fallot (ToF) | type of congenital heart disease, essentially a right-sided heart disease, with characteristic features of ventricular septal defect, right ventricular outflow tract obstruction, aortic dextroposition, and right ventricular hypertrophy |
This review focuses on mutations in the MT-ATP8 and MT-ATP6 genes (further named ATP8 and ATP6) encoding subunits 8 and a of ATP synthase, respectively, that were identified in patients with various disorders. We summarize what is known about their clinical and functional consequences. Based on recent high-resolution structures (Morales-Rios et al., 2015a; Zhou et al., 2015; Hahn et al., 2016; Guo et al., 2017), we define the topological locations of these mutations, which helps understand their impact on ATP synthase structure, function and assembly.
ATP synthase structure and function
Mitochondrial ATP synthase is a unique macromolecular rotary machine of ~625 kDa. It is composed of typically 17 different protein subunits (Figure 1) and organizes into a membrane-extrinsic F1 catalytic and membrane-embedded Fo domains, which are connected by a peripheral and central stalk (Allegretti et al., 2015; Morales-Rios et al., 2015a; Zhou et al., 2015). The matrix-oriented F1 is composed of a prominent (αβ)3 catalytic head into which the γδε central stalk rotor penetrates. The Fo c-ring typically consists of identical c subunits (subunit 9 in yeast). Together with subunit a, the c-ring shuttles protons across the membrane. The Fo domain further consists of subunits 8 (alias A6L), b (4 in yeast), f, d, F6 (h in yeast), and OSCP that together form the peripheral stalk connecting the catalytic head with the membrane stator. Three mitochondria-specific subunits, e, g, k, induce either directly or indirectly the formation of ATP synthase dimers (Hahn et al., 2016) that self-assemble in longer ribbons important for cristae formation (Parsons, 1963; Strauss et al., 2008).
Figure 1.
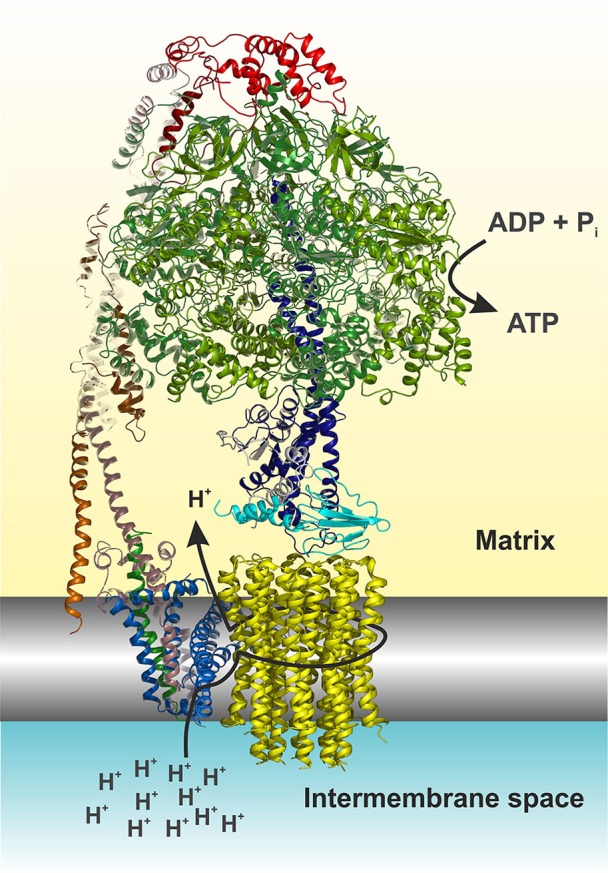
Cartoon representation of the yeast F1Fo ATP synthase. The view is horizontally to the membrane shown in grayscale. The structure was drawn according to (Hahn et al., 2016, PDB code 5FL7). For simplicity the structure is shown without subunits e, f, g, I, and a truncated subunit b. The figure was made in Pymol (The PyMOL Molecular Graphics System, Version 0.99, Schrödinger, LLC), using the following color code: α, forest; β, split pea; γ, density; δ, cyan; ε, white; OSCP, red; b (= 4 in yeast), dirty violet; d, orange; h, salmon; 8, green; a, blue; c-ring, yellow. The arrows indicate the path of protons (see also Figure 4) and nucleotide conversion. For details see text.
The ATP synthase harbors a unique rotary mechanism driven by the pmf to translocate ions through Fo, to generate rotation of its rotor and transmit torque into the F1 catalytic head where finally ATP is synthesized and released. Subunit a provides a pathway that involves a number of hydrophilic amino acids, which allows protons to enter from the IMS (Figure 1). Approximately in the middle of the membrane the proton can bind to a highly conserved acidic residue of subunit c helix 2 (cH2) (cE59 in H. sapiens) located at the outer surface of the c-ring. It has been suggested that the binding of a proton on this carboxylate residue disrupts a previously established electrostatic interaction of cE59 with a highly conserved, positively charged arginine residue in subunit a membrane helix 5 (aH5) (aR159 in H. sapiens; Vik and Antonio, 1994; Junge et al., 1997; Pogoryelov et al., 2010; Guo et al., 2017). This arginine acts as an electrostatic separator between the proton pathway from the IMS to the middle of the membrane and a second, spatially separated pathway that allows incoming protons still bound on the c-ring glutamate to be released into the matrix (Mitome et al., 2010). The operation direction of this process is primarily driven by the ion gradient that causes a ratchet type mechanism of the neutralized c-ring glutamate in the hydrophobic membrane, which is energetically unfavorable and does not allow the back stepping without externally applied force (Vik and Antonio, 1994; Junge et al., 1997). After an almost complete revolution of the c-ring, the glutamate is deprotonated in the aqueous exit channel (Pogoryelov et al., 2010). This channel is formed by hydrophilic residues of the c-ring/subunit a interface at the matrix side through which the protons can reach the matrix (n-Side) (Allegretti et al., 2015; Morales-Rios et al., 2015a; Zhou et al., 2015; Hahn et al., 2016; Guo et al., 2017). The c-ring is tightly bound to the central stalk subunits γδε, of which subunit γ protrudes into the F1 catalytic head, which induces cyclic conformational changes when rotating (Abrahams et al., 1994). Consequently, ADP and Pi are sequentially converted at the catalytic sites of the three subunits β, according to the binding change mechanism (Boyer, 1997). Cryo-EM structures of the bovine Bos taurus and yeasts Yarrowia lipolytica and Saccharomyces cerevisiae F1Fo ATP synthases, that are basically of the same subunit composition and structural construction as the human enzyme, have been described recently [15–17]. These structures show a very similar overall architecture and differ only with respect to the subunit c (9) stoichiometry (8 in mammals, 10 in yeasts), the loss of the dimerization domain subunits (e/g/k) during purification (yeast) and the non-essential and yeast specific subunits i and k (Zhou et al., 2015; Hahn et al., 2016; Guo et al., 2017). It therefore has become feasible to build reliable structural models of the membrane domain (Fo) of the eukaryotic, mitochondrial, ATP synthase (Figure 1), and to map the human disease-causing mutations at the molecular level within the ATP synthase structure and to pin-point their potentially adverse effects on the above-described mechanism.
Yeast and human cellular models of mtDNA diseases
Human cells contain up to thousands copies of mtDNA (Miller et al., 2003). Mutations in this DNA are highly recessive and usually co-exist with wild type mtDNA molecules, a situation referred to as heteroplasmy. A mutational load above 60% is usually required to induce a clinical phenotype (Stewart and Chinnery, 2015). Given the high mutational rate of the mitochondrial genome and the presence of numerous family or population-specific polymorphisms, it can be difficult to distinguish between a neutral mtDNA variant and a disease-causing mutation. Additionally, the effects of deleterious mtDNA mutations might be exacerbated by mtDNA nucleotide changes that are not pathogenic per se and by unknown factors in nuclear genetic background, i.e., the so-called modifier genes (Cai et al., 2008; Swalwell et al., 2008). These features make it difficult from patient's cells and tissues to precisely know how specific mtDNA mutations influence oxidative phosphorylation.
To better characterize the effects of mtDNA mutations, homoplasmic cell lines, i.e. with a 100% mutational load, in a defined nuclear genetic background are required. To this end King and Attardi (King and Attardi, 1989) developed an approach that used cybrid (cytoplasmic hybrid) cell lines obtained by fusing enucleated cytoplasts from patient's cells with cells lacking mtDNA (ρ0). This approach was used to evaluate the bioenergetics consequences of 11 ATP6 mutations (Trounce et al., 1994; Majander et al., 1997; Nijtmans et al., 2001; Carrozzo et al., 2004; Mattiazzi et al., 2004; Pallotti et al., 2004; Jonckheere et al., 2008; Sikorska et al., 2009; Aure et al., 2013; Blanco-Grau et al., 2013; Lopez-Gallardo et al., 2014; Hejzlarova et al., 2015; Wen et al., 2016). Another approach exploits unique features of S. cerevisiae. Mitochondria from this single-celled fungus and humans show many similarities (Steinmetz et al., 2002; Prokisch et al., 2004; Reinders et al., 2006; Pagliarini et al., 2008; Rhee et al., 2013), and mitochondrial genetic transformation can be achieved in this yeast in a highly controlled fashion, by the biolistic delivery into mitochondria of in-vitro-made mutated mtDNA fragments, followed by their integration into wild type mtDNA by homologous DNA recombination (Bonnefoy and Fox, 2001). Being unable to stably maintain heteroplasmy (Okamoto et al., 1998), it is easy to obtain yeast homoplasmic populations where all mtDNA molecules carry a mutation of interest. Owing to its good fermenting capacity, yeast models of human mitochondrial diseases can be kept alive when provided with sugars like glucose even when oxidative phosphorylation is completely inactivated (Baile and Claypool, 2013; Lasserre et al., 2015). This approach was used to investigate the impact on ATP synthase of nine ATP6 mutations identified in patients (Rak et al., 2007; Kucharczyk et al., 2009a,b,c, 2010, 2013; Kabala et al., 2014; Lasserre et al., 2015; Niedzwiecka et al., 2016; Wen et al., 2016).
Pathogenic mutations in ATP8 and ATP6
Subunits 8 and a are synthesized from a bi-cistronic mRNA unit (Figure 2). The two genes show a 46 nucleotide overlap. Thus, mutations in this unit can affect either subunit a or 8, or both. Currently, 36 different ATP8 and ATP6 mutations with a confirmed or suspected pathogenic character are recorded in MITOMAP database (Figure 3). We here review 22 additional mutations found in literature. The nucleotide and amino acid changes induced by these mutations, and what is known about their functional and clinical consequences is summarized in Table 2.
Figure 2.

ATP6 and ATP8 genes and sites of pathogenic mutations. The two coding sequences overlap, between positions 8527 and 8572 (cyan) of the human mitochondrial genome. Start and end nucleotide numbers are indicated. The number of mutations (m) identified in patients and their positions are indicated. These mutations are listed in Table 2.
Figure 3.

Sequence alignment of subunits a, b and 8 from a selected range of species. (A) Alignment of bacterial subunits b (b′ or b2 isoform) and mitochondrial subunits 8. The sequences of subunit b are from Rhodobacter sphaeroides (R.s.), Ruegeria sp. (R.sp), Roseobacter denitrificans (R.d.), Jannaschia sp. (J.sp) and Dinoroseobacter shibae (D.s.). The length of N- and C-termini extensions are given. The sequences of subunit 8 are from a selection of mammals and the yeast species Schizosaccharomyces pombe (S.p.), Podospora anserina (P.a.), Yarrowia lipolytica (Y.l.), Saccharomyces cerevisiae (S.c.) and Candida glabrata (C.g.). (B) Alignment of subunit a. Homo sapiens (H.s.), Bos taurus (B.t.). The sequences corresponding to overlapping ATP8 and ATP6 genes are marked in cyan as in Figure 2. Peptide sequences removed in the mature form of yeast subunit a are marked in yellow. At the top and bottom, the arrows mark the locations of the human mutations (Table 2) and the mutations modeled in S.c., respectively. The arrows are colored according to Figure 4. At the bottom, the secondary structural elements are drawn according to PSIPRED prediction (2D) and a cryo-EM structure (3D) (Hahn et al., 2016).
Table 2.
Pathogenic mutations in ATP8 and ATP6 genes, associated disease(s)/syndrome(s), number of cases (N), patient's age (year), heteroplasmy (H), pathogenicity (PG), and ATP synthase activities and mitochondrial morphology.
| mtDNA mutation, m. | Protein mutation | Disease(s) syndrome(s) | N | Age (y) | H % | ATP synthase | ROS/Mitochondria morphology | References | PG | |
|---|---|---|---|---|---|---|---|---|---|---|
| Activity | Assembly/Stability | |||||||||
| 8381A>G | 8T6A | MIDD, LVHT | 2 | 38,57 | 99 | nd | nd | nd/Abnormal | Perucca-Lostanlen et al., 2000; Finsterer et al., 2004 | U |
| 8393C>T | 8P10S | Epilepsy with brain pseudoatrophy | 1 | 10 | <99 | nd | nd | nd/nd | Galimberti et al., 2006 | U |
| 8403T>C | 8I13T | Episodic weakness | 1 | 8 | 99 | Normal | Normal | Higher/nd | Aure et al., 2013 | U |
| 8411A>G | 8M16V | Neurologic disorder with blindness | 1† | 10 | 97 | nd | nd | nd/nd | Mkaouar-Rebai et al., 2010 | U |
| 8463A>G | 8Y33C | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Sequeira et al., 2015 | U |
| 8472C>T | 8P36L | Autism | 3 | 4–8 | 100 | nd | nd | nd/nd | Piryaei et al., 2012 | U |
| 8481C>T | 8P39L | ToF | 1 | 1 | 100 | nd | nd | nd/nd | Tansel et al., 2014 | U |
| 8502A>T | 8N46I | MTLE-HS | 40 | Adults | 11–36 | nd | nd | nd/nd | Gurses et al., 2014 | S |
| 8510A>G | 8K49E | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Ueno et al., 2009 | U |
| 8519G>A | 8E52K | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Sequeira et al., 2015 | U |
| 8528T>C | 8W55R aM1T | Infantile cardiomyopathy | 5, 3† | 0.1–4 | 90–98 | Reduced | nd | nd/nd | Ware et al., 2009; Imai et al., 2016 | S |
| 8529G>A | 8W55X aM1I | Apical hypertrophic cardiomyopathy and neuropathy | 1 | 16 | 90 | Reduced | Defective | nd/nd | Jonckheere et al., 2008 | S |
| 8558C>T | 8P65S aA11V | LVHC | 1 | 0.2 | nd | nd | nd | nd/nd | Tang et al., 2010 | C |
| 8561C>G | 8P66A aP12R | Ataxia, neuropathy, diabetes mellitus | 2 | 59, 64 | 99 | Reduced | Defective | nd/nd | Kytovuori et al., 2016 | S |
| 8527A>G | aM1V | Neuromuscular Disorder | 1 | 7 | nd | nd | nd | nd/nd | Felhi et al., 2016 | U |
| 8597T>C | aI24T | MILS | 1 | 2 | 95 | nd | nd | nd/nd | Tsai et al., 2012 | U |
| 8611insC | aL29PfsX36 | Ataxia, Encephalopathy | 1 | 4 | 60–80 | Reduced | Defective | nd/Abnormal | Jackson et al., 2017 | S |
| 8618insT | aT33HfsX32 | NARP | 1 | 40 | 85 | nd | Defective | nd/nd | Lopez-Gallardo et al., 2009 | U |
| 8668T>C | aW48R | LHON | 1 | Adult | 99 | nd | nd | nd/nd | Kumar et al., 2010 | U |
| 8684C>T | aT53I | Autism associated Ovarian insufficiency LHON | 1 7 1 | 4–8 25 Adult | 100 | nd | nd | nd/nd | Piryaei et al., 2012 Venkatesh et al., 2010 Kumar et al., 2010 | U |
| 8697G>A | aM57I | Autism associated LHON | 5 | 4–8 | 100 | nd | nd | nd/nd | Kumar et al., 2010; Piryaei et al., 2012 | U |
| 8701A>G | aT59A | Autism associated | 1 | 4–8 | 100 | nd | nd | nd/nd | Piryaei et al., 2012 | U |
| 8719G>A | aG65X | Suspected myopathy | 1 | nd | <99 | nd | nd | nd/nd | Tang et al., 2013 | U |
| 8723G>A | aR66Q | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Ueno et al., 2009 | U |
| 8794C>T | aH90Y | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Sequeira et al., 2015 | U |
| 8836A>G | aM104V | LHON Autism associated | 1 3 | 11 4–8 | 100 | nd | nd | nd/nd | Abu-Amero and Bosley, 2006 Piryaei et al., 2012 | U |
| 8839G>C | aA105P | NARP | 1 | 57 | 21–88 | Normal | nd | nd/nd | Blanco-Grau et al., 2013 | S |
| 8843T>C | aI106T | Schizophrenia | 2 | nd | 100 | nd | nd | nd/nd | Ueno et al., 2009; Sequeira et al., 2015 | U |
| 8851T>C | aW109R | FBSN | 2 | 3 | 87–99 | Reduced | Defective | nd/Abnormal | De Meirleir et al., 1995; Honzik et al., 2013; Kucharczyk et al., 2013 | S |
| 8890A>G | aK122E | MS | 1 | 18 | 35–38 | nd | nd | nd/nd | Ye et al., 2013 | U |
| 8902G>A | aA126T | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Ueno et al., 2009 | U |
| 8932C>T | aP136S | Neuromuscular Disorder | 1 | 7 | 100 | Reduced | Defective | nd/nd | Petros et al., 2005; Felhi et al., 2016; Niedzwiecka et al., 2016 | S |
| 8945T>C | aM140T | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Ueno et al., 2009 | U |
| 8950G>A | aV142I | LHON plus dystonia | 1 | 23 | nd | Reduced | nd | nd/nd | Abu-Amero and Bosley, 2005 | U |
| 8969G>A | aS148N | MLASA Nephropathy | 1 1 | 6 14 | 60–90 | Reduced | Defective | Higher/nd | Burrage et al., 2014 Wen et al., 2016 | C |
| 8989G>C | aA155P | NARP | 1 | 53 | 33–94 | Reduced | nd | nd/nd | Duno et al., 2013; Wen et al., 2016 | S |
| 8993T>G | aL156R | NARP/MILS | 53, 10† | 0.1–53 | 13–99 | Reduced | Normal | Higher/Abnormal | Holt et al., 1990; Tatuch et al., 1992; Ciafaloni et al., 1993; Puddu et al., 1993; Trounce et al., 1994; Houstek et al., 1995; Uziel et al., 1997; Vazquez-Memije et al., 1998; Baracca et al., 2000, 2007; Garcia et al., 2000; Nijtmans et al., 2001; Carrozzo et al., 2004; Mattiazzi et al., 2004; Pallotti et al., 2004; Sakai et al., 2004; Enns et al., 2006; Morava et al., 2006; Rojo et al., 2006; Sgarbi et al., 2006; Cortes-Hernandez et al., 2007; Rak et al., 2007 | C |
| 8993T>C | aL156P | NARP/MILS | 35, 4† | 0.1–77 | 20–96 | Reduced | Defective | Higher/Normal | De Vries et al., 1993; Santorelli et al., 1994; Fujii et al., 1998; Vazquez-Memije et al., 1998; Vilarinho et al., 2001; Hurvitz et al., 2002; Pallotti et al., 2004; Morava et al., 2006; Baracca et al., 2007; Craig et al., 2007; Debray et al., 2007; Kucharczyk et al., 2009a; Kara et al., 2012; Aure et al., 2013; Martikainen et al., 2015 | C |
| 9011C>T | aA162V | LHON | 1 | 34 | 100 | nd | nd | nd/nd | Shidara and Wakakura, 2012 | U |
| 9016A>G | aI164V | LHON | 1 | Adult | 100 | nd | nd | nd/nd | Povalko et al., 2005 | U |
| 9025G>A | aG167S | MILS-like, NARP | 2, 1† | 0.2, nd | 100 | nd | nd | nd/nd | Lopez-Gallardo et al., 2014 | U |
| 9029A>G | aH168R | LHON-like | 1 | 38 | 95–99 | Reduced | nd | Higher/nd | Lopez-Gallardo et al., 2014 | S |
| 9032T>C | aL169P | NARP | 1 | 16 | 70–90 | Reduced | nd | Higher/nd | Lopez-Gallardo et al., 2014 | S |
| 9035T>C | aL170P | SCA | 21 | 4–48 | 90–99 | Reduced | nd | Higher/nd | Sikorska et al., 2009; Pfeffer et al., 2012 | C |
| 9055G>A | aA177T | Schizophrenia | 3 | nd | 100 | nd | nd | nd/nd | Sequeira et al., 2015 | U |
| 9058A>G | aT178A | LVHT | 1 | nd | nd | nd | nd | nd/nd | Tang et al., 2010 | U |
| 9071C>T | aS182L | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Ueno et al., 2009 | U |
| 9094C>T | aL190F | Ovarian insufficiency | 5 | 25 | nd | nd | nd | nd/nd | Venkatesh et al., 2010 | U |
| 9101T>C | aI192T | LHON | 1 | 21 | 100 | Reduced | nd | nd/nd | Lamminen et al., 1995; Majander et al., 1997 | S |
| 9127delAT | aI201PfsX2 | NARP | 1 | 18 | 10–82 | Reduced | nd | nd/nd | Mordel et al., 2017 | S |
| 9134A>G | aE203G | MS with cardiomyopathy | 1 | nd | nd | Reduced | nd | nd/nd | Honzik et al., 2012 | U |
| 9139G>A | aA205T | LHON | 2 | 30,45 | nd | nd | nd | nd/nd | La Morgia et al., 2008 | U |
| 9160T>C | aY212H | Schizophrenia | 1 | nd | 100 | nd | nd | nd/nd | Sequeira et al., 2015 | U |
| 9176T>G | aL217R | MILS | 6, 3† | 3–42 | 95–99 | Reduced | Defective | Higher/Abnormal | Thyagarajan et al., 1995; Dionisi-Vici et al., 1998; Makino et al., 1998; Okamoto et al., 1998; Jacobs et al., 2005; Kucharczyk et al., 2009b; Ronchi et al., 2011; Verny et al., 2011; Synofzik et al., 2012; Aure et al., 2013 | C |
| 9176T>C | aL217P | MILS, Periodic paralyzes, CMT, HSP | 19, 4† | 1–59 | 90–99 | Reduced | Defective | Higher/Abnormal | Thyagarajan et al., 1995; Dionisi-Vici et al., 1998; Makino et al., 1998; Okamoto et al., 1998; Jacobs et al., 2005; Kucharczyk et al., 2010; Ronchi et al., 2011; Verny et al., 2011; Synofzik et al., 2012; Aure et al., 2013 | C |
| 9185T>C | aL220P | Periodic paralyzes, Ataxia, MILS, CMT, MNS, SCA | 61, 4† | 2–58 | 73–99 | Reduced | Defective | nd/Abnormal | Moslemi et al., 2005; Castagna et al., 2007; Childs et al., 2007; Saneto and Singh, 2010; Pfeffer et al., 2012; Pitceathly et al., 2012; Aure et al., 2013; Brum et al., 2014; Kabala et al., 2014 | C |
| 9191T>C | aL222P | MILS | 1† | 2 | 90–94 | Reduced | nd | nd/Abnormal | Moslemi et al., 2005; Kabala et al., 2014 | S |
| 9205delTA | aSTOP elimination | Encephalopathy/Lactic acidosis | 3, 1† | Adult, nd | 98–99 | Reduced | nd | nd/Abnormal | Seneca et al., 1996; Jesina et al., 2004; Hejzlarova et al., 2015 | C |
–nr of died patients, nd, no data; PG, pathogenicity; C, confirmed; S, suspected; U, unknown; mutations not present in the MITOMAP list are underlined.
Mutations affecting only subunit 8
Ten mutations affecting only subunit 8 were identified in patients presenting with various disorders: epilepsy [m.8502A>T (8N46I)] (Gurses et al., 2014); LVHT or MIDD [m.8381A>G (8T6A)] (Perucca-Lostanlen et al., 2000; Finsterer et al., 2004); brain pseudo-atrophy, episodic weakness and neurological disorders [m.8393C>T (8P10S) (Galimberti et al., 2006), m.8403T>C (8I13T) (Aure et al., 2013), and m.8411A>G (8M16V) (Mkaouar-Rebai et al., 2010)], heart problems [m.8481C>T (8P39L)] (Tansel et al., 2014); schizophrenia [m.8463A>G (8Y33C), m.8510A>G (8K49E) and m.8519G>A (8E52K)] (Ueno et al., 2009; Sequeira et al., 2015); and autism [m.8472C>T (8P36L)] (Piryaei et al., 2012).
Mutations affecting both subunits 8 and a
Three mutations affecting both subunits 8 and a [m.8528T>C (8W55R + aM1T), m.8529G>A (8W55STOP + aM1I), m.8558C>T (8P65S + aA11V)] were identified in patients suffering from severe cardiomyopathies (Jonckheere et al., 2008; Ware et al., 2009; Tang et al., 2010; Imai et al., 2016). A fourth mutation affecting both proteins [m.8561C>G (8P66A + aP12R)] was detected in individuals with features of cerebellar ataxia, peripheral neuropathy and diabetes mellitus (Kytovuori et al., 2016).
Mutations affecting only subunit a
Most frequent mutations
Two mutations at the same amino acid position of subunit a, m.8993T>G (aL156R) and m.8993T>C (aL156P), were identified in numerous patients presenting with the NARP or MILS syndrome depending on the mutation load (Uziel et al., 1997; Jonckheere et al., 2012). The first one was consistently found to severely compromise mitochondrial ATP production, with deficits of up to 90%. While most studies concluded this was due to a block in proton translocation, some suggested a less efficient coupling or defects in the assembly/stability of ATP synthase (Tatuch et al., 1992; Trounce et al., 1994; Houstek et al., 1995; Vazquez-Memije et al., 1998; Garcia et al., 2000; Nijtmans et al., 2001; Carrozzo et al., 2004; Mattiazzi et al., 2004; Pallotti et al., 2004; Morava et al., 2006; Sgarbi et al., 2006; Baracca et al., 2007; Cortes-Hernandez et al., 2007). Although its pathogenic character is firmly established, the second mutation has less severe consequences on ATP synthase with a 70% drop in ATP production mainly because of a less efficient assembly or diminished stability of subunit a (Vilarinho et al., 2001; Morava et al., 2006; Craig et al., 2007; Kucharczyk et al., 2009a; Aure et al., 2013). In addition to bioenergetic deficits, the two mutations lead to an enhanced production of ROS and aberrant mitochondrial morphologies that may contribute to the disease process as well.
Similar diseases were associated with two mutations at amino acid position 217, m.9176T>G (aL217R), and m.9176T>C (aL217P). The first one is extremely detrimental with a block in subunit a assembly that leads to extreme clinical phenotypes when highly abundant in cells and tissues. The second one does not obviously compromise assembly of subunit a indicating that it affects the functioning of ATP synthase.
The m.9185T>C (aL220P) mutation was identified in individuals presenting with MILS, MNS, periodic paralyzes, spinocerebellar ataxia syndromes (SCA) or CMT (Castagna et al., 2007; Childs et al., 2007). Biochemical analyses revealed a substantial drop (50–90%) in ATP production, and study in yeast indicated that this mutation compromises the functioning of ATP synthase (Kabala et al., 2014).
Other, less frequent, mutations in subunit a
Eight additional mutations in subunit a [m.8597T>C (aI24T), m.8618insT, m.8839G>C (aA105P), m.8989G>C (aA155P), m.9025G>A (aG167S), m.9032T>C (aL169P), m.9127delAT (aI201PfsX2) and m.9191T>C (aL222P)] were identified in patients presenting with NARP or MILS disease. Although they were thus far found only in a limited number of individuals, biochemical investigations indicated that they have detrimental consequences for ATP synthase. For instance, the m.9191T>C was shown to dramatically affect incorporation of subunit a in the yeast enzyme (Kabala et al., 2014). These mutations are thus most likely pathogenic.
The m.8969G>A (aS148N) mutation was found in a patient presenting with MLASA (Burrage et al., 2014) and a 14-year-old Chinese girl diagnosed with a severe nephropathy (Wen et al., 2016). Biochemical investigations in yeast and human cells revealed a block in the transfer of protons through the Fo.
The m.8851T>C (aW109R) and m.8890A>G (aK122E) mutations were identified in patients presenting with FBSN and MS (De Meirleir et al., 1995; Honzik et al., 2013; Ye et al., 2013). Studies with yeast revealed that m.8851T>C leads to major drops (95%) in mitochondrial ATP synthesis owing to a block in Fo-mediated proton transfer (Kucharczyk et al., 2013). The m.9134A>G (aE203G) was identified in patient suffering from MS accompanied with cardiomyopathy (Honzik et al., 2012).
The m.9035T>C (aL170P) was identified at high (>90%) mutation load in 21 ataxia patients (Sikorska et al., 2009; Pfeffer et al., 2012). Cybrids carrying this mutation had reduced ATP levels (40–50% vs. controls) and produced 5–7 times more ROS than control cells. Another mutation m.8611insC (aL29PfsX36) was found in patient presenting ataxia with encephalomyopathy (Jackson et al., 2017).
Ten mutations [m.8668T>C (aW48R), m.8684C>T (aT53I), m.8697G>A (aM57I), m.8836A>G (aM104V), m.8950G>A (aV142I), m.9011C>T (aA162V), m.9016A>G (aI164V), m.9029A>G (aH168R), m.9101T>C (aI192T), and m.9139G>A (aA205T)] were identified in patients presenting with LHON, a disease caused by defects in the retinal ganglion cells and optic nerve that lead to blindness. Two of them (m.9029A>G and m.9101T>C) were shown to compromise oxidative phosphorylation by a yet-unknown mechanism (Lamminen et al., 1995; Lopez-Gallardo et al., 2014).
The m.8932C>T (aP136S) and m.8527A>G (aM1V) were identified in children with neuromuscular disorders (Felhi et al., 2016). The first one was also identified in prostatic cancer cells (Petros et al., 2005). In a yeast model of this mutation, ATP synthase assembly/stability was found substantially affected (Niedzwiecka et al., 2016).
The m.9205delTA (aSTOP elimination) mutation was identified in patients with a severe encephalopathy leading to premature death. Since the stop codon of ATP6 overlaps with the start codon of COX3, the expression of both genes is compromised, which results in a lower content in complex IV and ATP synthase (Jesina et al., 2004; Hejzlarova et al., 2015).
The m.8719G>A (aG65STOP) and m.9058A>G (aT178A) mutations were identified in patients presenting with LVHT (Tang et al., 2010, 2013). Their biochemical consequences are still unknown.
Ten mutations [m.8701A>G (aT59A), m.8723G>A (aR66Q), m.8794C>T (aH90Y), m.8843T>C (aI106T), m.8902G>A (aA126T), m.8945T>C (aM140T), m.9055G>A (aI177T), m.9071C>T (aS182L), m.9094C>T (aL190F), and m.9160T>C (aY212H)] were found in patients with autism or schizophrenia. Their consequences on ATP synthase has not yet been investigated.
It is puzzling that mutations that cluster in specific regions of ATP6 or ATP8 give rise to such a wide variety of clinical symptoms (Table 1). These phenotypic differences may be due to other unknown genetic variations in patients within mitochondrial or nuclear DNA that could exacerbate or attenuate the consequences on health of defects in ATP synthase subunits. Another important source of variability in the clinical outcome likely resides in the levels of heteroplasmy and different distributions of mtDNA mutations in cells and tissues. Furthermore, in addition to a lack of ATP, defects in ATP synthase may have multiple secondary effects, such as increased production of ROS and changes in upstream metabolic processes (Korshunov et al., 1997) that together will influence the disease process unpredictably.
Topology within the Fo of mutations in subunits a and 8
To define the topology of the ATP6 and ATP8 mutations identified in patients, we used the recently published structures of Y. lipolytica and S. cerevisiae ATP synthases (Hahn et al., 2016; Guo et al., 2017); the complete model of subunits a, 8, and the c-ring is shown in Figure 4. The amino acid alignments in Figure 3 establish the correspondences with human subunits a and 8 amino acids.
Figure 4.

Positions of human neurodegenerative disease-causing mutations in the structure of the yeast mitochondrial ATP synthase subunits a and 8 (human A6L). The views are from the matrix (A,B) and from the IMS (C,D) with detailed views in the IMS entry channel (B) and in the matrix exit channel (D). On (E,F), showing views along the membrane plane from outside the Fo stator and from the c-ring, respectively, the membrane borders are indicated as black lines. Subunits a, 8, and c are shown in blue, green, and yellow, respectively. The model is based on structural data from the Y. lipolytica and S. cerevisiae structures (Hahn et al., 2016; Guo et al., 2017). The positions of mutations, which have been found in human neurodegenerative diseases, are marked in white, red, blue, pink, orange and black for hydrophobic, negatively and positively charged, uncharged polar, proline and special residues, respectively. The mutations are labeled according to their positions in human subunit a listed in Table 2. The arrows indicate the path of protons in ATP synthesis direction. The conserved arginine (h.s. aR159, S.c. aR186) on helix aH5 is indicated by stick model; its orientation is randomly chosen. The figure was drawn with UCSF ChimeraX (Goddard et al., 2017).
Subunit a mutations
Helix 5 of subunit a (aH5) is kinked due to the presence of proline at position 153, a residue well-known for its propensity to bend or break alpha helices owing to its inability to participate fully in protein backbone hydrogen bonding. aP153 enables aH5 to follow the curvature of the c-ring and seal the two hydrophilic pockets that connect the a/c-ring interface to the intermembrane and matrix spaces. Five substitutions from hydrophobic alanine or leucine residues into proline are located on aH5 (aH5) in proximity to the essential aR159 residue (aA105P, aA155P and aL156P) that faces the proton binding glutamate of the c-ring, or close to aH168/aE203 in the proton entry channel (aL169P, aL170P). These mutations may distort or break aH5. Those at positions 155 and 156 at one helix turn from aR159 may compromise the ion translocation mechanism, for example by ion short circuiting (Mitome et al., 2010) or by preventing aR159 to interact with the c-ring glutamate due to its structurally shifted position. On aH6, the aL217P, aL220P, and aL222P mutations are on the edge of the exit channel close to the matrix. Their severe functional consequences possibly result from a change in the topology of the nearby aD224 residue that was suggested to be of critical importance for the exit of protons toward the mitochondrial matrix (Guo et al., 2017). Similarly, the aP136S change in the loop connecting aH4 and aH5 possibly alters the accessibility of protons in this region of subunit a.
The extremely detrimental consequences of aL217R on ATP synthase assembly/stability (Kucharczyk et al., 2009b) possibly results from the inability of subunit a to pack tightly owing to replacement of a hydrophobic residue with a bulkier and positively charged one within the membrane. The absence of proton conduction induced by the aL156R mutation at the a/c-ring interface (Rak et al., 2007), without any defect in ATP synthase assembly, may be caused by the inability of protons to exit from the ring or by electrostatic or steric hindrance that prevent rotation of the ring. Being located near the N-terminal side of aH5, the block in proton translocation induced by the aS148N mutation (Wen et al., 2016) possibly results from obstruction of the proton exit pore. The detrimental consequences of aH168R (Lopez-Gallardo et al., 2014) are not surprising considering its location in the p-side cleft in the proximity of the c-ring. This positive charge cuts off the connection of the p-side to the c-ring. A similar effect on the n-side of the membrane may result from the aW109R, where aH5 and aH6 diverge. A mutation at this position has an impact in proton translocation without impacting ATP synthase assembly/stability (Kucharczyk et al., 2013), indicating that this location of subunit a is close to the pathway along which protons are evacuated into the matrix. The clinical consequences of aK122E probably also result from a less efficient proton conduction toward the matrix.
While aM140T, aG167S, aA177T, aI192T, aA205T, and aY212H decrease the hydrophobicity of the a/c-ring interface, it is less obvious from our structural model to predict the consequences of other mutations that replace hydrophobic to non-charged hydrophilic residues (aI24T, aI106T, aA126T) or vice-versa (aT53I, aT59A, aT178A, aS182L) and those that lead to small hydrophobicity changes (aM57I, aM104V, aV142I, aA162V, aI164V, aL190F). However, meaningfully, most are within helices aH4-6 that are important for the movement of protons through Fo.
Subunit 8 mutations
The yeast subunit 8 has an overall kinked helical structure with a N-terminal transmembrane helix (8H1) and a short helix (8H2) exposed to the matrix. 8H1 is flanked by subunit i and aH1 (Hahn et al., 2016; Guo et al., 2017). At the distal stator side, 8H1 is exposed toward the dimer interface in a bent lipid bilayer region. At the base of the peripheral stalk, the C-terminus of 8H2 is nestled inside the helical domain composed by subunits b, d and f. In vertebrates, no secondary structures are predicted in the C-terminal half of subunit 8 and subunit f displays only 18% identity with its yeast homolog, which explains why they could not be modeled from the bovine ATP synthase cryo-EM density map (Zhou et al., 2015). Subunit 8 shows at its N-terminus a conserved 4 amino acid motif (MPQL, Figure 3). This motif and 8H1 share significant homologies with one of the two subunits b (isoform b′ or b2) from α-proteobacteria (Figure 3A), indicating that subunit 8 (A6L) is an evolutionary vestige of bacterial subunit b that remained mtDNA encoded (Hahn et al., 2016). Subunit 8 stabilizes in the membrane the helical N-terminal half of subunit a (Hahn et al., 2016). Four of the mutations identified in patients (8T6A, 8P10S, 8I13T, 8M16V) cluster at the N-terminal region of helix 8H1 in proximity to aH4, suggesting that they may affect the stability of subunits a and 8 (Hahn et al., 2016). The six other mutations identified in patients (8Y33C, 8P36L, 8P39L, 8N46I, 8K49E, 8E52K) are in the matrix-exposed helix 8H2. These mutations might affect the flexibility of the outer stalk, and thereby compromise the stability of Fo and/or, indirectly, the ion translocation mechanism or ATP synthase assembly process. This hypothesis is supported by the reduced functionality and stability of ATP synthase in these patients and by various studies on subunit b in the bacterial enzyme (Schneider and Altendorf, 1984, 1985; Wehrle et al., 2002; Greie et al., 2004).
ATP synthase dimers and mitochondrial morphology
The mitochondrial ATP synthase exists as dimers (Schagger and Pfeiffer, 2000; Paumard et al., 2002) that associate into rows that contribute to cristae formation (Davies et al., 2011; Hahn et al., 2016). The mutations in subunits 8 and a often correlate with pathological forms of mitochondria cristae as for example seen in the Leigh syndrome (Kucharczyk et al., 2009b). The defects caused by these mutations therefore not only affect the ATP synthase function but they can also affect the assembly process. The reduced amount, or lack thereof, of native and completely assembled ATP synthase dimers would certainly affect cristae formation, which is crucial for the accommodation of the OXPHOS respiratory chain complexes and the ATP synthase. This explains some of the pathologic forms of mitochondria found in the diseases and syndromes described in this review.
Conclusions
Diseases associated to mutations in the mitochondrial ATP6 and ATP8 genes are particularly challenging to study due to factors like heteroplasmy, complex inheritance, variable penetrance, and interactions with (e.g., nuclear) modifier genes, which makes it difficult to verify their pathogenicity. The possibility to create and keep alive homoplasmic strains of S. cerevisiae with defined mtDNA mutations in a controlled nuclear genetic background makes it possible to study their functional consequences. With the recently obtained cryo-EM structures of F1Fo ATP synthase from various mitochondrial origins it has become feasible to map mutations in ATP6 and ATP8 at the molecular level within the Fo. These “open eyes” provide the chance for a completely new level of understanding of how the mutations may affect ATP synthase structure, assembly, and mechanism. This knowledge also enables to evaluate the observed pathogenic forms of mitochondrial morphology that are associated with these syndromes on the structural level, from the mutation at the molecular level to its associated consequences at the macroscopic scale of the organelle. The recent technical advances enabling the structural analysis of macromolecular complexes by cryo-EM (Kühlbrandt, 2014), the advent of complete structures of ATP synthases (Allegretti et al., 2015; Morales-Rios et al., 2015b; Zhou et al., 2015; Hahn et al., 2016; Sobti et al., 2016; Guo et al., 2017) and the availability of genetically approachable systems like S. cerevisiae are just the first steps in these new shoes; they will considerably improve the comprehension of human diseases associated to defects in this key mitochondrial enzyme. We are still at the beginning of understanding these complex processes.
Author contributions
AD, AH, TM, DT-T, J-PdR, and RK discussed findings, analyzed literature and wrote the manuscript; AD, AH, and TM designed and made the Figures.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by grants from Association Française contre les Myopathies to J-PdR., the Agence Nationale de la Recherche (ANR-12-BSV8-024) to AD, the National Science Center of Poland (UMO-2016/23/B/NZ3/02098) to RK and by the Wellcome Trust to TM (WT110068/Z/15/Z).
References
- Abrahams J. P., Leslie A. G., Lutter R., Walker J. E. (1994). Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628. 10.1038/370621a0 [DOI] [PubMed] [Google Scholar]
- Abu-Amero K. K., Bosley T. M. (2005). Detection of mitochondrial respiratory dysfunction in circulating lymphocytes using resazurin. Arch. Pathol. Lab. Med. 129, 1295–1298. 10.1043/1543-2165(2005)129[1295:DOMRDI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Abu-Amero K. K., Bosley T. M. (2006). Mitochondrial abnormalities in patients with LHON-like optic neuropathies. Invest. Ophthalmol. Vis. Sci. 47, 4211–4220. 10.1167/iovs.06-0295 [DOI] [PubMed] [Google Scholar]
- Allegretti M., Klusch N., Mills D. J., Vonck J., Kühlbrandt W., Davies K. M. (2015). Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature 521, 237–240. 10.1038/nature14185 [DOI] [PubMed] [Google Scholar]
- Auré K., Dubourg O., Jardel C., Clarysse L., Sternberg D., Fournier E., et al. (2013). Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. Neurology 81, 1810–1818. 10.1212/01.wnl.0000436067.43384.0b [DOI] [PubMed] [Google Scholar]
- Baile M. G., Claypool S. M. (2013). The power of yeast to model diseases of the powerhouse of the cell. Front. Biosci. 18, 241–278. 10.2741/4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracca A., Barogi S., Carelli V., Lenaz G., Solaini G. (2000). Catalytic activities of mitochondrial ATP synthase in patients with mitochondrial DNA T8993G mutation in the ATPase 6 gene encoding subunit a. J. Biol. Chem. 275, 4177–4182. 10.1074/jbc.275.6.4177 [DOI] [PubMed] [Google Scholar]
- Baracca A., Sgarbi G., Mattiazzi M., Casalena G., Pagnotta E., Valentino M. L., et al. (2007). Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim. Biophys. Acta 1767, 913–919. 10.1016/j.bbabio.2007.05.005 [DOI] [PubMed] [Google Scholar]
- Blanco-Grau A., Bonaventura-Ibars I., Coll-Cantí J., Melià M. J., Martínez R., Martinez-Gallo M., et al. (2013). Identification and biochemical characterization of the novel mutation m.8839G>C in the mitochondrial ATP6 gene associated with NARP syndrome. Genes Brain Behav. 12, 812–820. 10.1111/gbb.12089 [DOI] [PubMed] [Google Scholar]
- Bonnefoy N., Fox T. D. (2001). Genetic transformation of Saccharomyces cerevisiae mitochondria. Methods Cell Biol. 65, 381–396. 10.1016/S0091-679X(01)65022-2 [DOI] [PubMed] [Google Scholar]
- Boyer P. D. (1997). The ATP synthase–a splendid molecular machine. Annu. Rev. Biochem. 66, 717–749. 10.1146/annurev.biochem.66.1.717 [DOI] [PubMed] [Google Scholar]
- Brum M., Semedo C., Guerreiro R., Pinto Marques J. (2014). Motor neuron syndrome as a new phenotypic manifestation of mutation 9185T>C in gene MTATP6. Case Rep. Neurol. Med. 2014:701761. 10.1155/2014/701761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrage L. C., Tang S., Wang J., Donti T. R., Walkiewicz M., Luchak J. M., et al. (2014). Mitochondrial myopathy, lactic acidosis, and sideroblastic anemia (MLASA) plus associated with a novel de novo mutation (m.8969G>A) in the mitochondrial encoded ATP6 gene. Mol. Genet. Metab. 113, 207–212. 10.1016/j.ymgme.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W., Fu Q., Zhou X., Qu J., Tong Y., Guan M. X. (2008). Mitochondrial variants may influence the phenotypic manifestation of Leber's hereditary optic neuropathy-associated ND4 G11778A mutation. J. Genet. Genomics 35, 649–655. 10.1016/S1673-8527(08)60086-7 [DOI] [PubMed] [Google Scholar]
- Carrozzo R., Rizza T., Stringaro A., Pierini R., Mormone E., Santorelli F. M., et al. (2004). Maternally-inherited Leigh syndrome-related mutations bolster mitochondrial-mediated apoptosis. J. Neurochem. 90, 490–501. 10.1111/j.1471-4159.2004.02505.x [DOI] [PubMed] [Google Scholar]
- Castagna A. E., Addis J., McInnes R. R., Clarke J. T., Ashby P., Blaser S., et al. (2007). Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9,185 of mitochondrial DNA. Am. J. Med. Genet. A 143A, 808–816. 10.1002/ajmg.a.31637 [DOI] [PubMed] [Google Scholar]
- Childs A. M., Hutchin T., Pysden K., Highet L., Bamford J., Livingston J., et al. (2007). Variable phenotype including Leigh syndrome with a 9185T>C mutation in the MTATP6 gene. Neuropediatrics 38, 313–316. 10.1055/s-2008-1065355 [DOI] [PubMed] [Google Scholar]
- Chinnery P. F. (2015). Mitochondrial disease in adults: what's old and what's new? EMBO Mol. Med. 10.15252/emmm.201505079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciafaloni E., Santorelli F. M., Shanske S., Deonna T., Roulet E., Janzer C., et al. (1993). Maternally inherited Leigh syndrome. J. Pediatr. 122, 419–422. 10.1016/S0022-3476(05)83431-6 [DOI] [PubMed] [Google Scholar]
- Cortés-Hernández P., Vázquez-Memije M. E., García J. J. (2007). ATP6 homoplasmic mutations inhibit and destabilize the human F1Fo-ATP synthase without preventing enzyme assembly and oligomerization. J. Biol. Chem. 282, 1051–1058. 10.1074/jbc.M606828200 [DOI] [PubMed] [Google Scholar]
- Craig K., Elliott H. R., Keers S. M., Lambert C., Pyle A., Graves T. D., et al. (2007). Episodic ataxia and hemiplegia caused by the 8993T→C mitochondrial DNA mutation. J. Med. Genet. 44, 797–799. 10.1136/jmg.2007.052902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M., Strauss M., Daum B., Kief J. H., Osiewacz H. D., Rycovska A., et al. (2011). Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, 14121–14126. 10.1073/pnas.1103621108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meirleir L., Seneca S., Lissens W., Schoentjes E., Desprechins B. (1995). Bilateral striatal necrosis with a novel point mutation in the mitochondrial ATPase 6 gene. Pediatr. Neurol. 13, 242–246. 10.1016/0887-8994(95)00184-H [DOI] [PubMed] [Google Scholar]
- De Vries D. D., Van Engelen B. G., Gabreëls F. J., Ruitenbeek W., Van Oost B. A. (1993). A second missense mutation in the mitochondrial ATPase 6 gene in Leigh's syndrome. Ann. Neurol. 34, 410–412. 10.1002/ana.410340319 [DOI] [PubMed] [Google Scholar]
- Debray F. G., Lambert M., Lortie A., Vanasse M., Mitchell G. A. (2007). Long-term outcome of Leigh syndrome caused by the NARP-T8993C mtDNA mutation. Am. J. Med. Genet. A 143A, 2046–2051. 10.1002/ajmg.a.31880 [DOI] [PubMed] [Google Scholar]
- Dimauro S., Schon E. A. (2003). Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 348, 2656–2668. 10.1056/NEJMra022567 [DOI] [PubMed] [Google Scholar]
- Dionisi-Vici C., Seneca S., Zeviani M., Fariello G., Rimoldi M., Bertini E., et al. (1998). Fulminant Leigh syndrome and sudden unexpected death in a family with the T9176C mutation of the mitochondrial ATPase 6 gene. J. Inherit. Metab. Dis. 21, 2–8. 10.1023/A:1005397227996 [DOI] [PubMed] [Google Scholar]
- Duno M., Wibrand F., Baggesen K., Rosenberg T., Kjaer N., Frederiksen A. L. (2013). A novel mitochondrial mutation m.8989G>C associated with neuropathy, ataxia, retinitis pigmentosa - the NARP syndrome. Gene 515, 372–375. 10.1016/j.gene.2012.12.066 [DOI] [PubMed] [Google Scholar]
- Enns G. M., Bai R. K., Beck A. E., Wong L. J. (2006). Molecular-clinical correlations in a family with variable tissue mitochondrial DNA T8993G mutant load. Mol. Genet. Metab. 88, 364–371. 10.1016/j.ymgme.2006.02.001 [DOI] [PubMed] [Google Scholar]
- Felhi R., Mkaouar-Rebai E., Sfaihi-Ben Mansour L., Alila-Fersi O., Tabebi M., Ben Rhouma B., et al. (2016). Mutational analysis in patients with neuromuscular disorders: detection of mitochondrial deletion and double mutations in the MT-ATP6 gene. Biochem. Biophys. Res. Commun. 473, 61–66. 10.1016/j.bbrc.2016.03.050 [DOI] [PubMed] [Google Scholar]
- Finsterer J., Stöllberger C., Schubert B. (2004). Acquired left ventricular hypertrabeculation/noncompaction in mitochondriopathy. Cardiology 102, 228–230. 10.1159/000081015 [DOI] [PubMed] [Google Scholar]
- Fujii T., Hattori H., Higuchi Y., Tsuji M., Mitsuyoshi I. (1998). Phenotypic differences between T→C and T→G mutations at nt 8993 of mitochondrial DNA in Leigh syndrome. Pediatr. Neurol. 18, 275–277. 10.1016/S0887-8994(97)00187-2 [DOI] [PubMed] [Google Scholar]
- Galimberti C. A., Diegoli M., Sartori I., Uggetti C., Brega A., Tartara A., et al. (2006). Brain pseudoatrophy and mental regression on valproate and a mitochondrial DNA mutation. Neurology 67, 1715–1717. 10.1212/01.wnl.0000242882.58086.9a [DOI] [PubMed] [Google Scholar]
- Garcia J. J., Ogilvie I., Robinson B. H., Capaldi R. A. (2000). Structure, functioning, and assembly of the ATP synthase in cells from patients with the T8993G mitochondrial DNA mutation. Comparison with the enzyme in Rho(0) cells completely lacking mtdna. J. Biol. Chem. 275, 11075–11081. 10.1074/jbc.275.15.11075 [DOI] [PubMed] [Google Scholar]
- Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., et al. (2017). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. 10.1002/pro.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greie J. C., Heitkamp T., Altendorf K. (2004). The transmembrane domain of subunit b of the Escherichia coli F1Fo ATP synthase is sufficient for H+ translocating activity together with subunits a and c. Eur. J. Biochem. 271, 3036–3042. 10.1111/j.1432-1033.2004.04235.x [DOI] [PubMed] [Google Scholar]
- Guo H., Bueler S. A., Rubinstein J. L. (2017). Atomic model for the dimeric Fo region of mitochondrial ATP synthase. Science 358, 936–940. 10.1126/science.aao4815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurses C., Azakli H., Alptekin A., Cakiris A., Abaci N., Arikan M., et al. (2014). Mitochondrial DNA profiling via genomic analysis in mesial temporal lobe epilepsy patients with hippocampal sclerosis. Gene 538, 323–327. 10.1016/j.gene.2014.01.030 [DOI] [PubMed] [Google Scholar]
- Hahn A., Parey K., Bublitz M., Mills D. J., Zickermann V., Vonck J., et al. (2016). Structure of a complete ATP synthase dimer reveals the molecular basis of inner mitochondrial membrane morphology. Mol. Cell 63, 445–456. 10.1016/j.molcel.2016.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejzlarová K., Kaplanová V., Nusková H., Kovárová N., Ješina P., Drahota Z., et al. (2015). Alteration of structure and function of ATP synthase and cytochrome c oxidase by lack of Fo-a and Cox3 subunits caused by mitochondrial DNA 9205delTA mutation. Biochem. J. 466, 601–611. 10.1042/BJ20141462 [DOI] [PubMed] [Google Scholar]
- Hejzlarova K., Mracek T., Vrbacky M., Kaplanova V., Karbanova V., Nuskova H., et al. (2014). Nuclear genetic defects of mitochondrial ATP synthase. Physiol. Res. 63(Suppl 1), S57–S71. [DOI] [PubMed] [Google Scholar]
- Holt I. J., Harding A. E., Petty R. K., Morgan-Hughes J. A. (1990). A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 46, 428–433. [PMC free article] [PubMed] [Google Scholar]
- Honzik T., Tesarova M., Magner M., Mayr J., Jesina P., Vesela K., et al. (2012). Neonatal onset of mitochondrial disorders in 129 patients: clinical and laboratory characteristics and a new approach to diagnosis. J. Inherit. Metab. Dis. 35, 749–759. 10.1007/s10545-011-9440-3 [DOI] [PubMed] [Google Scholar]
- Honzik T., Tesarova M., Vinsova K., Hansikova H., Magner M., Kratochvilova H., et al. (2013). Different laboratory and muscle biopsy findings in a family with an m.8851T>C mutation in the mitochondrial MTATP6 gene. Mol. Genet. Metab. 108, 102–105. 10.1016/j.ymgme.2012.11.002 [DOI] [PubMed] [Google Scholar]
- Houstek J., Klement P., Hermanska J., Houstkova H., Hansikova H., Van Den Bogert C., et al. (1995). Altered properties of mitochondrial ATP-synthase in patients with a T→G mutation in the ATPase 6 (subunit a) gene at position 8993 of mtDNA. Biochim. Biophys. Acta 1271, 349–357. 10.1016/0925-4439(95)00063-A [DOI] [PubMed] [Google Scholar]
- Hurvitz H., Naveh Y., Shoseyov D., Klar A., Shaag A., Elpeleg O. (2002). Transmission of the mitochondrial t8993c mutation in a new family. Am. J. Med. Genet. 111, 446–447. 10.1002/ajmg.10613 [DOI] [PubMed] [Google Scholar]
- Imai A., Fujita S., Kishita Y., Kohda M., Tokuzawa Y., Hirata T., et al. (2016). Rapidly progressive infantile cardiomyopathy with mitochondrial respiratory chain complex V deficiency due to loss of ATPase 6 and 8 protein. Int. J. Cardiol. 207, 203–205. 10.1016/j.ijcard.2016.01.026 [DOI] [PubMed] [Google Scholar]
- Jackson C. B., Hahn D., Schroter B., Richter U., Battersby B. J., Schmitt-Mechelke T., et al. (2017). A novel mitochondrial ATP6 frameshift mutation causing isolated complex V deficiency, ataxia and encephalomyopathy. Eur. J. Med. Genet. 60, 345–351. 10.1016/j.ejmg.2017.04.006 [DOI] [PubMed] [Google Scholar]
- Jacobs L. J., De Coo I. F., Nijland J. G., Galjaard R. J., Los F. J., Schoonderwoerd K., et al. (2005). Transmission and prenatal diagnosis of the T9176C mitochondrial DNA mutation. Mol. Hum. Reprod. 11, 223–228. 10.1093/molehr/gah152 [DOI] [PubMed] [Google Scholar]
- Jesina P., Tesarova M., Fornuskova D., Vojtiskova A., Pecina P., Kaplanova V., et al. (2004). Diminished synthesis of subunit a (ATP6) and altered function of ATP synthase and cytochrome c oxidase due to the mtDNA 2 bp microdeletion of TA at positions 9205 and 9206. Biochem. J. 383, 561–571. 10.1042/BJ20040407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere A. I., Hogeveen M., Nijtmans L. G., Van Den Brand M. A., Janssen A. J., Diepstra J. H., et al. (2008). A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. J. Med. Genet. 45, 129–133. 10.1136/jmg.2007.052084 [DOI] [PubMed] [Google Scholar]
- Jonckheere A. I., Smeitink J. A., Rodenburg R. J. (2012). Mitochondrial ATP synthase: architecture, function and pathology. J. Inherit. Metab. Dis. 35, 211–225. 10.1007/s10545-011-9382-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge W., Lill H., Engelbrecht S. (1997). ATP synthase: an electrochemical transducer with rotatory mechanics. Trends Biochem. Sci. 22, 420–423. 10.1016/S0968-0004(97)01129-8 [DOI] [PubMed] [Google Scholar]
- Kabala A. M., Lasserre J. P., Ackerman S. H., Di Rago J. P., Kucharczyk R. (2014). Defining the impact on yeast ATP synthase of two pathogenic human mitochondrial DNA mutations, T9185C and T9191C. Biochimie 100, 200–206. 10.1016/j.biochi.2013.11.024 [DOI] [PubMed] [Google Scholar]
- Kara B., Arikan M., Maras H., Abaci N., Cakiris A., Ustek D. (2012). Whole mitochondrial genome analysis of a family with NARP/MILS caused by m.8993T>C mutation in the MT-ATP6 gene. Mol. Genet. Metab. 107, 389–393. 10.1016/j.ymgme.2012.06.013 [DOI] [PubMed] [Google Scholar]
- King M. P., Attardi G. (1989). Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503. 10.1126/science.2814477 [DOI] [PubMed] [Google Scholar]
- Korshunov S. S., Skulachev V. P., Starkov A. A. (1997). High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416, 15–18. 10.1016/S0014-5793(97)01159-9 [DOI] [PubMed] [Google Scholar]
- Kucharczyk R., Ezkurdia N., Couplan E., Procaccio V., Ackerman S. H., Blondel M., et al. (2010). Consequences of the pathogenic T9176C mutation of human mitochondrial DNA on yeast mitochondrial ATP synthase. Biochim. Biophys. Acta 1797, 1105–1112. 10.1016/j.bbabio.2009.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharczyk R., Giraud M. F., Brethes D., Wysocka-Kapcinska M., Ezkurdia N., Salin B., et al. (2013). Defining the pathogenesis of human mtDNA mutations using a yeast model: the case of T8851C. Int. J. Biochem. Cell Biol. 45, 130–140. 10.1016/j.biocel.2012.07.001 [DOI] [PubMed] [Google Scholar]
- Kucharczyk R., Rak M., Di Rago J. P. (2009a). Biochemical consequences in yeast of the human mitochondrial DNA 8993T>C mutation in the ATPase6 gene found in NARP/MILS patients. Biochim. Biophys. Acta 1793, 817–824. 10.1016/j.bbamcr.2009.02.011 [DOI] [PubMed] [Google Scholar]
- Kucharczyk R., Salin B., Di Rago J. P. (2009b). Introducing the human Leigh syndrome mutation T9176G into Saccharomyces cerevisiae mitochondrial DNA leads to severe defects in the incorporation of Atp6p into the ATP synthase and in the mitochondrial morphology. Hum. Mol. Genet. 18, 2889–2898. 10.1093/hmg/ddp226 [DOI] [PubMed] [Google Scholar]
- Kucharczyk R., Zick M., Bietenhader M., Rak M., Couplan E., Blondel M., et al. (2009c). Mitochondrial ATP synthase disorders: molecular mechanisms and the quest for curative therapeutic approaches. Biochim. Biophys. Acta 1793, 186–199. 10.1016/j.bbamcr.2008.06.012 [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W. (2014). Biochemistry. The resolution revolution. Science 343, 1443–1444. 10.1126/science.1251652 [DOI] [PubMed] [Google Scholar]
- Kumar M., Tanwar M., Saxena R., Sharma P., Dada R. (2010). Identification of novel mitochondrial mutations in Leber's hereditary optic neuropathy. Mol. Vis. 16, 782–792. [PMC free article] [PubMed] [Google Scholar]
- Kytovuori L., Lipponen J., Rusanen H., Komulainen T., Martikainen M. H., Majamaa K. (2016). A novel mutation m.8561C>G in MT-ATP6/8 causing a mitochondrial syndrome with ataxia, peripheral neuropathy, diabetes mellitus, and hypergonadotropic hypogonadism. J. Neurol. 263, 2188–2195. 10.1007/s00415-016-8249-2 [DOI] [PubMed] [Google Scholar]
- La Morgia C., Achilli A., Iommarini L., Barboni P., Pala M., Olivieri A., et al. (2008). Rare mtDNA variants in Leber hereditary optic neuropathy families with recurrence of myoclonus. Neurology 70, 762–770. 10.1212/01.wnl.0000295505.74234.d0 [DOI] [PubMed] [Google Scholar]
- Lamminen T., Majander A., Juvonen V., Wikstrom M., Aula P., Nikoskelainen E., et al. (1995). A mitochondrial mutation at nt 9101 in the ATP synthase 6 gene associated with deficient oxidative phosphorylation in a family with Leber hereditary optic neuroretinopathy. Am. J. Hum. Genet. 56, 1238–1240. [PMC free article] [PubMed] [Google Scholar]
- Lasserre J. P., Dautant A., Aiyar R. S., Kucharczyk R., Glatigny A., Tribouillard-Tanvier D., et al. (2015). Yeast as a system for modeling mitochondrial disease mechanisms and discovering therapies. Dis. Model. Mech. 8, 509–526. 10.1242/dmm.020438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gallardo E., Emperador S., Solano A., Llobet L., Martin-Navarro A., Lopez-Perez M. J., et al. (2014). Expanding the clinical phenotypes of MT-ATP6 mutations. Hum. Mol. Genet. 23, 6191–6200. 10.1093/hmg/ddu339 [DOI] [PubMed] [Google Scholar]
- Lopez-Gallardo E., Solano A., Herrero-Martin M. D., Martinez-Romero I., Castano-Perez M. D., Andreu A. L., et al. (2009). NARP syndrome in a patient harbouring an insertion in the MT-ATP6 gene that results in a truncated protein. J. Med. Genet. 46, 64–67. 10.1136/jmg.2008.060616 [DOI] [PubMed] [Google Scholar]
- Lott M. T., Leipzig J. N., Derbeneva O., Xie H. M., Chalkia D., Sarmady M., et al. (2013). mtDNA variation and analysis using MITOMAP and MITOMASTER. Curr. Protoc. Bioinformatics 1.23.1–26. 10.1002/0471250953.bi0123s44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majander A., Lamminen T., Juvonen V., Aula P., Nikoskelainen E., Savontaus M. L., et al. (1997). Mutations in subunit 6 of the F1Fo-ATP synthase cause two entirely different diseases. FEBS Lett. 412, 351–354. 10.1016/S0014-5793(97)00757-6 [DOI] [PubMed] [Google Scholar]
- Makino M., Horai S., Goto Y., Nonaka I. (1998). Confirmation that a T-to-C mutation at 9176 in mitochondrial DNA is an additional candidate mutation for Leigh's syndrome. Neuromuscul. Disord. 8, 149–151. 10.1016/S0960-8966(98)00017-0 [DOI] [PubMed] [Google Scholar]
- Martikainen M. H., Gorman G. S., Goldsmith P., Burn D. J., Turnbull D. M., Schaefer A. M. (2015). Adult-onset myoclonus ataxia associated with the mitochondrial m.8993T>C “NARP” mutation. Mov. Disord. 30, 1432–1433. 10.1002/mds.26358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi M., Vijayvergiya C., Gajewski C. D., Devivo D. C., Lenaz G., Wiedmann M., et al. (2004). The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 13, 869–879. 10.1093/hmg/ddh103 [DOI] [PubMed] [Google Scholar]
- Miller F. J., Rosenfeldt F. L., Zhang C., Linnane A. W., Nagley P. (2003). Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 31:e61. 10.1093/nar/gng060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitome N., Ono S., Sato H., Suzuki T., Sone N., Yoshida M. (2010). Essential arginine residue of the Fo-a subunit in FoF1-ATP synthase has a role to prevent the proton shortcut without c-ring rotation in the Fo proton channel. Biochem. J. 430, 171–177. 10.1042/BJ20100621 [DOI] [PubMed] [Google Scholar]
- Mkaouar-Rebai E., Kammoun F., Chamkha I., Kammoun N., Hsairi I., Triki C., et al. (2010). A de novo mutation in the adenosine triphosphatase (ATPase) 8 gene in a patient with mitochondrial disorder. J. Child Neurol. 25, 770–775. 10.1177/0883073809344351 [DOI] [PubMed] [Google Scholar]
- Morales-Rios E., Montgomery M. G., Leslie A. G., Walker J. E. (2015a). Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 112, 13231–13236. 10.1073/pnas.1517542112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Rios E., Montgomery M. G., Leslie A. G., Garcia-Trejo J. J., Walker J. E. (2015b). Structure of a catalytic dimer of the α- and β-subunits of the F-ATPase from Paracoccus denitrificans at 2.3 A resolution. Acta Crystallogr. F Struct. Biol. Commun. 71, 1309–1317. 10.1107/S2053230X15016076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morava E., Rodenburg R. J., Hol F., De Vries M., Janssen A., Van Den Heuvel L., et al. (2006). Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am. J. Med. Genet. A 140, 863–868. 10.1002/ajmg.a.31194 [DOI] [PubMed] [Google Scholar]
- Mordel P., Schaeffer S., Dupas Q., Laville M. A., Gerard M., Chapon F., et al. (2017). A 2 bp deletion in the mitochondrial ATP 6 gene responsible for the NARP (neuropathy, ataxia, and retinitis pigmentosa) syndrome. Biochem. Biophys. Res. Commun. 494, 133–137. 10.1016/j.bbrc.2017.10.066 [DOI] [PubMed] [Google Scholar]
- Moslemi A. R., Darin N., Tulinius M., Oldfors A., Holme E. (2005). Two new mutations in the MTATP6 gene associated with Leigh syndrome. Neuropediatrics 36, 314–318. 10.1055/s-2005-872845 [DOI] [PubMed] [Google Scholar]
- Ng Y. S., Turnbull D. M. (2015). Mitochondrial disease: genetics and management. J. Neurol. 263, 179–191. 10.1007/s00415-015-7884-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzwiecka K., Kabala A. M., Lasserre J. P., Tribouillard-Tanvier D., Golik P., Dautant A., et al. (2016). Yeast models of mutations in the mitochondrial ATP6 gene found in human cancer cells. Mitochondrion 29, 7–17. 10.1016/j.mito.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Nijtmans L. G., Henderson N. S., Attardi G., Holt I. J. (2001). Impaired ATP synthase assembly associated with a mutation in the human ATP synthase subunit 6 gene. J. Biol. Chem. 276, 6755–6762. 10.1074/jbc.M008114200 [DOI] [PubMed] [Google Scholar]
- Okamoto K., Perlman P. S., Butow R. A. (1998). The sorting of mitochondrial DNA and mitochondrial proteins in zygotes: preferential transmission of mitochondrial DNA to the medial bud. J. Cell Biol. 142, 613–623. 10.1083/jcb.142.3.613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S. E., et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. 10.1016/j.cell.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotti F., Baracca A., Hernandez-Rosa E., Walker W. F., Solaini G., Lenaz G., et al. (2004). Biochemical analysis of respiratory function in cybrid cell lines harbouring mitochondrial DNA mutations. Biochem. J. 384, 287–293. 10.1042/BJ20040561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons D. F. (1963). Mitochondrial structure: two types of subunits on negatively stained mitochondrial membranes. Science 140, 985–987. 10.1126/science.140.3570.985 [DOI] [PubMed] [Google Scholar]
- Paumard P., Vaillier J., Coulary B., Schaeffer J., Soubannier V., Mueller D. M., et al. (2002). The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 21, 221–230. 10.1093/emboj/21.3.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucca-Lostanlen D., Narbonne H., Hernandez J. B., Staccini P., Saunieres A., Paquis-Flucklinger V., et al. (2000). Mitochondrial DNA variations in patients with maternally inherited diabetes and deafness syndrome. Biochem. Biophys. Res. Commun. 277, 771–775. 10.1006/bbrc.2000.3751 [DOI] [PubMed] [Google Scholar]
- Petros J. A., Baumann A. K., Ruiz-Pesini E., Amin M. B., Sun C. Q., Hall J., et al. (2005). mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 719–724. 10.1073/pnas.0408894102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G., Blakely E. L., Alston C. L., Hassani A., Boggild M., Horvath R., et al. (2012). Adult-onset spinocerebellar ataxia syndromes due to MTATP6 mutations. J. Neurol. Neurosurg. Psychiatr. 83, 883–886. 10.1136/jnnp-2012-302568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piryaei F., Houshmand M., Aryani O., Dadgar S., Soheili Z. S. (2012). Investigation of the Mitochondrial ATPase 6/8 and tRNA(Lys) Genes Mutations in Autism. Cell J. 14, 98–101. [PMC free article] [PubMed] [Google Scholar]
- Pitceathly R. D., Murphy S. M., Cottenie E., Chalasani A., Sweeney M. G., Woodward C., et al. (2012). Genetic dysfunction of MT-ATP6 causes axonal charcot-marie-tooth disease. Neurology 79, 1145–1154. 10.1212/WNL.0b013e3182698d8d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoryelov D., Krah A., Langer J. D., Yildiz Ö., Faraldo-Gómez J. D., Meier T. (2010). Microscopic rotary mechanism of ion translocation in the Fo complex of ATP synthases. Nat. Chem. Biol. 6, 891–899. 10.1038/nchembio.457 [DOI] [PubMed] [Google Scholar]
- Povalko N., Zakharova E., Rudenskaia G., Akita Y., Hirata K., Toyojiro M., et al. (2005). A new sequence variant in mitochondrial DNA associated with high penetrance of Russian Leber hereditary optic neuropathy. Mitochondrion 5, 194–199. 10.1016/j.mito.2005.03.003 [DOI] [PubMed] [Google Scholar]
- Prokisch H., Scharfe C., Camp D. G., II., Xiao W., David L., Andreoli C., et al. (2004). Integrative analysis of the mitochondrial proteome in yeast. PLoS Biol. 2:e160. 10.1371/journal.pbio.0020160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu P., Barboni P., Mantovani V., Montagna P., Cerullo A., Bragliani M., et al. (1993). Retinitis pigmentosa, ataxia, and mental retardation associated with mitochondrial DNA mutation in an Italian family. Br. J. Ophthalmol. 77, 84–88. 10.1136/bjo.77.2.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak M., Tetaud E., Duvezin-Caubet S., Ezkurdia N., Bietenhader M., Rytka J., et al. (2007). A yeast model of the neurogenic ataxia retinitis pigmentosa (NARP) T8993G mutation in the mitochondrial ATP synthase-6 gene. J. Biol. Chem. 282, 34039–34047. 10.1074/jbc.M703053200 [DOI] [PubMed] [Google Scholar]
- Reinders J., Zahedi R. P., Pfanner N., Meisinger C., Sickmann A. (2006). Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res. 5, 1543–1554. 10.1021/pr050477f [DOI] [PubMed] [Google Scholar]
- Rhee H. W., Zou P., Udeshi N. D., Martell J. D., Mootha V. K., Carr S. A., et al. (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331. 10.1126/science.1230593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo A., Campos Y., Sanchez J. M., Bonaventura I., Aguilar M., Garcia A., et al. (2006). NARP-MILS syndrome caused by 8993 T>G mitochondrial DNA mutation: a clinical, genetic and neuropathological study. Acta Neuropathol. 111, 610–616. 10.1007/s00401-006-0040-5 [DOI] [PubMed] [Google Scholar]
- Ronchi D., Bordoni A., Cosi A., Rizzuti M., Fassone E., Di Fonzo A., et al. (2011). Unusual adult-onset Leigh syndrome presentation due to the mitochondrial m.9176T>C mutation. Biochem. Biophys. Res. Commun. 412, 245–248. 10.1016/j.bbrc.2011.07.076 [DOI] [PubMed] [Google Scholar]
- Sakai Y., Kaga K., Kodama K., Higuchi A., Miyamoto J. (2004). Hearing evaluation in two sisters with a T8993G point mutation of mitochondrial DNA. Int. J. Pediatr. Otorhinolaryngol. 68, 1115–1119. 10.1016/j.ijporl.2004.03.015 [DOI] [PubMed] [Google Scholar]
- Saneto R. P., Singh K. K. (2010). Illness-induced exacerbation of Leigh syndrome in a patient with the MTATP6 mutation, m. 9185 T>C. Mitochondrion 10, 567–572. 10.1016/j.mito.2010.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santorelli F. M., Shanske S., Jain K. D., Tick D., Schon E. A., Dimauro S. (1994). A T→C mutation at nt 8993 of mitochondrial DNA in a child with Leigh syndrome. Neurology 44, 972–974. 10.1212/WNL.44.5.972 [DOI] [PubMed] [Google Scholar]
- Saraste M. (1999). Oxidative phosphorylation at the fin de siecle. Science 283, 1488–1493. 10.1126/science.283.5407.1488 [DOI] [PubMed] [Google Scholar]
- Schägger H., Pfeiffer K. (2000). Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19, 1777–1783. 10.1093/emboj/19.8.1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E., Altendorf K. (1984). Subunit b of the membrane moiety Fo of ATP synthase (F1Fo) from Escherichia coli is indispensable for H+ translocation and binding of the water-soluble F1 moiety. Proc. Natl. Acad. Sci. U.S.A. 81, 7279–7283. 10.1073/pnas.81.23.7279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E., Altendorf K. (1985). Modification of subunit b of the Fo complex from Escherichia coli ATP synthase by a hydrophobic maleimide and its effects on Fo functions. Eur. J. Biochem. 153, 105–109. 10.1111/j.1432-1033.1985.tb09274.x [DOI] [PubMed] [Google Scholar]
- Seneca S., Abramowicz M., Lissens W., Muller M. F., Vamos E., De Meirleir L. (1996). A mitochondrial DNA microdeletion in a newborn girl with transient lactic acidosis. J. Inherit. Metab. Dis. 19, 115–118. 10.1007/BF01799407 [DOI] [PubMed] [Google Scholar]
- Sequeira A., Rollins B., Magnan C., Van Oven M., Baldi P., Myers R. M., et al. (2015). Mitochondrial mutations in subjects with psychiatric disorders. PLoS ONE 10:e0127280. 10.1371/journal.pone.0127280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgarbi G., Baracca A., Lenaz G., Valentino L. M., Carelli V., Solaini G. (2006). Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem. J. 395, 493–500. 10.1042/BJ20051748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shidara K., Wakakura M. (2012). Leber's hereditary optic neuropathy with the 3434, 9011 mitochondrial DNA point mutation. Jpn. J. Ophthalmol. 56, 175–180. 10.1007/s10384-011-0106-3 [DOI] [PubMed] [Google Scholar]
- Sikorska M., Sandhu J. K., Simon D. K., Pathiraja V., Sodja C., Li Y., et al. (2009). Identification of ataxia-associated mtDNA mutations (m.4052T>C and m.9035T>C) and evaluation of their pathogenicity in transmitochondrial cybrids. Muscle Nerve 40, 381–394. 10.1002/mus.21355 [DOI] [PubMed] [Google Scholar]
- Skladal D., Halliday J., Thorburn D. R. (2003). Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 126, 1905–1912. 10.1093/brain/awg170 [DOI] [PubMed] [Google Scholar]
- Sobti M., Smits C., Wong A. S., Ishmukhametov R., Stock D., Sandin S., et al. (2016). Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states. Elife 5:e21598. 10.7554/eLife.21598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz L. M., Scharfe C., Deutschbauer A. M., Mokranjac D., Herman Z. S., Jones T., et al. (2002). Systematic screen for human disease genes in yeast. Nat. Genet. 31, 400–404. 10.1038/ng929 [DOI] [PubMed] [Google Scholar]
- Stewart J. B., Chinnery P. F. (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542. 10.1038/nrg3966 [DOI] [PubMed] [Google Scholar]
- Strauss M., Hofhaus G., Schroder R. R., Kuhlbrandt W. (2008). Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160. 10.1038/emboj.2008.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swalwell H., Blakely E. L., Sutton R., Tonska K., Elstner M., He L., et al. (2008). A homoplasmic mtDNA variant can influence the phenotype of the pathogenic m.7472Cins MTTS1 mutation: are two mutations better than one? Eur. J. Hum. Genet. 16, 1265–1274. 10.1038/ejhg.2008.65 [DOI] [PubMed] [Google Scholar]
- Synofzik M., Schicks J., Wilhelm C., Bornemann A., Schols L. (2012). Charcot-marie-tooth hereditary neuropathy due to a mitochondrial ATP6 mutation. Eur. J. Neurol. 19, e114–e116. 10.1111/j.1468-1331.2012.03812.x [DOI] [PubMed] [Google Scholar]
- Tang S., Batra A., Zhang Y., Ebenroth E. S., Huang T. (2010). Left ventricular noncompaction is associated with mutations in the mitochondrial genome. Mitochondrion 10, 350–357. 10.1016/j.mito.2010.02.003 [DOI] [PubMed] [Google Scholar]
- Tang S., Wang J., Zhang V. W., Li F. Y., Landsverk M., Cui H., et al. (2013). Transition to next generation analysis of the whole mitochondrial genome: a summary of molecular defects. Hum. Mutat. 34, 882–893. 10.1002/humu.22307 [DOI] [PubMed] [Google Scholar]
- Tansel T., Pacal F., Ustek D. (2014). A novel ATP8 gene mutation in an infant with tetralogy of Fallot. Cardiol. Young 24, 531–533. 10.1017/S1047951113000668 [DOI] [PubMed] [Google Scholar]
- Tatuch Y., Christodoulou J., Feigenbaum A., Clarke J. T., Wherret J., Smith C., et al. (1992). Heteroplasmic mtDNA mutation (T→G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am. J. Hum. Genet. 50, 852–858. [PMC free article] [PubMed] [Google Scholar]
- Thyagarajan D., Shanske S., Vazquez-Memije M., De Vivo D., Dimauro S. (1995). A novel mitochondrial ATPase 6 point mutation in familial bilateral striatal necrosis. Ann. Neurol. 38, 468–472. 10.1002/ana.410380321 [DOI] [PubMed] [Google Scholar]
- Trounce I., Neill S., Wallace D. C. (1994). Cytoplasmic transfer of the mtDNA nt 8993 T→G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc. Natl. Acad. Sci. U.S.A. 91, 8334–8338. 10.1073/pnas.91.18.8334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J. D., Liu C. S., Tsao T. F., Sheu J. N. (2012). A novel mitochondrial DNA 8597T>C mutation of Leigh syndrome: report of one case. Pediatr. Neonatol. 53, 60–62. 10.1016/j.pedneo.2011.11.012 [DOI] [PubMed] [Google Scholar]
- Tuppen H. A., Blakely E. L., Turnbull D. M., Taylor R. W. (2010). Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 1797, 113–128. 10.1016/j.bbabio.2009.09.005 [DOI] [PubMed] [Google Scholar]
- Ueno H., Nishigaki Y., Kong Q. P., Fuku N., Kojima S., Iwata N., et al. (2009). Analysis of mitochondrial DNA variants in Japanese patients with schizophrenia. Mitochondrion 9, 385–393. 10.1016/j.mito.2009.06.003 [DOI] [PubMed] [Google Scholar]
- Uziel G., Moroni I., Lamantea E., Fratta G. M., Ciceri E., Carrara F., et al. (1997). Mitochondrial disease associated with the T8993G mutation of the mitochondrial ATPase 6 gene: a clinical, biochemical, and molecular study in six families. J. Neurol. Neurosurg. Psychiatr. 63, 16–22. 10.1136/jnnp.63.1.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai S. B., Mootha V. K. (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. 10.1038/nature11707 [DOI] [PubMed] [Google Scholar]
- Vazquez-Memije M. E., Shanske S., Santorelli F. M., Kranz-Eble P., Devivo D. C., Dimauro S. (1998). Comparative biochemical studies of ATPases in cells from patients with the T8993G or T8993C mitochondrial DNA mutations. J. Inherit. Metab. Dis. 21, 829–836. 10.1023/A:1005418718299 [DOI] [PubMed] [Google Scholar]
- Venkatesh S., Kumar M., Sharma A., Kriplani A., Ammini A. C., Talwar P., et al. (2010). Oxidative stress and ATPase6 mutation is associated with primary ovarian insufficiency. Arch. Gynecol. Obstet. 282, 313–318. 10.1007/s00404-010-1444-y [DOI] [PubMed] [Google Scholar]
- Verny C., Guegen N., Desquiret V., Chevrollier A., Prundean A., Dubas F., et al. (2011). Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion 11, 70–75. 10.1016/j.mito.2010.07.006 [DOI] [PubMed] [Google Scholar]
- Vik S. B., Antonio B. J. (1994). A mechanism of proton translocation by F1Fo ATP synthases suggested by double mutants of the a subunit. J. Biol. Chem. 269, 30364–30369. [PubMed] [Google Scholar]
- Vilarinho L., Barbot C., Carrozzo R., Calado E., Tessa A., Dionisi-Vici C., et al. (2001). Clinical and molecular findings in four new patients harbouring the mtDNA 8993T>C mutation. J. Inherit. Metab. Dis. 24, 883–884. 10.1023/A:1013908728445 [DOI] [PubMed] [Google Scholar]
- Wallace D. C. (2010). Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 51, 440–450. 10.1002/em.20586 [DOI] [PubMed] [Google Scholar]
- Ware S. M., El-Hassan N., Kahler S. G., Zhang Q., Ma Y. W., Miller E., et al. (2009). Infantile cardiomyopathy caused by a mutation in the overlapping region of mitochondrial ATPase 6 and 8 genes. J. Med. Genet. 46, 308–314. 10.1136/jmg.2008.063149 [DOI] [PubMed] [Google Scholar]
- Wehrle F., Kaim G., Dimroth P. (2002). Molecular mechanism of the ATP synthase's Fo motor probed by mutational analyses of subunit a. J. Mol. Biol. 322, 369–381. 10.1016/S0022-2836(02)00731-3 [DOI] [PubMed] [Google Scholar]
- Wen S., Niedzwiecka K., Zhao W., Xu S., Liang S., Zhu X., et al. (2016). Identification of G8969>A in mitochondrial ATP6 gene that severely compromises ATP synthase function in a patient with IgA nephropathy. Sci. Rep. 6:36313. 10.1038/srep36313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T., Pagadala V., Mueller D. M. (2015). Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microb. Cell 2, 105–125. 10.15698/mic2015.04.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W., Chen S., Jin S., Lu J. (2013). A novel heteroplasmic mitochondrial DNA mutation, A8890G, in a patient with juvenileonset metabolic syndrome: a case report. Mol. Med. Rep. 8, 1060–1066. 10.3892/mmr.2013.1616 [DOI] [PubMed] [Google Scholar]
- Zeviani M., Carelli V. (2007). Mitochondrial disorders. Curr. Opin. Neurol. 20, 564–571. 10.1097/WCO.0b013e3282ef58cd [DOI] [PubMed] [Google Scholar]
- Zhou A., Rohou A., Schep D. G., Bason J. V., Montgomery M. G., Walker J. E., et al. (2015). Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. Elife 4:e10180. 10.7554/eLife.10180 [DOI] [PMC free article] [PubMed] [Google Scholar]