

Abstract
We report the discovery and medicinal chemistry optimization of a novel series of pyrazole-based inhibitors of human lactate dehydrogenase (LDH). Utilization of a quantitative high-throughput screening paradigm facilitated hit identification while structure-based design and multi-parameter optimization enabled the development of compounds with potent enzymatic and cell-based inhibition of LDH enzymatic activity. Lead compounds such as 63 exhibit low nM inhibition of both LDHA and LDHB, sub-micromolar inhibition of lactate production and inhibition of glycolysis in MiaPaCa2 pancreatic cancer and A673 sarcoma cells. Moreover, robust target engagement of LDHA by lead compounds was demonstrated using the Cellular Thermal Shift Assay (CETSA) and drug-target residence time was determined via SPR. Analysis of these data suggests that drug-target residence time (off-rate) may be an important attribute to consider for obtaining potent cell-based inhibition of this cancer metabolism target.
TOC GRAPHIC
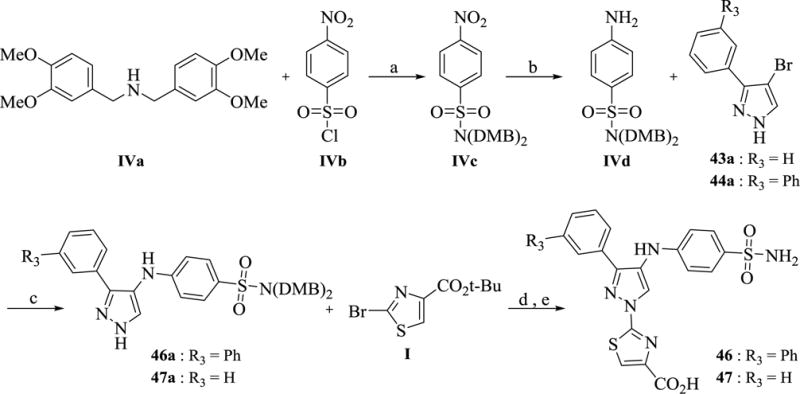
INTRODUCTION
Tumor cells are often dependent on glycolysis for adenosine 5′-triphosphate (ATP) biosynthesis, even in the presence of sufficient oxygen to support oxidative phosphorylation, a process termed aerobic glycolysis, and classically known as the “Warburg effect”.1 In such cancers, tumor cells exhibit a high rate of glycolysis, metabolizing glucose into pyruvate, which instead of entering mitochondria is reduced by lactate dehydrogenase (LDH) to lactate and excreted by the cells. This is in stark contrast to classic aerobic metabolism, in which cells demonstrate low rates of glycolysis, and instead rely on the oxidation of pyruvate in mitochondria for a comparatively greater energy payoff. Though aerobic glycolysis is an inefficient way to generate ATP, it has been proposed that rapidly proliferating cancer cells, have adapted this approach to facilitate the production of essential building blocks like nutrients such as amino acids, lipids and nucleotides to support rapid cell growth, rather than efficient ATP production.2 LDH is a key glycolytic enzyme that catalyzes the final step in the glycolytic pathway, reducing pyruvate to lactate, and regenerating NAD+ equivalents necessary for continued glycolysis. Expression of the LDHA gene is upregulated in many cancers, to support the high glycolytic activity in these cells.3,4 The LDH enzyme is a tetramer composed of M subunits coded for by the LDHA gene or H subunits ecoded for by the LDHB gene. In cancer cells, the enzyme composed of 4 M subunits known as LDH-5 is thought to predominate. Throughout this manuscript we will refer to the enzyme as LDHA. Reduction of LDH activity through knockdown or silencing of the LDHA gene has been shown to reduce tumor cell growth in vitro under hypoxic conditions and to suppress growth in tumor xenograft models.5 In addition, high levels of LDHA expression have been correlated with poor clinical outcome for a number of cancer types.6 Amongst cancers with unmet therapeutic need, glioblastoma,7 pancreatic8, and advanced stage and rare hereditary kidney cancers9 are all highly glycolytic, and thus represent potential opportunities for LDH inhibitors to provide clinical benefit.
However, despite its promise, LDHA has proven to be a relatively intractable drug target. The enzyme active-site has a highly mobile loop that caps the binding site for the small polar organic anion substrate (pyruvate or lactate) and an extended solvent exposed channel that binds cofactor. These features, combined with the high protein levels of LDH in cancer cells necessitate a small molecule inhibitor that binds with remarkable efficiency while simultaneously maintaining drug-like properties. Initial disclosures of LDH inhibitors emerged out of academic labs (e.g. FX-115b and NHI-210) with efforts from biotech11 and pharmaceutical companies, such as AstraZeneca (e.g. 1),12 emerging later. To date, no clinical-stage inhibitors of LDH have been reported; molecules from GlaxoSmithKline (GSK) (2).13 and Genentech (3)14 have shown modest cellular potency in vitro (e.g. inhibition of lactate production), but no appreciable in vivo activity, and do not appear to have progressed into clinical studies.
We designed and performed a quantitative high-throughput screening (qHTS) campaign, and utilized structure-based design and hit-to-lead optimization to discover novel compounds which are potent inhibitors of LDH enzyme activity, cellular lactate output and cancer cell line growth. Lead compounds from our work exhibit low nM inhibition of LDHA/LDHB and sub-μM inhibition of lactate production in MiaPaCa2 and A673 cells. Further, robust target engagement of LDHA with these lead compounds was demonstrated by Cellular Thermal Shift Assay (CETSA), and drug-target residence time was determined via SPR. Among these parameters, drug-target residence time (off-rate) appears to be a particularly strong predictor of cell-based inhibition of the target. In this report, we describe the discovery and medicinal chemistry optimization of a novel series of pyrazole-based LDH inhibitors. Compound 63 has proven to be a promising lead compound worthy of further optimization, given its sub-μM inhibition of cellular lactate production, demonstrated cellular target engagement, slow in vitro off-rate and good microsomal stability and aqueous solubility.
CHEMISTRY
The qHTS identified trifluoromethyl pyrazole compound 5 as a hit candidate which was evaluated via extensive SAR studies. Initial medicinal chemistry efforts focused on the pyrazole substitutents. The syntheses of 5 and related analogs 9, 12–15 were accomplished following slight modifications to a known literature method.15 As outlined in Scheme 1, commercially available trifluoromethyl-β-diketones were condensed with thiosemicarbazide to obtain key intermediates 5b, 9b, 12b-15b and and 5c, 9c, 12c-15c as a mixture of regioisomers, as reported previously.15 The unseparated mixture of the regioisomers was condensed with ethyl 3-bromo-2-oxopropanoate in the presence of sulfuric acid to obtain a ~50/50 mixture of regioisomers 5d, 9d, 12d-15d and 5e, 9e, 12e-15e, which were separated using reversed-phase chromatography. The regioisomers were distinguished by their difference in carbon and fluorine NMR as reported for similar compounds in the literature,16 and via LC/MS co-injection with a reference compound obtained from commercial source. The desired 3-arylsubstituted regioisomer is slightly less polar in nature and elutes as a second peak in reversed-phase HPLC. The resulting product was subsequently hydrolyzed with concentrated hydrochloric acid to obtain analogs 5, 9 and 12–15.
Scheme 1. Synthesis of 5, 9 and 12–15 a.
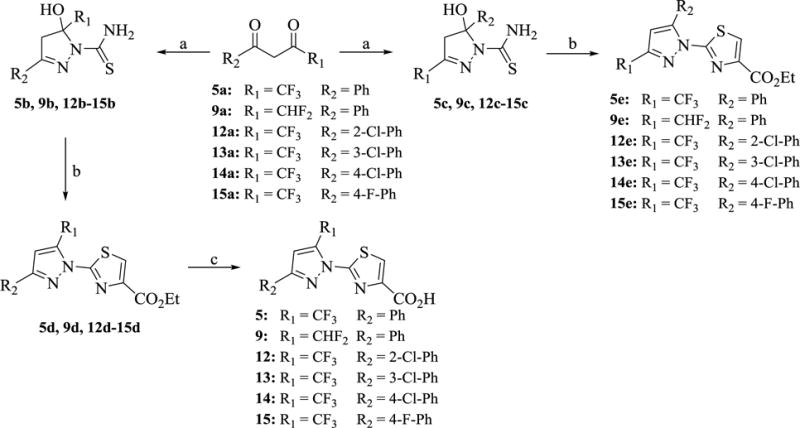
aReagents and conditions: (a) NH2CSNHNH2, EtOH, reflux, 12 h, 55–75% (b) i. BrCH2COCO2Et, EtOH, reflux, 1 h; ii. EtOH, H2SO4, reflux, 12 h, 24–35% (c) i. Reversed-phase chromatography separation of regioisomers ii. HCl, AcOH, 120 °C, 1 h.
The synthesis of the key precursors I, II and III along with analogs 7 and 10 is outlined in Scheme 2. Key intermediate tert-butyl 2-hydrazinylthiazole-4-carboxylate (II) was synthesized by esterification of the requisite 2-bromothiazole-4-carboxylic acid (Ia) to form the t-butyl ester (I). Subsequent displacement of the bromide with hydrazine at reflux provided II in 82% yield. Condensation of the acetylthiosemicarbazide (IIIa) with ethyl bromopyruvate (IIIb) in ethanol initially forms an acetyl derivative of ethyl 2-hydrazinylthiazole-4-carboxylate that is eventually cleaved in situ by the hydrogen bromide generated during the reaction to form ethyl 2-hydrazinylthiazole-4-carboxylate (III) as a HBr salt. Subsequent acid catalyzed reaction of II with benzoylacetonitrile in ethanol, followed by deprotection of the t-butyl ester group gave 7, or acetylation of the formed amino pyrazole intermediatee, followed by t-butyl deprotection provided 10 as shown in scheme 2.
Scheme 2. Synthesis of precursors (I, II, and III) and analogs 7 and 10 a.
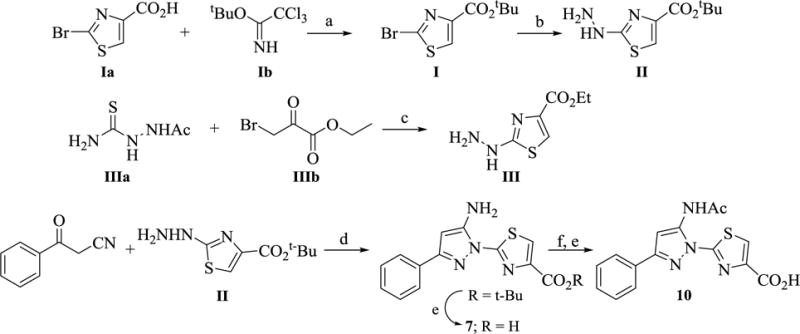
aReagents and conditions: (a) BF3·OEt2, CH2Cl2‒THF, 12 h, 88% (b) N2H4, EtOH, reflux, 2 h, 82% (c) EtOH, reflux, 5 h (d) EtOH, AcOH, reflux, 12 h, 77% (e) TFA, CH2Cl2 (f) Ac2O, Pyridine, 100 °C.
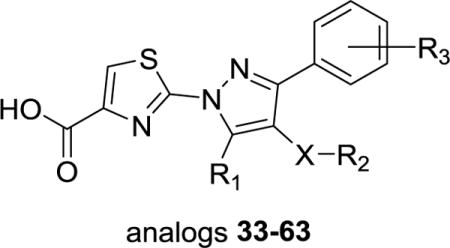
The synthetic route for hydroxypyrazole analogs 11 and 16–41 listed in Tables 1 and 2 is described in Scheme 3. Accordingly, direct condensation of the β-keto esters (11b, 16b-32b) in the presence of acetic acid with II and subsequent cleavage of the t-butylester group with TFA afforded analogs 11 and 16–32. For the synthesis of analogs 33–41, the β-keto esters (11b, 27b, 31b-32b and 36a) were alkylated with appropriately substituted benzyl bromides (33a-35a and 39a-41a) in the presence of sodium hydride in dioxane. The alkylated β-keto esters (33b-41b) are then heated with III under microwave irradiation in the presence of catalytic amount of tosic acid to obtain the cyclized esters. Interestingly, cyclization catalyzed by acetic acid in this case mostly produced a major unidentified byproduct. However, switching to tosic acid exclusively formed the desired 5-hydroxypyrazole derivative. Finally, analogs 33–41 were obtained by the LiOH hydrolysis of the corresponding ethyl esters in THF-methanol-water solvent. Commercially unavailable β-keto esters (21b & 27b-32b) and 4-(bromomethyl)benzenesulfonamide (33a) were prepared according to the literature method.17
Table 1.
LDHA inhibition of analogs (1, 5–32) with and without EDTAa
| ||||
|---|---|---|---|---|
|
| ||||
| Analog | R1 | R2 | LDHA (w/o EDTA) IC50 ± SD (μM) |
LDHA (w/EDTA) IC50 ± SD (μM) |
| 1 | NA | NA | 1.02 ± 0.07 | 1.34 ± 0.15 |
| 5 | CF3 | Ph | 22.2 ± 2.4 b | 28.8 ± 1.9 |
| 6 | Me | Ph | 15.4 ± 12b | >57 |
| 7 | NH2 | Ph | 26.7 ± 3 b | >57 |
| 8 | iPr | Ph | >57 | >57 |
| 9 | CHF2 | Ph | >57 | >57 |
| 10 | NHAc | Ph | 9.57 ± 1.8 b | >57 |
| 11 | OH | Ph | 0.144 ± 0.009 | >57 |
| 12 | CF3 | 2-Cl-Ph | >57 | >57 |
| 13 | CF3 | 3-Cl-Ph | >57 | >57 |
| 14 | CF3 | 4-Cl-Ph | 26.6 ± 0.1 | 27.7 ± 1.9 |
| 15 | CF3 | 4-F-Ph | 23.4 ± 5.7 | 32.3 ± 2.1 |
| 16 | OH | 4-F-Ph | 0.095 ± 0.001 | >57 |
| 17 | OH | 3-Cl-Ph | 0.168 ± 0.001 | >57 |
| 18 | OH | 2-F-Ph | 0.134 ± 0.001 | >57 |
| 19 | OH | 3-F-Ph | 0.162 ± 0.011 | >57 |
| 20 | OH | 3-CF3-Ph | 0.189 ± 0.001 | >57 |
| 21 | OH | 3-OCF3-Ph | 0.299 ± 0.001 | >57 |
| 22 | OH | 4-OMe-Ph | 0.229 ± 0.015 | >57 |
| 23 | OH | 2-pyridine | 1.02 ± 0.07 | >57 |
| 24 | OH | 4-pyridine | 0.669 ± 0.001 | >57 |
| 25 | OH | cyclohexyl | 1.10 ± 0.08 | >57 |
| 26 | OH | 1-naphthyl | 0.213 ± 0.024 | >57 |
| 27 | OH | 3,4-F-Ph | 0.150 ± 0.001 | >57 |
| 28 | OH | 3,4-Cl-Ph | 0.106 ± 0.001 | >57 |
| 29 | OH | 2,4-F-Ph | 0.084 ± 0.001 | >57 |
| 30 | OH | 3,5-F-Ph | 0.110 ± 0.007 | >57 |
| 31 | OH | (3-Ph)-Ph | 0.266 ± 0.001 | >57 |
| 32 | OH | 3-(2-F-Ph)-Ph | 0.095 ± 0.001 | >57 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay (n = 3) using a 22-dose response in 1536-well format.
Max. Response was less than 50% and thus IC50 values should be considered as lower confidence.
Table 2.
LDHA Inhibition of 33–63 with comparators 2 and 3a
| |||||
|---|---|---|---|---|---|
|
| |||||
| Analog | R1 | R2 | R3 | X | LDHA (w/EDTA) IC50 ± SD (μM) |
| 2 | NA | NA | NA | NA | 0.038 ± 0.004 |
| 3 | NA | NA | NA | NA | 0.424 ± 0.049 |
| 33 | OH | 4-(SO2NH2)-Ph | 3,4-F | CH2 | 0.672 ± 0.077 |
| 34 | OH | 3-(SO2NH2)-Ph | 3,4-F | CH2 | >57 |
| 35 | OH | Phenyl | 3,4-F | CH2 | >57 |
| 36 | OH | 4-(SO2NH2)-Ph | 3-Br | CH2 | 2.37 ± 0.001 |
| 37 | OH | 4-(SO2NH2)-Ph | 3-Ph | CH2 | 0.349 ± 0.023 |
| 38 | OH | 4-(SO2NH2)-Ph | 3-(2-F-Ph) | CH2 | 0.754 ± 0.087 |
| 39 | OH | 4-(SO2NHMe)-Ph | 3-Ph | CH2 | >57 |
| 40 | OH | 4-(CONH2)-Ph | 3-Ph | CH2 | >57 |
| 41 | OH | 4-(COOH)-Ph | 3-Ph | CH2 | 33.8 ± 5.5b |
| 42 | H | 4-(SO2NH2)-Ph | 3,4-F | CH2 | 0.095 ± 0.001 |
| 43 | H | 4-(SO2NH2)-Ph | H | CH2 | 0.176 ± 0.032 |
| 44 | H | 4-(SO2NH2)-Ph | 3-Ph | CH2 | 0.038 ± 0.001 |
| 45 | H | 4-(SO2NH2)-Ph | H | O | 0.124 ± 0.008 |
| 46 | H | 4-(SO2NH2)-Ph | 3-Ph | NH | 0.057 ± 0.004 |
| 47 | H | 4-(SO2NH2)-Ph | H | NH | 0.091 ± 0.006 |
| 48 | H | 4-(SO2NH2)-Ph | H | C(O)NH | >57 |
| 49 | H |
|
H | CH2 | 17.3 ± 8.1 |
| 50 | H |
|
H | CH2 | >57 |
| 51 | Me | 4-(SO2NH2)-Ph | H | CH2 | 0.042 ± 0.001 |
| 52 | CN | 4-(SO2NH2)-Ph | H | CH2 | 0.115 ± 0.130 |
| 53 | Ph | 4-(SO2NH2)-Ph | H | CH2 | 0.139 ± 0.009 |
| 54 | 3-pyr | 4-(SO2NH2)-Ph | H | CH2 | 0.349 ± 0.024 |
| 55 | 4-pyr | 4-(SO2NH2)-Ph | H | CH2 | 6.23 ± 0.79 |
| 56 | NH2 | 4-(SO2NH2)-Ph | 3-Ph | CH2 | 0.065 ± 0.011 |
| 57 | CF3 | 4-(SO2NH2)-Ph | 3-Ph | CH2 | 0.053 ± 0.001 |
| 58 |
|
4-(SO2NH2)-Ph | H | CH2 | 0.019 ± 0.001 |
| 59 |
|
4-(SO2NH2)-Ph | H | O | 0.065 ± 0.050 |
| 60 |
|
4-(SO2NH2)-Ph | 3-Ph | O | 0.110 ± 0.008 |
| 61 |
|
4-(SO2NH2)-Ph | 3-Ph | CH2 | 0.027 ± 0.001 |
| 62 |
|
4-(SO2NH2)-Ph | H | CH2 | 0.009 ± 0.001 |
| 63 |
|
4-(SO2NH2)-Ph | 3-Ph | CH2 | 0.032 ± 0.002 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay (n = 3) using a 22-dose response in 1536-well format.
Max. Response was less than 50% and thus IC50 values should be considered as lower confidence.
Scheme 3. Synthesis of 11, 16–32 and 33–41 a.
aReagents and conditions: (a) CH3CO2Et, LDA, THF, −78 °C, 2–6 h, 15–90% (b) II, AcOH, EtOH, reflux, 12 h, 12–70% (c) TFA, CH2Cl2, 1 h (d) NaH, Dioxane, 1 h, 0 °C, 60–75% (e) III, EtOH, TsOH, MW, 15 min, 46–86% f) LiOH, THF/MeOH/H2O, 1 h. bCommercially available.
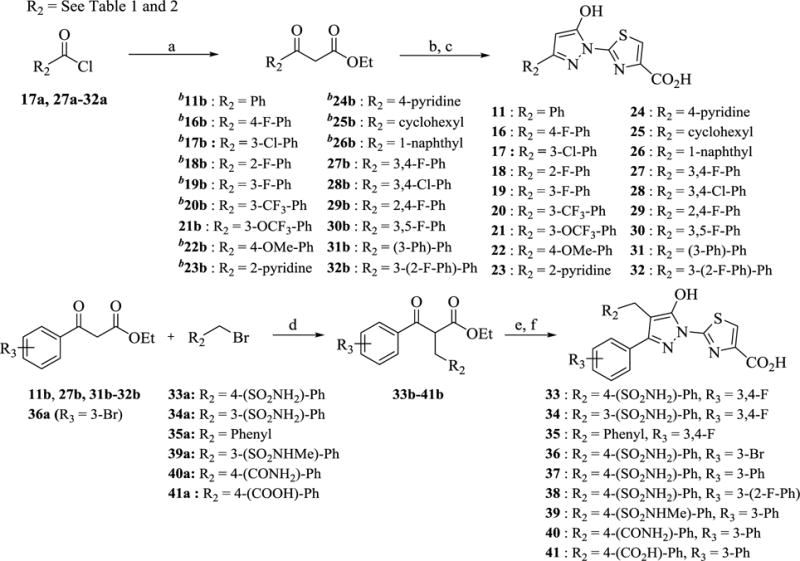
Scheme 4 illustrates a general strategy for the synthesis of analogs 43–45. Potassium carbonate-catalyzed reaction of ethyl 2-bromothiazole-4-carboxylate with the requisite commercially available bromo-pyrazole derivatives (43a-44a) in DMSO at 120 °C gave intermediates 43b-44b. The formed thiazole-containing intermediates 43b-44b were then converted to corresponding boronic acid pinacol esters 43c-44c by PdCl2(dppf) catalyzed cross coupling with bis (pinacolato)diboron using potassium acetate as base at 80 °C. Suzuki coupling of 43c-44c with 4-(bromomethyl)benzene sulfonamide (33a), following a standard protocol catalyzed by tetrakis(triphenylphosphine)palladium(0) in the presence of aqueous sodium carbonate base and subsequent ester hydrolysis using LiOH, provided analogs 43–44. Preparation of analog 45 commenced with potassium carbonate-assisted alkylation of commercially available phenacyl bromide (45a) with 4-hydroxybenzenesulfonamide to provide 45b in 92% yield. Intermediate 45b was dissolved in a solution of DMF-DMA and heated to form the formyl derivative in situ which was then refluxed with hydrazine in ethanol to give pyrazole derivative 45c in 27% yield. Finally, coupling of 45c with ethyl 2-bromothiazole-4-carboxylate using potassium carbonate in DMSO was followed by LiOH hydrolysis of the ester to provide analog 45.
Scheme 4. Synthesis of 43–45a.
aReagents and conditions: (a) ethyl 2-bromothiazole-4-carboxylate, K2CO3, 120 °C, 3–4 h, 27–78% (b) bis(pinacolato)diboron, PdCl2(dppf), AcOK, 1,4-dioxane, 95 °C, overnight 49 % (c) 4-(bromomethyl)benzenesulfonamide, 2N Na2CO3 (aq), Pd(Ph3P)4, tolune/EtOH (3/1), 80 °C, 2 h 64–95% (d) 1.5N LiOH (aq), THF, 2h (e) 4-hydroxybenzenesulfonamide, K2CO3, acetone, 20 h, 92% (f) 1,1-dimethoxy-N,N-dimethylmethanamine, 90 °C, overnight, then N2H4, EtOH, 60 °C, 4 h, 27%.
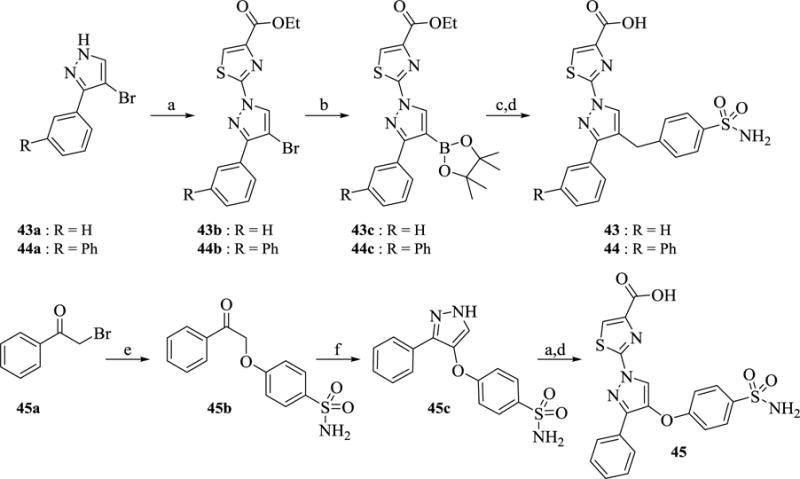
Access to analogs 46–47 was achieved using the synthetic route outlined in Scheme 5. The key precursor, 4-aminobenzene sulfonamide derivative IVd, was prepared starting from p-nitrobenzenesulfonyl chloride (IVb) by protecting the sulfonyl group with bis-(3,4-dimethoxybenzyl)amine (IVa), followed by nitro reduction using iron/ammonium chloride in ethanol. Subsequent Buchwald amination of IVd was carried out with 4-bromo-3-arylpyrazole (43a-44a) using t-BuBrettPhos and t-BuBrettPhos palladacycle catalyst system with LHMDS in THF to furnish the intermediates 46a-47a in good yield. The choice of LHMDS as the base and protection of the sulfonamide were critical for the success of the amination reaction. Several attempts to couple the amine with the unprotected sulfonamide were unsuccessful. Finally, potassium carbonate assisted coupling of 46a-47a with I in DMSO at 125 °C followed by deprotection of the tert-butyl ester afforded analog 46–47.
Scheme 5. Synthesis of analog 46 and 47 a.
aReagents and conditions: (a) iPr2NEt, CH2Cl2, 0 °C, 1 h, 96% (b) Fe, NH4Cl, EtOH, reflux, 94% (c) t-BuBrettPhos, t-BuBrettPhos-palladacycle, LHMDS, THF, 80 °C, 14 h, 62–83% (d) K2CO3, 125 °C, 14 h, 60% e) TFA, CH2Cl2, 100 °C, MW, 15 min.
A common synthetic strategy was used to synthesize analogs 48–50. As depicted in Scheme 6, potassium carbonate-assisted SNAr bromide displacement on I with 3-phenyl-1H-pyrazol-4-amine (Va) or 3-phenyl-1H-pyrazole-4-carbaldehyde (49a) in DMSO at elevated temperatures gave intermediates V and 49b respectively. Notably, the SNAr reaction of I with any 3,4-di-substituted pyrazoles was accomplished using the combination of potassium carbonate base, DMSO as a solvent, and an optimal temperature between 120–130 °C depending on the type of the substitution at 4-position of the pyrazole. As exemplified for intermediate V, the reaction proceeds at 120 °C to give the desired product in low yield. However, the product completely decomposes at higher temperatures, yet the reaction is sluggish at lower temperatures or using alternative conditions. Intermediates V and 49b were then subjected to amine coupling with either 4-sulfamoylbenzoic acid (48a) using HATU or reductive amination with 4-formylbenzenesulfonamide (50a) or 1-piperazinesulfonamide (VI) using sodium cyanoborohydride followed by TFA deprotection of the t-butyl ester group to obtain analogs 48–50.
Scheme 6. Synthesis of analogs 48–50 a.
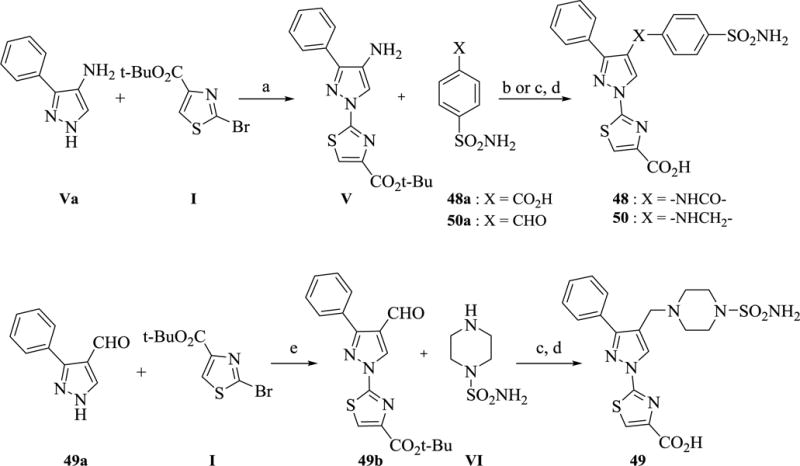
aReagents and conditions: (a) K2CO3, DMSO, 120 °C, 24 h, 25% (b) HATU, iPr2NEt, DMF, 60 °C, 4 h (c) NaBH3CN, AcOH, MeOH, (d) TFA, CH2Cl2, 1 h (e) K2CO3, DMSO, 125 °C, 3 h, 52%.
The syntheses of analogs 51–57 required an alternative and novel synthetic strategy, as our initial attempts to access these analogs from hydroxypyrazoles or alkylation of trifluoromethyl-β-diketones were unsuccessful. As described in Scheme 7, our synthesis began from LDA-catalyzed aroylation of acetonitrile using commercially available aroyl chlorides 42a and 56a at ‒78 °C to afford the requisite aroyl acetonitriles 42b and 56b. A three-component Hantzsch ester-catalyzed reductive alkylation of 42b, 51b (commercially available) and 56b with 4-formylbenzenesulfonamide (50a) at 60 °C in ethanol quickly provided intermediates 42c, 51c and 56c in good yield. Subsequent tosic acid-catalyzed cyclocondensation of the alkylated aroyl acetonitriles 42c, 51c and 56c with ethyl 2-hydrazinylthiazole-4-carboxylate (III) under microwave heating at 150 °C for 15 minutes in ethanol gave the critical intermediates 42d, 51d and 56d in 60–77% yield. Our initial attempts to convert the amine functionality in compound 42d to a bromide or chloride were unsuccessful due to undesired halogenation of the thiazole at 5-position. However, using a sequential diazotization-iodination procedure of the amines 42d, 51d and 56d in the presence of excess tosic acid with a premixed solution of KI/NaNO2 in acetonitrile furnished the key intermediates 42e, 51e and 56e in moderate yield. A fortuitous byproduct of this reaction was the des-amino (i.e. H-pyrazole) derivative (R3 = 3,4-F), which, after hydrolysis of the ethyl ester afforded compound 42. A portion of the intermediate 56e was hydrolyzed with lithium hydroxide and purified by HPLC to obtain compound 56. With intermediate 51e and 56e in hand, the iodide was then subjected to Suzuki coupling with the appropriate boronic acid using SiliaCat® DPP-Pd catalyst in the presence of aqueous Na2CO3 in DME under microwave irradiation conditions at 130 °C to provide the aryl (51: R1 = Me; 53: R1 = Ph, 54: R1 = 3-pyridine, 55: R1 = 4-pyridine) after ester hydrolysis with LiOH. For the synthesis of 52, the iodide was displaced with cyanide by heating with CuCN in DMSO at 160 °C. For 57, Hartwig’s trifluromethylator18 (1,10-Phenanthroline)(trifluoromethyl)copper(I) was used to install the requisite trifluoromethyl group. As with the the above mentioned analogs, the corresponding ethyl esters were hydrolyzed using LiOH to furnish analogs 52 and 57.
Scheme 7. Synthesis of analogs 51–57 a.
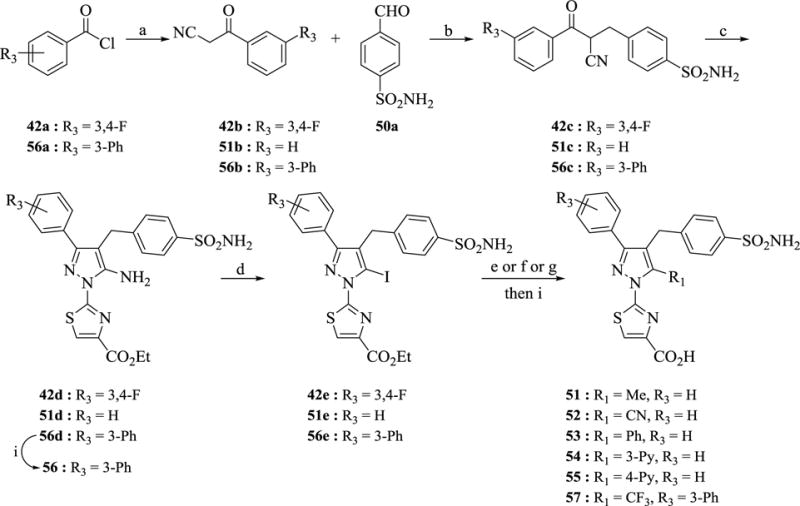
aReagents and conditions: (a) MeCN, LDA, −78 °C, 4 h, 78–97% (b) Hantzsch ester, L-Pro, EtOH, 60 °C, 0.5 h, 86–98% (c) III, TsOH, EtOH, 150 °C, MW, 15 min, 60–77 % (d) TsOH, NaNO2, KI, MeCN, 12 h, 36–47% (e) 52: CuCN, DMSO, 160 °C, 0.5 h, 78% (f) 51, 53–55: requisite boronic acid, SiliCat-DPP-Pd, Na2CO3, DME, MW, 130 °C, 0.5h (g) 57: (1,10-Phenanthroline)(trifluoromethyl)copper(I), DMF, 55 °C, 1 h, 97% (i) LiOH, THF-MeOH, 1 h.
A convergent synthesis via cross coupling of the iodide in intermediate 51e or 56e was desired for more expedient analog synthesis, however, that approach failed for a number of desired analogs, and more specifically with methylene cyclopropyl analogs 62–63. Therefore, a linear synthetic route was utilized, as shown in Scheme 8. Analogs 58 and 61 were synthesized using two different methods and rigorously analyzed to determine the correct regiochemistry of the respective products. The synthesis commenced from reaction of the requisite commercially available carboxylic acid with 1,2,3-benzotriazole to form N-acylbenzotriazole derivatives 58a and 62a. The next step involved coupling of the formed N-acylbenzotriazoles 58a and 62a to substituted acetophenones 59a and 61a via a magnesium bromide ethyl etherate-catalyzed soft enolization method in the presence of Hunig’s base to form 1,3-diketones 58b and 61b-63b in 60–69%. The 1,3-diketones 58b and 61b-63b were then efficiently alkylated with 4-(bromomethyl)benzene sulfonamide using cesium carbonate in DMSO at room temperature to generate intermediates 58c and 61c-63c. Intermediates 59c-60c were readily obtained from 58b-61b via chlorination with NCS followed by potassium carbonate assisted alkylation of the 4-hydroxybenzenesulfonamide. Tosic acid-catalyzed cyclocondensation of 2-alkylated 1,3-diketones 58c-63c with ethyl 2-hydrazineylthiazole-4-carboxylatehydrobromide III generated a mixture of both desired and undesired regioisomers. The ratio of the formation of the regioisomers varies depends on the nature of the substitution at R1. For example, the cyclization ratio of 58c-61c the ratio was 50:50, whereas it decreased to 10:90 desired to undesired for 62c-63c. The desired isomer is slightly less polar (as judged by LC/MS analysis) and thus elutes as the second peak with reversed-phase (C18) chromatography. Of note, we were unable to separate these isomers efficiently with normal phase silica gel chromatography. The unseparated mixtures were then hydrolyzed with lithium hydroxide in THF-methanol and separated in HPLC to give 58d-63d as first peak and 58–63 as second peak.
Scheme 8. Syntheses of analogs 58–63 a.
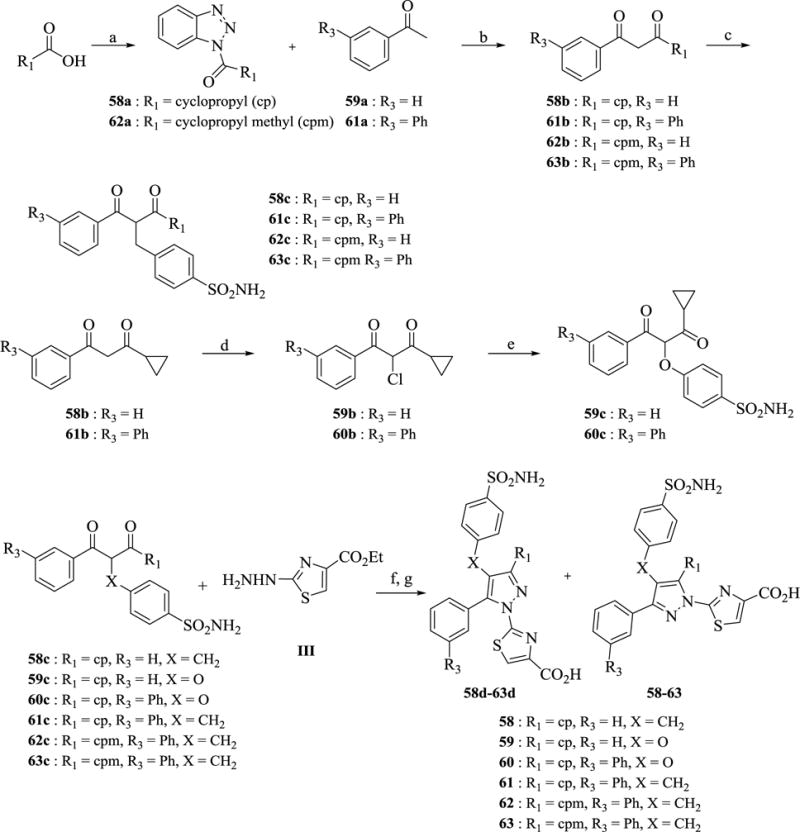
aReagents and conditions: (a) 1,2,3-Benzotriazole, SOCl2, CH2Cl2, 4 h, 91–100% (b) MgBr2·OEt, iPr2NEt, CH2Cl2, 12 h, 60–69% (c) 4-(bromomethyl)benzenesulfonamide, Cs2CO3, DMSO, 1 h, 55–83% (d) TMS-Cl, NCS, CH2Cl2, 0 °C-rt, 3 h, 73–94 % (e) 4-hydroxybenzenesulfonamide, K2CO3, DMSO, 50 °C, 1 h, 31–49% (f) TsOH, EtOH, reflux, 12 h, 77–83% (mixture) (g) LiOH, THF-MeOH, 1 h.
Analogs 64–68 were synthesized via functionalization at the corresponding ethyl esters at the thiazole ring as shown in scheme 9. Heating the intermediate 64a with ammonia in a sealed tube gave the amide analog 64 which, upon dehydration with trifluoroacetic anhydride (TFAA) in the presence of Hunig’s base in dichloromethane followed by reaction with sodium azide and ammonium chloride, gave the tetrazole analog 67. The intermediate 64a was obtained by the condensation of 37b and III as shown in the scheme 3. LAH reduction of the intermediate 66a furnished analog 66 in high yield. Trifluoromethylation of the intermediate 51e with (1,10-phenanthroline)(trifluoromethyl)copper(I) in DMF as shown in scheme 7 provided intermediate 66a. Manganese dioxide oxidation of the intermediate 68a and subsequent trifluromethyl addition to the aldehyde using TBAF and TMS-CF3 provided analog 64. The intermediate 68a was readily obtained from intermediate 43c using a Suzuki coupling with 4-(bromomethyl)benzenesulfonamideas shown in scheme 4.
Scheme 9. Syntheses of analogs 64–68 a.
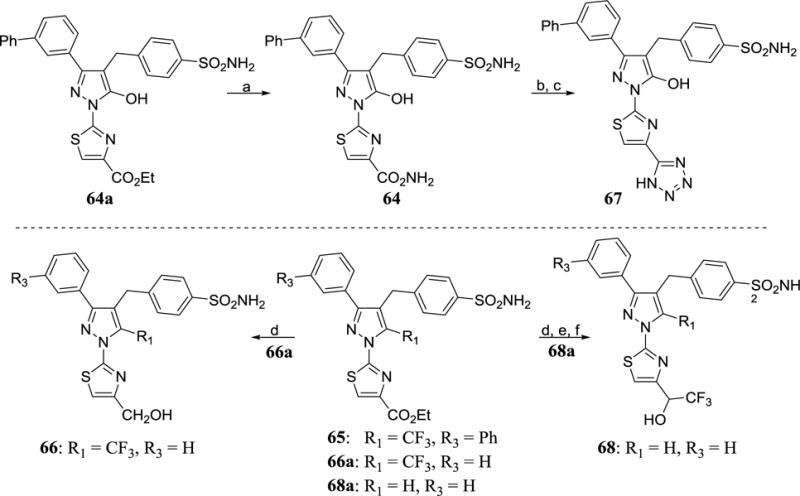
aReagents and conditions: (a) NH3, EtOH, 60 °C, 18 h, 90% (b) TFAA, iPr2NEt, CH2Cl2 (c) NaN3, NH4Cl, DMF, 125 °C, 2 h (d) LiAlH4, THF, 1 h (e) MnO2, CHCl3, 2 h (f) TMS-CF3, TBAF, THF, 4 h.
RESULTS AND DISCUSSION
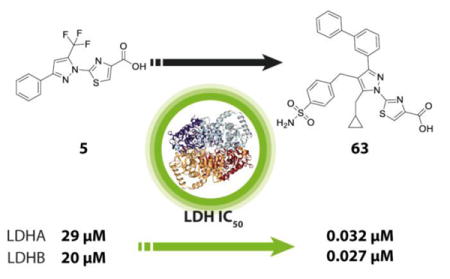
At the outset of our medicinal chemistry campaign, the leading molecule reported in the literature, denoted as 1 (Figure 1) was the result of a fragment-based screen and structure-guided optimization campaign by AstraZeneca researchers.12 While this compound had no appreciable cell-based activity (vide infra), it provided reproducible and robust biochemical inhibition of LDHA (IC50 = 1.02 μM, Table 1). Despite conducting our screen with concentrations ranging from 13 nM to 57 μM, very little inhibition was observed across a library of approximately 400,000 compounds, an observation in agreement with other screening efforts against this target. However, our qHTS screening paradigm19 enabled the identification of several compounds with modest potency (double digit μM) and partial efficacy (30–60% maximum response), but encouraging dose-dependent activity. One such compound, 2-(3-phenyl-5-(trifluoromethyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic acid (5) was thus identified as a putative LDH inhibitor and an attractive starting point for optimization based on its relatively small size (MW = 339 g/mol) presence of a carboxylic acid (could be docked to the pyruvate binding site) and chemical tractability. This compound has been previously reported as an EP1 receptor antagonist; the compound was found to have no appreciable CYP inhibition, and was stable in human plasma, suggesting good drug-like properties as a starting point for optimization.20 However, 5 exhibited only modest potency (22.2 μM, Table 1) and efficacy (40–60%). It is worth noting this compound would likely have been discarded as inactive in a traditional single-dose HTS screening paradigm, as it only displayed inhibition at concentrations greater (>20 μM) than those typically screened (e.g. 10 μM). However, the multiple dose qHTS format utilized at NCGC provides an opportunity to identify even modest inhibitors, making it an effective hit-identification platform for difficult targets like LDH.
Figure 1.
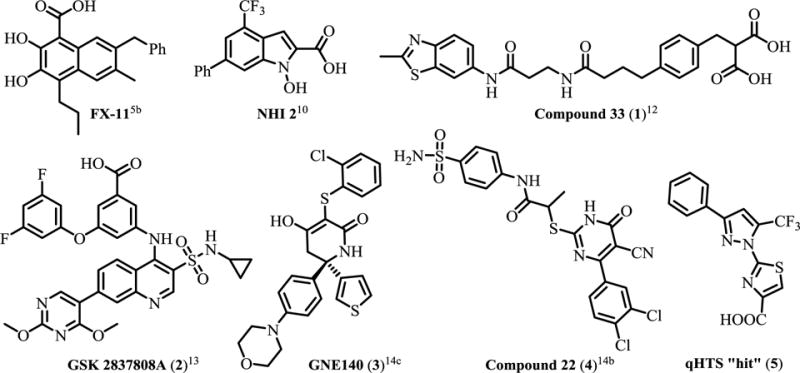
Representative previously described LDH inhibitors and qHTS “hit” 5.
Initial optimization efforts focused on systematic modifications to the CF3 group to explore variations in substituent size, lipophilicity, and polarity, as shown in Table 1. Given the weak potency and efficacy of these early analogs, tractable SAR was not easily discernible, but some changes were clearly tolerated. Replacement of CF3 (denoted as R1) with methyl (6), NH2 (7) or NHAc group (10) all led to compounds with similar potency and efficacy as 5. In contrast, replacement with an iPr (8) group or CHF2 (9) group resulted in a significant loss in potency (>57 μM). Encouragingly, a drastic improvement in potency (>100-fold) was observed when the CF3 group was replaced by OH, to provide the hydroxyl-pyrazole compound (11), with an IC50 of 144 nM. Additional hydroxyl analogs (16–32, Table 1) all exhibited submicromolar activity to further validate this SAR trend. Prior to the hydroxyl pyrazole discovery, we explored modifications to the phenyl group with representative examples (12–15) shown in Table 1. For these early analogs, a clear preference for halogens (e.g. Cl and F) at the 4-position emerged as exemplified by the potency difference between 12 and 13 (R2 = 2-Cl-Ph and 3-Cl-Ph, respectively), compared to 14 and 15 (R2 = 4-Cl-Ph and 4-F-Ph). Based on the improved potency of compound 11 (R1 = OH), we held this group constant and expanded our SAR efforts of the phenyl group (R2), with representative examples (16–32) highlighted in Table 1. Single halogen substitutions with either F or Cl substitution (16–19) were all well-tolerated, with the 4-F derivative (16) possessing the most potent activity (IC50 = 150 nM). Surprisingly, the preference for the 4-F group that we observed for the CF3-substituted pyrazoles did not translate to the hydroxypyrazole derivative, as compound 18 and 19 (R2 = 2-F-Ph and 3-F-Ph, respectively) had comparable potencies of 0.134 μM and 0.162 μM. Similar potency was also observed for larger substituents on 20 (R2 = 3-CF3-Ph) and 21 (R2 = 3-OCF3), which had IC50 values of 0.189 μM and 0.299 μM respectively. Incorporation of an electron donating group (22, R2 = 4-OMe-Ph) was tolerated, while replacement of the phenyl group with a 2-pyridine (23) or 4-pyridine (24) resulted in a modest loss of activity (IC50 values of 1.02 and 0.669 μM respectively). Changing the phenyl to a cyclohexyl group (25) gave potencies in line with the above-mentioned pyridine derivatives (IC50 = 1.1 μM), while the naphthyl derivative (26) had an IC50 value of 0.213 μM. Given data obtained from the preliminary SAR described above, we sought to further explore SAR of the pendant phenyl group at R2. Generally, bis-halogen substituted were well tolerated, as exemplified by analogs 27–30, with most compounds having comparable or improved potency (IC50 values ranging from 84 nM to 150 nM) relative to their mono-substituted counterparts. In an effort to explore the tolerance for larger hydrophobic groups, we synthesized several bi-phenyl derivatives, with representative examples being 31 (R2 = (3-Ph)-Ph) and 32 (R2 = 3-(2-F-Ph)-Ph), which had IC50 values of 266 nM and 95 nM, respectively. Though encouraged by the progress in improving the inhibitory potency this series in the enzymatic assay, we were concerned that none of these compounds demonstrated inhibition of lactate production in cellular assays (data not shown), despite biochemical potencies approaching 100 nM.
A majority of hydroxy-pyrazole based compounds reproducibly demonstrated non-classic dose-response curves. Namely, at most concentrations tested, these compounds yielded sigmoidal dose-response curves, often with 100% efficacy. However, at higher concentrations, inhibitory potency was being diminished.
One explanation for these aberrantly shaped IC50 curves could be that at higher concentrations, the compounds were not soluble and were precipitating out. But, these compounds have very good aqueous solubility, and no evidence of insolubility was observed. Alternatively, the compounds being tested might also contain a lower potency activator of LDHA that would overcome the inhibition at higher concentrations. Multiple studies have previously reported that several isoforms of LDHA, including human, demonstrate metal-binding activity, notably with divalent cations such as Mn2+, Co2+ and Zn2+. Incubation or complexation with certain metals has been reported to maintain or potentiate LDHA activity, while incubation with chelating agents such as EDTA has been found to reversibly inhibit activity of the yeast LDHA isoform.21 To test this hypothesis, the biochemical assay was adapted and rerun in the presence of 100 μM of EDTA to chelate any (trace) metal present in the assay. Notably, in the presence of EDTA, most of the hydroxyl-pyrazole derivatives (11, 16–32) did not just lose the aberrant portion of the IC50 curve, but instead lost almost all inhibitory activity, with many demonstrating IC50 values >57 μM (Table 1). In contrast to the hypothesis being tested, this result suggested that inhibition by the hydroxyl-pyrazole compounds either required or was potentiated by the presence of a trace metal. The activity of the original hit 5 and the CF3-pyrazole analogs 14 and 15 was unaffected by EDTA which suggested that inhibition by this chemotype requires binding to LDHA that can be enhanced by trace metals. Given the potential for metal coordination by the hydroxy-pyrazole, in combination with the thiazole nitrogen or sulfur and the carboxylic acid moiety, additional studies to characterize the source and influence of trace metals in this assay were conducted (see below for additional details). These experiments pointed to the assay buffer as the source of metal contamination, with trace amounts of Zn2+ being the most likely contaminant affecting the assay. Dialysis of individual assay components suggested that the trace metal was present in the commercial Tris buffer used for the assay, rather than in the stocks of LDHA enzyme or inhibitor (data not shown). Also, testing with several divalent cations demonstrated that activity of the hydroxyl-pyrazole inhibitors showed a dose-dependent potentiation with increasing metal concentration, and that the compounds were potentiated most strongly by Zn2+ (added as zinc chloride, data not shown). Metal impurities causing direct effects on LDHA has been reported previously, as Ward et al. found that the presence of silver resulted in inhibition of LDHA activity.12 Given these observations, we sought to obtain the crystal structure of this class of inhibitor bound to LDHA and carried our subsequent rounds of LDHA inhibition assays under two conditions to fully account for any metal coordination effects: in the presence of 100 μM EDTA, or in the presence of 100 μM ZnCl2.
The X-ray crystal structure of inhibitor 27 bound to hLDHA provided insight into the mechanism by which zinc enhanced inhibitor potency. The complex was obtained by transferring crystals grown with sodium malonate into a drop in which there was no malonate but which contained citrate. To this solution, 27 and ZnCl2 were added to a final concentration of 2 mM and allowed to soak for 3 days before crystals were harvested and flash frozen. The 1.95 Å crystal structure contained compound 27 bound to 3 of the 4 subunits of the LDHA tetramer, with malonate occupying the catalytic site of the remaining monomer. The inhibitor is anchored by a salt bridge interaction between its carboxylate and the guanidinium group of R168 (Figure 2A). A hydrogen bond also is observed between the carboxylate and the side chain of T247. The thiazole ring and the plane of N137 stack, and the distal difluorophenyl picks up hydrophobic interactions with the planes of P138 and Q99. The zinc is coordinated by H192, one water molecule, and three atoms in-plane from 27; the OH of the carboxylate, the NH of the thiazole, and the OH of the hydroxyl-pyrazole group providing a clear rationale for the additional potency conferred by zinc (Figure 2A, PDB: 5W8I). Our intial LDHA-inhibitor complexes were formed in the absence of NADH, however, the X-ray structure showed room below the inhibitor for occupancy by cofactor. Subsequent structures included NADH, which did not alter the position of the inhibitor but did lead to substantial increases in binding affinity as determined by SPR (vide infra).
Figure 2.
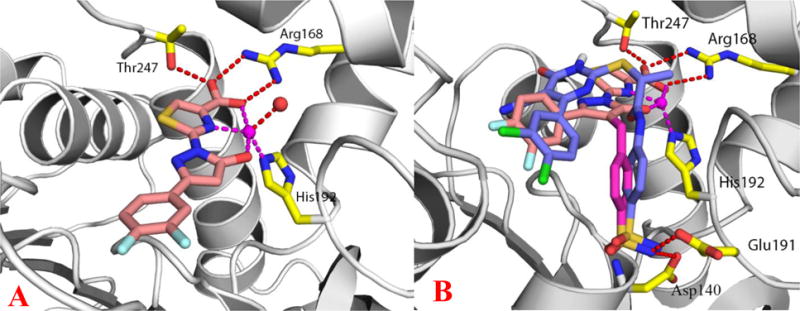
(A) Crystal structure of inhibitor 27 bound to LDHA in complex with zinc. The inhibitor is shown in sticks with salmon-colored carbons. The protein is shown in ribbon representation and the metal zinc is shown as a magenta sphere. A water molecule (red sphere) and protein residues R168, H192 and T247 (yellow-colored carbons) are coordinated with Zn or form H-bonding interactions with the inhibitor. PDB: 5W8I (B) Inhibitor 33 docked in the binding pocket of LDHA and overlaid with 4 (purple-colored carbons). The benzyl sulfonamide moiety shown as magenta-colored carbons.
With this experimental soaking method, we were then able to determine a 1.8 Å crystal structure of the weaker CF3-containing inhibitor 15 (Supplemental Figure 2, PDB: 5W8H). It was clear from the electron density in this structure that the binding mode of 15 is essentially the same as that of 27, with the carboxylate making the anchoring salt bridge interaction with R168 and the thiazole rings being superimposable between the two structures. Interestingly, the published LDHA inhibitor NHI-2 was predicted to adopt similar binding interactions in the active site, with the carboxylate interacting with R168 (R169 in ref. 10) and the trifluoromethyl pointing to H192 (H193 in ref. 10). The trifluoromethyl is well ordered in the crystal structure, but without the planar coordination enforced by zinc and with the increased steric bulk of the trifluoromethyl group (as compared to the hydroxyl), the pryazole is now tilted ~30° out of plane. In the absence of zinc, H192 now makes two hydrogen bonds directly to the carboxylate and thiazole nitrogen of 15.
Despite the apparent requirement of a coordinating zinc ion for optimal inhibitory potency, the similar binding orientation observed for 15, and the ability of 15 to adjust in order to fill the binding site and directly interact with H192 suggested that potent analogs could be designed that would bind without zinc.
A different LDHA inhibitor crystal structure published by Genentech (4, Figure 1, PDB: 4M49), revealed a comparable binding orientation as 27 in our series.14b and suggested an opportunity to design a hybrid molecule. The amino-phenyl sulfonamide substituent of 4 was reported to make numerous critical hydrogen bonds with the enzyme (i.e. Asp140, Glu191 and lle141). Indeed, molecular modeling overlay of the two structures (Figure 2B; purple = 4; tan = postulated benzyl sulfonamide analog of our chemotype) suggested that incorporation of a similar benzyl sulfonamide substituent onto the 4-position of the pyrazole ring could extend into the same pocket and pick up these same interactions without significantly disrupting the binding interactions of 27 (Figure 2B). Toward this end, compound 33 was synthesized, tested, and found to have an IC50 of 672 nM in the presence of EDTA (100 μM), as shown in Table 2. This compound represented the first sub-μM inhibitor of this chemical series that did not absolutely require zinc for potent inhibition. However, since this molecule retained the hydroxyl-pyrazole moiety, its activity could still be potentiated by zinc, leading to low nM inhibition (data not shown). Following this breakthrough, subsequent rounds of testing were conducted with EDTA in the assay buffer to ensure that metal-independent inhibition was driving SAR and further medicinal chemistry optimization.
Concurrently, GSK reported the discovery and biological characterization of 2, the first single/double-digit nM LDHA inhibitor with appreciable cell-based activity (lactate production assay), though the compound’s PK properties ultimately precluded its use in vivo.12 Given this, 2 was used as a benchmark compound for comparison purposes in both biochemical (Table 2) and cell-based assays (Table 5). Evaluation of parameters such as CLogP, Ligand Lipophilic Efficency (LLE) and Ligand Efficiency (LE) for prior art compounds 2 and 3 revealed an apparent preference for higher lipophilic character, with ClogP values of 7.75 and 4.79 for 2 and 3 respectively (see Supplemental Table 2). In comparison, compound 5 and further optimized analog 33 have ClogP values of 4.18 and 3.10 respectively. As a result, there is a marked improvement in LLE for 33 (3.07) over 5 (0.36) and prior art compounds 2 (-0.33) and 3 (1.58), as shown in Table S2. To further explore the SAR of our benzyl sulfonamide-containing scaffold, we obtained high-resolution (1.6 Å) crystal structures of 33 with (PDB: 5W8K) and without (PDB: 5W8J) the NADH cofactor (Figure 3A). As anticipated from the significantly improved inhibitory potency, the sulfonamide moiety formed critical hydrogen contacts with Asp140, Glu191 and I141. Compound 2 was less potent when inhibition assays were carried out with 10× higher NADH concentrations, whereas inhibition with our series of inhibitors was largely unaffected by this change. However, the crystal structure of the ternary complex with NADH and 33 suggested that inhibitor binding may be enhanced by the presence of NADH (Figure 3B). Results from surface plasmon resonance (SPR) studies (vide infra) substantiated this hypothesis.
Table 5.
Cell-based activity of representative analogs.
| A673 cells | MiaPaCa-2 cells | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Analog | Lactate inh. | Cytotoxicity | Long-term growth inh. | Lactate inh. | Cytotoxicity | Long-term growth inh. |
| IC50 ± SD (μM) a | IC50 ± SD (μM) a | IC50 (μM)c | IC50 ± SD (μM) a | IC50 ± SD (μM) a | IC50 (μM) c | |
| 1 | >57 | >57 | >20 | >57 | >57 | >20 |
| 2 | 14.5 ± 5.7 | >57 | 0.84 | 12.6 ± 3.6 | 28.4 ± 11.2b | 1.37 |
| 3 | 1.44 ± 0.09 | 2.63 ± 0.3 | - | 0.877 ± 0.059 | 1.24 ± 0.08 | - |
| 33 | >57 | >57 | - | >57 | >57 | - |
| 36 | >57 | >57 | - | >57 | >57 | - |
| 37 | 26.8 ± 3.1 | >57 | - | 27.7 ± 1.9 b | >57 | - |
| 38 | 30.0 ± 3.5b | >57 | - | 29.9 ± 0.1b | >57 | - |
| 42 | 30.8 ± 2.1b | >57 | - | >57 | >57 | - |
| 43 | 25.2 ± 19.4b | >57 | - | 33.5 ± 0.1b | >57 | - |
| 44 | 15.2 ± 3.2 | >57 | >20 | 9.88 ± 1.26 | >57 | >20 |
| 46 | 19.9 ± 4.5 | >57 | - | 7.24 ± 0.47 | >57 | - |
| 47 | 26.8 ± 3.1 | >57 | - | 22.4 ± 1.8 | >57 | - |
| 54 | 16.5 ± 6.2 | >57 | - | 26.6 ± 0.1b | >57 | - |
| 55 | >57 | >57 | - | >57 | >57 | - |
| 56 | 9.45 ± 0.01 | >57 | - | 17.7 ± 6.6 | >57 | - |
| 57 | 4.35 ± 1.21 | >57 | >20 | 4.80 ± 2.47 | >57 | >20 |
| 58 | 14.7 ± 3.6 | >57 | - | 17.5 ± 1.2 | >57 | - |
| 60 | 1.92 ± 0.43 | 25.8 ± 3.0 | - | 1.96 ± 0.13 | 28.3 ± 7.1 | - |
| 61 | 0.983 ± 0.335 | 13.9 ± 0.9 | 21.09 | 1.34 ± 0.15 | 18.9 ± 1.3 | 8.00 |
| 62 | 3.37 ± 0.39 | 24.8 ± 1.6 | >20 | 5.12 ± 0.33 | 24.9 ± 1.6 | >20 |
| 63 | 0.517 ± 0.088 | 2.90 ± 0.3 | 2.23 | 0.854 ± 0.059 | 3.96 ± 0.68 | 1.21 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay (n = 3).
Max. response was less than 50% and thus IC50 values should be considered as lower confidence.
IC50 values represent average of 2 assay replicates
Figure 3.
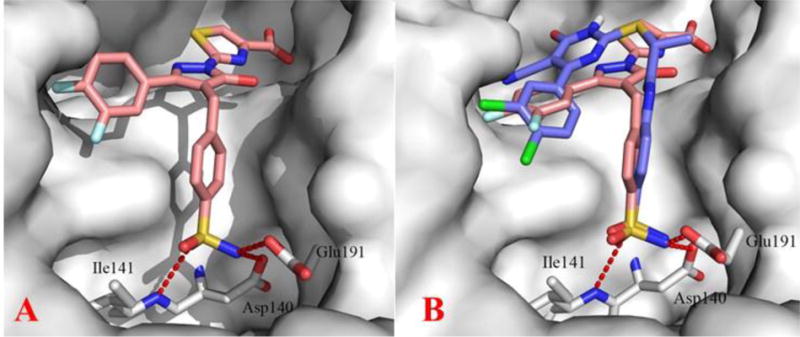
(A) Crystal structure of LDHA bound with 33. The inhibitor is shown in sticks and the protein is shown in surface representation. Key protein residues Asp140, Glu191 and lle141 form H-bonding interactions with the sulfonamide moiety as shown by dotted red lines. PDB: 5W8J (B) Overlay of inhibitor 33 (salmon) and 4 shown in purple (Figure 1) bound in the binding pocket of LDHA.
The importance of the sulfonamide and tolerance for modification of this region was evaluated via the synthesis of several analogs, with illustrative examples shown in Table 2. Moving the sulfonamide group to the 3-position of the phenyl ring (34), removing it completely (35), or adding a methyl to the amine (39) or an amide (40) resulted in loss of inhibitory activity (IC50 >57 μM). Moreover, repalcing the sulfonamide with a carboxylic acid (41) led to led to decrease in potency with IC50 value of ~34 μM. Given the tolerance and apparent benefit of bi-phenyl derivatives (Table 1), the corresponding 3-Ph (37) and 3-(2-F-Ph) (38) analogs were prepared and found to have IC50 values of 349 nM and 755 nM, respectively. As the hydroxy-pyrazole scaffold retains the ability to chelate Zn (and likely other metals), we sought to explore pyrazole substituents to identify analogs that alleviated this concern. The first series of analogs (42–50) removed the hydroxyl group altogether (R1 = H). Encouragingly, several of these compounds (e.g. 42) exhibited double digit nM IC50 values, comparable to the potency of (2). As observed previously, the bi-phenyl moiety (44; R3 = 3-Ph) was beneficial for improved inhibition in this series as well (IC50 = 38 nM). However, given the sufficient activity and ease of synthesis, the simple phenyl derivative (e.g. 43) was frequently used to explore further SAR. Accordingly, modification of the benzyl methylene group to either an oxygen (45) or nitrogen (46 and 47) was well tolerated, but the analagous amide (48) was not, with IC50 values of 124 nM, 57 nM, 91 nM, and >57 μM respectively. Moreover, replacement of the phenyl group with a piperazine moiety (49) led to a significant loss of potency (IC50 = 17.3 μM), as did incorporation of the 4-amino-phenyl sulfonamide group (50; IC50 = >57 μM). Taken together, these data support the notion that this region is critical for potent inhibition, and that tolerance for structural modification is limited.
Additional modifications to the pyrazole substitutent (R1 in Table 2) were explored next, as exemplified by analogs 51–63. Installation of a methyl group (51: IC50 = 0.042 μM and nitrile (52 IC50 = 0.115 μM) resulted in increased potency compared to the parent hydroxy-pyrazole derivative (33). This region did demonstrate some tolerance (albeit, with weaker potency) for larger hydrophobic groups, with compound 53 (R1 = Ph) possessing similar inhibition (IC50 = 0.139 μM). Interestingly, the 3-pyridine (54) and 4-pyridine (55) derivatives had significantly different potencies of 349 nM and 6.23 μM respectively, suggesting a deleterious interaction of the pyridine nitrogen when in the 4-position (or beneficial interation of the nitrogen in the 3-position). The amino derivative (56) was quite potent (IC50 = 65 nM), while incorporation of the original CF3 group led to a loss in activity (930 nM). Next, we incorporated a cyclopropyl group (58–61) and later a methylene cyclopropyl group (62–63) that were generally more potent than previous analogs, displaying potencies ranging from 9 to 110 nM. Extension of the cyclopropyl group with a methylene spacer enabled a potential cation-pi interaction between the pi-like character of the cyclopropyl group and an active-site tyrosine (Tyr238), as shown in Supplemental Figure 3, PDB: 5W8L. We also tested the respective regioisomers (61d and 63d) and neither of them showed any appreciable activity in enzymatic or cell-based assays (data not shown). Notably, while the biochemical potencies of the cyclopropyl and methylene cyclopropyl derivatives were not significantly different from other potent analogs, cellular assays provided a greater disparity in activity (vide infra).
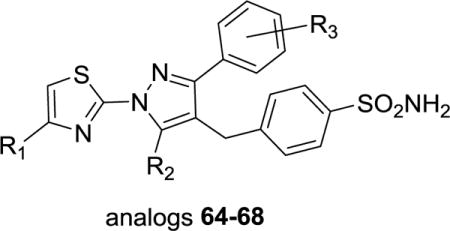
Thiazole carboxylic acid replacements were investigated. Efforts to replace the thiazole with any other ring structure, including oxazole, pyridine and other heterocycles, were all markedly detrimental to activity (data not shown). Finally, to determine the quantitative importance of the anchoring interaction between the carboxylic acid moiety and R168, we synthesized the corresponding amide (64), ethyl ester (65), primary alcohol (66), tetrazole (67) and secondary alcohol (68) derivatives. All showed a marked loss of potency (Table 3). The amide (64) and primary alcohol (66) analogs showed modest inhibition of 27.7 μM and 25.5 μM respectively, while 65, 67 and 68 were not inhibitory (IC50 = >57 μM), illustrating the requirement of the carboxylic acid moiety to maintain potent inhibition.
Table 3.
LDHA Inhibition of analogs 64–68a
| ||||
|---|---|---|---|---|
|
| ||||
| Analog | R1 | R2 | R3 | LDHA (w/EDTA) IC50 ± SD (μM) |
| 64 | CONH2 | OH | 3-Ph | 27.7 ± 1.9 |
| 65 | COOEt | CF3 | 3-Ph | >57 |
| 66 | CH2OH | CF3 | H | 25.5 ± 6.2b |
| 67 | tetrazole | OH | 3-Ph | >57 |
| 68 | CH(OH)CF3 | H | H | >57 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay (n ≥ 3) using a 22-dose response in 1536-well format.
Max. response was less than 50% and thus IC50 values should be considered as lower confidence.
Concurrently, Genentech published several papers describing the discovery and optimization of 3 (Figure 1), as a potent LDHA inhibitor.14c Compound 3 inhibited LDHA in our assay with an IC50 of 424 nM, substantially less potent than the reported values (IC50 < 10 nM). The discrepancy is likely due to differences in the assay conditions, and demonstrates the importance of conducting head-to-head comparisons of reference compounds when drawing conclusion about biological activity.
Selectivity of representative analogs was determined by assessing their biochemical activity against another LDH isozyme, LDHB, and two ‘off-target’ dehydrogenases, malate dehydrogenase (MDH) and isocitrate dehydrogenase 1 (IDH1). While there was negligible activity against both MDH and IDH1, most of the analogs revealed similar potency for LDHB when compared to LDHA (Table 4).
Table 4.
Selectivity against LDHB and other dehydrogenases.a
| Analog | LDHA IC50 (μM) |
LDHB IC50 (μM) |
MDH IC50 (μM) |
IDH1wt IC50 (μM) |
|---|---|---|---|---|
| 1 | 1.34 | 9.20 | >57 | >57 |
| 2 | 0.038 | 1.15 | 37.8 | >57 |
| 3 | 0.424 | 0.441 | >57 | >57 |
| 33 | 0.672 | 0.724 | >57 | >57 |
| 36 | 2.37 | 1.51 | 37.8b | >57 |
| 37 | 0.349 | 0.424 | >57 | 47.4b |
| 38 | 0.754 | 0.911 | 33.7 | >57 |
| 42 | 0.095 | 0.129 | >57 | >57 |
| 43 | 0.177 | 0.204 | >57 | >57 |
| 44 | 0.038 | 0.049 | 48.6 | 47.4 |
| 46 | 0.057 | 0.078 | 37.8b | >57 |
| 47 | 0.091 | 0.096 | >57 | >57 |
| 54 | 0.349 | 0.259 | >57 | >57 |
| 55 | 6.23 | 2.82 | >57 | >57 |
| 56 | 0.065 | 0.075 | 26.8b | >57 |
| 57 | 0.053 | 0.062 | 48.6 | 33.5b |
| 58 | 0.019 | 0.020 | 37.8 | >57 |
| 60 | 0.110 | 0.119 | >57 | 26.6 |
| 61 | 0.027 | 0.020 | 42.4 | 33.5 |
| 62 | 0.009 | 0.008 | >57 | 47.4 |
| 63 | 0.032 | 0.027 | 37.8b | 29.8 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the HTS assay (n = 3).
Max. response was less than 50% and thus IC50 values should be considered as lower confidence.
To determine the cellular activity of analogs, we tested them in a high-throughput fluorescence/absorbance-based lactate production assay (1536-well) using the A673 human sarcoma and MiaPaCa-2 human pancreatic cancer (see Table 5) cell lines in 22-point dose-response. Both cell lines demonstrate a highly glycolytic phenotype with robust production of lactate, making them an ideal choice for these assays. 2, which was reported to exhibit sub-μM inhibition of lactate production, demonstrated an IC50 of 14.5 μM, a result consistent across a number of cell lines. Though the reason for this discrepancy is unclear (difference in assay platforms may be one possibility), this observation underscored the need for novel inhibitors with greater efficacy in cells. Compound 3 exhibited better potency in this cell-based lactate production assay than did 2, with an IC50 of 1.44 μM. Compounds from our chemical series, e.g. 61 and 63, exhibited comparable potency in these cell lines. However, for many of the representative compounds shown in Table 5, the potent biochemical inhibition translated into little to no cell-based activity. To determine if cell permeability was a limiting factor, we measured the intracellular concentration of several inhibitors using LC-MS/MS analysis. The compounds appeared to reach appreciable intracellular levels (high μM), and thus well in excess of their respective biochemical IC50s values (data not shown). Differential lactate inhibition by compounds with comparable structures and presumably physicochemical properties (e.g. 44, R1 = H and 61 R1 = cyclopropyl) further suggested permeability was not the main barrier to cellular efficacy; these compounds demonstrated biochemical IC50 values of 38 nM and 27 nM respectively, yet 61, was approximately 15-fold more potent in the lactate production assay (0.983 μM (61) vs. 15.2 μM (44)). Moreover, the most potent compound in the lactate assay was compound 63 (IC50 = 0.517 μM), which possessed a methylene cyclopropyl group at R1 and 3-Ph group at R3, yet was less potent in the biochemical assay than the corresponding cyclopropyl derivative (61). The cytotoxicity of compounds against these glycolytic cell lines tracked reasonably well with inhibition of lactate production consistent with an on-target mechanism of cell killing. All compounds that demonstrated appreciable inhibition of lactate production (IC50 < 4 μM; 3, 60, 61, 62, 63) also exhibited modest cytotoxicity (IC50 1–30 μM) in a 48 hour Cell-Titer Glo assay in A673 and MiaPaCa-2 cells (see Table 5).
In an effort to probe this roughly 100 fold offset between biochemical and cellular potencies, we considered the concept of drug-target residence time which has been championed by Copeland and others.22 While this concept is usually brought up in the context of in vivo efficacy, it has been reported to impact cellular efficacy as well.23 Given the high concentration of intracellular LDH (estimated to be in the range of 2–17 μM),11,24 we reasoned that longer residence times might be necessary to achieve significant, sustained decreases in cellular LDH function. As noted above, this approach could also be beneficial in vivo, since drug concentration in systemic circulation decreases with time and LDH is an abundant, ubiquitously expressed protein. Thus, we obtained SPR data with representative compounds to examine this hypothesis.
For the initial study, four compounds (2, 33, 61 and 63) were chosen based on their biochemical potency and differential activity in the cell-based lactate output assay.25 Compounds from the internal chemical series displayed more potent binding affinities and longer off-rates (koff) in the presence of saturating NADH (500 μM) than without, supporting the hypothesis that arose from the initial crystallography studies. However, 2 was found to have greater binding affinity in the absence of NADH (KD = 1.7 nM versus 59 nM with NADH), in agreement with its reported NADH competitive mechanism of inhibition. Because inhibition of LDH in cells results in increased intracellular concentrations of NADH, a mechanism of inhibition that produces increased affinity for LDH and greater duration of target occupancy when NADH concentrations are elevated should be a more efficacious mechanism than a NADH-competitive one. The KD values for three representative compounds and a comparator (2, 33, 61 and 63) in the presence of NADH were 59 nM, 370 nM, 0.33 nM, and 0.11 nM respectively. Interestingly, the off-rates and corresponding residence times (τ) [calculated as 1/koff (s-1)] tracked well with the cellular potency [2 (τ = 8 secs), 33 (τ = 0.5 secs), 61 (τ = 470 secs) and 63 (τ = 1200 secs)]. While rational optimization of drug-target residence time has been reported,26 it can be difficult to τdelineate such structure-activity relationships, as minor structural changes may lead to surprisingly large differences in binding properties.27 As such, SPR analysis of binding kinetics will be incorporated into our future SAR efforts as an important optimization parameter and compound attribute, alongside biochemical potency, cell-based potency and ADME properties.
The Glycolysis Stress Test (GST) was performed on the Seahorse XF Analyzer in order to assess changes in glycolytic flux resulting from LDH inhibition (Figure 4). This assay measures the net glycolysis-dependent proton production in the cells and, as such, is a marker of the inhibition of glycolysis consequent on LDH inhibition. It is anticipated that, as LDH inhibition increases with the concentration of compound, the reserve biochemical capacity of the enzyme for glycolysis is exceeded, resulting in a depletion of NAD+ and, ultimately, inhibition of the entire pathway. The extracellular acidification rate (ECAR) was monitored in A673 cells before and after injection of five compounds (2, 3, 33, 61, and 63) over a range of concentrations. These data show a clear concentration-dependent decrease on the glycolytic flux (Figure 4A, 4B, and 4C), consistent with the anticipated inhibition of the proton efflux into the media. In addition, the capacity of the cell to switch from oxidative phosphorylation to glycolysis following injection of oligomycin, a specific inhibitor of ATP-synthase, is suppressed by all LDH inhibitors. This can be clearly seen from the quantification of % maximal ECAR (maximal glycolytic capacity) in A673 cells for a panel of five LDHA inhibitors, which shows that the inhibitory potency order is 63>61>33>3>2, which correlates with the cellular lactate output inhibition assay (see Table 5), with the exception of 33. As shown in Figure 4D, the leading compounds both 63 and 61 completely inhibit lactate production/extracellular acidification at concentrations of 2 and 5 μM, respectively, corroborating that the incorporation of bi-phenyl and cyclopropyl (61) or a methylene cyclopropyl (63) improved LDH inhibition in cell-based in vitro assays.
Figure 4.
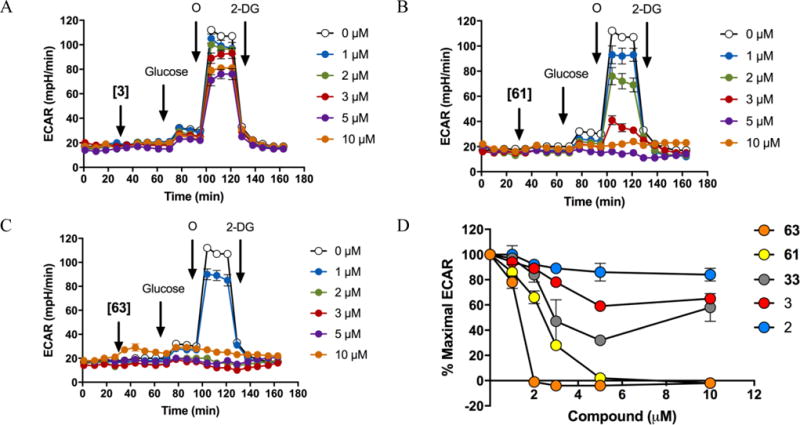
Decrease in glycolytic flux caused by LDH inhibitors in A673 cells. The Glycolysis Stress Test was performed in A673 cells, The Extracellular acidification rate (ECAR) over time; cellular basal ECAR was measured, then compounds (A) GNE140, (B) 61 or (C) 63 were injected in a dose-response manner, after 40 min, subsequent injections of glucose (10 mM), oligomycin (O at 1μg/mL; reaching maximal glycolytic capacity), and 2-deoxyglucose (2-DG at 50 mM; inhibition of glycolysis) were made. (D) Quantification of the maximal ECAR (% from control of maximal ECAR minus ECAR with 2-DG) of increasing concentrations of the five LDH inhibitors.
To obtain additional evidence of cellular target engagement of LDHA by our compounds, CETSA (Cellular Thermal Stabilization Assay), a technique which translates the principles of biochemical thermal shift assays to a cellular context, was adapted and optimized for LDHA in a number of cell lines.28 Accordingly, a panel of representative compounds were tested in A673 cells, spanning a wide range of potencies (Supplemental Figure 1A–E). Cellular binding and stabilization of LDHA was observed with a number of top LDHA inhibitors (Table 6) at concentrations as low as 100 nM. Compounds 62 and 63 were particularly potent at stabilizing LDHA in A673 cells. Given its relatively direct readout on target engagement, we examined the relationship of CETSA-based activity to other measures of LDHA inhibitor potency. Though the compounds with the most potent activity against LDHA in the biochemical assay were also among the most potent in the CETSA assays, no clear correlation emerged between the cellular stabilization of LDHA via CETSA and either biochemical or cellular activities of these molecules. Interestingly, 1 demonstrated cellular stabilization of LDHA, despite showing no activity in any prior cell-based LDHA assays. Conversely, 57 demonstrated potent inhibition of LDHA and moderate inhibition of cellular lactate, yet displayed comparatively little stabilization of LDHA via CETSA, and no toxicity or growth inhibition of either cell line tested. As such, CETSA-measured LDHA-binding was utilized as one marker of activity in a panel of many.
Table 6.
CETSA LDHA stabilization in A673 cells.
| Analog | LDHA inh. | Lactate inh. | CETSA: Cellular LDHA stabilization | ||
|---|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | EC50 (μM)a | Max stab.b (norm’d) | AUCc (norm’d) | |
| 1 | 1.020 | >57 | 1.60 | 64.8 | 62.08 |
| 2 | 0.038 | - | 8.18 | 52.4 | 43.22 |
| 3 | 0.424 | 12.60 | 1.26 | 79.0 | 72.96 |
| 5 | 22.200 | - | >100 | 8.5 | 13.84 |
| 33 | 0.672 | >57 | 3.53 | 47.9 | 53.66 |
| 44 | 0.038 | 9.88 | 2.56 | 100.0 | 89.77 |
| 57 | 0.053 | 4.80 | >100 | 41.6 | 26.76 |
| 61 | 0.027 | 1.34 | 0.25 | 63.0 | 70.66 |
| 62 | 0.009 | 5.12 | 0.07 | 70.8 | 100.00 |
| 63 | 0.032 | 0.85 | 0.17 | 69.9 | 81.95 |
EC50 values represent the half maximal (50%) concentration (the concentration needed to stabilize half of the detectable LDHA protein at a set melting temperature) as determined by CETSA.
Maximum stabilization values represent the highest amount of stabilized LDHA detected for each sample, as measured by Western blot band intensity. These values are scaled against the band with the greatest amount of stabilized LDHA (maximum response of 44) to provide a relative measure of target binding.
Area-under-the-curve (AUC) values demonstrate the total amount of LDHA stabilized by each compound across all concentrations, scaled against the AUC of the compound with the greatest total amount of LDHA stabilization (62), again to provide a relative measure of LDHA engagement.
Having developed compounds with potent inhibition of LDH (<20 nM) and lactate production (1–2 μM), and with robust target engagement, as assessed via SPR and CETSA, we investigated the cytotoxicity of these agents against MiaPaCa-2 and A673 cells. Initial efforts utilized a 48 hr cytotoxicity assay using ATP content as a viability surrogate (CellTiter-Glo), and longer-term effects on cell growth were studied by monitoring confluence over 5 days using an Incucyte ZOOM (see Table 5). Early analogs had minimal to no effect on cell proliferation, whereas 3 and 63 demonstrated suppression of cell growth over time (Table 5, 63Figure 5A). Dose-response treatment of MiaPaCa-2 cells with 63 showed effects on cellular proliferation at concentrations as low as 250 nM, and with nearly complete arrest of cell growth at 20 μM (Table 5, and Figure 5B). Area-under-the-curve (AUC) analysis was conducted on MiaPaCa2 and A673 dose-response data to enable comparison of LDH inhibitors (Figures 5C & 5D, respectively); 3 and 63 both exhibited single digit μM activity in both A673 and MiaPaCa-2 cells. While inhibition of cell proliferation itself was not exceptionally potent, it was encouraging that cytotoxicity was positively correlated with target potency (enzymatic and cell-based), suggesting that the effects were target-mediated.
Figure 5.
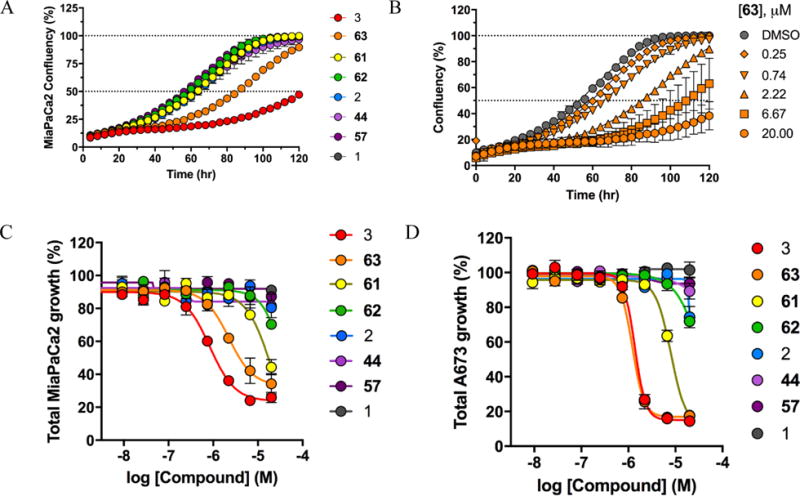
Incucyte long-term cellular growth data. (A) Cellular confluency of MiaPaCa-2 cells treated with LDHA inhibitors at 2.22 μM over 120 hours. (B) Dose-response relative growth of MiaPaCa-2 cells treated with 63 for 120 hours. Relative growth of (C) MiaPaCa-2 and (D) A673 cells treated with LDHA inhibitors for 120 hours.
To help define the utility of these initial compounds for proof-of-concept animal studies, we performed in vivo PK and in vitro ADME studies on representative inhibitors 61 and 63 (Table 7). Tier 1 ADME profiling was comprised of rat liver microsome (RLM) stability, PAMPA permeability and aqueous solubility (pH 7.4). Both compounds exhibited high microsomal stabilities of >30 minutes (data not shown), the highest estimable T1/2 from a single measurement at 15 minutes.29 Owing to the presence of a carboxylic acid moiety, PAMPA permeability was low (1–8 × 10–6 cm/sec), while solubility was high (>82 μg/mL, the maximum concentration tested in the assay). Time-course studies revealed that compounds were also stable in mouse and human liver microsomes (T1/2 >200 min for both compounds and species) and hepatocytes (mouse: T1/2 ~ 240 min) (Table 7). Given the presence of both carboxylic acid and sulfonamide moieties in the lead molecules, the potential to form glucuronide conjugates exists. As such, we tested microsomal stability in the presence of NADPH/UDPGA substrates. Encouragingly, neither 61 nor 63 showed any evidence of glucuronidation when incubated in either mouse or human liver microsomes, suggesting phase II metabolism may not be a significant issue for this chemotype (data not shown). This observation was further supported by the long half-life in mouse hepatocytes.
Table 7.
Representative ADME data for 61 and 63a
| T1/2 (min) | T1/2 (min) | (10–6 cm/s) | (μg/mL) | T1/2 (min) | ||
|---|---|---|---|---|---|---|
|
|
||||||
| Analog | MLM | HLM | Mouse Hepatocytes | PAMPA | Kinetic Solubility | Plasma Stability |
| 61 | 321 | 414 | 268 | 1 | >82 | >120 |
| 63 | 444 | 206 | 239 | 8 | >82 | >120 |
Aqueous kinetic solubility (PBS buffer) and PAMPA permeability were conducted at NCATS. Mouse plasma stability studies were conducted at Pharmaron Inc and involved 5 time points. The microsomal stability data [mouse liver microsomes (MLM), human liver microsomes (HLM) and mouse hepatocytes] were conducted at QuintaraBio and represent the stability in the presence of NADPH and UDPGA. The parent compound was monitored at 5 time points over 90 minutes.
As a result of the encouraging ADME data of lead compounds, the in vivo PK properties of lead compounds were determined. Both compounds showed clearance values that far exceed hepatic blood flow (HBF) in mouse species (90 mL/min/kg), with in vivo clearances of 294 and 227 mL/min/kg for 61 and 63, respectively (Table 8). These data, coupled with the long half-life in mouse liver microsomes suggested a mechanism other than liver metabolism was contributing significantly to compound elimination in vivo. Potential explanations for clearance much greater than hepatic blood flow include compound instability in plasma, renal clearance, sequestration in erythrocytes and/or high volume of distribution. These compounds were demonstrably stable in mouse plasma, with no degradation observed over 120 minutes (Table 7). Notably, compound 63 did exhibit a high volume of distribution, possibly accounting for some of the observed high clearance values. However, further studies are required to determine whether renal clearance or erythrocyte sequestration is a contributing factor, and on-going medicinal chemistry optimization around this chemotype may lead to compounds with improved PK properties for use in animal studies. Encouragingly, despite their high clearance and poor PAMPA permeability, appreciable systemic exposure was achieved in the range of cellular IC50 values following PO dosing at 50 mg/kg. Passive absorption was undoubtedly impacted by the presence of a carboxylic acid moiety, yet bioavailability (F) was determined to be ~50% for both compounds, suggesting that analogs with reduced clearance may lead to satisfactory exposure following oral dosing. Given the SAR for this series, which demonstrated the critical importance of the carboxylic acid moiety, future efforts to improve PK properties will focus on other areas of the molecule.
Table 8.
Pharmacokinetic Profiles of 61 and 63 in CD1 micea
| 61 | 63 | |||
|---|---|---|---|---|
|
|
||||
| Parameter | IV | PO | IV | PO |
| Cl (L/h/kg) | 227 | ‒ | 294 | ‒ |
| T1/2 (h) | 0.85 | 1.94 | 2.98 | 3.75 |
| Cmax (μM) | 1.45b | 1.20 | 0.74 b | 1.78 |
| Tmax (h) | ‒ | 0.25 | ‒ | 0.25 |
| AUClast (μM·h) | 0.26 | 3.2 | 0.19 | 2.5 |
| Vss (L/kg) | 5.6 | ‒ | 27 | ‒ |
| F(%) | ‒ | 49 | ‒ | 50 |
values calculated from drug concentration in plasma following IV (2 mg/kg) and PO (50 mg/kg) dosing. n = 3, 8-time points taken over 24 hours. Compounds 61 and 63 were formulated as solution in PBS buffered saline with 1.1 eq. NaOH (final pH 7–8).
Cmax = C0 (t = 0) for IV administration. All pharmacokinetic studies were conducted at Pharmaron Inc.
CONCLUSION
LDH has long been implicated in the pathogenesis of cancer and many other diseases, and thus has received considerable attention from both pharmaceutical companies and the academic scientific community. Despite multiple efforts to discover potent and drug-like inhibitors of LDHA, few viable inhibitors emerged until the recent pioneering work by GSK and Genentech. These published inhibitors possess potent biochemical activity, yet their cellular effects remain modest and pronounced in vivo activity remains elusive. Herein, we describe the discovery and medicinal chemistry optimization of a potent and cell-active pyrazole-based inhibitor of LDH. The original “hit” molecule 5 was identified via a qHTS campaign, and medicinal chemistry optimization was aided by both crystallography efforts and a battery of biological assays. As noted by other groups, despite obtaining potent biochemical inhibition of LDH, early representatives in this series had very modest cellular activity (e.g. inhibition of lactate production). However, through utilization of target engagement assays (CETSA) and biophysical characterization (SPR), we gained insight into the drivers of cellular potency, with the target residency times of these analogs being a key determinant. To that end, we identified compound 63 as a chemical probe for LDH, and a promising lead compound worthy of further optimization, given its sub-μM inhibition of lactate production, robust cellular target engagement, inhibition of glycolytic flux and favorable off-rate in SPR studies. While 63 possesses some favorable ADME attributes (e.g. microsomal stability, solubility), additional optimization will be required to achieve a PK profile suitable for use in in vivo efficacy studies. Current efforts are focused on implementing the lessons learned so far, and outlined herein, to guide development of compounds with better cellular potency and PK properties. Results from these additional efforts, along with further biological characterization of the compounds, will be reported in due course.
EXPERIMENTAL SECTION
General Methods for Chemistry
All air or moisture sensitive reactions were performed under positive pressure of nitrogen or argon with oven-dried glassware. Anhydrous solvents and bases such as dichloromethane, N,N-dimethylforamide (DMF), acetonitrile, ethanol, DMSO, dioxane Hunig’s base and triethylamine were purchased from Sigma-Aldrich. Palladium catalysts were purchased from Strem chemicals and used as such. SiliaCat® Heterogeneous Catalyst DPP-Pd (Catalog # R390–100) was purchased from SiliCycle Inc. Preparative purification was performed on a Waters semi-preparative HPLC system using a Phenomenex Luna C18 column (5 micron, 30 × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10% to 50% acetonitrile over 8 minutes was used during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA). Method 1: A 7-minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8-minute run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50 °C. Method 2: A 3-minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5-minute run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 micron, 3 × 100 mm) was used at a temperature of 50 °C. Purity determination was performed using an Agilent Diode Array Detector for both Method 1 and Method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical shifts are reported in ppm with undeuterated solvent (DMSO-d6 at 2.49 ppm) as internal standard for DMSO-d6 solutions. All of the analogs tested in the biological assays have purity greater than 95%, based on both analytical methods. High resolution mass spectrometry was recorded on Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
General procedure for the synthesis of acybenzotriazole derivatives (Method A)
To a solution of 1H-benzo[d][1,2,3]triazole (23.8 g, 200 mmol, 4 eq) in CH2Cl2 (150 mL) was added thionyl chloride (3.65 mL, 49.9 mmol, 1 eq) and stirred at room temperature for 0.5 h. Appropriate alkyl carboxylic acid (49.9 mmol, 1 eq) was then added slowly and stirred for another 2 h. The reaction mixture was filtered and the filter cake was washed with CH2Cl2 (50 mL). The filtrate was neutralized with bicarbonate solution slowly and stirred for 30 minutes then transferred to a separatory funnel. The organic layer was washed with bicarbonate solution then with brine, dried over MgSO4, filtered, and concentrated. The residue was purified directly on a flash system using 120 g silica column eluting with 0–20 % ethyl acetate in hexanes over 15 column volumes. The first peak was collected and dried to get an oil or solid.
General procedure for the synthesis of 1-aryl-3-substituted propane-1,3-diones (Method B)
A mixture of acetophenone 59a or 61a (51.0 mmol, 1 eq), magnesium bromide diethyl etherate (32.9 g, 127 mmol, 2.5 eq) and acybenzotriazole derivative 58a or 62a (76 mmol, 1.5 eq in CH2Cl2 (100 mL) was added Hunig’s base (26.7 mL, 153 mmol, 3 eq) slowly (cooling is necessary for large scale) then stirred at rt for 12 h. The reaction mixture was cooled in an ice bath and quenched with 1 M HCl. The product was extracted with CH2Cl2 and the organic layer was subsequently washed brine. After drying the organic layer with MgSO4, the crude product was purified on a ISCO flash system using 220 g gold column eluting with 0–30 % ethyl acetate over 20 column volumes in hexanes to afford yellow oil after removing the solvent in 60–69% yield.
General procedure for the alkylation of 1-aryl-3-substituted propane-1,3-diones (Method C)
1-Aryl-3-substituted propane-1,3-dione (35.9 mmol, 1 eq) and cesium carbonate (14.05 g, 43.1 mmol, 1.2 eq) in DMSO (25 mL) was stirred at room temperature for 10 minutes. 4-(Bromomethyl)- benzenesulfonamides (10.78 g, 43.1 mmol, 1.2 eq) was added in one portion and further stirred at room temperature for another 1–2 h. The resulting mixture was diluted with large excess ethyl acetate and filtered through celite to remove any solid impurities. The filtrate was washed with saturated ammonium chloride 3 times and then with brine. The organic layer was dried with Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified directly on flash system using 220 g silica column eluting with 20–60% ethyl acetate in hexanes over 16 column volumes to afford pure products 58c, 61c-63c in 55–83 % yield.
General procedure for the cyclization of 1-aryl-2, 3-disubstituted propane-1,3-diones and hydrolysis (Method D)
A mixture of appropriate 1-aryl-2, 3-disubstituted propane-1,3-dione (2.24 mmol, 1 eq), ethyl 2-hydrazinylthiazole-4-carboxylate hydrogen bromide III (0.600 g, 2.24 mmol, 1 eq) and tosic acid (0.425 g, 2.24 mmol, 1 eq) in ethanol (10 mL) was heated in microwave for 15 minutes at 150 °C. The excess solvent removed using forced air and the residue was taken in dichloromethane. The crude suspension was then directly loaded to a 100 g silica column fitted to a flash system and eluted with 20–40% ethyl acetate in hexanes to get a mixture of regioisomers in 77–83% yield. This mixture of products was taken as such in a THF-methanol (2:1) mixture and treated with 1.5 M solution (5 eq) of aqueous lithium hydroxide. The reaction mixture was stirred for 1 hour, the solvent was removed and acidified with 1 molar hydrochloric acid then extracted with ethyl acetate. After evaporation of organic layer, the crude material was taken in DMSO and the regioisomers were separated on a preparative HPLC. The desired isomer was eluted as a second peak.
(1H-Benzo[d][1,2,3]triazol-1-yl)(cyclopropyl)methanone (58a)
This compound was prepared using Method A starting from cyclopropane carboxylic acid in 100% yield. LC-MS Retention Time: (Method 2) = 3.256 min, m/z (M+H)+ = 188.
1-Cyclopropyl-3-phenylpropane-1,3-dione (58b)
This compound was prepared using Method B starting from 58a and 59a in 60% yield. LC-MS Retention Time: (Method 2) = 3.52 min, m/z (M+H)+ = 189.
4-(2-Benzoyl-3-cyclopropyl-3-oxopropyl)benzenesulfonamide (58c)
This compound was prepared using Method C starting from 58b in 61% yeild. LC-MS Retention Time: (Method 2) = 3.32 min, m/z (M+H)+ = 358.
2-(5-Cyclopropyl-3-phenyl-4-(4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic acid (58)
This compound was prepared using general Method D starting from 58c and III. LC-MS Retention Time: (Method 1) = 4.905 min and (Method 2) = 3.323 min; 1H NMR (400 MHz, DMSO-d6) δ 13.17 (s, 1H), 8.29 (s, 1H), 7.72 – 7.67 (m, 2H), 7.52 – 7.46 (m, 2H), 7.42 – 7.32 (m, 3H), 7.30 – 7.23 (m, 4H), 4.12 (s, 2H), 2.23 (tt, J = 8.5, 5.6 Hz, 1H), 1.00 – 0.91 (m, 2H), 0.68 – 0.60 (m, 2H); HRMS (ESI) m/z (M+H)+ calcd. for C23H21N4O4S; 481.0999 found 481.0992.
2-(5-Cyclopropyl-3-phenyl-4-(4-sulfamoylphenoxy)-1H-pyrazol-1-yl)thiazole-4-carboxylic acid (59)
This compound was prepared using general Method D starting from 59c and III. LC-MS Retention Time: (Method 1) = 5.119 min and (Method 2) = 3.257 min; 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H), 8.35 (s, 1H), 7.83 – 7.71 (m, 4H), 7.45 – 7.31 (m, 3H), 7.29 – 7.14 (m, 4H), 2.63 (tt, J = 8.5, 5.5 Hz, 1H), 0.99 – 0.88 (m, 2H), 0.92 – 0.80 (m, 2H); HRMS (ESI) m/z (M+H)+ calcd. for ; C22H19N4O5S; 483.0791 found 483.0809.
2-(3-([1,1′-Biphenyl]-3-yl)-5-cyclopropyl-4-(4-sulfamoylphenoxy)-1H-pyrazol-1-yl)thiazole-4-carboxylic acid (60)
This compound was prepared using general Method D starting from 60c and III. LC-MS Retention Time: (Method 1) = 5.823 min and (Method 2) = 3.496 min; 1H NMR (400 MHz, DMSO-d6) δ 13.17 (s, 1H), 8.36 (s, 1H), 7.93 (td, J = 1.8, 0.5 Hz, 1H), 7.85 – 7.71 (m, 3H), 7.66 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 7.56 – 7.32 (m, 6H), 7.30 – 7.21 (m, 4H), 2.73 – 2.61 (m, 1H), 1.02 – 0.90 (m, 2H), 0.93 – 0.83 (m, 2H); HRMS (ESI) m/z (M+H)+ calcd. for; C28H23N4O5S2; 559.1104 found 559.1127.
1-([1,1′-Biphenyl]-3-yl)-3-cyclopropylpropane-1,3-dione (61b)
This compound was prepared using Method B starting from 58a and 61a in 66% yield. LC-MS Retention Time: (Method 2) = 3.85 min, m/z (M+H)+ = 265.
4-(2-Benzoyl-3-cyclopropyl-3-oxopropyl)benzenesulfonamide (61c)
This compound was prepared using Method C starting from 61b in 63% yield. LC-MS Retention Time: (Method 2) = 3.46 min, m/z (M+H)+ = 434.
2-(3-([1,1′-Biphenyl]-3-yl)-5-cyclopropyl-4-(4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic acid (61)
This compound was prepared using general Method D starting from 61c and III. LC-MS Retention Time: (Method 1) = 5.636 min and (Method 2) = 3.556 min; 1H NMR (400 MHz, DMSO-d6) δ 13.09 (s, 1H), 8.31 (s, 1H), 7.78 – 7.72 (m, 2H), 7.67 (ddd, J = 7.5, 1.9, 1.3 Hz, 1H), 7.60 (td, J = 1.8, 0.6 Hz, 1H), 7.57 – 7.45 (m, 2H), 7.40 (d, J = 4.3 Hz, 4H), 7.36 – 7.30 (m, 3H), 7.28 (s, 2H), 4.17 (s, 2H), 2.28 (tt, J = 8.6, 5.6 Hz, 1H), 1.09 – 0.85 (m, 2H), 0.77 – 0.54 (m, 2H); HRMS (ESI) m/z (M+H)+ calcd. for ; C29H25N4O4S; 557.1312 found 557.1320.
1-(1H-Benzo[d][1,2,3]triazol-1-yl)-2-cyclopropylethan-1-one (62a)
This compound was prepared using Method A from 2-cyclopropylacetic acid in 91% yield. LC-MS Retention Time: (Method 2) = 3.53 min, m/z (M+H)+ = 202.
4-Cyclopropyl-1-phenylbutane-1,3-dione (62b)
This compound was prepared using Method B starting from 62a and 59a in 63% yield. LC-MS Retention Time: (Method 2) = 3.76 min, m/z (M+H)+ = 203.
4-(2-Benzoyl-4-cyclopropyl-3-oxobutyl)benzenesulfonamide (62c)
This compound was prepared using Method C starting from 62b in 55% yield. LC-MS Retention Time: (Method 2) = 3.35 min, m/z (M+H)+ = 372.
2-(5-(Cyclopropylmethyl)-3-phenyl-4-(4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic acid (62)
This compound was prepared using general Method D starting from 62c and III. LC-MS Retention Time: (Method 1) = 5.199 min and (Method 2) = 3.495 min; 1H NMR (400 MHz, DMSO-d6) δ 13.15 (s, 1H), 8.29 (s, 1H), 7.74 – 7.66 (m, 2H), 7.60 – 7.53 (m, 2H), 7.44 – 7.36 (m, 3H), 7.32 – 7.25 (m, 4H), 4.14 (s, 2H), 3.15 (d, J = 6.9 Hz, 2H), 1.22 – 1.05 (m, 1H), 0.38 – 0.28 (m, 2H), 0.26 – 0.17 (m, 2H); HRMS (ESI) m/z (M+H)+ calcd. for C24H23N4O4S2; 495.1155 found 495.1174.
1-([1,1′-Biphenyl]-3-yl)-4-cyclopropylbutane-1,3-dione (63b)
This compound was prepared using Method B starting from 62a and 61a in 69% yield. LC-MS Retention Time: (Method 2) = 4.02 min, m/z (M+H)+ = 279.
4-(2-([1,1′-Biphenyl]-3-carbonyl)-4-cyclopropyl-3-oxobutyl)benzenesulfonamide (63c)
This compound was prepared using Method C starting from 63b in 83% yield. LC-MS Retention Time: (Method 2) = 3.59 min, m/z (M+H)+ = 448.
2-(3-([1,1′-Biphenyl]-3-yl)-5-(cyclopropylmethyl)-4-(4-sulfamoylbenzyl)-1H-pyrazol-1-yl)thiazole-4-carboxylic acid (63)
This compound was prepared using general Method D starting from 63c and III. LC-MS Retention Time: (Method 1) = 6.08 min and (Method 2) = 3.521 min; 1H NMR (400 MHz, DMSO-d6) δ 13.11 (s, 1H), 8.28 (s, 1H), 7.75 – 7.69 (m, 2H), 7.68 – 7.62 (m, 2H), 7.59 (dt, J = 7.7, 1.4 Hz, 1H), 7.52 – 7.45 (m, 1H), 7.45 – 7.37 (m, 4H), 7.35 – 7.29 (m, 3H), 7.27 (s, 2H), 4.16 (s, 2H), 3.16 (d, J = 6.9 Hz, 2H), 1.20 – 1.03 (m, 1H), 0.37 – 0.28 (m, 2H), 0.24 – 0.18 (m, 2H); HRMS (ESI) m/z (M+H)+ calcd. for ; C30H27N4O4S; 571.1468 found 571.1471.
Cell-Lines
The MiaPaCa-2 and A673 cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines were cultured in DMEM (Invitrogen 11965118) culture medium supplemented with 10 % fetal bovine serum and 100 units/mL Penicillin, 100 μg/mL Streptomycin and maintained in a 37° C, 5% CO2/95% humidified air incubator.
Biochemical assays
LDHA biochemical assay: Briefly, 3 μL of human lactate dehydrogenase 5 (#A38558H, Meridian Life Science, Inc., Memphis, TN) in LDH assay buffer (200 mM Tris HCl pH 7.4, 100 μM EDTA and 0.01% Tween-20) was added to a black solid bottom 1536-well assay plate (Greiner Bio-One) using a BioRAPTR FRD dispenser (Beckman Coulter, Brea, CA). A 1536-well pintool dispenser outfitted with 20 nL pins (Wako Automation, San Diego, CA) was used to transfer 23 nL of DMSO-solubilized compound (both library and vehicle controls) to each 1536-well assay plate. Following compound transfer, 1 μL of substrate solution containing NADH and sodium pyruvate (Sigma-Aldrich, St. Louis, MO) in LDH assay buffer was dispensed via BioRAPTR FRD to initiate the reaction. Final concentrations in the 4 μL reaction volume were 2 nM LDHA enzyme, 0.06 mM NADH and 0.2 mM sodium pyruvate. Following a 5 minute incubation period at room temperature, 1 μL of detection reagent (C. kluyveri diaphorase (Sigma-Aldrich) and resazurin sodium salt (Sigma-Aldrich) in LDH assay buffer) was added to a total volume of 5 μL. Final concentrations of detection reagents were 0.133 mg/mL diaphorase and 37 μM resazurin. Plates were immediately transferred to a ViewLux microplate imager (PerkinElmer, Waltham, MA), and any resulting resorufin fluorescence was measured (ex540, em590 nm) at 0 and 20 minutes. Fluorescence was normalized using enzyme-free and DMSO-treated control wells on each plate.
LDHB biochemical assay: Human lactate dehydrogenase 1 (#A38155H, Meridian Life Science, Inc., Memphis, TN) was assayed as described above for LDHA. Final concentrations in the 4 μL reaction volume were 2 nM LDHB enzyme, 130 μM NADH and 160 μM sodium pyruvate.
MDH biochemical assay: Briefly, 3 μL of MDH solution (containing 13.33 IU/mL malate dehydrogenase from porcine heart, 0.2 mM NAD, 0.067mg/mL diaphorase and 0.067 mM resazurin in MDH assay buffer (50 mM Tris pH 8.0, 5 mM MgCl2, 0.01% Brij 3)) was added to a black solid bottom 1536-well assay plate (Greiner Bio-One) using a BioRAPTR FRD dispenser (Beckman Coulter, Brea, CA). A 1536-well pintool dispenser outfitted with 20 nL pins (Wako Automation, San Diego, CA) was used to transfer 23 nL of DMSO-solubilized compound (Cherrypick plates) to each 1536-well assay plate. Following compound transfer, plates were incubated in room temperature for 10 min. 1 μL of substrate solution containing malic acid (160 μM) was dispensed via BioRAPTR FRD to initiate the reaction. Plates were immediately transferred to a ViewLux microplate imager (PerkinElmer, Waltham, MA), and any resulting resorufin fluorescence was measured (ex540, em590 nm) at 0 and 5 min. Well fluorescence was normalized using enzyme-free and DMSO-treated control wells on each plate, and changes in fluorescence (ΔRFU) were calculated using the difference in fluorescent signal for each well at 5 versus 0 minutes.
IDH1 biochemical assay: IDH1 protocol was performed as previously described.30 WT IDH1 (3 μL) in assay buffer (20 mM Tris pH 7.5, 10 mM MgCl2, 150 mM NaCl, and 0.05% protease-free BSA) was added to the black solid bottom 1,536-well assay plate using a BiorapTR flying reagent dispenser (BiorapTR; Beckman Coulter). A pintool (Kalypsys) was used to transfer 23 nL of compound solution (library and control) to the 1,536-well assay plates, and plates were spun down at 1,000 rpm for 1 min. After 30 min of incubation at room temperature, 1 μL of substrate buffer was added to initiate the reaction at final concentrations of 0.045 μg/mL enzyme, 2 mM BME, 240 μM isocitrate, 180 uM NADP+, 60 μg/mL diaphorase, and 37.5 μM resazurin. The plate was rapidly transferred to a ViewLux (PerkinElmer) and the fluorescence product resorufin was measured (excitation = 525 nm, emission = 598 nm) in kinetic mode. The plates were read continuously from t = 0 to t = 5 min.
Lactate production assays
A673 and MiaPaCa-2 cells were cultured as described above, and plated in 1536-well black clear bottom tissue culture plates using a Multidrop Combi peristaltic dispenser (ThermoFisher, Waltham, MA) at a density of 500 cells/well in 4 μL of DMEM (Invitrogen 31053036) culture medium. A 1536-well pintool dispenser outfitted with 20 nL pins (Wako Automation, San Diego, CA) was used to transfer 23 nL of compound in DMSO to the 1536-well assay plates. After 2 hr incubation at 37 °C, 2 μL of reconstituted Lactate Reaction Mix (BioVision K607–100) was dispensed into each well using a BioRAPTR FRD dispenser (Beckman Coulter, Brea, CA). Plates were incubated at room temperature for 30 minutes, transferred to a ViewLux microplate imager (PerkinElmer, Waltham, MA) and the fluorescence (Ex/Em 525/598 nm) and absorbance (573 nm) were measured accordingly.
Cellular Thermal Shift Assay (CETSA)
The cellular thermal shift assay and the isothermal dose response was run as previously described.27 Additional details are described in supplemental methods.
Cytotoxicity Assay
A673 and MiaPaCa-2 cells were cultured as described above, and plated in 1536-well white solid tissue culture plates using a Multidrop Combi peristaltic dispenser (ThermoFisher, Waltham, MA) at a density of 500 cells/well in 5 μL of DMEM (Invitrogen 11965118) culture medium supplemented with 10 % fetal bovine serum and 100 units/mL Penicillin, 100 μg/mL Streptomycin. A 1536-well pintool dispenser outfitted with 20 nL pins (Wako Automation, San Diego, CA) was used to transfer 23 nL of compound in DMSO to the 1536-well assay plates. After 48 hr incubation at 37 °C, 2.5 μL of CellTiter-Glo (Promega) was dispensed into each well using a BioRAPTR FRD dispenser (Beckman Coulter, Brea, CA). Plates were incubated at room temperature for 10 minutes, transferred to a ViewLux microplate imager (PerkinElmer, Waltham, MA) and the ATP-coupled luminescence was measured using a 1 second exposure.
Incucyte cellular proliferation assay
A673 and MiaPaCa-2 cells were cultured as described above, and plated in 384-well black clear bottom tissue culture plates using an Multidrop Combi peristaltic dispenser (ThermoFisher, Waltham, MA) at a density of 1000 cells/well in 40 μL of DMEM (11965118) culture medium supplemented with 10 % fetal bovine serum and 100 units/mL Penicillin, 100 μg/mL Streptomycin. DMSO-solubilized compounds were added using a 384-well pintool dispenser (Wako Automation, San Diego, CA), and plates were incubated and measured in an IncuCyte ZOOM live cell analysis system (Essen Bioscience, Ann Arbor, MI), in a cell culture environment at 37 °C containing 5% CO2. Cell confluency was assessed using high definition phase contrast every 4 hours, for a total of 120 hours. Area under the curve and dose response curve analyses was performed using Prism (Graphpad Software).
Glycolytic Stress Test assay
A673 cells were cultured in Dulbecco’s Modified Eagle’s Medium (ATCC Catalog No. 302002) supplemented with fetal bovine serum (10%). The cells were plated into a XF96 cell culture microplate in the above medium and maintained in a 5% CO2 incubator at 37˚C for 24h prior to the experiments. The day of the assay, compounds are diluted to the appropriate concentration in freshly prepared assay media (Seahorse basic DMEM with 2 mM Glutamine, pH 7.4 at 37 °C). The media in the plate with cells was then changed to assay media and maintained in a non-CO2 incubator at 37 ˚C for 1h prior to the assay. The Seahorse XF Glycolysis Stress Test (GST) was conducted by injecting the LDH inhibitors, then, at 40 min, subsequent injections of glucose (10 mM final concentration), oligomycin (1 μg/mL final concentration), and 2-deoxyglucose (2-DG; 50 mM final concentration) as described in.31
PAMPA permeability assay
The stirring double-sink PAMPA method patented by pION Inc. (Billerica, MA) was employed to determine the permeability of compounds via PAMPA passive diffusion. The PAMPA lipid membrane consisted of an artificial membrane of a proprietary lipid mixture and dodecane (Pion Inc.), optimized to predict gastrointestinal tract (GIT) passive diffusion permeability, was immobilized on a plastic matrix of a 96 well “donor” filter plate placed above a 96 well “acceptor” plate. A pH 7.4 solution was used in both donor and acceptor wells. The test articles, stocked in 10 mM DMSO solutions, were diluted to 0.05 mM in aqueous buffer (pH 7.4), and the concentration of DMSO was 0.5% in the final solution. During the 30-min permeation period at room temperature, the test samples in the donor compartment were stirred using the Gutbox technology (Pion Inc.) to reduce the unstirred water layer. The test article concentrations in the donor and acceptor compartments were measured using an UV plate reader (Nano Quant, Infinite 200 PRO, Tecan Inc., Männedorf, Switzerland). Permeability calculations were performed using Pion Inc. software and were expressed in units of 10–6 cm/s.
Kinetic solubility test assay
Pion’s patented μSOL assay for kinetic solubility determination was used. In this assay, the classical saturation shake-flask solubility method was adapted to a 96-well microtiter plate format and a co-solvent method with n-propanol as the reference compound was utilized. Test compounds were prepared in 10 mM DMSO solutions (45 μL), and diluted with the co-solvent to a final drug concentration of 150 μM in the aqueous solution (pH 7.4). Samples are incubated at room temperature for 6 hrs to achieve equilibrium. The samples were then filtered to remove any precipitate formed. The concentration of the compound in the filtrate was measured by UV absorbance. The reference drug concentration of 17 μM was used for quantitation of unknown drug concentration in filtrate. Spectroscopically pure 1-propanol was used as a cosolvent to suppress precipitation in the reference solutions. The kinetic solubility (μg/mL) was calculated with using the μSOL Evolution software.
Rat liver microsome stability assay
See reference 30 for details.
Mouse Pharmacokinetic Studies
Studies were conducted by Pharmaron. Male CD1 mice (sourced from Si Bei Fu Laboratory Animal Technology Co. Ltd), approximately 6–8 weeks of age and a weight of approximately 20–30 g were dosed with 61 and 63 at 2 mg/kg (IV) and 50 mg/kg (PO). The formulation (0.1 M NaoH in PBS Buffered saline, adjusted with 1N HCl to pH 7–8.5) was prepared on the day of dosing or directly prior to dosing. Each cohort had 3 mice and plasma was collected at 5 min, 15 min, 30 min, 1h, 2h, 4h, 8h, 12h, and 24h post dose for IV & 15 min 30 min, 1h, 2h, 4h, 8h, 12h and 24h for PO. Approximately 0.025 mL blood was collected via the dorsal metatarsal vein at each time point. Blood samples were then transferred into plastic micro centrifuge tubes containing Heparin-Na as anti-coagulant. Samples were then centrifuged at 4,000 g for 5 minutes at 4 °C to obtain plasma. Plasma samples were then stored in polypropylene tubes, quickly frozen and kept at −75 °C until analyzed by LC/MS/MS. The following pharmacokinetic parameters were measured T1/2, C0, Cmax, Tmax, CL, Vd, AUClast and F. Animals were also monitored during the in-life phase by once daily cageside observations, any adverse clinical signs are noted as part of the PK report.
Use of Animal Subjects
All animal studies included as part of this manuscript were performed in accordance with institutional guidelines as defined by Institutional Animal Care and Use Committee (IACUC).
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. GR, KRB, BTM, DJM, XH, SMY, TDL, DMC, JK, KP, EJK, DKL, HZ, MDH, AS, AJ and DJM also gratefully acknowledge funding by the Intramural Research Program, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH). The authors wish to thank Sam Michael and Richard Jones for automation support; Paul Shinn, Misha Itkin, Zina Itkin and Danielle van Leer for the assistance with compound management; William Leister, Heather Baker, Elizabeth Fernandez and Christopher Leclair for analytical chemistry and purification support; Kimloan Nguyen for in vitro ADME data. We would also like to thank Genentech for the generous donation of GNE140 for our studies, David Myszka at Biosenor Tools for conducting the SPR experiments and Pharmaron Inc. for conducting the pharmacokinetic studies.
Abbreviations
- LDH
Lactate dehydrogenase
- qHTS
quantitative high-throughput screening
- CETSA
Cellular Thermal Shift assay
- SPR
Surface Plasmon Resonance
- SAR
structure-activity relationships
- TFA
trifluoroacetic acid
- LHMDS
Lithium bis(trimethylsiyl)amide
- HATU
(1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate)
- MW
microwave
- TBAF
Tetra-n-butylammonium fluoride
- NCGC
NIH Chemical Genomics Center
- EDTA
Ethylenediaminetetraacetic acid
Footnotes
Ancillary Information
Supporting Information: Additional supplemental figures, experimental procedures and spectroscopic data (1H NMR, LC/MS and HRMS) for representative compounds. Molecular formula strings are also available. This material is available free of charge via the internet at http://pubs.acs.org.
PDB ID Codes: 5W8I (27), 5W8H (15), 5W8J (33), 5W8K (33), 5W8L (63). Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions: Ganesha Rai and Kyle R. Brimacombe contributed equally to this work.
Conflicts of Interest
The authors declare no competing financial interest
References
- 1.Warburg O, Posener K, Negelein E. Ueber den stoffwechsel der tumoren. Biochem Z. 1924;152:319–344. [Google Scholar]
- 2.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu Y, Liao M, Liu R, Chen J, Feng H, Fu Z. Overexpression of lactate dehydrogenase-A in human intrahepatic cholangiocarcinoma: its implication for treatment. World J Surg Oncol. 2014;12:78. doi: 10.1186/1477-7819-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rong Y, Wu W, Ni X, Kuang T, Jin D, Wang D, Lou W. Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumour Biol. 2013;34:1523–1530. doi: 10.1007/s13277-013-0679-1. [DOI] [PubMed] [Google Scholar]
- 5.(a) Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]; (b) Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sheng SL, Liu JJ, Dai YH, Sun XG, Xiong XP, Huang G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012;279:3898–3910. doi: 10.1111/j.1742-4658.2012.08748.x. [DOI] [PubMed] [Google Scholar]; (d) Wang ZY, Loo TY, Shen JG, Wang N, Wang DM, Yang DP, Mo SL, Guan XY, Chen JP. LDH-A silencing suppresses breast cancer tumorigenicity through induction of oxidative stress mediated mitochondrial pathway apoptosis. Breast Cancer Res Treat. 2012;131:791–800. doi: 10.1007/s10549-011-1466-6. [DOI] [PubMed] [Google Scholar]; (e) Yao F, Zhao T, Zhong C, Zhu J, Zhao H. LDHA is necessary for the tumorigenicity of esophageal squamous cell carcinoma. Tumour Biol. 2012;34:25–31. doi: 10.1007/s13277-012-0506-0. [DOI] [PubMed] [Google Scholar]
- 6.(a) Giatromanolaki A, Sivridis E, Gatter KC, Turley H, Harris AL, Koukourakis MI. Lactate dehydrogenase 5 (LDH-5) expression in endometrial cancer relates to the activated VEGF/VEGFR2(KDR) pathway and prognosis. Gynecol Oncol. 2006;103:912–918. doi: 10.1016/j.ygyno.2006.05.043. [DOI] [PubMed] [Google Scholar]; (b) Kolev Y, Uetake H, Takagi Y, Sugihara K. Lactate dehydrogenase-5 (LDH-5) expression in human gastric cancer: association with hypoxia-inducible factor (HIF-1alpha) pathway, angiogenic factors production and poor prognosis. Ann Surg Oncol. 2008;15:2336–2344. doi: 10.1245/s10434-008-9955-5. [DOI] [PubMed] [Google Scholar]; (c) Koukourakis MI, Giatromanolaki A, Sivridis E, Bougioukas G, Didilis V, Gatter KC, Harris AL. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br J Cancer. 2003;89:877–885. doi: 10.1038/sj.bjc.6601205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simona D, Giacomelli C, Zappelli E, Granchi C, Trincavelli ML, Minutolo F, Martini C. Lactate dehydrogenase-A inhibition induced human glioblastoma multiforme stem cell differentiation and death. Sci Rep. 2015;5:15556. doi: 10.1038/srep15556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blum R, Kloog Y. Metabolism addition in pancreatic cancer. Cell Death Dis. 2014;5:e1065. doi: 10.1038/cddis.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shuch B, Linehan MW, Srinivasan R. Aerobic glycolysis: A novel target in kidney cancer. Expert Rev Anticancer Ther. 2013;13:711–719. doi: 10.1586/era.13.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, Lanza A, Betti L, Giannaccini G, Lucachhini A, Funel N, León LG, Giovannetti E, Peters GJ, Palchaudhuri R, Calvaresi EC, Hergenrother PJ, Minutolo F. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents againsts cancer cells. J Med Chem. 2011;24:1599–1612. doi: 10.1021/jm101007q. [DOI] [PubMed] [Google Scholar]
- 11.Kohlmann A, Zech SG, Li F, Zhou T, Squillance RM, Commodore L, Greenfield MT, Lu X, Miller DP, Huang WS, Qi G, Thomas RM, Wang Y, Zhang S, Dodd R, Liu S, Xu R, Xu Y, Miret JJ, Rivera V, Clackson T, Shakesphere WC, Zhu X, Dalgarno DC. Fragment growing and linking lead to novel nanomolar lactate dehydrogenase inhibitors. J Med Chem. 2013;56:1023–1040. doi: 10.1021/jm3014844. [DOI] [PubMed] [Google Scholar]
- 12.Ward RA, Brassington C, Breeze AL, Caputo A, Critchlow S, Davies G, Goodwin L, Hassall G, Greenwood R, Holdgate GA, Mrosek M, Norman RA, Pearson S, Tart J, Tucker JA, Vogtherr M, Whittaker D, Wingfield J, Winter J, Hudson K. Design and synthesis of novel lactate dehydrogenase A inhibitors by fragment-based lead generation. J Med Chem. 2012;55:3285–3306. doi: 10.1021/jm201734r. [DOI] [PubMed] [Google Scholar]
- 13.Billiard J, Dennison JB, Briand J, Annan RS, Chai D, Colón M, Dodson CS, Gilbert SA, Greshock J, Jing J, Lu H, McSurdy-Freed JE, Orband-Miller LA, Mills GB, Quinn CJ, Schneck JL, Scott GF, Shaw AN, Waitt GM, Wooster RF, Duffy KJ. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013;1:19. doi: 10.1186/2049-3002-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Fauber BP, Dragovich PS, Chen J, Corson LB, Ding CZ, Eigenbrot C, Giannetti AM, Hunsaker T, Labadie S, Liu Y, Liu Y, Malek S, Peterson D, Pitts K, Sideris S, Ultsch M, VanderPorten E, Wang J, Wei BQ, Yen I, Yue Q. Identification of 2-amino-5-aryl-pyrazines as inhibitors of human lactate dehydrogenase. Bioorg Med Chem Lett. 2013;23:5533–5539. doi: 10.1016/j.bmcl.2013.08.060. [DOI] [PubMed] [Google Scholar]; (b) Dragovich PS, Fauber BP, Corson LB, Ding CZ, Eigenbrot C, Ge H, Giannetti AM, Hunsaker T, Labadie S, Liu Y, Malek S, Pan B, Peterson D, Pitts K, Purkey HE, Sideris S, Ultsch M, VanderPorten E, Wei B, Xu Q, Yen I, Zhang H, Zhang Z. Identification of substituted 2-thio-6-oxo-1,6-duhydropyrimidines as inhibitors of human lactate dehydrogenase. Bioorg Med Chem Lett. 2013;23:3186–3194. doi: 10.1016/j.bmcl.2013.04.001. [DOI] [PubMed] [Google Scholar]; (c) Boudreau A, Purkey HE, Hitz A, Robarge K, Peterson D, Labadie S, Kwong M, Hong R, Gao M, Del Nagro C, Pusapati R, Ma S, Salphati L, Pang P, Zhou A, Lai T, Li Y, Chen Z, Wei B, Yen I, Sideris S, McCleland M, Firestein R, Corson L, Vanderbilt A, Williams S, Daemen A, Belvin M, Eigenbrot C, Jackson PK, Malek S, Hatzivassiliou G, Sampath D, Evangelista M, O’Brien T. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol. 2016;12:779–786. doi: 10.1038/nchembio.2143. [DOI] [PubMed] [Google Scholar]
- 15.Aggarwal R, Kumar R, Kumar S, Garg G, Mahajan R, Sharma J. Synthesis and antibacterial activity of some 5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazol-1-thiocarboxamides, 3-trifluoromethylpyrazol-1-thiocarboxamides and 4-aryl-2-(5(3)-trifluoromethyl-1-pyrazolyl)thiazoles. J Fluorine Chem. 2011;132:965–972. [Google Scholar]
- 16.a) Denisova AB, Glukhareva TV, Andronnikova GP, Mokrushin VS, Dehaen W, Luyten I, Sosnovskikh VY, Van Meervelt L, Bakulev VA. Regioselectivity of the Synthesis of 2-pyrazolinylthiazoles by reacting 2-hydrazinothiazoles with unsymmetrical β-diketones. J Chem Res Synopses. 2001:12–13. [Google Scholar]; J Chem Res Miniprint. 2001:133–147. [Google Scholar]; b) Singh SP, Kumar D, Batra H, Naithani R, Rozas I, Elguero J. The reaction between hydrazines and b-dicarbonyl compounds: proposal for a mechanism. Can J Chem. 2000;78:1109–112. [Google Scholar]; c) Denisova AB, Sosnovskikh VY, Dehaen W, Toppet S, Meervelt LV, Bakulev VA. The regioselectivity of the formation of 2-pyrazolylthiazoles and their precursors from the reaction of 2-hydrazinothiazoles with 4,4,4-trifluoro-1-hetaryl-1,3-butanediones. J Fluorine Chem. 2002;115:183–192. [Google Scholar]
- 17.(a) Hornback JM, Poundstone ML, Vadlamani B, Graham SM, Gabay J, Patton ST. Photochemical cyclization of o-methylphenyl 1,3-diketones. J Org Chem. 1988;53:5597–5601. [Google Scholar]; (b) Wasserman HH, Ennis DS, Vu CB, Schulte G, Munk ME, Madison M, Velusamy KV. Synthesis and characterization of pyrroplinonecarboxylates formed by reaction of vicinal tricarbonyl derivatives with aldehyde schiff bases. Heterocycles. 1993;35:975–995. [Google Scholar]; (c) Hu X, Sun J, Wang HG, Manetsch R. Bcl-XL-templated assembly of its own protein–protein interaction modulator from fragments decorated with thio acids and sulfonyl azides. J Am Chem Soc. 2008;130:13820–13821. doi: 10.1021/ja802683u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morimoto H, Tsubogo T, Litvinas ND, Hartwig JF. A broadly applicable copper reagent for trifluoromethylations and perfluoroalkylations of aryl iodides and bromides. Angew Chem Int Ed. 2011;50:3793–3798. doi: 10.1002/anie.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inglese J, Auld DS, Jadhav A, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activites in large chemical libraries. Proc Natl Acad Sci USA. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atobe M, Naganuma K, Kawanishi M, Morimoto A, Kashahara K, Ohashi S, Suzuki H, Hayashi T, Miyoshi S. Hit-to-lead optimization of 2-(1H-pyrazol-1-yl)-thiazole derivatives as a novel class of EP1 receptor antagonist. Bioorg Med Chem Lett. 2013;23:6064–6067. doi: 10.1016/j.bmcl.2013.09.032. [DOI] [PubMed] [Google Scholar]
- 21.(a) Morpeth FF, Massey V. Metal binding to D-lactate dehydrogenase. Biochemistry. 1982;21:1318–1323. doi: 10.1021/bi00535a033. [DOI] [PubMed] [Google Scholar]; (b) Marti HH, Jung HH, Pfeilschifter J, Bauer C. Hypoxia and cobalt stimulate lactate dehydrogenase (LDH) activity in vascular smooth muscle cells. Pfluegers Arch. 1994;429:216–222. doi: 10.1007/BF00374315. [DOI] [PubMed] [Google Scholar]; (c) Cremona T. The lactic dehydrogenase of yeast. J Biol Chem. 1963;239:1457–1465. [PubMed] [Google Scholar]
- 22.(a) Copeland RA, Pompliano DL, Meek TD. Drug‒Target residence time and its implication for lead optimization. Nat Rev Drug Discovery. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]; (b) Copeland RA. The drug‒target residence time model: a 10-year retrospective. Nat Rev Drug Discovery. 2016;15:87–95. doi: 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- 23.Berezov A, Zhang HT, Greene MI, Murali R. Disabling Erb B receptors with rationally designed exocyclic mimetics of antibodies: Structure-function analysis. J Med Chem. 2001;44:2565–2574. doi: 10.1021/jm000527m. [DOI] [PubMed] [Google Scholar]
- 24.Itzhak DN, Tyanova S, Cox J, Borner GH. Global, quantitative and dynamic mapping of protein subcellular localization. eLife. 2016;5:e16950. doi: 10.7554/eLife.16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.GNE140 had not yet been disclosed when these studies were initially conducted.
- 26.(a) Chang A, Schiebel J, Yu W, Bommineni GR, Pan P, Baxter MV, Khanna A, Sotriffer CA, Kisker C, Tonge PJ. Rational optimization of drug-target residence time: Insights from inhibitor binding to the S. aureus Fabl enzyme-product complex. Biochemistry. 2013;52:4217–4228. doi: 10.1021/bi400413c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cusack KP, Wang Y, Hoemann MZ, Marjanovic J, Heym RG, Vasudevan A. Design strategies to address kinetics of drug binding and residence time. Bioorg Med Chem Lett. 2015;25:2019–2027. doi: 10.1016/j.bmcl.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 27.(a) Copeland RA. Conformational adaptation in drug-target interactions and residence time. Future Med Chem. 2011;3:1491–1501. doi: 10.4155/fmc.11.112. [DOI] [PubMed] [Google Scholar]; (b) Van Aller GS, Pappalardi MB, Ott HM, Diaz E, Brandt M, Schwartz BJ, Miller WH, Dhanak D, McCabe MT, Verma SK, Creasy CL, Tummino PJ, Kruger RG. Long residence time inhibition of EZH2 in activated polycomb repressive complex 2. ACS Chem Biol. 2014;9:622–629. doi: 10.1021/cb4008748. [DOI] [PubMed] [Google Scholar]
- 28.(a) Molina DM, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues Using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]; (b) Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundbäck T, Nordlund P, Molina DM. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 29.Di L, Kerns EH, Gao N, Li SQ, Huang Y, Bourassa JL, Huryn DM. Experimental design on single-time-point high-throughput microsomal stability assay. J Pharm Sci. 2004;93:1537–1544. doi: 10.1002/jps.20076. [DOI] [PubMed] [Google Scholar]
- 30.Davis MI, Shen M, Simeonov A, Hall MD. Diaphorase coupling protocols for red-shifting dehydrogenase assays. Assay Drug Dev Technol. 2016;14:207–212. doi: 10.1089/adt.2016.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hill BG, Benavides GA, Lancaster JR, Ballinger S, Dell’Italia L, Jianhua Z, Darley-Usmar VM. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.