

Abstract
Background
Tensin2 is a focal adhesion-localized multidomain protein expressed in various tissues, and its dysfunction leads to alterations in podocytes. However, these podocyte-related manifestations are dependent on murine strain. Tensin2 dysfunction results in susceptible strains developing podocyte foot process effacement and massive albuminuria, whereas podocytes in resistant strains remain almost intact. In our previous studies, quantitative trait loci analysis and congenic analysis using resistant C57BL/6J and susceptible ICGN mice identified a modifier locus associated with podocyte injury caused by tensin2 dysfunction on chromosome 2. However, the effect of this modifier locus on chromosome 2 is insufficient to explain the resistance of C57BL/6J mice to tensin2 dysfunction, indicating the existence of other modifier genes.
Results
Whereas previous studies focused on the severity of chronic kidney disease, the present study focused on podocyte injury. We performed a genome-wide linkage analysis of backcrosses between two tensin2-deficient mouse strains, B6.ICGN-Tns2nph and FVB.ICGN-Tns2nph, and detected a novel major modifier locus on chromosome 10. The combined effect of the C57BL/6J alleles of the two loci on chromosomes 2 and 10 reduced the urinary albumin excretion caused by tensin2 dysfunction to a level comparable to that of C57BL/6J mice.
Conclusions
These data indicate that the resistance to podocyte injury caused by tensin2 dysfunction is mainly produced by the effects of the modifier genes on the two loci. The identification of these modifier genes is expected to help elucidate the mechanism underlying podocyte injury.
Electronic supplementary material
The online version of this article (10.1186/s12863-018-0611-1) contains supplementary material, which is available to authorized users.
Keywords: Tensin2, Albuminuria, Podocyte, Linkage analysis
Background
Tensin2 (Tns2), a member of the tensin family that includes Tns1, Tns3 and Tns4, is a focal adhesion-localized multidomain protein that possesses protein tyrosine phosphatase (PTP) and Src homology 2 (SH2)-phosphotyrosine binding (PTB) domains [1]. Tns2 mRNA is detected in the brain, heart, kidney, skeletal muscle, liver, lung, colon, small intestine and other tissues [2–6]. Stable expression of Tns2 in HEK293 cells alters cell morphology and reduces actin filaments. Overexpression of Tns2 in HEK293 cells reduces cell proliferation in a functional PTP domain-dependent manner and inhibits cell migration [7]. In contrast, stable expression of Tns2 protein with its C terminus fused to green fluorescent protein in HEK293 cells promotes cell migration [2]. In a podocyte cell line, it was shown that Tns2 knockdown enhances actin stress fiber formation and cell migration [8]. Taken together, these studies suggest that Tns2 function is involved in the reorganization of the actin cytoskeleton.
However, in vivo, Tns2 dysfunction only leads to apparent abnormalities in the kidney glomerulus [4, 8]. Newborn ICGN mice, carrying a Tns2-null mutation (hereafter nph), showed normal glomerular development, whereas podocyte foot process effacement and abnormal thickening of the glomerular basement membrane (GBM) were observed by 14 days after birth [9]. GBM thickening, which begins during glomerular maturation in ICGN mice, is considered to be a result of the abnormal extracellular matrix accumulation caused by podocytes but not by endothelial cells [9]. As observed in chronic kidney disease, ICGN mice then develop glomerular injury, including mesangial expansion, and massive albuminuria followed by tubulointerstitial injury with age [10, 11].
The podocyte-related manifestations of Tns2 dysfunction also depend on murine strain. For example, in response to nph mutation, albuminuria or podocyte foot process effacement was observed in FVB/NJ (FVB) and DBA/2J, but not in C57BL/6J (B6) and 129+Ter/SvJcl mice [12–16]. This disparity among strains indicates the presence of modifier genes. A comparison of the differences in the genetic backgrounds between the Tns2-deficiency resistant and susceptible strains can help in identifying these modifier genes, which may also help in elucidating the mechanism underlying Tns2 function in podocytes. Indeed, we previously performed quantitative trait loci (QTL) analysis and congenic analysis using resistant B6 and susceptible ICGN mice, and identified a modifier locus associated with podocyte injury caused by the nph mutation, Tpir, on chromosome 2 [17, 18]. ICGN.B6 congenic strains carrying the B6 allele of Tpir showed alleviated podocyte foot process effacement, GBM thickening and albuminuria, although the degree of improvement was inferior to that in the B6 genetic background itself, which shows almost no podocyte alteration [18]. This result indicates the existence of other modifier genes. The following points may explain why our previous QTL analysis failed to find the other significant loci: (1) our previous analysis focused on the severity of chronic kidney disease, and evaluated the traits related to the late-phase of chronic kidney disease in 16-week-old mice, such as tubulointerstitial injury, renal anemia and renal failure, but not albuminuria; (2) the quantitative traits, except for the histological scores, varied greatly in the ICGN mice themselves; and (3) the glomerular injury score focused on mesangial matrix accumulation, which is primarily caused by abnormal extracellular matrix metabolism in mesangial cells [19]. A further problem is that complete genetic information on the ICGN mouse genome remains unavailable.
In this study, we focused on podocyte injury and undertook a genome-wide linkage analysis of backcrosses between two Tns2-deficient mouse strains, B6.ICGN-Tns2nph (B6-Tns2nph) and FVB.ICGN-Tns2nph (FVB-Tns2nph) to identify modifier loci that may have been missed in previous studies.
Methods
Animals
B6-Tns2nph and FVB-Tns2nph congenic mice were generated as described previously [12, 15]. B6 x FVB F1 hybrid Tns2nph (F1) mice were obtained by mating female B6-Tns2nph mice to male FVB-Tns2nph mice. N2 backcross Tns2nph progenies were then obtained by mating female F1 mice to male FVB-Tns2nph mice (Fig. 1a). The SPF room was air-conditioned at 22 ± 4 °C, maintained at 40–60% relative humidity, and mice were maintained under a 12 h light-dark cycle. A standard laboratory diet, CE-2 (CLEA Japan, Tokyo, Japan), and tap water were provided ad libitum.
Fig. 1.
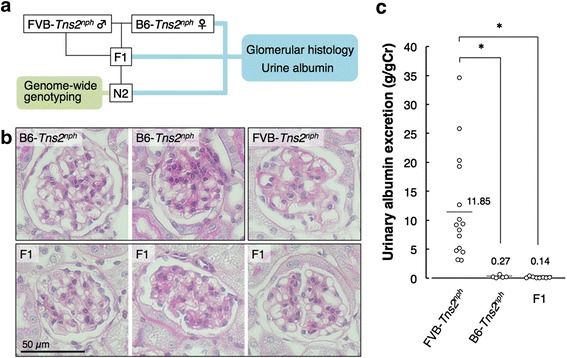
Glomerular histology and urinary albumin excretion in Tns2-deficient B6, FVB and F1 mice. (a) Schema of the study. b Representative glomerular structures in PAS staining. In B6-Tns2nph mice, most glomeruli were normal (left panel), while some glomeruli showed a slight increase in mesangial matrix (right panel). In FVB-Tns2nph mice, many glomeruli showed marked expansion of the mesangial matrix, marked thickening of the GBM and abnormal dilation of the capillary lumen. In F1 mice, there were normal glomeruli (left panel), glomeruli with mesangial expansion (middle panel) and glomeruli with GBM thickening (right panel). c Urinary albumin excretion in FVB-Tns2nph (n = 15), B6-Tns2nph (n = 5) and F1 (n = 8) mice. Values represent the mean. Asterisk, P < 0.01
Genotyping
Genome-wide genotyping of the Tns2nph N2 mice was performed using 99 polymorphic markers (Additional file 1: Table S1). The Tyr locus was genotyped by coat color, and the other loci were genotyped using polymerase chain reaction (PCR)-based method. Genomic deoxyribonucleic acid was extracted from ear tissue obtained during ear punching for identification. PCR was performed with the following thermal cycling: initial denaturation at 95 °C for 3 min; 10 cycles of 95 °C for 10 s, 65 °C for 30 s (reduced by − 1 °C/cycle), and 72 °C for 10 s; and 30 cycles of 95 °C for 10 s, 55 °C for 30 s, 72 °C for 10 s. Newly designed PCR primers are listed in the Additional file 1: Table S2.
Measurement of urinary albumin excretion
Urine samples were collected from 8-week-old mice by gentle manual compression of the abdomen. To measure albumin, urine samples were diluted with sample buffer containing 2% sodium dodecyl sulfate (SDS), 5% ß-mercaptoethanol, 10% glycerol, 60 mM Tris-HCL (pH 6.8) and bromophenol blue, and heated at 95 °C for 5 min. Samples containing 1 μL of urine were applied to 10% SDS-polyacrylamide gel electrophoresis. As a positive control, 5 μg of bovine serum albumin was loaded simultaneously. The gel was fixed and stained with Coomassie brilliant blue (CBB; Nacalai Tesque, Kyoto, Japan) according to manufacturer’s instructions, and scanned using a standard commercial scanner. CBB-stained urinary albumin was quantified by the ImageJ gel analysis program (http://imagej.net/). Urinary creatinine was measured using a creatinine colorimetric assay kit (Cayman chemical, Michigan, USA) according to manufacturer’s instructions. The urinary albumin excretion was normalized against the urinary creatinine. Urine collection was performed twice over a three-day period, and the measured urinary albumin excretion was averaged for each mouse.
Histology
Kidneys from 8-week-old mice were fixed with 4% paraformaldehyde (PFA) at 4 °C overnight. The PFA-fixed paraffin sections (2 μm thick) were subjected to normal histological processes and stained with periodic acid-Schiff (PAS) solution.
Marker-trait association test
N2 mice were divided into two groups, affected and not affected, according to their urinary albumin excretion. With reference to the distribution of urinary albumin excretion observed in FVB-Tns2nph and B6-Tns2nph mice, we defined the N2 mice with urinary albumin excretion larger than the minimum value in FVB-Tns2nph mice as the FVB type (affected with albuminuria) and the N2 mice with urinary albumin excretion less than the maximum value in B6-Tns2nph mice as the B6 type (not affected with albuminuria). Another way of division was to define the upper 30% or 40% or 50% of N2 mice as the FVB type and the lower 30% or 40% or 50% of N2 mice as the B6 type. A chi-square test was applied to detect marker-trait associations.
Statistics
Data are expressed as means ± standard deviation. Student’s t-test and Dunnett’s multiple comparison test were used for comparisons of two or more than two independent groups, respectively. A p value < 0.05 was considered statistically significant. Dunnett’s multiple comparison test was carried out using GraphPad Prism 5 software (MDF, Tokyo, Japan).
Results
Phenotype of Tns2-deficient B6, FVB and (B6 x FVB) F1 mice
Consistent with previous reports [12–15], FVB-Tns2nph mice showed albuminuria (urinary albumin excretion, 11.8 ± 9.2 g/gCr) and severe glomerular injuries, such as marked expansion of the mesangial matrix, marked thickening of the GBM and abnormal dilation of capillary lumen, whereas B6-Tns2nph mice did not show either albuminuria (urinary albumin excretion, 0.26 ± 0.25 g/gCr) or glomerular injury, apart from a slight increase in the mesangial matrix in some glomeruli (Fig. 1b, c). To assess the mode of inheritance of traits for resistance to Tns2 dysfunction, F1 mice were subjected to similar analyses at 8 weeks. The structures of some glomeruli in the F1 mice were nearly normal (Fig. 1b); however, unlike that in B6-Tns2nph mice, a focal thickening of the GBM was also observed in F1 mice (Fig. 1b), suggesting that F1 mice exhibited an intermediate phenotype for glomerular injury caused by Tns2 dysfunction. On the other hand, the urinary albumin excretion of F1 mice (0.14 ± 0.13 g/gCr) was approximately equal to that of B6-Tns2nph mice (Fig. 1c), suggesting that resistance to albuminuria caused by Tns2 dysfunction is inherited as a dominant trait in F1 mice. Although the glomerular injuries may afford a sensitive reflection of the severity of renal disease caused by Tns2 dysfunction, we considered that urinary albumin excretion is a better indicator of resistance to Tns2 dysfunction than is glomerular injury, which is complicated and difficult to quantify.
Genome-wide linkage analysis in N2 backcross mice
As F1 mice as well as B6-Tns2nph mice exhibited a suppressive phenotype, we backcrossed the F1 mouse onto the FVB-Tns2nph mouse to identify the modifier loci related to resistance to Tns2 dysfunction. We then performed genome-wide genotyping of the resultant N2 mice (n = 174) and subsequent phenotyping for albuminuria. N2 mice showed a continuous spectrum of values for urinary albumin excretion and did not simply segregate into the B6- and FVB-Tns2nph types according to whether or not they were affected with albuminuria (Fig. 2). Therefore, with reference to the distribution of urinary albumin excretion observed in FVB-Tns2nph (3.12–34.6 g/gCr) and B6-Tns2nph mice (0.052–0.69 g/gCr), we defined the N2 mice with urinary albumin excretion larger than 3.1 g/gCr (59 of 174) as the FVB type (affected) and the N2 mice with urinary albumin excretion less than 0.7 g/gCr (89 of 174) as the B6 type (not affected). In the N2 mice as defined above, marker-trait associations were detected on chromosome 10. The genetic markers rs259766385 and rs239646663 on chromosome 10 showed a statistical significant relationship with albuminuria (Table 1). In addition, the analyses using different phenotype definitions (e.g., the upper 40% and lower 40% of N2 mice) found suggestive signals on chromosome 2 (Table 1). Furthermore, the combined rs259766385 genotypes and the genetic markers on chromosome 2 provided a more significant p value in comparison with the chi-square test for each marker (Table 2). Table 2 shows a comparison between the two combined genotypes; one in which both the markers are homogeneic, the other in which at least one of the markers is heterogeneic. In Fig. 3, the variations in urinary albumin excretion were compared among the four combined genotypes for rs234188972 on chromosome 2 and rs259766385 on chromosome 10. Urinary albumin excretion in both groups with allelic heterogeneity at rs259766385 (F/B and B/B groups) was significantly lower than that in the group carrying FVB/FVB genotypes at the two markers (F/F group) (Fig. 3). The median value for urinary albumin excretion in the group carrying B6/FVB and FVB/FVB genotypes at rs234188972 and rs259766385, respectively (B/F group, 0.71 g/gCr), was approximately equal to those of the F/B and B/B groups (0.52 and 0.32 g/gCr, respectively) and appeared lower than that of the F/F group (2.4 g/gCr), although no significant difference was detected between the B/F and F/F groups (Fig. 3).
Fig. 2.
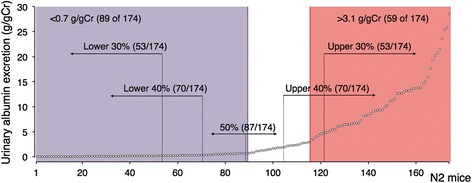
Distribution of urinary albumin excretion in N2 mice. Vertical lines illustrated in the figure represent the borders of the phenotype definitions for marker-trait association test. n = 174
Table 1.
Relationship between albuminuria and genotype for genetic markers on chromosomes 2 and 10
| Chromosome | Marker | Position (cM) | χ2 (P) | Student-T (P)c | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Min/Maxa | 50%b | 40%b | 30%b | Min/Max | 50% | 40% | 30% | |||
| 2 | D2Mit398 | 61.76 | 0.19 | 0.062 | 0.052 | 0.20 | 0.48 | 0.47 | 0.40 | 0.31 |
| 2 | rs214164633 | 65.66 | 0.16 | 0.080 | 0.075 | 0.20 | 0.26 | 0.25 | 0.24 | 0.18 |
| 2 | rs234188972 | 70.98 | 0.18 | 0.067 | 0.062 | 0.24 | 0.14 | 0.16 | 0.14 | 0.10 |
| 2 | rs247793542 | 75.41 | 0.64 | 0.36 | 0.30 | 0.84 | 0.25 | 0.23 | 0.26 | 0.24 |
| 2 | rs228447345 | 81.91 | 0.65 | 0.28 | 0.44 | 1 | 0.42 | 0.45 | 0.49 | 0.49 |
| 10 | D10Mit298 | 2.78 | 0.58 | 0.42 | 0.60 | 0.48 | 0.43 | 0.47 | 0.36 | 0.29 |
| 10 | D10Mit3 | 16.53 | 0.010 | 0.053 | 0.086 | 0.04 | 0.0099 | 0.0084 | 0.012 | 0.013 |
| 10 | D10Mit197 | 35 | 0.049 | 0.059 | 0.059 | 0.10 | 0.0078 | 0.011 | 0.012 | 0.014 |
| 10 | rs259766385 | 40.66 | 0.0022 | 0.0039 | 0.0068 | 0.0065 | 0.00038 | 0.00098 | 0.00074 | 0.00061 |
| 10 | rs239646663 | 43.64 | 0.0043 | 0.0099 | 0.0040 | 0.019 | 0.00053 | 0.00076 | 0.0010 | 0.0015 |
| 10 | D10Mit233 | 61.58 | 0.054 | 0.092 | 0.042 | 0.079 | 0.14 | 0.17 | 0.20 | 0.15 |
aThe N2 mice with urinary albumin excretion larger than 3.1 g/gCr, the minimum value in FVB-Tns2nph mice, were defined as the FVB type (affected) and the N2 mice with urinary albumin excretion less than 0.7 g/gCr, the maximum value in B6- Tns2nph mice, were defined as the B6 type (not affected)
bThe upper X% and lower X% of N2 mice in urinary albumin excretion were defined as the FVB type (affected) and the B6 type (not affected), respectively
cUrinary albumin excretion in N2 mice was compared between those with B6/FVB and FVB/FVB genotypes at the genetic markers
Table 2.
Relationship between albuminuria and combined genotypes of chromosomes 2 and 10 (rs259766385)
| Chromosome | Marker | Position (cM) | χ2 (P) | Student-T (P)c | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Min/Maxa | 50%b | 40%b | 30%b | Min/Max | 50% | 40% | 30% | |||
| 2 | D2Mit398 | 61.76 | 0.003 | 0.0019 | 0.0049 | 0.014 | 0.055 | 0.078 | 0.075 | 0.027 |
| 2 | rs214164633 | 65.66 | 0.0013 | 0.0012 | 0.0027 | 0.046 | 0.0016 | 0.0045 | 0.0028 | 0.0010 |
| 2 | rs234188972 | 70.98 | 0.0013 | 0.00090 | 0.0032 | 0.0097 | 0.00021 | 0.0015 | 0.00083 | 0.00025 |
| 2 | rs247793542 | 75.41 | 0.0063 | 0.0054 | 0.012 | 0.024 | 0.0020 | 0.0070 | 0.0042 | 0.0013 |
| 2 | rs228447345 | 81.91 | 0.0039 | 0.0021 | 0.012 | 0.10 | 0.0089 | 0.028 | 0.025 | 0.015 |
aThe N2 mice with urinary albumin excretion larger than 3.1 g/gCr, the minimum value in FVB-Tns2nph mice, were defined as the FVB type (affected) and the N2 mice with urinary albumin excretion less than 0.7 g/gCr, the maximum value in B6- Tns2nph mice, were defined as the B6 type (not affected)
bThe upper X% and lower X% of N2 mice in urinary albumin excretion were defined as the FVB type (affected) and the B6 type (not affected), respectively
cUrinary albumin excretion in the homogeneic genotype, in which both markers were FVB/FVB genotype, were compared with those in the heterogeneic genotype, in which at least one of the markers was B6/FVB genotype
Fig. 3.
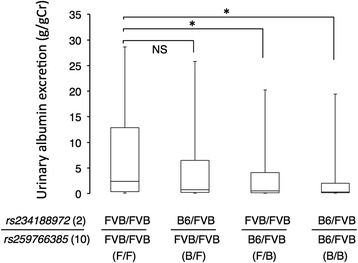
Effects of modifier loci on urinary albumin excretion in N2 mice. Urinary albumin excretion of N2 mice was divided by the four combined genotypes of rs234188972 on chromosome 2 and rs259766385 on chromosome 10. The numbers of groups were 47, 37, 47 and 43 for F/F, B/F, F/B and B/B, respectively. Bars represent the maximum and minimum values. Values represent the median. Asterisk, P < 0.05. NS, not significant
Discussion
In this study, to identify the modifier loci in podocyte alterations caused by Tns2 dysfunction, we assessed the relationship between albuminuria phenotype and genotype in backcrosses between resistant B6-Tns2nph and susceptible FVB-Tns2nph mice. Initially, we demonstrated that F1 mice exhibited an intermediate phenotype for glomerular injury between resistant B6 and susceptible FVB types, while little of no urinary albumin was detected in both the F1 mice and B6-Tns2nph mice. The results indicated that heterozygous alleles at the modifier loci show an intermediate phenotype for podocyte injury, leading to an increase in albumin leakage through the glomerular filtration barrier. However, F1 mice showed a resistant B6-type phenotype for albuminuria, probably due to the proximal tubular reabsorption of albumin [20].
Subsequently, F1 mice were backcrossed to FVB-Tns2nph mice. The N2 backcrosses showed a wide spectrum of urinary albumin excretion ranging from B6 type to FVB type, and including intermediate values. These values indicated that some N2 mice exhibited podocyte and glomerular injuries that were more severe than those of F1 mice but milder than those of FVB-Tns2nph mice. These injuries are expected to lead to greater albumin leakage, which cannot be reabsorbed by the proximal tubules, resulting in a continuous spectrum of values for urinary albumin excretion in N2 mice. These results also indicate that there are multiple modifier loci for podocyte injury caused by Tns2 dysfunction. However, a QTL analysis of urinary albumin excretion failed to identify any modifier loci (data not shown). We assumed that this is because that the intended modifier effects are hindered due to a wide spectrum of values for urinary albumin excretion in affected mice themselves (see Fig. 1c). Therefore, we simply segregated N2 mice into two groups, affected and not affected, and examined the specific relationship between genotype and phenotype using chi-square test. Then, a marker-trait association test suggested the existence of two loci, a significant locus on chromosome 10 and a suggestive locus on chromosome 2, whose peak marker rs234188972 is included in Tpir (57.65–78.72 cM). B6 homozygous alleles of Tpir exerted a protective effect against podocyte alterations and reduced urinary albumin excretion in affected mice to approximately 50% [18]. In this study, B6 heterozygous alleles of the suggestive locus on chromosome 2 were observed to partially alleviate podocyte injury, based on the results shown in Fig. 3. We, therefore, speculated that a causative gene at the suggestive locus is identical to Tpir, and we designated this suggestive locus as Tpir1.
As the protective effect of the B6 allele of Tpir was insufficient in comparison with that of the B6 genetic background itself, our previous study predicted the existence of an as yet unidentified major modifier locus [18], which actually corresponds to the significant locus identified on chromosome 10. B6 heterozygous alleles of the significant locus markedly reduced urinary albumin excretion in affected mice. Furthermore, B6 heterozygous alleles of both Tpir1 and the significant locus exerted a protective effect on albuminuria at levels close to that of the B6 genetic background. We, therefore, designated the significant locus as Tpir2. No genes located around Tpir1 and Tpir2 are known to be involved in nephropathy.
The genomic region of chromosome 10 at 38.74–44.34 cM including rs259766385, the peak marker of Tpir2, consists of multiple syntenic regions on human chromosomes 12, 19, 21 and 22. It is notable that the syntenic regions on human 12q23.2 – q23.3 and 19p13.3, and 21q22.3 include the genetic markers associated with urinary albumin excretion (PAH, rs2374688, D19S591) and glomerular filtration rate (rs2839235) (Fig. 4) [21, 22]. It is interesting that the multiple genetic markers associated with urinary albumin excretion or glomerular filtration rate on the different human chromosomes are located on a single syntenic 5.6-cM interval (38.74–44.34 cM) of mouse chromosome 10, albeit including a gene-rich region (39.72-cM) that encompasses 233 genes. This suggests that there might be multiple modifier genes on Tpir2. The Tpir2 genes located on syntenic human 12q23.2 – q23.3, 19p13.3 and 21q22.3 are shown in the Additional file 1: Table S3.
Fig. 4.
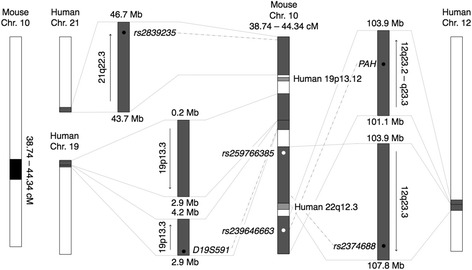
Schematic representation of the genomic region of mouse chromosome 10 at 38.74–44.34 cM and the homologous regions in human. The genomic regions of mouse chromosome 10 at 38.74–44.34 cM are syntenic to human 12q23.2 – q23.3, 19p13.3, 19p13.12, 21q22.3 and 22q13.3. White dots represent mouse genetic markers used in this study: rs259766385 and rs239646663 are the peak markers of Tpir2. Black dots represent human genetic markers: rs2374688 and rs2839235 are the single nucleotide polymorphisms associated with urinary albumin excretion and glomerular filtration rate in human, respectively [21]; and PAH and D19S591 (also known as GATA44F10) are the genetic markers associated with urinary albumin excretion in patients with essential hypertension [22]. Chr., chromosome
Conclusions
Our present analysis identified two genetic regions associated with podocyte injury induced by Tns2 dysfunction, Tpir1 and Tpir2, which are on chromosomes 2 and 10, respectively. Tpir1, which is considered to be identical to Tpir, the genetic region identified by our previous study, is insufficient to explain the resistance of the B6 genetic background to Tns2 dysfunction, while Tpir2 is a major modifier locus for this resistance. The combined protective effect of the B6 alleles of Tpir1 and Tpir2 was comparable to that of the B6 genetic background itself. The identification of these modifier genes is expected to help elucidate the mechanism underlying podocyte injury.
Additional file
Table S1. Genomic positions of polymorphic genetic markers between B6 and FVB strains. Table S2. Sequences of the PCR primers used to identify polymorphic indel mutations between B6 and FVB strains. Table S3. Genes located on syntenic human 12q23.2 – q23.3, 19p13.3 and 21q22.3. (XLSX 28 kb)
Acknowledgements
We wish to thank our lab members for their helpful discussions.
Funding
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Glant-in-Aid for Scientific Research, KAKENHI (16K16606) to HS and KAKENHI (20302614) to NS. The funders had no role in the design of the study and collection, analysis, decision to publish, interpretation of data or preparation of the manuscript.
Availability of data and materials
All the data supporting the conclusions of the study are included in the manuscript and the additional file.
Author’s contributions
HS and NS conceived and designed the work. YT and SO performed the experiments and analyzed the data. HS and YT drafted the manuscript. NS revised the manuscript. All authors reviewed the manuscript. All authors have read and approved the final version of the manuscript.
Abbreviations
- CBB
Coomassie brilliant blue
- CKD
Chronic kidney disease
- cM
Centimorgan
- GBM
Glomerular basement membrane
- gCr
Gram creatinine
- PAS
Periodic acid-Schiff
- PCR
Polymerase chain reaction
- PFA
Paraformaldehyde
- PTB
Phosphotyrosine binding
- PTP
Protein tyrosine phosphatase
- QTL
Quantitative trait loci
- Rho-GAP
Rho GTPase-activating protein
- SDS
Sodium dodecyl sulfate
- SH2
Src homology 2
Ethics approval and consent to participate
All research was conducted according to the Regulations for the Care and Use of Laboratory Animals of Kitasato University. The animal experimentation protocol was approved by the President of Kitasato University based on the judgment made by the Institutional Animal Care and Use Committee of Kitasato University (Approval ID: No. 15–167).
Competing interests
The authors declare no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12863-018-0611-1) contains supplementary material, which is available to authorized users.
Contributor Information
Yuki Takahashi, Email: dv17004@st.kitasato-u.ac.jp.
Hayato Sasaki, Phone: +81-176-24-9496, Email: hsasaki@vmas.kitasato-u.ac.jp.
Shiori Okawara, Email: vm12027x@st.kitasato-u.ac.jp.
Nobuya Sasaki, Email: nobsasa@vmas.kitasato-u.ac.jp.
References
- 1.Haynie DT. Molecular physiology of the tensin brotherhood of integrin adaptor proteins. Proteins. 2014;82(7):1113–1127. doi: 10.1002/prot.24560. [DOI] [PubMed] [Google Scholar]
- 2.Chen H, Duncan IC, Bozorgchami H, Lo SH. Tensin1 and a previously undocumented family member, tensin2, positively regulate cell migration. Proc Natl Acad Sci U S A. 2002;99(2):733–738. doi: 10.1073/pnas.022518699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hafizi S, Alindri F, Karlsson R, Dahlbäk B. Interaction of Axl receptor tyrosine kinase with C1-TEN, a novel C1 domain-containing protein with homology to tensin. Biochem Biophys Res Commun. 2002;299(5):793–800. doi: 10.1016/S0006-291X(02)02718-3. [DOI] [PubMed] [Google Scholar]
- 4.Cho AR, Uchio-Yamada K, Torigai T, Miyamoto T, Miyoshi I, Matsuda J, Kurosawa T, Kon Y, Asano A, Sasaki N, et al. Deficiency of the tensin2 gene in the ICGN mouse: an animal model for congenital nephrotic syndrome. Mamm Genome. 2006;17(5):407–416. doi: 10.1007/s00335-005-0167-z. [DOI] [PubMed] [Google Scholar]
- 5.Nishino T, Sasaki N, Chihara M, Nagasaki K, Torigoe D, Kon Y, Agui T. Distinct distribution of the tensin family in the mouse kidney and small intestine. Exp Anim. 2012;61(5):525–532. doi: 10.1538/expanim.61.525. [DOI] [PubMed] [Google Scholar]
- 6.Hong SY, Shih YP, Sun P, Hsieh WJ, Lin WC, Lo SH. Down-regulation of tensin2 enhances tumorigenicity and is associated with a variety of cancers. Oncotarget. 2016;7(25):38143–38153. doi: 10.18632/oncotarget.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hafizi S, Ibraimi F, Dahlbäk B. C1-TEN is a negative regulator of the Akt/PKB signal transduction pathway and inhibits cell survival, proliferation, and migration. FASEB J. 2005;19(8):971–973. doi: 10.1096/fj.04-2532fje. [DOI] [PubMed] [Google Scholar]
- 8.Marusugi K, Nakano K, Sasaki H, Kimura J, Yanobu-Takanashi R, Okamura T, Sasaki N. Functional validation of tensin2 SH2-PTB domain by CRISPR/Cas9-mediated genome editing. J Vet Med Sci. 2016;78(9):1413–1420. doi: 10.1292/jvms.16-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogura A, Fujimura H, Asano T, Koura M, Naito I, Kobayashi Y. Early ultrastructural glomerular alterations in neonatal nephrotic mice (ICGN strain) Vet Pathol. 1995;32(3):321–323. doi: 10.1177/030098589503200317. [DOI] [PubMed] [Google Scholar]
- 10.Uchio K, Manabe N, Tamura K, Miyamoto M, Yamaguchi M, Ogura A, Yamamoto Y, Miyamoto H. Decreased matrix metalloproteinase activity in the kidneys of hereditary nephrotic mice (ICGN strain) Nephron. 2000;86(2):145–151. doi: 10.1159/000045733. [DOI] [PubMed] [Google Scholar]
- 11.Mizuno S, Kurosawa T, Matsumoto K, Mizuno-Horikawa Y, Okamoto M, Nakamura T. Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J Clin Invest. 1998;101(9):1827–1834. doi: 10.1172/JCI1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasaki H, Marusugi K, Kimura J, Kitamura H, Nagasaki K, Torigoe D, Agui T, Sasaki N. Genetic background-dependent diversity in renal failure caused by the tensin2 gene deficiency in the mouse. Biomed Res. 2015;36(5):323–330. doi: 10.2220/biomedres.36.323. [DOI] [PubMed] [Google Scholar]
- 13.Uchio-Yamada K, Sawada K, Tamura K, Katayama S, Monobe Y, Yamamoto Y, Ogura A, Manabe N. Tenc1-deficient mice develop glomerular disease in a strain-specific manner. Nephron Exp Nephrol. 2013;123(3–4):22–33. doi: 10.1159/000354058. [DOI] [PubMed] [Google Scholar]
- 14.Uchio-Yamada K, Monobe Y, Akagi K, Yamamoto Y, Ogura A, Manabe N. Tensin2-deficient mice on FVB/N background develop severe glomerular disease. J Vet Med Sci. 2016;78(5):811–818. doi: 10.1292/jvms.15-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishino T, Sasaki N, Nagasaki K, Ahmad Z, Agui T. Genetic background strongly influences the severity of glomerulosclerosis in mice. J Vet Med Sci. 2010;72(10):1313–1318. doi: 10.1292/jvms.10-0144. [DOI] [PubMed] [Google Scholar]
- 16.Nishino T, Sasaki N, Nagasaki K, Ichii O, Kon Y, Agui T. The 129 genetic background affects susceptibility to glomerulosclerosis in tensin2-deficient mice. Biomed Res. 2012;33(1):53–56. doi: 10.2220/biomedres.33.53. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki H, Sasaki N, Nishino T, Nagasaki K, Kitamura H, Torigoe D, Agui T. Quantitative trait loci for resistance to the congenital nephropathy in tensin 2-deficient mice. PLoS One. 2014;9(6):e99602. doi: 10.1371/journal.pone.0099602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sasaki H, Kimura J, Nagasaki K, Marusugi K, Agui T, Sasaki N. Mouse chromosome 2 harbors genetic determinants of resistance to podocyte injury and renal tubulointerstitial fibrosis. BMC Genet. 2016;17(1):69. doi: 10.1186/s12863-016-0378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003;14(5):1358–1373. doi: 10.1097/01.ASN.0000065640.77499.D7. [DOI] [PubMed] [Google Scholar]
- 20.Birn H, Christensen EI. Renal albumin absorption in physiology and pathology. Kidney Int. 2006;69(3):440–449. doi: 10.1038/sj.ki.5000141. [DOI] [PubMed] [Google Scholar]
- 21.Hwang SJ, Yang Q, Meigs JB, Pearce EN, Fox CS. A genome-wide association for kidney function and endocrine-related traits in the NHLBI's Framingham heart study. BMC Med Genet. 2007;8(Suppl 1):S10. doi: 10.1186/1471-2350-8-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freedman BI, Beck SR, Rich SS, Heiss G, Lewis CE, Turner S, Province MA, Schwander KL, Arnett DK, Mellen BG, et al. A genome-wide scan for urinary albumin excretion in hypertensive families. Hypertension. 2003;42(3):291–296. doi: 10.1161/01.HYP.0000087890.33245.41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genomic positions of polymorphic genetic markers between B6 and FVB strains. Table S2. Sequences of the PCR primers used to identify polymorphic indel mutations between B6 and FVB strains. Table S3. Genes located on syntenic human 12q23.2 – q23.3, 19p13.3 and 21q22.3. (XLSX 28 kb)
Data Availability Statement
All the data supporting the conclusions of the study are included in the manuscript and the additional file.