

Abstract
Background
There is a worldwide interest for sustainable and environmentally-friendly ways to produce fuels and chemicals from renewable resources. Among them, the production of acetone, butanol and ethanol (ABE) or Isopropanol, Butanol and Ethanol (IBE) by anaerobic fermentation has already a long industrial history. Isopropanol has recently received a specific interest and the best studied natural isopropanol producer is C. beijerinckii DSM 6423 (NRRL B-593). This strain metabolizes sugars into a mix of IBE with only low concentrations of ethanol produced (< 1 g/L). However, despite its relative ancient discovery, few genomic details have been described for this strain. Research efforts including omics and genetic engineering approaches are therefore needed to enable the use of C. beijerinckii as a microbial cell factory for production of isopropanol.
Results
The complete genome sequence and a first transcriptome analysis of C. beijerinckii DSM 6423 are described in this manuscript. The combination of MiSeq and de novo PacBio sequencing revealed a 6.38 Mbp chromosome containing 6254 genomic objects. Three Mobile Genetic Elements (MGE) were also detected: a linear double stranded DNA bacteriophage (ϕ6423) and two plasmids (pNF1 and pNF2) highlighting the genomic complexity of this strain. A first RNA-seq transcriptomic study was then performed on 3 independent glucose fermentations. Clustering analysis allowed us to detect some key gene clusters involved in the main life cycle steps (acidogenesis, solvantogenesis and sporulation) and differentially regulated among the fermentation. These putative clusters included some putative metabolic operons comparable to those found in other reference strains such as C. beijerinckii NCIMB 8052 or C. acetobutylicum ATCC 824. Interestingly, only one gene was encoding for an alcohol dehydrogenase converting acetone into isopropanol, suggesting a single genomic event occurred on this strain to produce isopropanol.
Conclusions
We present the full genome sequence of Clostridium beijerinckii DSM 6423, providing a complete genetic background of this strain. This offer a great opportunity for the development of dedicated genetic tools currently lacking for this strain. Moreover, a first RNA-seq analysis allow us to better understand the global metabolism of this natural isopropanol producer, opening the door to future targeted engineering approaches.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4636-7) contains supplementary material, which is available to authorized users.
Keywords: Clostridium beijerinckii, DSM6423, Genome, RNA-seq transcriptome, Clustering, IBE fermentation, Isopropanol
Background
The use of petrochemical derived fuels and chemicals such as olefins cause severe damages to the environment and is not sustainable in the long term, due to the limited nature of fossil oil. Approaches based on the conversion of renewable biomass into chemicals of interest may represent a sustainable alternative to replace, at least partially, these petroleum-based chemicals. As an example, isopropanol can be added into fuel blends [1] or converted into olefins using chemical dehydration steps [2]. Isopropanol is a commercial C3 alcohol used as solvent or antifreeze, which can also be used as a precursor of propylene, one of the main platform molecules used in the chemical industry. Very few microorganisms have been described as natural isopropanol producers and almost none of them has been thoroughly studied. Solventogenic Clostridia have been mainly studied for their ability to produce a mixture of acetone, butanol and ethanol (ABE) from a variety of substrates (including C6 and C5 sugars), and a few strains are also able to further reduce acetone into Isopropanol, yielding an Isopropanol, Butanol and Ethanol (IBE) mixture. C. acetobutylicum ATCC 824 and C. beijerinckii NCIMB 8052 are recognized as model organisms for ABE production, while C. beijerinckii DSM6423 (formerly NRRL B-593) is the best known IBE producer. The genome of C. beijerinckii NCIMB 8052 was sequenced a few years ago and consists in a 6.0 Mb chromosome [3], which is 50% larger than that of C. acetobutylicum ATCC 824 [4]. Unlike these strains, the assembled genome of C. beijerinckii DSM 6423 (NRRL B-593) is not available, even if it has been sequenced through a Hiseq approach recently, together with 29 other Clostridial strains, in order to perform a comparative genomic analysis of saccharolytic strains belonging to the genus of Clostridium [5].
Transcriptional analysis is mandatory to gain knowledge about gene regulation and thus determine strategies for strain improvement. To date, transcriptomic analyses of solventogenic Clostridia have focused on ABE strains [6]. Most of these transcriptomic analyses were performed using DNA microarray methods and other hybridization techniques exhibiting a relatively low dynamic range for the detection of transcriptional levels due to background, saturation and poor sensitivity for gene expression. RNA-seq has now supplemented these approaches because of its capacity to provide a more accurate quantification and a larger dynamic range of expression levels. In addition, it has very low background noise because DNA sequence reads can be unambiguously mapped to unique regions along the genome [7]. The genome-wide transcriptional dynamics of C. beijerinckii NCIMB 8052 over a batch fermentation process was investigated using these technique [8].
This study presents the complete genome sequencing of the currently most studied natural isopropanol producing strain, together with RNA-seq analyses covering its whole fermentation metabolism for glucose conversion into IBE. Comparative analysis with the model organisms highlights some transcriptomic regulations.
Methods
Bacterial culture and bioreactor scale fermentations
Laboratory stocks of C. beijerinckii DSM 6423 spores were stored in sterile H2O at − 20 °C. Spores were heat shocked in boiling water for 1 min, then inoculated at a 2% inoculum level into 50 mL medium containing per liter: yeast extract, 5.0 g; KH2PO4, 0.75 g; K2HPO4, 0.75 g; MgSO4·7H2O, 0.4 g; MnSO4·H2O, 0.01 g; FeSO4·7H2O, 0.01 g; NaCl, 1.0 g; asparagine, 2.0 g; (NH4)2SO4, 2.0 g; cysteine, 0.125 g; and glucose 12.5 g. The medium was incubated at 37 °C ± 1 °C for 24 h in an anaerobic environment under N2:CO2:H2 (volume ratio of 85:10:5) atmosphere. Subsequently, the actively growing culture was inoculated at 5% into a fermentation medium (GAPES [9]) containing per liter: yeast extract, 5.0 g; KH2PO4, 1.0 g; K2HPO4, 0.76 g; NH4Ac, 3.0 g; MgSO4·7H2O, 1.0 g; FeSO4·7H2O, 0.1 g; p-aminobenzoic acid, 0.1 g; and glucose, 60 g; in a 500 mL bioreactor system as described previously [10]. Temperature was controlled at 37 °C ± 1 °C. A stirring at 350 rpm was employed for mixing. Cell density and product concentration were monitored through the course of fermentation.
Experimental design
In order to get 3 biological replicates, the same procedure was repeated 3 times on 3 different weeks. Each week, a fresh preculture was used to inoculate 2 identical bioreactors. Samples were taken over the early exponential, late exponential and stationary phases (samples at 3, 6, 8, 11, and 24 h). Following centrifugation of the samples, cell pellets were immediately frozen in liquid nitrogen and supernatants were used for HPLC analysis. For each timepoint, the RNA samples of the 2 bioreactors were pooled, leading to 1 RNA sample per bioreactor and timepoint.
Culture growth and fermentation products analysis
Culture growth was measured by following optical density at 600 nm (OD600) in the fermentation broth using an Ultrospec 2000 (Pharmacia Biotech) spectrophotometer. Metabolites were determined in clear supernatants of samples taken from the fermentation. Sugars, solvents, and organic acids were determined by HPLC using a gel permeation/size exclusion column (Shodex Ionpack KC-811) coupled to a refractometer and UV detector as described earlier [10, 11].
Genomic DNA extraction and sequencing
Genomic DNA of C. beijerinckii DSM6423 was purified using the GenElute bacterial genomic DNA kit (Sigma-Aldrich, Saint-Louis, USA). Concentration of gDNA was determined using Qbit spectrophotometer (Thermofisher) and quality checked on 0.5% agar gel. Mi-seq analysis was performed by the I2BC plat-form (Saclay, France) using the following protocol. Genomic DNA samples was fragmented using a “‘COVARIS S2” sonicator, to a mean size of 700 bp. DNA Libraries were constructed from 1 μg of this fragmented DNA, according to the “Illumina DNA sample prep protocol” (End-repair, A-tailing, ligation, PCR enrichment), on a Beckman SPRI-TE automate and using reagents from the “SPRIworks Fragment library system I” kit (Beckman Coulter). The final product were gel purified to achieve a library mean size of 700 bp. Library quality was assessed on an agilent Bioanalyzer instrument (Agilent) and sequenced on a Illumina MiSeq instrument, using a Paired-end 2 × 250 bp protocol, with a MiSeq Reagent Kit v2 500 cycle (MS-102-2003, Illumina), according to the constructor recommendations. The run resulted in a 80× coverage of the genome of C. beijerinckii and the different reads, spanning 250 nucleotides each, were assembled into DNA fragments called contigs. De novo sequencing was performed using the PacBio RSII platform (GATC, Germany).
Contigs were uploaded to the MicroScope bioinformatics platform where the putative genes were automatically annotated through a specific computer workflow [12]. MicroScope is a Microbial Genome Annotation & Analysis Platform developed by the Genoscope institute (Evry, France) [13]. Sequenced strains and RNAseq results can be uploaded and automatically analyzed on this platform.
RNA extractions and RNA-seq protocols
The extraction was performed as previously described [8]. Briefly, cell lysis and RNA isolation was done with the TRIzol Plus RNA purification kit (AMbion). 5 mL TRIzol reagent was added to the frozen pellet followed by 1 mL chloroform after cell lysis. After shacking and centrifugation, the upper phase was mixed with an equal volume of 70% ethanol before transfer to PureLink RNA Spin Cartridges furnished with the kit. Kit instructions allowed to obtain purified total RNA which was further processed by cleanup with the RNeasy mini kit (Qiagen). DNA was eliminated with DNAse I treatment (AM1906, Invitrogen). 20 μL RNA were treated with 2 μL DNAse in 50 μL nuclease free water for 30 min at 37 °C before addition of another 1.5 μL DNAse and 30 min extra incubation. Treatment was done twice and was followed by RNA cleanup with the RNeasy kit. A full quality assessment of the RNA, quantification using Nanodrop, PCR to check for residual DNA and migration on 1% agarose gel were done before depletion of ribosomal RNA with MicrobExpress kit (ThermoFischer). Depleted RNA samples were quantified and stored at − 80 °C before being sent for analysis. DNA libraries from RNA samples and Hi-seq single read sequencing were performed by the Imagif platform (I2BC, Orsay, France).
RNA-seq sequencing data analysis
Clean reads of each sample were mapped to the reference genome of the previously sequenced C. beijerinckii DSM 6423, with predicted protein encoding genes and rRNA using the TAMARA (Transcriptome Analysis based on MAssive sequencing of RnAs) tool provided on the MicroScope platform [12]. Table 1 shows statistics about the number of total and mapped reads.
Table 1.
Summary of RNA-Seq sequencing and data analysis results
| Replicate | Time point (h) | Total read number | Reliable reads | Reads mapped on genomic objects (except rRNA) | Number of genes with detectable expression |
|---|---|---|---|---|---|
| A | 3 | 19,158,892 | 17,799,816 | 7,024,274 | 5944 |
| B | 3 | 11,574,716 | 10,099,244 | 3,266,941 | 5829 |
| C | 3 | 15,203,792 | 12,936,903 | 4,403,183 | 5851 |
| A | 6 | 16,007,459 | 14,775,252 | 5,341,913 | 5876 |
| B | 6 | 13,731,477 | 11,723,511 | 4,255,219 | 5884 |
| C | 6 | 13,472,975 | 11,754,891 | 3,741,547 | 5861 |
| A | 9 | 11,050,565 | 9,196,745 | 2,854,832 | 5895 |
| B | 9 | 14,381,148 | 12,261,616 | 3,777,589 | 5906 |
| C | 9 | 11,873,306 | 10,118,493 | 3,235,171 | 5880 |
| A | 11 | 13,663,240 | 11,531,714 | 3,327,568 | 5887 |
| B | 11 | 10,195,910 | 8,792,217 | 2,208,510 | 5789 |
| C | 11 | 19,046,554 | 16,439,661 | 4,596,207 | 5917 |
| A | 24 | 13,578,705 | 11,371,420 | 1,153,741 | 5203 |
| B | 24 | 21,453,820 | 17,646,890 | 1,871,469 | 5606 |
| C | 24 | 9,805,052 | 8,262,117 | 1,046,981 | 5121 |
Transcriptomic high throughput sequencing data were analyzed using a bioinformatic pipeline implemented in the Microscope platform [12]. In a first step, the RNA-Seq data quality was assessed by including reads trimming. In a second step, reads were mapped onto the C. beijerinckii DSM6423 genome sequence (previously submitted for sequencing) using the SSAHA2 package [14] that combines the SSAHA searching algorithm (sequence information is encoded in a perfect hash function) aiming at identifying regions of high similarity, and the cross-match sequence alignment program [15], which aligns these regions using a banded Smith-Waterman-Gotoh algorithm [16]. An alignment score equal to at least half of the read is required for a hit to be retained. To lower false positives discovery rate, the SAMtools (v.0.1.8, [17]) are then used to extract reliable alignments from SAM formatted files. The number of reads matching each genomic object harbored by the reference genome is subsequently computed with the Bioconductor GenomicFeatures package [18]. If reads matching several genomic objects, the count number is weighted in order to keep the same total number of reads. Finally, the Bioconductor-DESeq package [19] with default parameters is used to analyze raw counts data and test for differential expression between conditions. Moreover, the quality of the RNA-seq data was strengthened by checking the relative stability of housekeeping genes such as gyrA and rpoB across time points.
Results
Genome assembly
Genome sequence of C. beijerinckii DSM6423 was obtained through PacBio and MiSeq sequencing. The PacBio de novo sequencing yielded 17 unitigs. Unitigs were further combined in four scaffolds when assembled with the 412 contigs obtained through MiSeq sequencing. Junctions between unitigs were confirmed by PCR, and the 4 scaffolds were PCR gap-closed, yielding a curated 6,383,364-bp long circular chromosome, with an average GC content of 29.81% (Fig. 1). Automatic functional annotation resulted in the prediction of 6052 protein genes and 202 RNA genes on this chromosome.
Fig. 1.

Circular representation of Clostridium beijerinckii DSM6423 genome. CGView representation [51] of the circular genome of the DSM6423 strain and its extrachromosomic elements (plasmids: pNF1, pNF2 and double stranded linear phage ɸ6423). Circles display (from the outside): (1) GC percent deviation (GC window - mean GC) in a 1000-bp window. (2) Predicted CDSs transcribed in the clockwise direction. (3) Predicted CDSs transcribed in the counterclockwise direction. Genes displayed in (2) and (3) are color-coded according different categories: red and blue: MaGe validated annotations orange: MicroScope automatic annotation with a reference genome purple: Primary/Automatic annotations. (4) GC skew (G + C/G-C) in a 1000-bp window. (5) rRNA (blue), tRNA (green), misc_RNA (orange), Transposable elements (pink) and pseudogenes (grey)
Three Mobile Genetic Elements (MGE) were also identified: two natural plasmids named pNF1 and pNF2, and one bacteriophage, named ɸ6423 (Fig. 1). Plasmid pNF1 is 10,278-bp long and contains a gene sharing homology with both the bacteriocin gene from Clostridium butyricum MIYAIRI 588 [20] and the closticin 574 gene from Clostridium tyrobutyricum ADRIAT 932 [21]. The last 83 aminoacids of the gene product shared 81.9% and 64.6% pairwise identity with the corresponding peptides possessing bacteriocin activity from C. butyricum MIYAIRI 588 and C. tyrobutyricum ADRIAT 932, respectively. Plasmid pNF2 is 4282-bp long and carries four predicted ORFs, one of them being potentially involved in plasmid replication. Phage ɸ6423 have a linear double-stranded DNA genome of 16,762 bp, with 143 bp inverted terminal repeats. To our knowledge, this is the first time such a bacteriophage is described in the Clostridium genus.
Identification of Regions of Genomic Plasticity (RGPs) was performed on the DSM6423 chromosome in order to identify potentially horizontally transferred genes (HGT) which are gathered in genomic regions. In total, 47 RGPs were found in the genome of DSM 6423 when compared with that of C. beijerinckii NCIMB 8052. One of these RGPs contains the specific secondary alcohol dehydrogenase adh gene whose product allows the conversion of acetone into isopropanol. This gene is located in a 23-kb genomic island having no equivalent in the NCIMB 8052 strain and harboring 28 coding sequences in which 6 metabolic genes and one transposase were predicted (Additional file 1). There is a high probability that this part of the genome was acquired through horizontal transfer. This NADP(H)-linked, zinc-containing secondary alcohol dehydrogenase (sADH) is a tetrameric protein having the highest catalytic efficiency on acetone [22, 23]. Interestingly, This sADH has a significant sequence identity with other class I alcohol dehydrogenases such as the tetrameric enzymes from Entamoeba histolytica and bacterial secondary-alcohol dehydrogenase from Thermoanaerobacter brockii which is an exceptionally high degree of identity between enzymes from such distantly related organisms.
C. beijerinckii cultivation in bioreactor
To further improve genetic knowledge on C. beijerinckii DSM6423, a transcriptomic study was performed. A RNA-Seq approach was chosen in order to have a timelapse study of the metabolism of the strain throughout the fermentation process. Three independent fermentations were carried out in bioreactors on three different weeks, showing good reproducibility (Fig. 2).
Fig. 2.

Fermentation profile of Clostridium beijerinckii DSM6423 on glucose. C. beijerinckii DSM 6423 was cultivated in bioreactors in GAPES medium. a pH, b biomass followed by OD600, c acids and d solvents. Values are the mean and standard deviation of the 6 biological replicates. See Additional file 3 for details on the biological replicates
Acid re-assimilation and solvent production appeared to start around 6-8 h. This was concomitant with the characteristic loss of mobility of the cells and the appearance of cigar–shape cells as observed under the microscope (Additional file 2).
The final solvent profile obtained corresponded well to those described earlier for this strain [24, 25]. Moreover, the biphasic pH is characteristic of the solvantogenic strains such as the well-known C. acetobutylicum ATCC824 [26], even if the latter is able tolerate more acidic pH conditions. In the presence of sodium acetate in the medium, C. beijerinckii DSM6423 starting its switch to solvantogenesis at a higher pH level (5,2) than C. acetobutilicum ATCC824 (pH 4,2 [27]). Survase et al. [24] studied the time course of a batch fermentation on glucose and showed that solvent production started at the end of the exponential phase, at approximately 6–9 h, which is consistent with our observations. Their batch culture showed a maximum concentration of 2.16 g/L (36.8% of total solvents) of isopropanol and 3.71 g/L (63.2% of total solvents) of butanol after 24 h fermentation with no further significant increase and the maximum solvent yield was 0.30 g/g glucose consumed with 33.8% of substrate conversion. Interestingly, the Isopropanol/Butanol ratio was progressively growing along the solvantogenic phase (Fig. 2a). This may be due to the presence of sodium acetate in the fermentation medium to prevent a severe pH drop linked to acids transport and assimilation [28]. This behavior is in contradiction with those observed for a batch culture of C. beijerinckii DSM6423 with a control of the pH (close to 4.8) using NaOH [24]. In that case, a constant I/B ratio was maintained during the ten first hours of solvantogenesis. Interestingly, in the case of C. acetobutylicum, [29] a significant increase of ABE production but no modification of the A/B ratio was observed when ammonium acetate is used to control the pH. This tend to confirm a specific behavior of C. beijerinckii DSM6423.
During our batch fermentation we obtained 3 g/L isopropanol and 5.3 g/L butanol after 30 h fermentation time with 28.8% substrate conversion (Additional file 3). De Vrije et al. reported end concentrations of 10.7 g/L total IBE, with 3.2 g/L isopropanol and 6.9 g/L of butanol after 48 h of fermentation when the strain was grown on a mix of glucose and xylose [30].
For each triplicate, samples were collected for RNA-Seq analyses at 3 h (acidogenesis phase, exponential growth), 6 h (acidogenesis to solventogenesis switch), 8 h and 11 h (solventogenesis) and 24 h (late solventogenesis) after inoculation. The 15 resulting RNA samples were sequenced and analyzed using the reconstructed genome of DSM6423.
RNA seq analysis
Highly expressed genes
Determination of the most highly expressed genes was performed using RPKM values during the whole fermentation experiment (Additional file 4). About 15% of these genes were coding for proteins of unknown functions, which represent 34% of the coding sequenced predicted in the genome. As expected, the most expressed genes after 3 h of fermentation are coding for ribosomal proteins, consistently with the protein synthesis activity during exponential growth phase and cell division. A set of genes involved in cell motility, such as the flagellin coding protein, was also found to be highly expressed, consistently with the high mobility of the Clostridium cells at this time (Additional file 2). Other highly expressed genes at 3 h were involved in transcription, translation, protein folding and of course Isopropanol/Butanol/Ethanol production. Concerning the mobile genetic elements discovered in C. beijerinckii DSM6423, 3 highly expressed genes were located on the bacteriophage Φ6423 and only one, corresponding to a cell wall binding protein, on the natural plasmid pNF1.
Differentially expressed genes
In order to investigate the transcriptional regulations during the fermentation, the most differentially expressed genes were selected by comparing their expression at a given time point (6 h, 8 h, 11 h and 24 h) to their expression at the previous time point (3 h, 6 h, 8 h and 11 h respectively) (Fig. 3, a). Thresholds were selected at |log2(fold change)| > 1.5 and adjusted p-value< 0.001, which resulted in a list of 1008 significantly differentially expressed genes (16% of the genome). Interestingly, the majority of these genes (683 genes) showed a different level of gene expression between 3 h and 6 h, which corresponds to the switch from acidogenesis to solventogenesis. Most of the genes (938 genes, 93%) were regulated once during the fermentation, 69 genes were regulated 2 times, and only 1 gene appeared to be regulated 3 times during the fermentation. This tends to demonstrate that each time point corresponded to a specific physiological state during the cultivation As suggested by the number of regulated genes, the main transcriptomic regulations occur between 3 and 11 h, with only 31 genes being specifically regulated between 11 and 24 h. Therefore, further clustering analysis focused on the 977 genes regulated between 3 and 11 h.
Fig. 3.
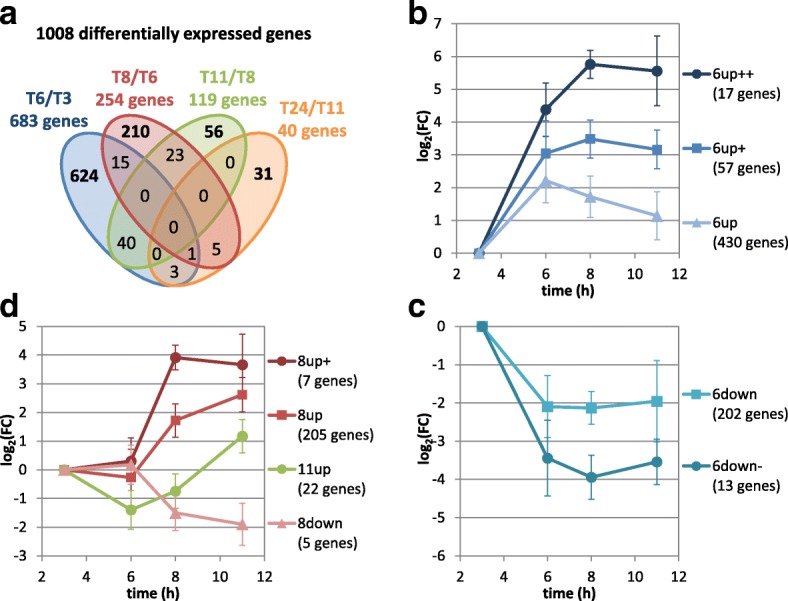
Global transcriptomic analysis of C. beijerinckii DSM6423 fermentation on glucose. a Venn Diagram showing the number of genes regulated in various physiological time points. b to d kinetic expression profiles of various clusters of genes: genes up-regulated at 6 h (B), genes down-regulated at 6 h (c), and genes regulated at 8 h or 11 h (d)
Kinetic analysis of transcription regulations
In order to highlight the transcriptional regulation along the fermentation, we calculated the transcription profiles of each gene, using the 3 h sample as reference. Clustering was performed using CAST algorithm [31] with a threshold of 0.88 and revealed 8 clusters containing 953 genes. Three clusters (504 genes) correspond to genes up-regulated at 6 h (Fig. 3b), 2 clusters (215 genes) to genes down regulated at 6 h (Fig. 3c) and 3 clusters (234 genes) to genes up-regulated at 8 h or 11 h (Fig. 3d).
Up-regulated genes at 6 h are divided in 3 different clusters (Additional file 5). A first cluster (6up++) contains 17 genes up-regulated at 6 h and 8 h and involved in the Panthothenate and CoA biosynthetic pathway, oxidative stress or solvantogenesis (butanol production). A second cluster of 57 genes (6up+), strictly up-regulated at 6 h, comprises a set of genes related to sporulation, including the predicted anti-sigma F factor antagonist (SpoIIAA and SpoIIAB), and a putative operonic structure linked to acetone production (CIBE_4606 to 4609).
The third cluster (6up) comprises 430 genes that are up-regulated at 6 h and then down-regulated. This cluster include genes that are not regulated after 6 h, notably the anti-sigma factors regulating sporulation, an acetolactacte synthase and several genes encoding for enzymes involved in ferrous storage (ferritin). Some genes of this cluster are directly down-regulated at 8 h, including genes involved in solvantogenesis (isopropanol production), responses to the heat shock effects and suffer assimilation. Finally, several genes of this cluster are down-regulated at 11 h including sigma factors related to the initiation of sporulation and a RNA polymerase sigma factor 28. The σ28 is a minor sigma factor responsible for the initiation of transcription of a number of genes involved in motility and flagellar synthesis [32].
The 235 genes that were down-regulated at 6 h were divided in two different clusters. The main one (6down) contained a gene cluster involved in the mobility of the bacterial cells, in particular motA and motB. This is in agreement with the loss of mobility and the change in cell shape characterizing the onstart of the solventogenic phase (Additional file 2). The second cluster of genes, down-regulated at 6 h then 8 h (6down-) corresponded to genes coding for ribosomal proteins, oxidative stress response proteins and a high number of putative membrane transporters. This might suggest some major changes in the membrane transport system for the re-assimiliation of acids linked to solventogenesis.
The set of genes up-regulated after 8 h was divided in 3 different clusters including a first one (8up, 205 genes) mainly comprising genes involved in solvantogenesis. The second cluster (8up+) of genes was strictly up-regulated at 8 h and included genes linked to oxidative stress and the latter phases of sporulation. The last cluster (11up) corresponded to a set of 22 genes up-regulated at 11 h and mainly linked to sporulation.
A really small cluster of genes were down regulated at 8H (8down). Among them, one gene is coding for a member of the 2-oxoacid oxidoreductases, a family of enzymes that oxidatively decarboxylate different 2-oxoacids to form their CoA derivatives. Moreover, a putative operonic structure classified in this cluster is potentially involved in ferrous iron transport (CIBE_5053 to 55).
Main transcriptomic regulations
Central metabolism
In order to better understand the transcriptional regulation occurring in C. beijerinckii DSM6423 cells during fermentation, the transcriptomic data allowed us to confirm the role of some predicted metabolic genes coming from the automatic annotation. Analysis revealed 22 genes potentially involved in glycolysis, with multiples candidates (“isozymes”) for some key reactions. A majority of these genes was highly transcribed throughout the fermentation tending to demonstrate their implication in this central pathway.
Transcriptomics analysis of the whole metabolic pathways also revealed a significant expression level of predicted operons encoding enzymes involved in the production of acids and the conversion of acetyl-CoA to butyryl-CoA (Fig. 4). No regulation was observed for operonic structures predicted to be involved in acetate (CIBE_1400 and 1401) or butyrate (CIBE_0223 and 0224) production. This might be explained by the specific medium used in these experiment, containing 3 g/L of sodium acetate. Unlike what has been observed in C. acetobutylicum (Schreiber & Dürre, 2015), the gene coding for the phosphoglycerate mutase (pgm, CIBE_0772) is predicted to be a part of the operon comprising genes coding for the glyceraldehyde-3-phosphate dehydrogenase (gap), the phospho-glycerate kinase (pgk) and the triosephosphate isomerase (respectively gap, pgk and tpi, CIBE_0769 to 0771; Fig. 4).
Fig. 4.

Main genes and predicted operonic structures involved in the central metabolism of in C. beijerinckii DSM6423. a glycolysis; b acids and solvents production). Number of isozymes, predicted by Microscope tool (Genoscope, Evry, France) are indicated in brackets
As observed in other Clostridium strains, genes dedicated to the production of butyryl-CoA from acetoacetyl-CoA seems to be organized in an operon (CIBE_0339 to 0343, Fig. 4) and are not regulated in our fermentation conditions. More interestingly, the 3 genes (CIBE_5186, CIBE _2196 and CIBE _1684) apparently involved in the pyruvate decarboxylation into acetyl-CoA do not appear to be regulated in the same way. The first one was highly and constantly transcribed throughout the fermentation whereas the two others were slightly transcribed (30× less) and further induced at 8 h and 11 h. This might suggest a fine-tuned regulation of the acetyl CoA production along the fermentation.
Another operon containing CIBE_4606 to CIBE_4609 gene, coding for an alcohol dehydrogenase, two CoA transferases and an acetoacetate decarboxylase, are equivalent to the ald-ctfA-ctfB-adc operon of C. beijerinckii NCIMB 8052 [3]. Expression of this operon appeared stable throughout the fermentation. Two genes encoding ctfA and ctfB analogs (CIBE_3149 and 3150) were then identified and appeared to be overexpressed at 8 and 11 h.
More surprisingly, a high level of transcription was already observed at 3 h for the predicted operons involved in butanol (CIBE_2622 to CIBE_2624), acetone (CIBE_4606 to CIBE_4609) or isopropanol (CIBE_3468 to CIBE_3470; Fig. 4) production. Expression of this latter operon was even up-regulated at 6 h. This specificity confirm the distinct transcriptomic regulation of the solvantogenesis between C. beijerinckii and C. acetobutylicum strains. In C. acetobutylicum ATCC824, the corresponding genes adhE (CA_P0162), ctfA (CA_P0163) and ctfB (CA_P0164) are located on the mega-plasmid pSOL1 and only induced during solvantogenesis, while adc (CA_P0165) is organized in a monocistronic operon in the opposite direction [33, 34]. The switch between acidogenic and solventogenic phases appears to be less pronounced in the case of C. beijerinckii DSM6423, compared to other solventogenic clostridia, such as C. acetobutylicum ATCC824. In total, more than 20 genes predicted as encoding alcohol dehydrogenases were detected in the genome of the DSM 6423 strain and most of them were expressed from the beginning of the fermentation.
Sporulation
It is well established that spo0A is the master regulator for sporulation events in Bacillus and in Clostridia [35, 36]. Phosphorylated Spo0A has been reported to induce the expression of several targets, including the sol operon and multiple sporulation sigma factor genes in C. acetobutylicum ATCC 824 [37]. As described previously, the solventogenic phase started at 6 h and may be linked to the initiation of the sporulation phase. This was confirmed by the overexpression at 6 h of a set of 12 genes including the predicted genes coding for Spo0A (CIBE_2041; Additional file 5), the sigma factors σF (CIBE_0994) and σG (CIBE_1357) but also the anti-anti-sigma factor SpoIIAA and the anti-sigma factor SpoIIAB (CIBE_0992 and 0993, respectively). In C. acetobutylicum ATCC824, a multicomponent system involving SpoIIAA and SpoIIAB activates Spo0A through phosphorylation, which further stimulates the expression of sigF [38]. The overexpression at 6 h of the predicted anti-sigma factor SpoIIAB and the specific regulation of the SpoIVB predicted gene (repressed at 6 h then up-regulated at 8 h; Additional file 5) suggest that the initiation of the sporulation and solventogenesis are simultaneous.
An increase in the expression of some other sporulation-related genes, such as spoIVA and spoIVB was also detected at 8 h. In Bacillus subtilis, SpoIVB protein is the key determinant for inter-compartmental signaling of pro-σK processing [39, 40]. Pro-σK is the inactive form of the final transcription factor σK acting in the mother cell compartment of the sporulating cell [41]. The σK regulon is then involved in the final stages of spore maturation including spore coat biosynthesis and release of the spore from the sporangial cell. This is in accordance with the observation that these genes are up-regulated at 8 h.
The specific regulation of the motility locus
Loss of motility usually comes with the transition from exponential to stationary growth phases and therefore when switching from acidogenesis to solventogenesis. Clostridia cells stop moving and lose their rod shape to gain the characteristic cigar shape instead. Expression of motility genes, such as motA and motB, and of a high number of genes coding flagellar proteins was repressed at 6 h, corresponding to the start of the solventogenic phase. This is in accordance with previous transcriptomics results observed in Clostridium beijerinckii NCIMB8052 [8], in Clostridium acetobutylicum [42] and more generally in Clostridia and Bacilli [43]. Most genes in the flagellar/chemotaxis cluster were down-regulated at the beginning of the solvantogenesis, which is related in our strain with the onset of sporulation. Similarly, the flagellar motility coding gene flgC is strongly expressed during the early exponential phase and further down regulated at 6 h, at the onset of the stationary phase.
Discussion
C. beijerinckii is a prominent solvent-producing bacterium with great potential as a microbial cell factory for the biofuel and chemical industries, in particular for isopropanol production. This strain has already been used for advanced bioprocesses, and increased productivities were obtained by cell immobilization, in-situ product removal and use of glucose/xylose mixes as substrate [44]. Recently, mutagenesis and genome shuffling have been applied to this strain to generate mutants with improved tolerance to isopropanol [11]. However, a complete genome characterization of this strain was still lacking.
Although transcriptional analysis is essential to understand gene functions and regulation and thus elucidate proper strategies for further strain improvement, limited information was available for the natural isopropanol producer C. beijerinckii DSM 6423. The genome sequencing of DSM 6423 yielded a 6,4 Mbp chromosome, two plasmids and one bacteriophage. These mobile genetic elements could be used as a chassis for the development of genetic tools dedicated to this strain.
The genome-wide transcriptional dynamics of C. beijerinckii NCIMB8052 revealed an overexpression of the glycolysis genes throughout the fermentation, especially during the acidogenesis phase. The expression of genes involved in this first part of the metabolism was then down-regulated at the beginning of the well-known metabolic shift from acidogenesis to solventogenesis. According to Wang et al. [8], out of the > 20 genes encoding alcohol dehydrogenase in C. beijerinckii NCIMB8052, Cbei_1722 and Cbei_2181 were highly up-regulated at the onset of solventogenesis, corresponding to their key roles in primary alcohol production. In our fermentation experiments, the expression pattern of glycolysis genes was similar than in C. beijerinckii NCIMB8052, with high expression levels throughout the whole fermentation [8]. This seems to be specific of C. beijerinckii strains as a time course transcriptional analysis of C. acetobutylicum ATCC824 showed higher expression during stationary phase for most of the glycolysis genes [35].
Studies concerning C. acetobutylicum, with genome-wide gene expression analysis of the switch between acidogenesis and solventogenesis [42] or solvent stress response using butanol [6] led to new insights into the physiological role of several genes or operons involved in solvent formation. Concerning C. beijerinckii DSM 6423, using the MicroScope web-platform to identify the genes involved in the main metabolic pathways and their duplications, we were able to detect at least 4 putative operonic structures related to acidogenesis and solvantogenesis. One of them, CIBE_4606 to 4609, corresponds to the sol operon of C. beijerinckii NCIMB8052 where ald, ctfAB and adc are organized in an operon and co-regulated [8]. The structure of other important gene operons involved in metabolic pathways for acid and solvent production in C. beijerinckii NCIMB 8052, including pta-ack, ptb-buk, hbd-etfA-etfB-crt operons was also confirmed.
Most sporulation genes in C. beijerinckii NCIMB8052 demonstrated similar temporal expression patterns to those observed in B. subtilis and C. acetobutylicum. Sporulation sigma factor genes sigE and sigG exhibited accelerated and stronger expression in C. beijerinckii NCIMB8052, which is consistent with the more rapid forespore and endspore development in this strain. A previous study involving insertional inactivation of spo0A indicated that Spo0A does control the formation of solvents, spores and granulose in C. beijerinckii NCIMB8052 [45]. A spo0A mutant of C. beijerinckii NCIMB8052 also showed an asporogenous and non-septating phenotype [46]. Moreover, in C. acetobutylicum ATCC 824, spo0H and spo0A are both constitutively expressed at constant levels throughout the growth cycle [36]. In our strain, production of solvents was detected already at 6 h after inoculation of the fermenters and initiation of sporulation was concomitant with the mid to late solventogenic phase (8 to 11 h). There is a slight down-regulation of spo0A at 8 h, similar to what Wang et al. observed with NCIMB8052 [8], and microscopic studies indicated that sporulation was both enhanced and accelerated due to spo0A overexpression compared to parental strains. It is therefore not surprising to see sporulation genes, such as spoIVA, highly expressed starting at 11 h.
The expression of motility genes, such as motA and motB, and flagellar protein coding flgC was repressed at 6 h, which corresponds to the onstart of the solventogenic phase in the studied fermentation. In Salmonella enterica, electrostatic interactions between the stator protein MotA and the rotor protein FliG are important for bacterial flagellar motor rotation [47]. The Salmonella flagellar motor, which is embedded within the cell membranes, is powered by proton motive force. According to the study, five flagellar proteins, MotA, MotB, FliG, FliM, and FliN, are involved in motor performance. Therefore repression of the motAB genes expression in C. beijerinckii might be linked to loss of motility during phase transition. Also, the flagellar motility related gene flgC is strongly expressed in DSM 6423 during the early exponential phase and down regulated at 6 h at the onset of the stationary phase. Also noticeable is the overexpression of chaperonins groEL and groES at 8 h. The groESL genes are involved in stress response of Clostridium acetobutylicum and play a role in solvent tolerance [27]. The toxicity of butanol in particular limits its formation in microbial fermentations. Over-expression of groESL, grpE and htpG, significantly improved butanol tolerance of C. acetobutylicum [48].
Conclusions
C. beijerinckii DSM6423 is the most well-known natural isopropanol producer and harbors the secondary alcohol dehydrogenase gene that was cloned in heterologous strains to allow acetone conversion into isopropanol by C. acetobutylicum [10, 49, 50]. Improving the natural production of this strain through a targeted approach requires the full sequencing of its genome, together with a transcriptomic analyses. Such analyses were carried out in this study and provide useful data to better understand the genetic background and physiology of this strain.
Notably, this work described a complete genomic study of a natural IBE producer including a first genome physical map of a natural IBE producer. This first analysis highlighted several genetic and metabolic particularities including a specific genomic event occurred on this strain to produce isopropanol. A better understanding of the metabolic pathways and various genes involved opens the door for future targeted approaches to make of this strain an efficient microbial cell factory for isopropanol or IBE production.
Additional files
Region of Genome Plasticity (RGP) analysis. (PDF 56 kb)
Microscopic observation of C. beijerinckii DSM6423 cells during glucose fermentation. (PDF 105 kb)
Details on the 6 biological replicates of C. beijerinckii DSM6423 glucose fermentation. (PDF 48 kb)
List of the most highly transcribed genes during C. beijerinckii DSM6423 glucose fermentation at 3 h. (PDF 79 kb)
Clusters of differential expressed genes in C. beijerinckii DSM6423 glucose fermentation. Nine hundred seventy-seven genes differentially expressed at T6 versus T3, or at T8 versus T6 or T11 versus T8 were clustered using CAST algorithm. This table shows the genes included in each cluster, with their ID and their functional annotation, and with their expression using expression at T3 as reference. (PDF 126 kb)
Acknowledgements
This work has benefited from the facilities and expertise of the High-throughput Sequencing Platform of I2BC (Imagif, Orsay, France). We also thank David Roche and Françoise Fayolle-Guichard for their contributions in the design of the fermentation protocol dedicated to the RNA seq analysis and Isabelle Martin-Verstraete (Institut Pasteur, France) for fruitful discussions.
Availability of data and materials
The DSM6423 full genome sequence was deposited on the European Nucleotide Archive (ENA) under the accession number PRJEB11626 (https://www.ebi.ac.uk/ena/data/view/PRJEB11626).
The DSM6423 RNA seq data analyzed during this study were deposited on NCBI BioProject Database under the Accession Number GSE100024 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100024).
Abbreviations
- ABE
Acetone Butanol Ethanol
- HGT
Horizontal Gene Transfert
- IBE
Isopropanol Butanol Ethanol
- MGE
Mobile Genetic Element
- RGPs
Regions of Genomic Plasticity
- RPKM
Reads per kilo base per million mapped reads
- sADH
Secondary Alcohol Dehydrogenase
Authors’ contributions
HMG prepared the DSM6423 RNA samples from IBE Fermentation. FB-M and EJ performed clustering analysis of the RNA seq data. AB and BC were involved in the IBE fermentation and the extraction of the gDNA and FW performed the physical map of C. beijerinckii DSM6423. NLF and AMLC supervised the study and contributed in writing the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4636-7) contains supplementary material, which is available to authorized users.
The original version of this article was revised: The images that were first incorrectly published as figures 2, 3 and 4 have been now been correctly published as figures 3, 4 and 2 respectively.
A correction to this article is available online at https://doi.org/10.1186/s12864-018-4799-2.
Change history
6/1/2018
Following the publication of this article [1], the authors noticed that Figs. 2, 3 and 4 were in the incorrect order and thus had incorrect captions.
Contributor Information
Hadrien Máté de Gérando, Email: hadrien-felix@hotmail.com.
François Wasels, Email: francois.wasels@ifpen.fr.
Angélique Bisson, Email: angelique.bisson@ifpen.fr.
Benjamin Clement, Email: benjamin.clement@ifpen.fr.
Frédérique Bidard, Email: frederique.bidard-michelot@ifpen.fr.
Etienne Jourdier, Email: etienne.jourdier@ifpen.fr.
Ana María López-Contreras, Email: ana.lopez-contreras@wur.nl.
Nicolas Lopes Ferreira, Phone: +33 147 527 284, Email: nicolas.lopes-ferreira@ifpen.fr.
References
- 1.Li Y, Meng L, Nithyanandan K, Lee TH, Lin Y, Lee C-fF, Liao S. Combustion, performance and emissions characteristics of a spark-ignition engine fueled with isopropanol-n-butanol-ethanol and gasoline blends. Fuel. 2016;184:864–872. doi: 10.1016/j.fuel.2016.07.063. [DOI] [Google Scholar]
- 2.Zhang D, Al-Hajri R, Barri SAI, Chadwick D. One-step dehydration and isomerisation of n-butanol to iso-butene over zeolite catalysts. Chem Commun. 2010;46:4088–4090. doi: 10.1039/c002240c. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Li X, Mao Y, Blaschek HP. Single-nucleotide resolution analysis of the transcriptome structure of Clostridium beijerinckii NCIMB 8052 using RNA-Seq. BMC Genomics. 2011;12:479. doi: 10.1186/1471-2164-12-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nölling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, et al. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol. 2001;183:4823–4838. doi: 10.1128/JB.183.16.4823-4838.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poehlein A, Solano JDM, Flitsch SK, Krabben P, Winzer K, Reid SJ, et al. Microbial solvent formation revisited by comparative genome analysis. Biotechnol Biofuels. 2017;10:58. doi: 10.1186/s13068-017-0742-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwarz KM, Kuit W, Grimmler C, Ehrenreich A, Kengen SW. A transcriptional study of acidogenic chemostat cells of Clostridium acetobutylicum – cellular behavior in adaptation to n-butanol. J Biotechnol. 2012;161:366–377. doi: 10.1016/j.jbiotec.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Li X, Mao Y, Blaschek HP. Genome-wide dynamic transcriptional profiling in Clostridium beijerinckii NCIMB 8052 using single-nucleotide resolution RNA-Seq. BMC Genomics. 2012;13:102. doi: 10.1186/1471-2164-13-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gapes JR, Nimcevic D, Friedl A. Long-term continuous cultivation of Clostridium beijerinckii in a two-stage chemostat with on-line solvent removal. Appl Environ Microbiol. 1996;62:3210–3219. doi: 10.1128/aem.62.9.3210-3219.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collas F, Kuit W, Clément B, Marchal R, López-Contreras AM, Monot F. Simultaneous production of isopropanol, butanol, ethanol and 2,3-butanediol by Clostridium acetobutylicum ATCC 824 engineered strains. AMB Express. 2012;2:45. doi: 10.1186/2191-0855-2-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Máté de Gérando H, Fayolle-Guichard F, Rudant L, Millah SK, Monot F, Lopes Ferreira N, López-Contreras AM. Erratum to: improving isopropanol tolerance and production of Clostridium beijerinckii DSM 6423 by random mutagenesis and genome shuffling. Appl Microbiol Biotechnol. 2017;101:7769. doi: 10.1007/s00253-017-8505-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, et al. MicroScope—an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 2013;41:D636–D647. doi: 10.1093/nar/gks1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MicroScope : Microbial Genome Annotation & Analysis Platform. https://www.genoscope.cns.fr/agc/microscope/home/. Accessed 18 Nov 2016.
- 14.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces UsingPhred. I. accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 16.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakanishi S, Tanaka M. Sequence analysis of a bacteriocinogenic plasmid of Clostridium butyricum and expression of the bacteriocin gene in Escherichia coli. Anaerobe. 2010;16:253–257. doi: 10.1016/j.anaerobe.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Kemperman R, Kuipers A, Karsens H, Nauta A, Kuipers O, Kok J. Identification and characterization of two novel Clostridial Bacteriocins, Circularin A and Closticin 574. Appl Environ Microbiol. 2003;69:1589–1597. doi: 10.1128/AEM.69.3.1589-1597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ismaiel AA, Zhu CX, Colby GD, Chen JS. Purification and characterization of a primary-secondary alcohol dehydrogenase from two strains of Clostridium beijerinckii. J Bacteriol. 1993;175:5097–5105. doi: 10.1128/jb.175.16.5097-5105.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korkhin Y, Kalb A, Peretz M, Bogin O, Burstein Y, Frolow F. NADP-dependent bacterial alcohol dehydrogenases: crystal structure, cofactor-binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J Mol Biol. 1998;278:967–981. doi: 10.1006/jmbi.1998.1750. [DOI] [PubMed] [Google Scholar]
- 24.Survase SA, Jurgens G, van Heiningen A, Granström T. Continuous production of isopropanol and butanol using Clostridium beijerinckii DSM 6423. Appl Microbiol Biotechnol. 2011;91:1305–1313. doi: 10.1007/s00253-011-3322-3. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed I, Ross RA, Mathur VK, Chesbro WR. Growth rate dependence of solventogenesis and solvents produced by Clostridium beijerinckii. Appl Microbiol Biotechnol. 1988;28:182–187. doi: 10.1007/BF00694309. [DOI] [Google Scholar]
- 26.Green EM, Boynton ZL, Harris LM, Rudolph FB, Papoutsakis ET, Bennett GN. Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiology. 1996;142:2079–2086. doi: 10.1099/13500872-142-8-2079. [DOI] [PubMed] [Google Scholar]
- 27.Alsaker KV, Paredes C, Papoutsakis ET. Metabolite stress and tolerance in the production of biofuels and chemicals: gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicum. Biotechnol Bioeng. 2010;105:1131–1147. doi: 10.1002/bit.22628. [DOI] [PubMed] [Google Scholar]
- 28.Hüsemann MH, Papoutsakis ET. Effects of propionate and acetate additions on solvent production in batch cultures of Clostridium acetobutylicum. Appl Environ Microbiol. 1990;56:1497–1500. doi: 10.1128/aem.56.5.1497-1500.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu Y, Hu S, Chen J, Shao L, He H, Yang Y, et al. Ammonium acetate enhances solvent production by Clostridium acetobutylicum EA 2018 using cassava as a fermentation medium. J Ind Microbiol Biotechnol. 2009;36:1225–1232. doi: 10.1007/s10295-009-0604-1. [DOI] [PubMed] [Google Scholar]
- 30.de Vrije T, Budde M, van der Wal H, Claassen PA, López-Contreras AM. “In situ” removal of isopropanol, butanol and ethanol from fermentation broth by gas stripping. Bioresour Technol. 2013;137:153–159. doi: 10.1016/j.biortech.2013.03.098. [DOI] [PubMed] [Google Scholar]
- 31.Ben-Dor A, Shamir R, Yakhini Z. Clustering gene expression patterns. J Comput Biol. 1999;6:281–297. doi: 10.1089/106652799318274. [DOI] [PubMed] [Google Scholar]
- 32.Daughdrill GW, Chadsey MS, Karlinsey JE, Hughes KT, Dahlquist FW. The C-terminal half of the anti-sigma factor, FlgM, becomes structured when bound to its target, σ28. Nat Struct Biol. 1997;4:285–291. doi: 10.1038/nsb0497-285. [DOI] [PubMed] [Google Scholar]
- 33.Gerischer U, Dürre P. Cloning, sequencing, and molecular analysis of the acetoacetate decarboxylase gene region from Clostridium acetobutylicum. J Bacteriol. 1990;172:6907–6918. doi: 10.1128/jb.172.12.6907-6918.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cornillot E, Nair RV, Papoutsakis ET, Soucaille P. The genes for butanol and acetone formation in Clostridium acetobutylicum ATCC 824 reside on a large plasmid whose loss leads to degeneration of the strain. J Bacteriol. 1997;179:5442–5447. doi: 10.1128/jb.179.17.5442-5447.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alsaker KV, Papoutsakis ET. Transcriptional program of early sporulation and stationary-phase events in Clostridium acetobutylicum. J Bacteriol. 2005;187:7103–7118. doi: 10.1128/JB.187.20.7103-7118.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dürre P, Hollergschwandner C. Initiation of endospore formation in Clostridium acetobutylicum. Molecular biology and pathogenesis of the clostridia. 2004. pp. 69–74. [DOI] [PubMed] [Google Scholar]
- 37.Alsaker KV, Spitzer TR, Papoutsakis ET. Transcriptional analysis of spo0A overexpression in Clostridium acetobutylicum and its effect on the cell’s response to butanol stress. J Bacteriol. 2004;186:1959–1971. doi: 10.1128/JB.186.7.1959-1971.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tracy BP, Jones SW, Papoutsakis ET. Inactivation of σE and σG in Clostridium acetobutylicum illuminates their roles in clostridial-cell-form biogenesis, granulose synthesis, solventogenesis, and spore morphogenesis. J Bacteriol. 2011;193:1414–1426. doi: 10.1128/JB.01380-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez M, Cutting S, Stragier P. Transcription of spoIVB is the only role of σ(G) that is essential for Pro-σ(K) processing during spore formation in Bacillus subtilis. J Bacteriol. 1995;177:4825–4827. doi: 10.1128/jb.177.16.4825-4827.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cutting S, Driks A, Schmidt R, Kunkel B, Losick R. Forespore-specific transcription of a gene in the signal transduction pathway that governs Pro-σK processing in Bacillus subtilis. Genes Dev. 1991;5:456–466. doi: 10.1101/gad.5.3.456. [DOI] [PubMed] [Google Scholar]
- 41.Kroos L, Zhang B, Ichikawa H, Yu Y-T. Control of σ factor activity during Bacillus subtilis sporulation. Mol Microbiol. 1999;31:1285–1294. doi: 10.1046/j.1365-2958.1999.01214.x. [DOI] [PubMed] [Google Scholar]
- 42.Grimmler C, Janssen H, Krauβe D, Fischer R-J, Bahl H, Dürre P, et al. Genome-wide gene expression analysis of the switch between acidogenesis and solventogenesis in continuous cultures of Clostridium acetobutylicum. J Mol Microbiol Biotechnol. 2011;20:1–15. doi: 10.1159/000320973. [DOI] [PubMed] [Google Scholar]
- 43.Paredes CJ, Alsaker KV, Papoutsakis ET. A comparative genomic view of clostridial sporulation and physiology. Nat Rev Microbiol. 2005;3:969 EP. doi: 10.1038/nrmicro1288. [DOI] [PubMed] [Google Scholar]
- 44.Pyrgakis KA, de Vrije T, Budde MA, Kyriakou K, López-Contreras AM, Kokossis AC. A process integration approach for the production of biological iso-propanol, butanol and ethanol using gas stripping and adsorption as recovery methods. Advances Biorefinery Engineering Food Supply Chain Waste Valorisation. 2016;116:176–194. [Google Scholar]
- 45.Ravagnani A, Jennert KCB, Steiner E, Grünberg R, Jefferies JR, Wilkinson SR, et al. Spo0A directly controls the switch from acid to solvent production in solvent-forming clostridia. Mol Microbiol. 2000;37:1172–1185. doi: 10.1046/j.1365-2958.2000.02071.x. [DOI] [PubMed] [Google Scholar]
- 46.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. The ClosTron: mutagenesis in Clostridium refined and streamlined. J Microbiol Methods. 2010;80:49–55. doi: 10.1016/j.mimet.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 47.Morimoto YV, Nakamura S, Hiraoka KD, Namba K, Minamino T. Distinct roles of highly conserved charged residues at the MotA-FliG interface in bacterial flagellar motor rotation. J Bacteriol. 2013;195:474–481. doi: 10.1128/JB.01971-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mann MS, Dragovic Z, Schirrmacher G, Lütke-Eversloh T. Over-expression of stress protein-encoding genes helps Clostridium acetobutylicum to rapidly adapt to butanol stress. Biotechnol Lett. 2012;34:1643–1649. doi: 10.1007/s10529-012-0951-2. [DOI] [PubMed] [Google Scholar]
- 49.Lee J, Jang Y-S, Choi SJ, Im JA, Song H, Cho JH, et al. Metabolic engineering of Clostridium acetobutylicum ATCC 824 for isopropanol-butanol-ethanol fermentation. Appl Environ Microbiol. 2012;78:1416–1423. doi: 10.1128/AEM.06382-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dusséaux S, Croux C, Soucaille P, Meynial-Salles I. Metabolic engineering of Clostridium acetobutylicum ATCC 824 for the high-yield production of a biofuel composed of an isopropanol/butanol/ethanol mixture. Metab Eng. 2013;18:1–8. doi: 10.1016/j.ymben.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 51.Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2005;21:537–539. doi: 10.1093/bioinformatics/bti054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Region of Genome Plasticity (RGP) analysis. (PDF 56 kb)
Microscopic observation of C. beijerinckii DSM6423 cells during glucose fermentation. (PDF 105 kb)
Details on the 6 biological replicates of C. beijerinckii DSM6423 glucose fermentation. (PDF 48 kb)
List of the most highly transcribed genes during C. beijerinckii DSM6423 glucose fermentation at 3 h. (PDF 79 kb)
Clusters of differential expressed genes in C. beijerinckii DSM6423 glucose fermentation. Nine hundred seventy-seven genes differentially expressed at T6 versus T3, or at T8 versus T6 or T11 versus T8 were clustered using CAST algorithm. This table shows the genes included in each cluster, with their ID and their functional annotation, and with their expression using expression at T3 as reference. (PDF 126 kb)
Data Availability Statement
The DSM6423 full genome sequence was deposited on the European Nucleotide Archive (ENA) under the accession number PRJEB11626 (https://www.ebi.ac.uk/ena/data/view/PRJEB11626).
The DSM6423 RNA seq data analyzed during this study were deposited on NCBI BioProject Database under the Accession Number GSE100024 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100024).