

Key Points
GTPase RAB1B is a direct transcriptional target of RUNX1 in MK/platelets and is downregulated in RUNX1 haplodeficiency.
RUNX1 downregulation is associated with defective RAB1B-related ER-to-Golgi transport and alterations in α-granule vWF.
Abstract
Patients with RUNX1 haplodeficiency have thrombocytopenia, platelet dysfunction, and deficiencies of α-granules and dense granules. Platelet expression profiling of a patient with a heterozygous RUNX1 mutation (c.969-323G>T) revealed decreased RAB1B, which encodes a small G protein. RAB GTPases regulate vesicle trafficking, and RAB1B is implicated in endoplasmic reticulum (ER)-to-Golgi transport in nonhematopoietic cells, but its role in megakaryocytes (MK) is unknown. We addressed the hypothesis that RAB1B is a transcriptional target of RUNX1 and that RAB1B regulates ER-to-Golgi transport in MK cells. Chromatin immunoprecipitation studies and electrophoretic mobility shift assay using phorbol 12-myristate 13-acetate (PMA)–treated human erythroleukemia cells revealed RUNX1 binding to RAB1B promoter region RUNX1 consensus sites, and their mutation reduced the promoter activity. RAB1B promoter activity and protein expression were inhibited by RUNX1 siRNA and enhanced by RUNX1 overexpression. These indicate that RAB1B is a direct RUNX1 target, providing a mechanism for decreased RAB1B in patient platelets. Vesicle trafficking from ER to Golgi in PMA-treated human erythroleukemia cells was impaired along with Golgi disruption on siRNA downregulation of RUNX1 or RAB1B. The effects of RUNX1 knockdown were reversed by RAB1B reconstitution. Trafficking of von Willebrand factor (vWF), an α-granule MK synthesized protein, was impaired with RUNX1 or RAB1B downregulation and reconstituted by ectopic RAB1B expression. Platelet vWF was decreased in patients with RUNX1 mutations. Thus, ER-to-Golgi transport, an early critical step in protein trafficking to granules, is impaired in megakaryocytic cells on RUNX1 downregulation, secondary to decreased RAB1B expression. Impaired RAB1B mediated ER-to-Golgi transport contributes to platelet α-granule defects in RUNX1 haplodeficiency.
Visual Abstract
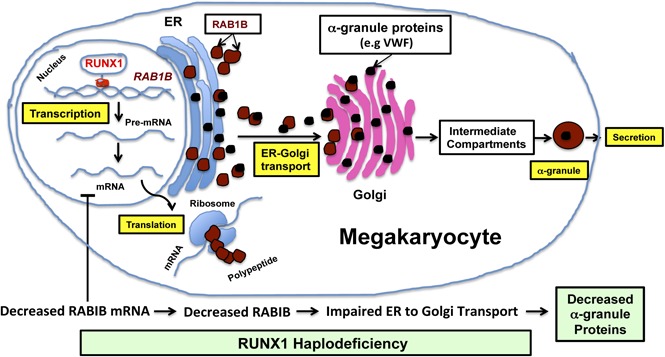
Introduction
RUNX1 (also called AML1 or CBFA2) is a hematopoietic transcription factor that plays a major role in definitive hematopoiesis, megakaryopoiesis, and platelet production.1,2 RUNX1 haplodeficiency is associated with familial thrombocytopenia, platelet dysfunction, α-granule and dense granule deficiencies, impaired secretion of granule contents, and predisposition to acute leukemia.1,3-5 Little is known regarding the mechanisms leading to deficiencies in the granules and their cargoes in patients with RUNX1 haplodeficiency. It is likely that multiple mechanisms contribute to the platelet functional defects.4 In previous studies in a patient with RUNX1 haplodeficiency, we reported the presence of thrombocytopenia, abnormalities in α and dense granules, and impaired aggregation, secretion, phosphorylation of pleckstrin and myosin light chain, and activation of GPIIb-IIIa on platelet receptor activation.6-9 Platelet expression profiling of this patient using Affymetrix microarrays showed that several genes were downregulated,7 and we have shown that some of the genes are direct transcriptional targets of RUNX1.8,10-13 Profiling studies further showed downregulation of RAB1B,7 which encodes the Ras-related small GTPase RAB1B, which is intimately involved in vesicle transport of proteins from endoplasmic reticulum (ER) to the Golgi compartment, a central organelle in the secretory pathways.14-17 This is a critical step in the trafficking of proteins synthesized in the ER to their final location in specific platelet granules. In eukaryotic cells, the major players in the secretory and endocytic pathways leading to formation of distinct vesicles/granules bearing different cargoes include the ER, the ER-Golgi intermediate compartment, the Golgi and its subcompartments, and the different classes of endosomes, lysosomes, and regulated secretory granules.18 Rab proteins regulate specific steps in membrane trafficking, including in vesicle formation and movement, and in membrane fusion.18 These GTPases shuttle between GTP-bound (active) and GDP-bound (inactive) forms to regulate their binding to downstream effectors and execute cellular functions.
Little is currently known regarding the role of RAB1B in platelets or megakaryocytes (MK). Previous studies implicating RAB1B in ER-to-Golgi trafficking have been performed in HeLa cells14,15,17 or CHO cells.16 Neither of these is of hematopoietic origin, and HeLa cells do not express RUNX1.19 On the basis of the findings in our patient with RUNX1 haplodeficiency, we pursued the hypothesis that RAB1B is a transcriptional target of RUNX1 and that downregulation of RAB1B or RUNX1 is associated with defective ER-to-Golgi transport in megakaryocytic cells. These studies provide evidence that RUNX1 regulates via RAB1B, which is essential for ER-to-Golgi transport, an early event in protein trafficking that governs α-granule formation and contents. Our studies show that RUNX1 haplodeficiency alters trafficking of vWF and platelet vWF levels.
Patients and methods
Patient information
The initial patient (P1) studied has thrombocytopenia and abnormal platelet function associated with a single point mutation (c.969-323G>T) in intron 3 at the splice acceptor site for exon 4, leading to a frame shift with premature termination in the conserved RUNT homology domain of RUNX1.9 Details of the platelet function abnormalities6,7,9 and expression profiling studies in this patient7 have been previously described.
Studies were performed in 2 patients from a second family (8-year-old male, P2, and his 3-year-old sister, P3) with thrombocytopenia and a RUNX1 mutation (c.508+1G>A). The maternal grandmother and great uncle had history of acute myeloid leukemia. This patient had abnormal agonist-induced aggregation and secretion on laboratory testing. The studies on the patients and control subjects were approved by institutional review boards of the Lewis Katz School of Medicine at Temple University and the Children's Hospital of Philadelphia, and were performed after obtaining the informed consent. The studies were conducted following the guidelines of the Helsinki Declaration.
Immunofluorescence studies
Platelets from the patients and healthy controls or human erythroleukemia (HEL) cells were treated as described for each study and then seeded on coverslips precoated with human plasma fibronectin and fixed and imaged by epifluorescence and confocal microscopy as described.8 Images were acquired on a Nikon E1000 microscope or Leica TCS SP5 confocal microscope, using a 63×/1.40 n.a. oil immersion objective at room temperature and Q Capture or Leica imaging software, respectively. Postacquisition processing and analysis was performed with Adobe Photoshop and ImageJ, and was limited to image cropping and brightness/contrast adjustments applied to all pixels per image simultaneously. Fluorophores used were fluorescein isothiocyanate or Cy3.
Cell culture
HEL cells from American Type Cell Culture (Rockville, MD) were grown and induced in RPMI-1640 medium as described.20
Real-time PCR
Total RNAs from platelets isolated from whole blood of healthy donors and the patient8 were subjected to first-strand cDNA synthesis using Superscript III (Applied Biosystems) and amplified by real-time polymerase chain reaction (PCR) by SYBR Green PCR mix, using primers (supplemental Table 1) for RAB1B, RAB1A, and GAPDH (0.1 μM each). The parameters used for real-time PCR were as follows: 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds, 55°C for 20 seconds, and 72°C for 20 seconds, using a Master Cycler Real-Time PCR system (Eppendorf, Hauppauge, NY), and relative abundances were calculated by the ΔΔCT method, using GAPDH as the reference gene.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed on HEL cells (1 × 108) treated with phorbol 12-myristate 13-acetate (PMA; 10 nM) for 24 hours, using the ChIP-It kit (Active Motif, Carlsbad, CA) and antibodies as described.10 Chromatin samples were immunoprecipitated with anti-RUNX1 antibody (sc-8563x) or with normal immunoglobulin G (IgG; sc-2028; Santa Cruz, Dallas, TX). Immunoprecipitated samples were analyzed by PCR, using the primers shown in supplemental Table 2. Amplification was performed using Go Taq Green Master Mix with 1 cycle at 95°C for 2 minutes, followed by 32 cycles of 95°C for 30 seconds, 62°C for 45 seconds, and 72°C for 60 seconds.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSA) were performed using HEL cell nuclear extracts as described.20 RAB1B promoter region −785/−1 (from ATG) has 4 RUNX1 consensus sites. Oligonucleotides encompassing these sites (RAB1B site 1 probe −445/−426, site 2 probe −630/−611, site 3 probe −659/−640, and site 4 probe −778/−759) were labeled with infrared dye 700 (LI-COR Biosciences, Lincoln, NE; supplemental Table 3). Supershift assays were performed with anti-RUNX1 antibody (sc-8563x) or normal IgG (sc-2028; Santa Cruz). Gels were visualized on Odyssey Infrared Imaging System (LI-COR Biosciences).
Promoter-reporter assays
The RAB1B wild-type (WT) promoter (−785/+15) and constructs with specific mutations in RUNX1 consensus sites were generated by directional PCR method, using gene-specific primers incorporated with restriction sites (Xho I at 5′ side and Hind III at 3′ side). The PCR products were cloned into the TOPO TA cloning vector. The recombinant TOPO TA plasmid was digested with appropriate restriction enzymes, and the promoter region was subcloned between the same restriction sites of the pGL3-Basic promoter. The sequences were confirmed by DNA sequencing on the ABI Prism 377 (Applied Biosystems).
Promoter-reporter constructs were transfected individually along with a standard Renilla luciferase (in 50:1 ratio) into HEL cells (2 × 106), using Lipofectamine 2000 (Thermo Fisher Scientific, Philadelphia, PA). The primer sequences of WT construct and its mutants are shown in supplemental Table 4. Dual-luciferase assays were performed as described.20 Promoter activity was expressed as the ratio of firefly luciferase activity to Renilla luciferase activity, relative to that of the promoter-less vector. All experiments were performed 3 times in triplicate.
RUNX1 downregulation and overexpression
HEL cells (2 × 106) were cotransfected with WT RAB1B luciferase construct, along with control siRNA or RUNX1 siRNA (100 nM each; Santa Cruz) to study the effect of downregulation of these genes. To study the effect of RUNX1 overexpression, cells (2 × 106) were cotransfected with RAB1B WT luciferase construct or its mutants, along with RUNX1 expression vector, RUNX1-pCMV6-XL4, or its empty expression vector, pCMV6-XL4 (1 μg each; Origene Technologies, Rockville, MD). pRL-TK (Renilla luciferase) plasmid was also used in each transfection as an internal standard, and dual-luciferase assays were performed as described here. HEL cell lysates were prepared and immunoblotted using antibodies against RUNX1, RAB1B, and actin or GAPDH.10
Immunoblotting
Human platelets and HEL were lysed in a M-Per protein extraction reagent (Pierce Biotechnology) supplemented with a protease inhibitor cocktail (Active Motif, Carlsbad, CA). Cell lysates (20 μg) were subjected to 4% to 20% Mini-PROTEAN TGX gels (Bio-Rad, Hercules, CA), and the protein was transferred to PVDF membrane (Millipore, Billerica, MA) and was probed with antibodies against RAB1B (sc-599), RUNX1 (sc-8563 or sc- 365644), actin (sc-1616R; Santa Cruz, CA), or von Willebrand Factor (vWF; ab6994; Abcam, Cambridge, MA). The blots were probed with IR Dye-labeled secondary antibodies (LI-COR Biosciences), using the Odyssey Infrared Imaging System (LI-COR Biosciences).
ER-to-Golgi transport studies
ER-to-Golgi transport was studied using vesicular stomatitis virus G (VSVG) envelope glycoprotein tagged with enhanced GFP, as described by Presley et al21 with modifications. PMA-treated (10 nM, 24 hours) HEL cells (1 × 106) were nucleofected with VSVG-GFP and E2-Crimson-GALT-CFP plasmids (1 μg each), along with control siRNA or RAB1B siRNA or RUNX1 siRNA (100 nM each), or RUNX1 siRNA + 2 μg of RAB1B expression plasmid (Origene Technologies, Rockville, MD), using Amaxa cell line Nucleofector kit V in a nucleofector 2b device (Lonza Company). These cells were resuspended in the medium supplemented with 10 nM PMA and incubated at 37°C with 5% CO2. After 24 hours, cells were resuspended by centrifugation in fresh medium with 10 nM PMA, transferred onto fibronectin-coated plates, and incubated at 40°C for 12 to 15 hours, followed by incubation at 32°C for 30 minutes. The cells were then fixed and imaged by confocal microscopy, followed by calculation of the Pearson’s correlation coefficient from at least 100 cells per condition, as described.8,22 VSVG cDNA sequence was isolated by restriction digestion from pEGFP-VSVG vector (Addgene 11912, deposited by Jennifer Lippincott-Schwartz) purchased from Addgene (a plasmid repository; Cambridge, MA) and cloned into pEGFP-C3 vector (Takara Bio, Mountain View, CA). GALT-CFP plasmid was prepared as follows: E2-Crimson-tagged β1, 4 galactosyltransferase 1 (GalT) with KDEL ER targeting sequence deleted was created in the pECFP vector by subcloning the E2-Crimson cassette in place of enhanced cyan fluorescent protein cassette in the GALT-CFP plasmid (Addgene 11937, a gift from Jennifer Lippincott-Schwartz)
Bioinformatics
RAB1B sequence (accession no. NM_030981.2) was obtained from the National Center for Biotechnology Information gene database. Potential binding sites for transcription factors were analyzed using the computer program TFBIND (http://tfbind.hgc.jp).
Statistical analysis for in vitro studies
Results of the in vitro studies are expressed as mean ± standard error of the mean (SEM). Differences were compared using the Student t test.
Results
Decreased platelet RAB1B expression in RUNX1-haplodeficiency
Platelet expression profiling revealed that RAB1B is decreased in the patient with RUNX1 haplodeficiency (fold change, 0.318; P = .025) compared with platelets from 6 healthy control participants, using Affymetrix U133 Gene Chips.7 This was validated by quantitative PCR (Figure 1A). Immunofluorescence studies using anti-RAB1B antibodies showed that RAB1B protein expression was decreased in platelets from our first patient, P1, compared with healthy participants (Figure 1B). Studies in 2 siblings, patients P2 and P3, from an unrelated family with a RUNX1 mutation also showed platelet RAB1B mRNA was decreased (Figure 1C), with decreased platelet RAB1B protein on immunofluorescence studies (Figure 1D). On immunoblotting, platelet RAB1B was lower in the patients compared with control subjects (Figure 1E).
Figure 1.

RAB1B expression in platelets from the patients with RUNX1 haplodeficiency and healthy control subjects. (A) Platelets RAB1B mRNA levels in the index (P1) patient and 5 normal subjects (N1-N5) by quantitative PCR. Shown are mRNA levels normalized to GAPDH. (B) Immunofluorescence studies of platelet RAB1B expression in the index patient P1 and a normal subject. Platelets were labeled with RAB1B polyclonal antibody, detected with fluorescein isothiocyanate (pseudocolored red)–conjugated secondary antibody, and imaged on a Nikon E1000 microscope. Corrected total cell immunofluorescence is shown ± SEM. (C) Platelet RAB1B mRNA in 2 siblings (P2 and P3) from a second family with RUNX1 mutation and 5 normal subjects (N1-N5). (D) Platelet RAB1B immunofluorescence in patient P2 and 2 normal subjects. Bar, 10 μm (scale also applies to panel B). Corrected total cellular fluorescence is shown ± SEM. The P values represent comparisons of the patient with each of the control subjects. (E) Platelet RAB1B by immunoblotting in 3 patients (P1-P3) and 5 healthy subjects (N1-N5). Also shown is the ratio of RAB1B to GAPDH.
RUNX1 regulates RAB1B
RAB1B promoter sequence revealed 4 RUNX1 consensus binding sites within 800 bp from the ATG codon: at −440/−435 (site 1), −622/−617 (site 2), −660/−655 (site 3), and −770/−765 (site 4; Figure 2A). The ChIP assays were performed on HEL cell chromatin, using an anti-RUNX1 antibody. PCR amplification of the immunoprecipitated chromatin showed enrichment of promoter regions with site 1 and site 4 by anti-RUNX1 antibody, and no enrichment was noted with normal IgG. The promoter fragments containing site 2 and site 3 were not enriched (Figure 2A). These results indicate that RUNX1 binds in vivo to RAB1B promoter.
Figure 2.

Characterization of RUNX1 sites in the RAB1B upstream region. (A) RAB1B upstream region showing 4 consensus sites for RUNX1. Shown below is PCR amplification of the immunoprecipitates of HEL cells with control IgG (lane 1) and RUNX1 antibodies (lane 2). Amplification of the input or total DNA (lane 3) and genomic DNA (gDNA, lane 4) served as positive controls. Samples were analyzed by PCR using primers for the RAB1B region and GAPDH. Shown are representative of 3 experiments. (B) EMSA using WT nucleotide probes encompassing RUNX1 consensus site 1 (−426/−445; left) and site 4 (−759/−778; right) in RAB1B promoter and nuclear extracts from PMA-treated HEL cells. (a, left) EMSA using site 1 probe (lanes 1-5): lane 1, no extract; lane 2, protein binding to the probe; lane 3, loss of binding by competition with unlabeled probe; lane 4, no loss of binding by competition with normal IgG; and lane 5 competition with RUNX1 antibody and inhibition of binding. (b, right) EMSA using probe with site 4 (lanes 1-5); similar results were obtained as with the probe with site 1. Shown are representative of 3 experiments. (C) Luciferase reporter studies on RAB1B promoter in PMA-treated HEL cells. Luciferase activity with WT construct with RUNX1 sites 1-4 (solid triangles) and constructs with RUNX1 binding sites mutated (open triangles). Mutations in the sites 1 and 4 decreased promoter activity, but not the mutations in the sites 3 and 4, suggesting sites 1 and 4 are functional. The mean ± SEM is shown for 3 independent experiments in triplicates. P values are for comparisons against WT promoter.
EMSA were performed using HEL nuclear extracts and labeled RAB1B oligos containing each of the RUNX1 consensus sites (supplemental Table 2). Because of the proximity of sites 2 and 3 (Figure 2A), a single probe encompassing both sites was used in these studies. These studies revealed that RUNX1 was present in complex with consensus sites 1 and 4 (Figure 2B), but not with sites 2 and 3 (data not shown), a result similar to that with ChIP analysis. In studies with the oligo containing site 1 (Figure 2B), there was protein binding (Figure 2B, lane 2), which was competed with excess unlabeled oligo (lane 3). There was no alteration in binding by normal IgG (lane 4), but it was inhibited by RUNX1 antibodies (lane 5). Similar results were observed in studies of oligo with site 4. Together, the ChIP and EMSA studies revealed RUNX1 binding to sites 1 and 4. In promoter-reporter studies using constructs with RAB1B promoter region (Figure 2C), mutations in site 1 or site 4 resulted in marked reduction in promoter activity, but no change was noted in reporter activities with specific mutations in sites 2 or 3. Together, these findings indicate that RUNX1 binds to sites 1 and 4 in the RAB1B promoter region, and that these sites are functional.
RUNX1 overexpression in HEL cells increased RAB1B protein expression (Figure 3A) and RAB1B promoter activity of the WT construct (Figure 3B). The increase in promoter activity was abolished when RUNX1 binding sites 1 and 4 (but not sites 2 and 3) were mutated individually (Figure 3B). No increase in promoter activity was noted on RUNX1 overexpression when sites 1 and 4 were mutated. These findings are consistent with the conclusion that sites 1 and 4, but not sites 2 and 3, are functional. RUNX1 siRNA inhibited RAB1B protein expression (Figure 3C) and promoter activity (Figure 3D). Overall, these findings indicate that RAB1B is regulated by RUNX1.
Figure 3.

Effect of RUNX1 overexpression and RUNX1 siRNA downregulation on RAB1B protein expression and promoter activity. (A) Immunoblot analysis of RUNX1, RAB1B, and actin on overexpression of RUNX1 in HEL cells. Bars shown are an average of 3 independent experiments. P values shown are for comparison with control (pCMV6 vector alone). (B) Effect of RUNX1 overexpression on RAB1B promoter activity. WT construct and mutant constructs with sites 1-4 individually mutated were cotransfected with RUNX1-pCMV6 expression vector (green bars), empty vector pCMV6 (blue bars), or neither (red bars) in HEL cells. Reporter activity was measured at 48 hours. Bar graphs show activity as mean ± SE of 3 independent experiments in triplicate. (C) Immunoblot analysis of lysates from HEL cells transfected with RUNX1 siRNA showing inhibition of RAB1B protein. Bars shown are average of 3 independent experiments. P values are for comparison with control vector. (D) Effect of RUNX1 siRNA on RAB1B promoter activity. RAB1B WT promoter construct was cotransfected with control siRNA or RUNX1 siRNA in HEL cells. Reporter activity was measured at 48 hours. Bar graphs show activity as mean ± SEM of 3 independent experiments in triplicate. RUNX1 siRNA reduced RAB1B protein and RAB1B promoter activity.
RUNX1 downregulation impairs RAB1B-related ER-to-Golgi trafficking
We investigated effects of RUNX1 downregulation on ER-to-Golgi transport in megakaryocytic HEL cells. RUNX1 downregulation revealed impaired ER-to-Golgi trafficking and dissolution of Golgi (Figure 4). HEL cells were cotransfected with vesicle protein VSVG-GFP and Golgi marker E2-Crimson GalT, along with scrambled control or RUNX1 siRNA, followed by temperature shift assay to drive synchronous ER-to-Golgi transport of VSVG-GFP.21 In control siRNA-transfected cells, most of the VSVG-GFP colocalized with GalT (Pearson's correlation coefficient [r] = 0.609 ± 0.035), indicating accumulation of VSVG in Golgi structures. On RUNX1 knockdown, VSVG colocalization with GalT was lost (r = 0.378 ± 0.033; P < .05), and Golgi structures were disrupted (Figure 4A-B). Identical results were obtained on downregulation of RAB1B (Figure 4A-B), with loss of colocalization with GalT (r = 0.374 ± 0.026; P < .05). Reconstitution of RAB1B in RUNX1-depleted cells restored the Golgi apparatus and the colocalization of VSVG (r = 0.711 ± 0.046; no significant difference compared with control; Figure 4B). In Figure 4C, the immunoblots show the corresponding protein levels of the RUNX1, RAB1B, and actin in control cells, with siRNA knockdown of RUNX1 or RAB1B, and after ectopic expression of RAB1B in RUNX1-depleted cells. In control experiments, RAB1B knockdown decreased transcript levels of RAB1B, but not RAB1A (supplemental Figure 1). Together, these studies indicate that in megakaryocytic cells, loss of RAB1B impairs ER-to-Golgi vesicular transport and Golgi stability, which was also noted with RUNX1 downregulation (Figure 4). The similarity in the effects with RAB1B and RUNX1 knockdown on vesicle transport and Golgi stability, and their reversal with RAB1B reconstitution, suggests that RAB1B downregulation is the cause of the impaired ER-to-Golgi transport noted on RUNX1 downregulation.
Figure 4.

Regulation of ER-to-Golgi transport by RUNX1 and RAB1B in HEL cells. (A) PMA-treated HEL cells cotransfected with VSVG-GFP and E2 Crimson-GalT, along with siRNAs and plasmid constructs as indicated, were seeded on coverslips, kept at 40°C for 16 hours, and then transferred to 32°C for 30 minutes and fixed. VSVG, green; GalT, red; DAPI, blue. Yellow arrowheads indicate areas of VSVG in Golgi structures, indicated by colocalization with GalT, which appears as yellow. Bar, 10 μm. VSVG and GalT were colocalized intact in the cells transfected with control siRNA (Pearson’s correlation coefficient r = 0.609 ± 0.035; mean ± SEM). In cells transfected with RUNX1 siRNA (r = 0.378 ± 0.033; P < .05) or RAB1B siRNA (r = 0.374 ± 0.026; P < .05), Golgi was disrupted and there was no accumulation of VSVG-GFP, as RAB1B facilitates VSVG transport. (B) Resumption of VSVG transport by reconstituted RAB1B in HEL cells after RUNX1 siRNA downregulation (r = 0.711 ± 0.046; P = NS compared with control). Bar, 10 μm. (C) Immunoblot analysis showing reconstitution of RAB1B protein by ectopic RAB1B expression in RUNX1-depleted HEL cells.
RUNX1 downregulation impairs RAB1B-related VWF trafficking
MK synthesize vWF, a protein present in platelet α-granules, and several α-granule proteins are decreased in patients with RUNX1 haplodeficiency.4 vWF is synthesized and dimerized in the ER with multimer formation occurring in the trans- and post-Golgi compartments23,24 before being localized to the α-granules. We postulated that vWF trafficking would be impaired because of Golgi disruption by RUNX1 or RAB1B depletion, leading to defective vWF protein maturation and decreased vWF in granules. In studies performed with temperature shift as described in “Patients and methods.” vWF localized to compact Golgi structures, as indicated by colocalization with GalT in control siRNA-transfected cells (Pearson correlation coefficient r = 0.793 ± 0.018; Figure 5A). On RUNX1 knockdown, the vWF concentration in the Golgi was lost (r = 0.478 ± 0.072; P < .05), associated with disrupted Golgi structures (Figure 5A). Identical results were obtained on downregulation of RAB1B (r = 0.429 ± 0.082; P < .05). With ectopic RAB1B expression in RUNX1-depleted cells, Golgi structures were restored associated with enrichment of vWF in the Golgi (r = 0.665 ± 0.048; no significant difference compared with control). On immunoblot analysis, bands were strikingly decreased on downregulation of RAB1B (lane 2) or RUNX1 (lane 3). Ectopic RAB1B expression in RUNX1-depleted cells partially reconstituted the vWF bands (lane 4). These findings suggest that vWF trafficking in HEL cells is regulated by RAB1B, and that it is impaired with RUNX1 downregulation. In line with these findings, immunoblotting of platelet lysates from patients with RUNX1 haplodeficiency showed that VWF was decreased relative to platelets from normal donors (Figure 5C).
Figure 5.

Effect of siRNA depletion of RAB1B and RUNX1 on vWF trafficking in HEL cells. (A) PMA-treated HEL cells were transfected with siRNAs and expression plasmid, as indicated and as described in the legend for Figure 4. In control siRNA-transfected cells, vWF (green) colocalized with the Golgi (E2-Crimson-GalT, red; Pearson correlation coefficient r = 0.793 ± 0.018). On treatment with RUNX1 siRNA (r = 0.478 ± 0.072; P < .05; compared with control) or RAB1B siRNA (r = 0.429 ± 0.082; P < .05), Golgi structures were disrupted and the colocalization with vWF was lost. RAB1B expression in RUNX1 siRNA-depleted cells reconstituted the Golgi structures along with colocalization of vWF (r = 0.665 ± 0.082; P = NS), similar to that seen in control cells. Bar, 10 μm. (B) Immunoblot analysis of vWF, RUNX1, RAB1B, and actin in HEL cells treated as described earlier. Downregulation of RAB1B or RUNX1 decreased vWF; reconstitution of RAB1B in RUNX1-depleted cells restored the vWF. (C) Platelet vWF by immunoblotting in 3 patients with RUNX1 mutations and 5 normal subjects. Also shown is the ratio of vWF to GAPDH as loading control.
Discussion
These studies provide the first evidence that platelet RAB1B expression is downregulated in patients with RUNX1 mutation (Figure 1), and that RAB1B is regulated in megakaryocytic HEL cells by RUNX1 (Figures 2 and 3). The latter provides a mechanism for the decreased platelet RAB1B in patients with RUNX1 haplodeficiency. Rab proteins regulate specific steps in vesicle transport in the secretory and endocytic pathways, and this is driven by their cellular locations.18 RAB1B has been shown in HeLa cells14,15,17 and CHO cells16 to regulate ER-to-Golgi vesicle transport,17,21,25 and loss of RAB1B expression resulted in decreased ER-to-Golgi vesicle trafficking with disruption of Golgi. However, cell biological roles of RAB1B in MK cells have not been explored. Moreover, HeLa cells do not express RUNX1.19 Our studies provide the first evidence that RAB1B regulates ER-to-Golgi vesicle transport in megakaryocytic cells. RAB1B downregulation impaired ER-to-Golgi vesicle trafficking and induced Golgi dissolution (Figure 4). Identical findings were observed on RUNX1 downregulation with reversal of the findings on ectopic expression of RAB1B in RUNX1-depleted cells (Figures 4B and 5), indicating that the defective ER-to-Golgi transport with RUNX1 downregulation was secondary to decreased RAB1B expression. This does not exclude a contributory effect of other genes also downregulated in RUNX1 haplodeficiency.7
ER-to-Golgi trafficking is a critical and early step in the transport of proteins synthesized in the ER to the specific granules.26-28 Defect in granule number or content (of both α and dense granules) is a hallmark of RUNX1 haplodeficiency.4 Platelet α-granules are endowed with numerous proteins, many, but not all, of which are synthesized in the MK. Deficiencies of α-granule proteins such as platelet factor-4 and β-thromboglobulin have been documented in patients with RUNX1 mutations.4,11 We show that vWF is also decreased. In MK, small vesicles budding from the trans-Golgi network are transported from MK to pro-platelets via multivesicular bodies.26-28 Multiple mechanisms contribute to the handling and trafficking of various proteins to specific platelet granules. We postulate that impaired ER-to-Golgi transport resulting from decreased RAB1B expression (arising from RUNX1 mutations) is an important mechanism causing defective protein cargo trafficking early in the process that leads to granule biogenesis in RUNX1 haplodeficiency, particularly for α-granule proteins that are synthesized in the MK.
Our studies show that RUNX1 or RAB1B downregulation impairs handling of vWF (Figure 5), a protein synthesized by MK and present in α-granules. Several RAB proteins, but not RAB1B, to our knowledge, have been implicated in the handling of vWF in endothelial cells.29 Our studies implicate RAB1B in vWF trafficking in megakaryocytic HEL cells (Figure 5A). The lack of full reconstitution of vWF on RAB1B re-expression in RUNX1-depleted cells is consistent with the conclusion that RAB1B is necessary, but not sufficient, to normalize vWF trafficking. This may be related to other genes also being downregulated with RUNX1 downregulation.7 In line with these findings, platelet VWF was decreased in patients with RUNX1 mutations (Figure 5C). Of note, in our patient, the platelet vWF transcript level was not decreased on expression profiling,7 the latter being the reason for selecting vWF for the studies on. RAB1B-regulated protein trafficking. In addition, plasma vWF was normal, suggesting a differential regulation of vWF between MK and endothelial cells. These findings are in line with the conclusion that the abnormal platelet/MKα-granule vWF in RUNX1 haplodeficiency results from downregulation of genes involved in protein trafficking in MK, rather than RUNX1 regulation of the vWF gene.
Impaired roles of RAB proteins, specifically RAB1B, have been recognized in some human neurodegenerative diseases: Alzheimer’s disease30,31 and Parkinson’s disease.32,33 RAB1B regulates transport of β-amyloid precursor protein from ER-to-Golgi.30,31 In Parkinson’s disease models, α-synuclein blocks ER-to-Golgi trafficking, and RAB1 rescues the neuron loss.32,33 Interestingly, Golgi fragmentation, noted in our studies (Figure 4), is also reported in neurodegenerative diseases.33
In summary, our studies show that RAB1B is regulated by transcription factor RUNX1 in megakaryocytic cells/platelets and is downregulated in platelets from RUNX1 haplodeficiency patients. RUNX1 downregulation is associated with defective ER-to-Golgi transport that is reversed, at least in part, by re-expression of RAB1B. ER-to-Golgi transport is an important step in the trafficking of proteins synthesized in the MK to specific granules, and a defect in this process likely contributes to the defects in protein-bearing platelet α-granules. These findings provide new insights into the mechanisms leading to granule deficiencies in RUNX1 haplodeficiency.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This study was supported by research funding from National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL109568 and R01HL13736 (A.K.R.), and R01HL137207 (L.E.G.).
Authorship
Contribution: G.J. performed the research, analyzed and interpreted data, and wrote the paper; L.E.G. designed and performed the research, analyzed and interpreted the data with respect to the immunofluorescence studies, and contributed to the writing of the manuscript; G.M. performed some of the measurements; M.P.L. performed studies in establishing the RUNX1 mutation in 2 of the patients and revised the manuscript; A.K.R. conceived, designed, and performed the research, interpreted data, and wrote the paper; and all authors have read and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: A. Koneti Rao, Sol Sherry Thrombosis Research Center, Section of Hematology, Lewis Katz School of Medicine at Temple University, 3420 N Broad St, 204 MRB, Philadelphia, PA 19140; e-mail: koneti@temple.edu.
References
- 1.Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017;129(15):2070-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonifer C, Levantini E, Kouskoff V, Lacaud G. Runx1 structure and function in blood cell development. Adv Exp Med Biol. 2017;962:65-81. [DOI] [PubMed] [Google Scholar]
- 3.Song WJ, Sullivan MG, Legare RD, et al. . Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166-175. [DOI] [PubMed] [Google Scholar]
- 4.Songdej N, Rao AK. Hematopoietic transcription factor mutations: important players in inherited platelet defects. Blood. 2017;129(21):2873-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Bruijn MF, Speck NA. Core-binding factors in hematopoiesis and immune function. Oncogene. 2004;23(24):4238-4248. [DOI] [PubMed] [Google Scholar]
- 6.Gabbeta J, Yang X, Sun L, McLane MA, Niewiarowski S, Rao AK. Abnormal inside-out signal transduction-dependent activation of glycoprotein IIb-IIIa in a patient with impaired pleckstrin phosphorylation. Blood. 1996;87(4):1368-1376. [PubMed] [Google Scholar]
- 7.Sun L, Gorospe JR, Hoffman EP, Rao AK. Decreased platelet expression of myosin regulatory light chain polypeptide (MYL9) and other genes with platelet dysfunction and CBFA2/RUNX1 mutation: insights from platelet expression profiling. J Thromb Haemost. 2007;5(1):146-154. [DOI] [PubMed] [Google Scholar]
- 8.Mao GF, Goldfinger LE, Fan DC, et al. . Dysregulation of PLDN (pallidin) is a mechanism for platelet dense granule deficiency in RUNX1 haplodeficiency. J Thromb Haemost. 2017;15(4):792-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun L, Mao G, Rao AK. Association of CBFA2 mutation with decreased platelet PKC-θ and impaired receptor-mediated activation of GPIIb-IIIa and pleckstrin phosphorylation: proteins regulated by CBFA2 play a role in GPIIb-IIIa activation. Blood. 2004;103(3):948-954. [DOI] [PubMed] [Google Scholar]
- 10.Jalagadugula G, Mao G, Kaur G, Goldfinger LE, Dhanasekaran DN, Rao AK. Regulation of platelet myosin light chain (MYL9) by RUNX1: implications for thrombocytopenia and platelet dysfunction in RUNX1 haplodeficiency. Blood. 2010;116(26):6037-6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aneja K, Jalagadugula G, Mao G, Singh A, Rao AK. Mechanism of platelet factor 4 (PF4) deficiency with RUNX1 haplodeficiency: RUNX1 is a transcriptional regulator of PF4. J Thromb Haemost. 2011;9(2):383-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jalagadugula G, Mao G, Kaur G, Dhanasekaran DN, Rao AK. Platelet protein kinase C-θ deficiency with human RUNX1 mutation: PRKCQ is a transcriptional target of RUNX1. Arterioscler Thromb Vasc Biol. 2011;31(4):921-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaur G, Jalagadugula G, Mao G, Rao AK. RUNX1/core binding factor A2 regulates platelet 12-lipoxygenase gene (ALOX12): studies in human RUNX1 haplodeficiency. Blood. 2010;115(15):3128-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez H, García IA, Sampieri L, Alvarez C. Spatial-temporal study of Rab1b dynamics and function at the ER-Golgi interface. PLoS One. 2016;11(8):e0160838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE. GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J Cell Biol. 1992;119(4):749-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plutner H, Cox AD, Pind S, et al. . Rab1b regulates vesicular transport between the endoplasmic reticulum and successive Golgi compartments. J Cell Biol. 1991;115(1):31-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvarez C, Garcia-Mata R, Brandon E, Sztul E. COPI recruitment is modulated by a Rab1b-dependent mechanism. Mol Biol Cell. 2003;14(5):2116-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513-525. [DOI] [PubMed] [Google Scholar]
- 19.Martinez M, Hinojosa M, Trombly D, et al. . Transcriptional auto-regulation of RUNX1 P1 promoter. PLoS One. 2016;11(2):e0149119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jalagadugula G, Dhanasekaran DN, Kim S, Kunapuli SP, Rao AK. Early growth response transcription factor EGR-1 regulates Galphaq gene in megakaryocytic cells. J Thromb Haemost. 2006;4(12):2678-2686. [DOI] [PubMed] [Google Scholar]
- 21.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389(6646):81-85. [DOI] [PubMed] [Google Scholar]
- 22.Wurtzel JG, Kumar P, Goldfinger LE. Palmitoylation regulates vesicular trafficking of R-Ras to membrane ruffles and effects on ruffling and cell spreading. Small GTPases. 2012;3(3):139-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6(1):217-246. [DOI] [PubMed] [Google Scholar]
- 24.de Wit TR, van Mourik JA. Biosynthesis, processing and secretion of von Willebrand factor: biological implications. Best Pract Res Clin Haematol. 2001;14(2):241-255. [DOI] [PubMed] [Google Scholar]
- 25.García IA, Martinez HE, Alvarez C. Rab1b regulates COPI and COPII dynamics in mammalian cells. Cell Logist. 2011;1(4):159-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heijnen HF, Debili N, Vainchencker W, Breton-Gorius J, Geuze HJ, Sixma JJ. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood. 1998;91(7):2313-2325. [PubMed] [Google Scholar]
- 27.Youssefian T, Cramer EM. Megakaryocyte dense granule components are sorted in multivesicular bodies. Blood. 2000;95(12):4004-4007. [PubMed] [Google Scholar]
- 28.Richardson JL, Shivdasani RA, Boers C, Hartwig JH, Italiano JE Jr. Mechanisms of organelle transport and capture along proplatelets during platelet production. Blood. 2005;106(13):4066-4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nightingale T, Cutler D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J Thromb Haemost. 2013;11(Suppl 1):192-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang S, Li Y, Zhang X, Bu G, Xu H, Zhang YW. Trafficking regulation of proteins in Alzheimer’s disease. Mol Neurodegener. 2014;9(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dugan JM, deWit C, McConlogue L, Maltese WA. The Ras-related GTP-binding protein, Rab1B, regulates early steps in exocytic transport and processing of beta-amyloid precursor protein. J Biol Chem. 1995;270(18):10982-10989. [DOI] [PubMed] [Google Scholar]
- 32.Cooper AA, Gitler AD, Cashikar A, et al. . Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313(5785):324-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rendón WO, Martínez-Alonso E, Tomás M, Martínez-Martínez N, Martínez-Menárguez JA. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson’s disease. Histochem Cell Biol. 2013;139(5):671-684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.