

Key Points
Cell-free DNA and circulating tumor cells are related to TMTV in follicular lymphoma at diagnosis.
Cell-free DNA levels, in addition to TMTV, define high-risk follicular lymphoma patients.
Abstract
Outcomes for follicular lymphoma (FL) have greatly improved, but most patients will ultimately relapse. High total metabolic tumor volume (TMTV), computed from baseline 18F-fluorodeoxyglucose–positron emission tomography (PET), is associated with shorter progression-free survival (PFS), but circulating tumor cells (CTCs) and cell-free DNA (cfDNA) may also reflect tumor burden and be of prognostic value. The aim of our study was to correlate CTCs and cfDNA with TMTV in FL at diagnosis and to determine their prognostic values. We retrospectively analyzed 133 patients (with previously untreated FL and a baseline PET) from 2 cohorts with either a baseline plasma sample (n = 61) or a bcl2-JH-informative peripheral blood (PB) sample (n = 68). Quantification of circulating bcl2-JH+ cells and cfDNA was performed by droplet digital polymerase chain reaction. A significant correlation was found between TMTV and both CTCs (P < .0001) and cfDNA (P < .0001). With a median 48-month follow-up, 4-year PFS was lower in patients with TMTV > 510 cm3 (P = .0004), CTCs >0.0018 PB cells (P = .03), or cfDNA >2550 equivalent-genome/mL (P = .04). In comparison with TMTV alone, no additional prognostic information was obtained by measuring CTCs. In contrast, Cox multivariate analysis, including cfDNA and TMTV, showed that both cfDNA and TMTV remained predictive of outcome. In conclusion, CTCs and cfDNA correlate with TMTV in FL, and all 3 influence patient outcome. PFS was shorter for patients with high cfDNA and TMTV, suggesting that these parameters provide relevant information for tumor-tailored therapy.
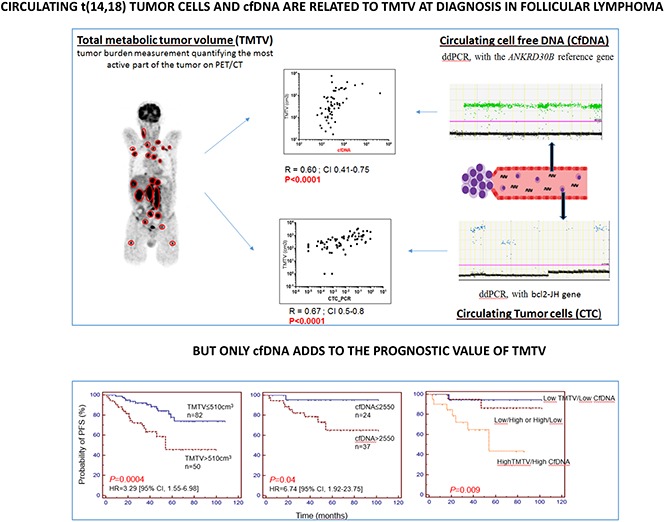
Visual Abstract
Introduction
Follicular lymphoma (FL) is the most common indolent lymphoma subtype in Western countries.1 It is characterized by prolonged survival, with median survival times exceeding 10 years,2 but its prognosis varies widely among individuals. Currently, depending on prognostic factors, treatment decisions range from a watch-and-wait approach to anti-CD20 directed immunotherapy alone or in combination with chemotherapy.3 The current pretherapy prognosticators are the Follicular Lymphoma International Prognostic Index (FLIPI)4 and the more recently described FLIPI2.5 Both include an evaluation of tumor burden using the Ann Arbor staging system and assessments of the number of nodal sites, the longest diameter of the largest involved node (LoDLIN), or both.
Positron emission tomography/computed tomography (PET/CT) is now recommended for initial evaluation and staging in all 18F-fluorodeoxyglucose (FDG)–avid non-Hodgkin lymphomas, including FL.6-8 In comparison with CT, PET/CT displays higher sensitivity for identifying nodal and extranodal disease, without loss of specificity.9 Quantitative tumor burden parameter, namely total metabolic tumor volume (TMTV), can be obtained from fluorodeoxyglucose–positron emission tomography (FDG-PET)/CT at staging, resulting in hematologists’ growing interest in this evaluation method.10 A high tumor burden FL with a baseline TMTV >510 cm3 has recently been reported as being associated with shorter progression-free survival (PFS) and, to a lesser extent, with reduced overall survival (OS).11
Besides tissue tumor burden, circulating tumor cells (CTCs) may also be useful predictors of outcome. CTCs are detected in the vast majority of FL patients at diagnosis. When these cells are in sufficient numbers, they can be identified by routine blood smear examinations or flow cytometry.12 For lower levels of CTCs, the t(14;18)(q32;q21) translocation, present in 80% to 90% of FL,13 can be used as a marker of disease and is routinely detected by polymerase chain reaction (PCR).14 In both cases, patients with high levels of CTCs have a poor outcome.12,15-17
Circulating cell-free DNA (cfDNA) in plasma has emerged as a new and sensitive oncology biomarker. A fraction of this cfDNA is derived from tumor cells (ie, circulating cell-free tumor DNA [ctDNA]).18 More recently, the presence of ctDNA has been reported in diffuse large B-cell lymphoma (DLBCL)19-25 and to a lesser extent in FL.26 In patients with Hodgkin lymphoma and DLBCL, an elevated plasma ctDNA level has been shown to be associated with inferior failure-free survival.27
The aim of our study was to analyze in FL at diagnosis the relationship between total metabolic tumor burden evaluated by PET/CT and the circulating tumor burden evaluated either by the number of CTCs or by the plasmatic ctDNA load and to determine their respective or combined prognostic values.
Methods
Study design
Two clinical cohorts were studied. Both included consecutive patients with previously untreated grades 1 to 3A FL, treated between 2007 and 2014, with a baseline PET/CT suitable for TMTV calculation. The first cohort included patients who had pretreatment peripheral blood (PB) sampling for routine bcl2-JH PCR detection, mostly treated in the Henri Mondor University Hospital in Créteil, France. The second cohort included patients who had pretreatment plasma samples stored at −80°C, mostly treated in the Dijon University Hospital. Histopathology diagnosis of FL was routinely performed by local pathologists, and results were confirmed according to the World Health Organization 2008 classification1 by an expert hematopathologist from within the French Lymphopath network.28 Patients with transformed FL at diagnosis were excluded.
Patients’ clinical data (including baseline clinical and biological characteristics, FLIPI score, first-line treatment, response according to International Working Group criteria,29 date of first event, and outcome) were retrospectively collected. TMTV was computed from baseline PET/CT scans, as has been previously reported.11
The study was conducted according to the institutional ethical guidelines and in accordance with the Declaration of Helsinki. The retrospective collection and anonymous analysis of data were approved by our institutional review board.
Materials
Genomic DNA was prospectively extracted at diagnosis from 2 mL of total PB, using a Qiasymphony automated extraction device (Qiagen, Courtaboeuf, France), or from a diagnostic biopsy sample, using a phenolchloroform precipitation procedure. Samples were stored at −20°C until reanalysis (Research collection, Assistance Publique Hôpitaux de Paris, Créteil, France).
Frozen aliquots of 400 to 900 μL of plasma, collected at diagnosis and stored at Dijon Hospital, were transferred to the Laboratoire d'Immunologie Biologique, Henri Mondor University Hospital, Créteil, France. The cfDNA was extracted using the QIAamp Circulating Nucleic Acid Kit on a Qiasymphony automated device according to the manufacturer’s instructions. The DNA was eluted into 75 μL of buffer in the machine, and 60 µL were recovered and stored at 4°C until further processing.
Quantification of bcl2-JH translocated alleles in peripheral blood
The patient’s specific bcl2 breakpoint was routinely identified at diagnosis by using Biomed-2 multiplex PCR runs.14 When no translocation was detected in PB, tumor DNA was analyzed if available.
To quantify the t(14,18) translocation with the JH primer/probe couple commonly used for immunoglobulin heavy chain gene allele-specific oligonucleotide (ASO IgH) real-time quantitative PCR quantification in the Euro–minimal residual disease (EU-MRD) protocol,30 we first determined the JH segment involved in the patient’s translocation. DNA was amplified with a modified Biomed-2 PCR protocol in which the consensus JH primer was substituted by a mix of JH4, JH5, and JH6 primers labeled with HEX, FAM, and AT550 fluorescent dye, respectively. PCR products were loaded onto 3130 Genetic Analyzers (Applied Biosystems, Foster City, CA) (supplemental Figure 1). The JH segments involved in translocation were revealed by the color of the bcl2-JH amplified fragment. When no visible PCR product was obtained, the same procedure was repeated using JH1-, JH2-, and JH3-labeled primers.
A droplet digital PCR (ddPCR) assay31,32 targeting the bcl2-JH translocation was then designed and performed using the QX200 Droplet Digital PCR system (Bio-Rad Laboratories, Hercules, CA). A mix of bcl2 primers corresponding to tube A (2 major breakpoint region [MBR] primers), tube B (4 3′MBR primers), or tube C (2 minor cluster region [mcr] primers and 1 5′mcr primer) of Biomed-2 PCR was used as 5′ primer. The 3′ JH primer/probe couples were used according to the JH segment joined to the bcl2 gene,33 and BHQ-1 quenchers were used instead of TAMRA for the JH probe (Eurofins Genomics, Les Ulis, France). Computed results were analyzed by QuantaSoft (version 1.7; Bio-Rad) and were interpreted according to current international guidelines (D. Drandi, unpublished data) (for details, see the supplemental Methods). When quantitative criteria were reached, the 3 replicates were fused and analyzed on a single 750-ng metawell using the QuantaSoft program for quantification. The albumin gene was used as the standard to estimate the total number of analyzed cells (1 cell is 2 albumin copies), as is done routinely in the EU-MRD procedure.30 Results were obtained as a ratio of (bcl2-JH copy number)/(reference gene copy number/2) and expressed as the ratio of tumor cells in PB cells. Before concluding that a PB sample was negative, a patient’s tumoral sample (either bone marrow or tumor biopsy) was tested to assess the amplificability of the patient’s translocation.
Quantification of plasma cell-free DNA by ddPCR
The ddPCR assay31 was performed using the QX200 Droplet Digital PCR system (Bio-Rad Laboratories, Hercules, CA), and computed results were analyzed with QuantaSoft (version 1.7; Bio-Rad).
The ANKRD30B gene was used as the reference gene to quantify the cell-free circulating equivalent genome (Eqg) using the PrimePCR ddPCR copy number assay kit (Bio-Rad) (supplemental Figure 2A). The 20-μL ddPCR reaction included 10 μL of 2× ddPCR Master Mix (Bio-Rad), 1 μL of 20× primers and probe (final concentrations: 500 nmol/L and 200 nmol/L), and 7.5 µL cfDNA.
The quantity of cfDNA in plasma was expressed as the equivalent genome per milliliter (Eqg/mL) by measuring as follows: (absolute number of reference gene (ANKRD3) copies in 7.5 µL of cfDNA eluate) (10% of eluted volume) × 10/(volume of extracted plasma) × 1000.
Screening and quantification of t(14,18) translocated DNA in cfDNA
The initial screening involved three PCRs. Each PCR contained all 5′ bcl2 primers mixed with one of the JH4, JH5, JH6 3′ primer/probe couples. For negative samples, a second PCR screening round was performed using the JH1, JH2, and JH3 primers. In the absence of a sample allowing the detectability of the patient’s translocation to be checked using the Biomed-2 technique, a negative sample after the screening procedure was considered as noninterpretable.
To quantify the proportion of cfDNA containing the t(14;18) in plasma samples (supplemental Figure 2B), we performed simultaneous quantification of ANKRD30B reference gene levels as described above.
Assessment of TMTV on PET/CT
PET/CT scans were performed 60 minutes after injection of FDG, either on a Gemini GXL16 scanner (Philips, Da Best, The Netherlands) or on a Biograph 2 scanner (Siemens, Erlangen, Germany). Quality control of image reconstruction was ensured by measuring the maximum standardized uptake value (SUVmax) in a spherical volume of interest (VOI) in the liver (median SUVmax = 2.71; extremes, 1.55 to 4.21) and in the mediastinum (median SUVmax = 1.86; extremes, 0.97 to 2.92). Image volumes were imported in a DICOM format onto an Imagys workstation (Keosys, Saint-Herblain, France) and analyzed by a senior nuclear medicine physician blinded to patient outcome. In a subset of 61 patients, images were analyzed by a second observer to assess interobserver reproducibility of TMTV measurements (Lin’s concordance correlation ρC = 0.95). Lymphoma lesions were identified visually as areas of increased uptake outside areas of physiological uptake. CT fusion display was used to ensure that the metabolic volumes did not spill over into anatomical structures. The TMTV was obtained by summing the metabolic volumes of all individual nodal and extranodal lesions, using the 41% SUVmax threshold method, as has been previously described11,34 and following European guidelines.35
Statistical analyses
X-tile analysis36 and receiver-operating characteristic (ROC) analysis were performed to determine optimal cutoff values of biological and imaging variables of tumor load for survival prediction, as has been previously described.11
Population characteristics were compared using the Fischer exact test for categorical variables and the Mann-Whitney test for continuous variables. The prognostic value of biological and imaging variables was assessed using PFS (calculated from the date of diagnosis until progression, relapse, or death from any cause) as the end point, with censoring at the time of the last follow-up. Survival curves were obtained from Kaplan-Meier estimates and compared using the log-rank test. Multivariate analysis was performed using the Cox proportional hazards model to assess the independence of prognostic variables. P < .05 was considered significant. Statistical analyses were conducted using X-tile 3.6.1 software (Yale University, New Haven, CT) and MedCalc 12.2.1.0 (MedCalc Software, Ostend, Belgium).
Results
Patient characteristics at diagnosis
Peripheral blood DNA and plasma samples were available at diagnosis for 91 and 61 patients, respectively. Nineteen patients had both PB DNA and plasma samples (Figure 1). A bcl2-JH translocation was detected in 68 out of 91 patients (75%), either in PB (n = 43) or in a diagnostic biopsy (n = 25). Breakpoints were located inside the MBR, mcr, and 3′MBR regions in 47, 17, and 4 patients, respectively. Altogether, 133 patients had a TMTV calculation from diagnostic PET/CT.
Figure 1.

Patients and cohorts. A total of 133 patients with assessment of TMTV at diagnosis were included. They were treated at Dijon University Hospital (n = 61; green rectangle) or at Henri Mondor University Hospital in Créteil (n = 72). A plasma sample was available for cfDNA extraction for all the patients treated at the Dijon hospital (n = 61; green rectangle). A DNA sample extracted from a peripheral blood sample at diagnosis was available for 72 patients treated at the hospital in Creteil and for 19 patients treated at the Dijon hospital (n = 91; blue rectangle). Out of these 91 samples, 68 (54 + 14) were bcl2-JH informative (75%). Two groups of patients were therefore analyzed; the first included 61 patients with cfDNA and TMTV, and the second group included 68 patients with interpretable peripheral blood and TMTV.
Clinical characteristics of the 133 patients with a TMTV calculation, including 61 patients with cfDNA and 68 patients with quantifiable CTC, are given in Table 1.
Table 1.
Patient characteristics according to TMTV and circulating parameters
| Patients with TMTV, n = 133 | TMTV > 510, n = 50/133 | Patients with cfDNA analyses, n = 61 | Patients with cfDNA > 2550, n = 37/61 | Patients with evaluable* CTCs, n = 68 | Patients with CTCs > 0.0018, n = 42/68 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n (missing) | % | n (missing) | % | n (missing) | % | n (missing) | % | n (missing) | % | n (missing) | % | |
| Median age (range), y | 60 (28-75) | 61 (31-77) | 62 (34-77) | 64 (41-77) | 59 (33-75) | 58 (33-75) | ||||||
| Age >60 y | 64 | 48 | 26 | 52 | 33 | 54 | 23 | 62 | 28 | 41 | 17 | 40 |
| Female sex | 68 | 51 | 22 | 44 | 32 | 52 | 16 | 43 | 33 | 49 | 17 | 40 |
| Ann Arbor stage III-IV | 126 | 95 | 50 | 100 | 58 | 95 | 36 | 97 | 67 | 99 | 42 | 100 |
| No. of involved areas >4 | 83 (3) | 64 | 42 (2) | 87† | 46 | 75 | 32 | 86† | 44 | 65 | 32 | 76 |
| Bone marrow biopsy (+) | 78 (5) | 61 | 40 (1) | 81† | 39 (1) | 65 | 26 (1) | 72 | 41 (3) | 63 | 36 (1) | 88† |
| LodLIN (cm) >6 | 47 (5) | 37 | 20 | 40 | 19 | 31 | 13 | 35 | 20 (1) | 30 | 13 (1) | 32 |
| LDH >Nl | 20 | 15 | 09 | 18 | 13 | 21 | 10 | 27 | 11 | 16 | 8 | 19 |
| β2-microglobulin >Nl | 68 (13) | 57 | 38 (4) | 83† | 35 | 57 | 27 | 73 | 33 (12) | 59 | 26 (6) | 72 |
| FLIPI score 3-4 | 61 | 46 | 31 | 62 | 34 | 56 | 25 | 68* | 31 | 46 | 20 | 48 |
| FLIPI2 score 3-4 | 55 (6) | 43 | 30 (2) | 62* | 28 (1) | 47 | 23 (1) | 64* | 28 (5) | 44 | 21 (2) | 52 |
| Median SUVmax (range) | 8.8 (2.4-27.6) | 8.36 (3.41-27.6) | 9.63 (2.4-21.8) | 10.6 (2.38-18.3) | 9.0 (4.99-27.6) | 8.31 (4.99-27.6) | ||||||
| Median TMTV, cm3 (range) | 354 (14-7796) | 855 (520-7796) | 392 (17-7795) | 724 (17-7795) | 413 (32-3432) | 658 (32-3432) | ||||||
| First-line treatment | ||||||||||||
| R chemo‡ | 48 | 36 | 19 | 38 | 17 | 28 | 9 | 24 | 23 | 34 | 13 | 31 |
| R chemo‡ + M | 60 | 45 | 19 | 38 | 29 | 47 | 16 | 43 | 27 | 40 | 18 | 43 |
| Chemo-free§ | 4 | 3 | — | — | 2 | 3 | 1 | 3 | 1 | 1 | — | — |
| Chemo-free§ + M | 21 | 16 | 12 | 24 | 13 | 21 | 11 | 30 | 17 | 25 | 11 | 26 |
Chi-squared comparison of parameters between patients with high levels and total analyzed population. Values are “n (missing)” and “%” of patients except as noted.
LDH, lactate dehydrogenase; M, Revlimid maintenance; R, Revlimid.
P < .05.
P < .01.
R chemo: PRIMA-like R-CHOP R (6 cycles of R-CHOP [rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone] plus 2 cycles of rituximab): n = 84. R2-CHOP (addition of lenalidomide [Revlimid; Celgene] to the R-CHOP regimen): n = 15. Other R-chemo schedule: n = 9.
Chemo-free: R2 (oral lenalidomide 20 mg for 21 days in a 28-day cycle, with rituximab 375 mg/m2 given intravenously on day 1 of each cycle for at least 6 cycles): n = 23; Revlimid alone (n = 2).
Depending on the cohort, median age at diagnosis was between 59 and 62 years, and 46% to 56% of patients presented with intermediate/high FLIPI scores. Most patients (95%) had Ann Arbor stage III/IV FL, with involvement of the bone marrow (61%). Two patients had a tumor mass that was no longer detectable on PET after tumor biopsy. In the remaining patients, median SUVmax and TMTV were 8.8 (range, 2.4-27.6) and 354 cm3 (range, 17-7796 cm3), respectively. Median TMTV was similar in both cohorts.
Quantitative relationship between circulating and tissue biomarkers of tumor burden
With a median value of 7 bcl2/JH (+) cells in 103 PB cells (7/103), 64 out of the 68 evaluable patients (94%) had detectable CTCs at diagnosis (range, 5/105 to 9/101).
Between 400 and 900 µL of plasma were analyzed from 61 patients. Circulating cfDNA was detected in all samples. The median number of Eqg was 2600/mL plasma, but a high level of heterogeneity was observed with a range from 650 to 410 400 (Figure 2A). Targeted bcl2-JH PCR and screening procedures were performed in all 61 patients. No bcl2-JH copy was detected in 38 patients. Among these patients, bcl2-JH translocation amplificability was demonstrated in only 3 patients with available PB samples. As a consequence, only these 3 negative results were taken into account in further correlation studies. With a median number of 120 cp/mL, a bcl2-JH translocation was detected in 23 patients (range, 8 to 13 200 cp/mL). Regardless of the absolute number of bcl2-JH circulating copies, tumor DNA never constituted more than 10% of the total cfDNA (Figure 2B).
Figure 2.

Circulating total and bcl2-JH cell-free DNA in FL at diagnosis. cfDNA was extracted from 400 to 900 µL of plasma. (A) The Eqg per mL of plasma was evaluated by ddPCR quantification of a reference gene (ANKRD30B gene, centromeric localization on chromosome 18). Each spot represents a patient. The vertical logarithmic scale highlights the large interpatient variability in the total amounts of cfDNA. (B) The number of circulating bcl2-JH segments were quantified by ddPCR in 26 patients. The red circles correspond to the number of bcl2-JH allele copies and the blue circles to the number of Eqg.
The relationship between “circulating” biomarkers (either CTCs or cfDNA) and TMTV is shown in Figure 3. There were significant correlations between TMTV and circulating tumor markers, both for CTCs (R = 0.67, P < .0001) (Figure 3A) and cfDNA (R = 0.6, P < .0001) (Figure 3B). Accordingly, when compared with patients with low TMTV, patients with TMTV >510 cm3 had higher CTCs (median, 0.08 [0.00086-0.96] vs 0.00075 [0.00-0.4], P = .0001) (Figure 3C) and cfDNA (median, 3975 [1320-410 400] vs 2440 [650-9080], P = .0006) (Figure 3D) levels.
Figure 3.

Quantitative relationship between circulating biomarkers and TMTV at diagnosis. TMTV was calculated from initial PET/CT in all 133 patients studied and compared with the number of circulating tumor cells evaluated by ddPCR (CTCs) on peripheral blood cell samples from 68 patients (A), or with the total cfDNA from 61 patients (Eqg/mL) (B). The distributions of the number of CTCs (C) and cfDNA levels (D) according to low or high TMTV are also shown. ****P < .0001; ***P < .001.
Treatments and clinical outcome
The majority of patients (n = 108) received immunochemotherapy with (n = 60) or without (n = 48) maintenance. Induction immunochemotherapy regimens were administered according to the PRIMA37 or R2CHOP38 schedules in 84 and 15 patients, respectively. Nine additional patients received other forms of Revlimid chemotherapy. Two patients received rituximab alone, while 23 patients received rituximab combined with lenalidomide, followed by rituximab maintenance in the setting of a randomized phase 3 trial (RELEVANCE NCT01476787) (Table 1). The overall response rate was 97% (129/133 patients). With an end date of July 2016, the median follow-up of living patients was 48 months (range, 6-108 months). Disease progression occurred in 32 patients, with a median time to progression of 22 months (supplemental Figure 3A) (range, 1-62 months; PFS follow-up information was not available for 1 patient). Eleven patients died after a median period of 37 months (range, 8-92 months), including 9 patients from lymphoma, 1 from suicide, and 1 from lung cancer (supplemental Figure 3B).
Prognostic value of TMTV and circulating biomarkers of tumor burden
X-tile and ROC analyses were performed to determine optimal cutoff values of TMTV, CTCs, and cfDNA for PFS predictions. With a median follow-up of 48 months, patients with a TMTV of ≤510 cm3 showed significantly improved PFS in comparison with patients with a high TMTV, with 4-year PFS estimates of 84% (95% confidence interval [CI], 75% to 94%) vs 59% (95% CI, 42% to 75%), respectively (P = .0004; Figure 4A).
Figure 4.

Progression-free survival according to tissue or circulating tumor burden biomarkers. Kaplan-Meier estimates of PFS based on TMTV (A), CTC (B), cfDNA (C), and TMTV and cfDNA combined (D). Of the 21 patients with combined high or low levels, 17 had high cfDNA levels and a low TMTV, and 4 had low cfDNA levels and a high TMTV. H, high level; HR, hazard ratio; L, low level.
Patients with low levels of or no CTCs (≤1.8 × 10−3 PB cells) showed significantly improved PFS in comparison with patients with high CTC levels, with 4-year estimates of 86% (95% CI, 71% to 100%) vs 58% (95% CI, 37% to 78%), respectively (P = .03; Figure 4B). Similarly, patients with low levels of total cfDNA (≤2550 Eqg/mL of plasma) showed significantly improved PFS, 96% (95% CI, 87% to 100%) vs 73% (95% CI, 56% to 90%), respectively (P = .04; Figure 4C). By comparing Cox models, including FLIPI alone against FLIPI with a marker, we found that all 3 parameters were independent of FLIPI (log-likelihood ratio test: TMTV P = .002654; CTC P = .01976; cfDNA P = .02354).
In a Cox multivariate analysis including CTCs and TMTV as continuous variables, only CTCs tended to remain of prognostic value (P = .09). By contrast, both cfDNA (P = .0034) and TMTV (P = .0001) remained predictive of outcome when used as covariates. Accordingly, PFS was shorter in the 20 patients with a high level of both cfDNA and TMTV, with a 4-year PFS estimate of 65% (Figure 4D).
Impact of maintenance therapy on circulating biomarkers and TMTV at diagnosis
As was expected, maintenance was associated with improved PFS in our FL cohort (supplemental Figure 4).
The benefits of maintenance therapy were also observed in the subset of patients with high TMTV (n = 50) and high CTC (n = 42), but there was no evidence of this therapy being of benefit to patients with high cfDNA (supplemental Figure 5).
We next focused on the 82 patients who received maintenance treatment (1 patient was lost to follow-up). High total cfDNA levels and TMTV were associated with shorter PFS (4-year PFS of 100% vs 74% [P = .05] and 88% vs 80% [P = .02], respectively) (Figure 5A,C). By contrast, CTCs had no impact on PFS in these patients (Figure 5B).
Figure 5.

Impact of maintenance therapy on the prognostic value of tumor burden biomarkers at diagnosis. Kaplan-Meier estimates of PFS in patients with maintenance treatment based on TMTV (A), CTC (B), cfDNA (C), and TMTV and cfDNA combined (D). Of the 16 patients with combined high or low levels, 13 had high cfDNA levels and a low TMTV, and 3 had low cfDNA levels and a high TMTV.
When combined, cfDNA and TMTV still allowed identification of 3 groups of patients: 1 with a low level of cfDNA and a low TMTV (n = 12); 1 with either a high level of cfDNA or a high TMTV (n = 16); and 1 with high levels of both parameters (n = 15), with 100%, 80%, and 72% 4-year PFS estimates, respectively (P = .06) (Figure 5D).
Discussion
We report here, for the first time, the relationship between circulating and tissue tumor burden evaluations at diagnosis in patients with untreated FL and their respective prognostic values. Circulating components were evaluated using 2 parameters, CTCs or cfDNA, and TMTV was used to evaluate tissue tumor mass.
Our patient cohorts set up in 2 French university hospitals (median patient age of around 60 years, 75% PFS and 92% OS at 4 years, and high FLIPI scores) had clinical and biological presentations that were similar to those of FL patients requiring treatment included in previous reports.37,39 The PRIMA randomized study37 has shown that maintenance with rituximab significantly prolongs remission, and maintenance is now a standard of care in the first-line treatment of FL.3 Because this standard of care evolved over the time course of our study, approximately half of our patients received rituximab maintenance. As recently published by a team from the MD Anderson Cancer Center,40 we also found that maintenance had a positive impact on PFS and OS in a “true-life” cohort. Our analysis of the prognostic value of tumor burden (evaluated by assessing TMTV, CTCs, and cfDNA levels) in patients who had received maintenance therapy suggested that CTC levels were no longer useful as prognostic indicators in maintenance-treated FL patients. Similar findings were also reported in a follow-up PRIMA study.41 In contrast, TMTV and cfDNA retained their prognostic value in these patients. However, because the number of patients eligible to be included in this part of our study was small, further analyses on large cohorts are required to confirm these findings.
Total metabolic tumor volume is a measure of the viable fraction of tumors and may provide a better estimate of tumor burden than would anatomical imaging for measures of tumor bulk and Ann Arbor staging.34 As was previously shown in a clinical trial,11 we found that baseline TMTV was of prognostic value in our FL series. Moreover, the best cutoff determined by X-tile and ROC analyses in our cohort was identical (510 cm3) to the threshold identified in the previous series,11 highlighting the robustness of this parameter as a prognostic factor.
Circulating tumor cells and TMTV were highly correlated and were predictive of outcome in univariate analysis but did not exhibit independent prognostic value in multivariate analysis. This suggests that CTCs are essentially a reflection of the tissue tumor mass, which is the basis of the concept for studying residual disease in the blood. This concept has been validated by several studies.42,43 However, Luminari et al44 recently showed that PET and MRD at the end of induction treatment are not completely correlated to each other in FL patients and can be used as complementary techniques to better predict PFS, suggesting that the circulating and solid components at diagnosis were not equivalent with regard to therapeutic efficacy.
We found that there was a strong relationship between total cfDNA levels and TMTV. In a subset of patients, we were able to quantify ctDNA by quantification of bcl2-JH translocated copies. As is reported in most solid tumors,45 we showed that tumor DNA represents a small fraction of total cfDNA (<10%), even in patients with high levels of cfDNA. This highlights the fact that FDG avidity is also related not only to tumor cells. The best illustration of this finding is observed in Hodgkin lymphoma (HL), in which the tumor tissue comprises a few neoplastic cells (accounting for less than 1% of the total cell count found in biopsy specimens) surrounded by a large population of nonneoplastic cells. Therefore, the very high metabolic activity observed in HL is probably partly related to FDG uptake by the surrounding reactive cells in the tumor microenvironment.46
In contrast to CTCs, cfDNA remained independent of TMTV as a predictor of PFS. On one hand, FL prognosis in the rituximab era has been shown to be partly linked to the characteristics of the microenvironment47 and mainly linked to the presence of nonlymphoid cells.48,49 Our results suggest that the nontumor cfDNA could also come from the tumor, enlarging the concept of liquid biopsy45 to the microenvironment compartment. On the other hand, using next-generation sequencing of clonal rearranged VDJ, we have shown that the distribution of subclones differed between the tumor and plasma samples.26 Thus, even if the global quantity of cfDNA and TMTV are related, circulating DNA may reflect selected cell populations of prognostic value. It remains to be determined whether the prognostic value of cfDNA is more strongly related to the microenvironment than to FL tumor cells themselves.
The prognostic values of TMTV and cfDNA differed depending on the predicted outcomes. TMTV was a better predictor of poor outcome with a 4-year estimate of 59% in patients with TMTV >510 cm3, whereas cfDNA was a better predictor of good outcome with a 4-year estimate of 96% in patients with cfDNA ≤2550. When combining both biomarkers, we identified a group of parameters associated with a very good prognosis (low TMTV + low cfDNA, 4-year PFS = 94%) and a group associated with a worse prognosis (high TMTV + high cfDNA, 4-year PFS = 65%), stressing the complementarity between tissue and circulating markers of tumor burden.
We have shown that all 3 tumor burden–associated parameters are independent of FLIPI. Unfortunately, because of the low number of events in our cohort, statistical rules did not allow for a multivariate analysis to be performed with more than 2 parameters. However, TMTV and cfDNA values are not intended to replace the FLIPI and are more likely to be used to refine the tumor mass parameters of FLIPI, replacing, for example, the number of lymph node areas and the stage. Larger cohorts are needed to validate this hypothesis.
In conclusion, our results suggest that total cfDNA at diagnosis is not a simple reflection of metabolic tumor burden and is of prognostic value in FL, independently of TMTV. Future prospective studies will need to confirm whether this noninvasive diagnostic tool is of additional value for establishing individual prognostic outcomes.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Emma Pilling for language editing services and Marielle Romet (Santé Active Edition) for general editing services.
This work was supported by the Association pour la Recherche Thérapeutique, Génétique et Immunologique dans les Lymphomes, and by the CALYM Carnot Institute (Consortium for the Acceleration of Innovation and Its Transfer in the Lymphoma Field).
Authorship
Contribution: M.-H.D.-L., E.I., R.-O.C., C.H., and J.D. contributed to the study design, performed the research, analyzed data, and wrote the paper; A.v.d.G., A.B.-R., J.D., and C.C.-B. provided data and edited the manuscript; J.D., G.L., B.V., P.R., I.N., A.B.-F., S.H., and I.B. provided data; and E.I. and J.-P.J. performed statistical analyses.
Conflict-of-interest disclosure: M.-H.D.-L. received funding for travel/accommodation from Baxter, Janssen, and Celgene. R.-O.C. received honoraria from Roche, Takeda, and Gilead; research funding from Roche and Gilead; and had a consulting role for Roche, Takeda, Gilead, AbbVie, Bristol-Myers Squibb, and Merck. C.H. had a consulting role for Roche and received research funding from Roche and Amgen. The remaining authors declare no competing financial interests.
Correspondence: Marie-Hélène Delfau-Larue, Hematologic and Immunologic Biology Department, Assistance Publique Hôpitaux de Paris, GH Mondor 51 av du Marechal Delattre de Tassigny, 94000 Creteil, France; e-mail: marie-helene.delfau@hmn.aphp.fr.
References
- 1.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization; 2008. [Google Scholar]
- 2.Bachy E, Houot R, Morschhauser F, et al. ; Groupe d’Etude des Lymphomes de l’Adulte (GELA). Long-term follow up of the FL2000 study comparing CHVP-interferon to CHVP-interferon plus rituximab in follicular lymphoma. Haematologica. 2013;98(7):1107-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghielmini M, Vitolo U, Kimby E, et al. ; Panel Members of the 1st ESMO Consensus Conference on Malignant Lymphoma. ESMO Guidelines consensus conference on malignant lymphoma 2011 part 1: diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL) and chronic lymphocytic leukemia (CLL). Ann Oncol. 2013;24(3):561-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solal-Céligny P, Roy P, Colombat P, et al. Follicular lymphoma international prognostic index. Blood. 2004;104(5):1258-1265. [DOI] [PubMed] [Google Scholar]
- 5.Federico M, Bellei M, Marcheselli L, et al. Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol. 2009;27(27):4555-4562. [DOI] [PubMed] [Google Scholar]
- 6.Cheson BD, Fisher RI, Barrington SF, et al. ; United Kingdom National Cancer Research Institute. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrington SF, Mikhaeel NG, Kostakoglu L, et al. Role of imaging in the staging and response assessment of lymphoma: consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol. 2014;32(27):3048-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luminari S, Biasoli I, Arcaini L, et al. The use of FDG-PET in the initial staging of 142 patients with follicular lymphoma: a retrospective study from the FOLL05 randomized trial of the Fondazione Italiana Linfomi. Ann Oncol. 2013;24(8):2108-2112. [DOI] [PubMed] [Google Scholar]
- 9.Meignan M. VI. FDG-PET as a biomarker in lymphoma: from qualitative to quantitative analysis. Hematol Oncol. 2015;33(Suppl 1):38-41. [DOI] [PubMed] [Google Scholar]
- 10.Meignan M, Sasanelli M, Casasnovas RO, et al. Metabolic tumour volumes measured at staging in lymphoma: methodological evaluation on phantom experiments and patients. Eur J Nucl Med Mol Imaging. 2014;41(6):1113-1122. [DOI] [PubMed] [Google Scholar]
- 11.Meignan M, Cottereau AS, Versari A, et al. Baseline metabolic tumor volume predicts outcome in high-tumor-burden follicular lymphoma: a pooled analysis of three multicenter studies [published online ahead of print 22 August 2016]. J Clin Oncol. doi:10.1200/JCO.2016.66.9440. [DOI] [PubMed] [Google Scholar]
- 12.Sarkozy C, Baseggio L, Feugier P, et al. Peripheral blood involvement in patients with follicular lymphoma: a rare disease manifestation associated with poor prognosis. Br J Haematol. 2014;164(5):659-667. [DOI] [PubMed] [Google Scholar]
- 13.Yunis JJ, Oken MM, Kaplan ME, Ensrud KM, Howe RR, Theologides A. Distinctive chromosomal abnormalities in histologic subtypes of non-Hodgkin’s lymphoma. N Engl J Med. 1982;307(20):1231-1236. [DOI] [PubMed] [Google Scholar]
- 14.Evans PA, Pott C, Groenen PJ, et al. Significantly improved PCR-based clonality testing in B-cell malignancies by use of multiple immunoglobulin gene targets. Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia. 2007;21(2):207-214. [DOI] [PubMed] [Google Scholar]
- 15.Rambaldi A, Carlotti E, Oldani E, et al. Quantitative PCR of bone marrow BCL2/IgH+ cells at diagnosis predicts treatment response and long-term outcome in follicular non-Hodgkin lymphoma. Blood. 2005;105(9):3428-3433. [DOI] [PubMed] [Google Scholar]
- 16.Galimberti S, Luminari S, Ciabatti E, et al. Minimal residual disease after conventional treatment significantly impacts on progression-free survival of patients with follicular lymphoma: the FIL FOLL05 trial. Clin Cancer Res. 2014;20(24):6398-6405. [DOI] [PubMed] [Google Scholar]
- 17.Zohren F, Bruns I, Pechtel S, et al. Prognostic value of circulating Bcl-2/IgH levels in patients with follicular lymphoma receiving first-line immunochemotherapy. Blood. 2015;126(12):1407-1414. [DOI] [PubMed] [Google Scholar]
- 18.Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armand P, Oki Y, Neuberg DS, et al. Detection of circulating tumour DNA in patients with aggressive B-cell non-Hodgkin lymphoma. Br J Haematol. 2013;163(1):123-126. [DOI] [PubMed] [Google Scholar]
- 20.Roschewski M, Dunleavy K, Pittaluga S, et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: a correlative biomarker study. Lancet Oncol. 2015;16(5):541-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bohers E, Viailly PJ, Dubois S, et al. Somatic mutations of cell-free circulating DNA detected by next-generation sequencing reflect the genetic changes in both germinal center B-cell-like and activated B-cell-like diffuse large B-cell lymphomas at the time of diagnosis. Haematologica. 2015;100(7):e280-e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camus V, Sarafan-Vasseur N, Bohers E, et al. Digital PCR for quantification of recurrent and potentially actionable somatic mutations in circulating free DNA from patients with diffuse large B-cell lymphoma. Leuk Lymphoma. 2016;57(9):2171-2179. [DOI] [PubMed] [Google Scholar]
- 23.Scherer F, Kurtz DM, Newman AM, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8(364):364ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurtz DM, Green MR, Bratman SV, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood. 2015;125(24):3679-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi D, Diop F, Spaccarotella E, et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood. 2017;129(14):1947-1957. [DOI] [PubMed] [Google Scholar]
- 26.Sarkozy C, Huet S, Carlton VEH, et al. The prognostic value of clonal heterogeneity and quantitative assessment of plasma circulating clonal IG-VDJ sequences at diagnosis in patients with follicular lymphoma. Oncotarget. 2017;8(5):8765-8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hohaus S, Giachelia M, Massini G, et al. Cell-free circulating DNA in Hodgkin’s and non-Hodgkin’s lymphomas. Ann Oncol. 2009;20(8):1408-1413. [DOI] [PubMed] [Google Scholar]
- 28.Laurent C, Baron M, Amara N, et al. Impact of expert pathologic review of lymphoma diagnosis: study of patients from the French Lymphopath Network. J Clin Oncol. 2017;35(18):2008-2017. [DOI] [PubMed] [Google Scholar]
- 29.Cheson BD, Pfistner B, Juweid ME, et al. ; International Harmonization Project on Lymphoma. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579-586. [DOI] [PubMed] [Google Scholar]
- 30.van der Velden VHJ, Cazzaniga G, Schrauder A, et al. ; European Study Group on MRD detection in ALL (ESG-MRD-ALL). Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21(4):604-611. [DOI] [PubMed] [Google Scholar]
- 31.Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604-8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drandi D, Kubiczkova-Besse L, Ferrero S, et al. Minimal residual disease detection by droplet digital PCR in multiple myeloma, mantle cell lymphoma, and follicular lymphoma: a comparison with real-time PCR. J Mol Diagn. 2015;17(6):652-660. [DOI] [PubMed] [Google Scholar]
- 33.van Dongen JJM, Langerak AW, Brüggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257-2317. [DOI] [PubMed] [Google Scholar]
- 34.Sasanelli M, Meignan M, Haioun C, et al. Pretherapy metabolic tumour volume is an independent predictor of outcome in patients with diffuse large B-cell lymphoma. Eur J Nucl Med Mol Imaging. 2014;41(11):2017-2022. [DOI] [PubMed] [Google Scholar]
- 35.Boellaard R, Delgado-Bolton R, Oyen WJG, et al. ; European Association of Nuclear Medicine (EANM). FDG PET/CT: EANM procedure guidelines for tumour imaging: version 2.0. Eur J Nucl Med Mol Imaging. 2015;42(2):328-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10(21):7252-7259. [DOI] [PubMed] [Google Scholar]
- 37.Salles G, Seymour JF, Offner F, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011;377(9759):42-51. [DOI] [PubMed] [Google Scholar]
- 38.Tilly H, Morschhauser F, Salles G, et al. Phase 1b study of lenalidomide in combination with rituximab-CHOP (R2-CHOP) in patients with B-cell lymphoma. Leukemia. 2013;27(1):252-255. [DOI] [PubMed] [Google Scholar]
- 39.Dupuis J, Berriolo-Riedinger A, Julian A, et al. Impact of [(18)F]fluorodeoxyglucose positron emission tomography response evaluation in patients with high-tumor burden follicular lymphoma treated with immunochemotherapy: a prospective study from the Groupe d’Etudes des Lymphomes de l’Adulte and GOELAMS. J Clin Oncol. 2012;30(35):4317-4322. [DOI] [PubMed] [Google Scholar]
- 40.Cheah CY, Chihara D, Ahmed M, et al. Factors influencing outcome in advanced stage, low-grade follicular lymphoma treated at MD Anderson Cancer Center in the rituximab era. Ann Oncol. 2016;27(5):895-901. [DOI] [PubMed] [Google Scholar]
- 41.Sarkozy C, Seymour JF, Ferme C, et al. Rituximab maintenance obviates the poor prognosis associated with circulating lymphoma cells in patients with follicular lymphoma. Blood. 2014;123(17):2740-2742. [DOI] [PubMed] [Google Scholar]
- 42.Corradini P, Astolfi M, Cherasco C, et al. Molecular monitoring of minimal residual disease in follicular and mantle cell non-Hodgkin’s lymphomas treated with high-dose chemotherapy and peripheral blood progenitor cell autografting. Blood. 1997;89(2):724-731. [PubMed] [Google Scholar]
- 43.Metzner B, Pott C, Müller TH, et al. Long-term clinical and molecular remissions in patients with follicular lymphoma following high-dose therapy and autologous stem cell transplantation. Ann Oncol. 2013;24(6):1609-1615. [DOI] [PubMed] [Google Scholar]
- 44.Luminari S, Galimberti S, Versari A, et al. Positron emission tomography response and minimal residual disease impact on progression-free survival in patients with follicular lymphoma. A subset analysis from the FOLL05 trial of the Fondazione Italiana Linfomi. Haematologica. 2016;101(2):e66-e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meignan M, Itti E, Gallamini A, Younes A. FDG PET/CT imaging as a biomarker in lymphoma. Eur J Nucl Med Mol Imaging. 2015;42(4):623-633. [DOI] [PubMed] [Google Scholar]
- 47.Dave SS, Wright G, Tan B, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351(21):2159-2169. [DOI] [PubMed] [Google Scholar]
- 48.Blaker YN, Spetalen S, Brodtkorb M, et al. The tumour microenvironment influences survival and time to transformation in follicular lymphoma in the rituximab era. Br J Haematol. 2016;175(1):102-114. [DOI] [PubMed] [Google Scholar]
- 49.Xerri L, Huet S, Venstrom JM, et al. ; a LYSA study. Rituximab treatment circumvents the prognostic impact of tumor-infiltrating T-cells in follicular lymphoma patients. Hum Pathol. 2017;64:128-136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.