

Abstract
Boa is a neotropical genus of snakes historically recognized as monotypic despite its expansive distribution. The distinct morphological traits and color patterns exhibited by these snakes, together with the wide diversity of ecosystems they inhabit, collectively suggest that the genus may represent multiple species. Morphological variation within Boa also includes instances of dwarfism observed in multiple offshore island populations. Despite this substantial diversity, the systematics of the genus Boa has received little attention until very recently. In this study we examined the genetic structure and phylogenetic relationships of Boa populations using mitochondrial sequences and genome-wide SNP data obtained from RADseq. We analyzed these data at multiple geographic scales using a combination of phylogenetic inference (including coalescent-based species delimitation) and population genetic analyses. We identified extensive population structure across the range of the genus Boa and provide multiple lines of support for three widely-distributed clades roughly corresponding with the three primary land masses of the Western Hemisphere. We also find both mitochondrial and nuclear support for independent origins and parallel evolution of dwarfism on offshore island clusters in Belize and Cayos Cochinos Menor, Honduras.
Keywords: Bayesian species delimitation, Boidae, population genomics, population structure, RADseq
Graphical Abstract
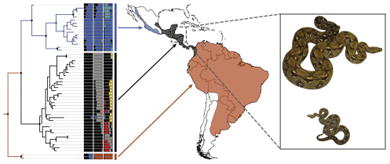
1. Introduction
Widespread, generalist species are powerful model systems for understanding how diverse ecological factors may drive regional patterns of species divergence and diversification (e.g., Brouat et al., 2004; Fields et al., 2015; Hull et al., 2008). The snake family Boidae includes several examples of such systems, with species occupying wide distributions and encompassing a broad range of latitudes, altitudes, and ecosystems (Henderson et al., 1995). Modern Boid snake distributions are the result of numerous vicariance events associated with the fragmentation of Gondwana, and thus these snakes have been cited as a classic example of the role that plate tectonics plays in shaping species distributions (Bauer, 1993; Laurent, 1979; Noonan and Chippindale, 2006a, b; Rage, 1988, 2001). Recent studies have also examined the phylogenetic relationships among certain Boid lineages, and collectively have identified evidence for previously unrecognized diversity (Colston et al., 2013; Hynková et al., 2009; Reynolds et al., 2014; Suárez-Atilano et al., 2014).
Boa constrictor, the sole species historically comprising the monotypic genus Boa, occurs almost continuously from southern South America through northern Mexico. Multiple studies have placed Boa constrictor as sister to the Neotropical clade containing Corallus, Eunectes, and Epicrates (Burbrink, 2005; Noonan and Chippindale, 2006a). Numerous subspecies have been described, yet there have been substantial differences in taxonomic recognition among studies. Mainland subspecies include B. c. amarali (Bolivia, Paraguay, and southern Brazil; Stull, 1932), B. c. constrictor (South America; Linnaeus, 1758), B. c. eques (Piura, Peru; Eydoux and Souleyet, 1842), B. c. imperator (Central and North America; Daudin and Sonnini, 1803), B. c. longicauda (Tombes, Peru; Price and Russo, 1991), B. c. melanogaster (Ecuador; Langhammer, 1985), B. c. occidentalis (Argentina and Bolivia; Philippi, 1873), and B. c. ortonii (northwest Peru; Cope, 1877). In addition to mainland taxa, multiple island populations have been identified as distinct subspecies, including B. c. nebulosa (Lazell, 1964) from Dominica, B. c. orophias (Linnaeus, 1758) from St. Lucia, B. c. sabogae (Barbour, 1906) from the Pearl Islands of Panama, and B. c. sigma (Smith, 1943) from the Tres Marías islands of Mexico. These subspecies are mostly recognized based on approximate geographic range and morphological traits (O’Shea, 2007). The Argentine boa (B. c. occidentalis), for instance, tends to be dark-colored or black, with white patterning; this color combination is quite distinct from other subspecies. Striking color morphs are also found among island subspecies (e.g., hypomelanism in B. c. sabogae) and populations. Much of the diversity in B. constrictor color and pattern morphs is known, mostly anecdotally, from the pet trade, where these snakes are popular. Moreover, while mainland B. c. imperator in Central and Northern America are long and large-bodied, several Central American islands consist of populations composed entirely of dwarfed individuals (e.g., Cayos Cochinos and Crawl Cay). Limited work with these populations (i.e., common garden experiments) and knowledge from the pet trade indicates that the dwarfed phenotype is heritable and apparently coincides with a shift towards arboreality likely driven by selection imposed by the availability of migratory birds, a primary food source for the snakes on these small islands (Boback and Carpenter, 2007; Boback, 2005, 2006).
Despite examples of morphologically and geographically distinct B. constrictor populations, population-level analyses of the species have been entirely lacking until recently. Hynková et al. (2009) used data from the mitochondrial cytochrome B locus and found evidence of two major clades, one restricted to South America and one comprising populations in Central and North America. Reynolds et al. (2014) used multiple mitochondrial and nuclear genes from two invasive Puerto Rican samples (also examined in the context of mainland populations by Reynolds et al. (2013)) to further examine the genus Boa. This resulted in the splitting of B. constrictor (sensu lato) into two species: B. constrictor from South America and B. imperator from Central and North America. Suárez-Atilano et al. (2014) identified two additional distinct clades in Northern-Central America using dense sampling and data from two genes (mitochondrial cytochrome b and nuclear ornithine decarboxylase) and 10 microsatellites. Given these suggestions of unrecognized species within the genus, and the recently variable taxonomy of the group, we refer to all populations in the genus Boa (B. constrictor, sensu lato) as the Boa complex hereafter. Despite this recent progress, major gaps in our knowledge of the diversification of the Boa complex remain, as previous studies have lacked robust population-level sampling across the entire distribution, and from Central American island populations in particular. Furthermore, conclusions from previous studies were also limited to relatively small sets of molecular markers and were based largely on mitochondrial gene sequences.
Here we explore population genetic boundaries, population structure, and phylogenetic relationships across the Boa complex, with a focus on Northern-Central American populations that remain taxonomically unresolved, including expanded sampling from multiple dwarfed island populations. We used both mitochondrial and nuclear SNP datasets to address four major aims: (1) to characterize the degree of congruence between genetic markers (mitochondrial versus nuclear) in defining lineages of Boa; (2) to determine the number of species that should be recognized within the genus Boa; (3) to understand the fine-scale population structure and genetic diversity existing among Boa lineages and quantify levels of gene flow that may exists between major Boa clades; and (4) to investigate the potential for independent origins of dwarfism in a number of Boa island lineages.
2. Materials and Methods
2.1 Population sampling and DNA extraction
We extracted DNA from seventy-seven Boa samples that were obtained from one of three sources: (1) preserved tissues from vouchered specimens at the University of Texas at Arlington Amphibian and Reptile Diversity Research Center; (2) blood or scale samples obtained from wild-caught individuals (and progeny) from Belize that are maintained in a colony at Dickinson College; and (3) shed skin samples from commercial breeders with confident provenance (see Supplementary Tables S1–S2 for details). DNA was extracted from blood or tissue using either a Zymo Research Quick-gDNA Miniprep kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s protocol or a standard phenol-chloroform-isoamyl alcohol extraction.
2.2 Mitochondrial locus amplification and sequencing
Primers L14910 and H16064 (Burbrink et al., 2000) were used to amplify the mitochondrial cytochrome b gene (cyt-b; 1112 bp). Cycling conditions included 40 cycles with a 45°C annealing temperature and standard Taq polymerase (New England BioLabs Inc., Ipswitch, MA, USA). PCR products were visualized using gel electrophoresis and purified using Agencourt AMPure XP beads (Beckman Coulter, Inc., Irving, TX, USA) according to manufacturer’s protocols. Sanger sequencing reactions were conducted using ABI BigDye, and visualized on an ABI 3730 capillary sequencer (Life Technologies, Grand Island, NY, USA) using the amplification primers.
Forward and reverse sequence chromatographs for individual samples were aligned and quality trimmed using Geneious 6.1.6 (Biomatters Ltd., Auckland, NZ). New sequences were combined with previously published cyt-b sequences for Boa (Hynková et al., 2009; Suárez-Atilano et al., 2014; see Supplementary Table S2 for full details on sampling) and outgroup species obtained from GenBank (see Supplementary Table S3). Mitochondrial nucleotide sequences for all samples were aligned using Muscle v. 3.8.31 (Edgar, 2004), with manual adjustments and trimming to exclude samples with sequence lengths shorter than 500 bp. We also excluded samples with uncertain localities from GenBank based upon descriptions in Hynková et al. (2009). The samples included in individual analyses described below are indicated in Supplementary Table S4.
2.3 RADseq library preparation and sequencing
Forty-nine samples from North and Central American and two samples from South American populations were sequenced using double digest Restriction-site Associated DNA sequencing (RADseq hereafter), using the protocol of Peterson et al. (2012). SbfI and Sau3AI restriction enzymes were used to digest genomic DNA, and double-stranded adapters containing unique barcodes and unique molecular identifiers (UMIs; eight consecutive random nucleotides prior to the ligation site) were ligated to digested DNA per sample. Following adapter ligation, samples were pooled into groups of eight and were size selected for fragments ranging from 590 to 640bp using the Blue Pippin (Sage Science, Beverly, MA, USA); this size range was chosen to target roughly 20,000 loci, based on preliminary estimates from an in silico digestion of the Boa constrictor reference genome (Bradnam et al., 2013). Sub-pools were pooled again based on quantification of samples on a Bioanalyzer (Agilent, Santa Clara, CA, USA) using a DNA 7500 chip. Final pools were sequenced using 100 bp paired-end reads on an Illumina HiSeq 2500 (Illumina Inc., San Diego, CA, USA).
2.4 RADseq data analysis and variant calling
Raw Illumina reads from RADseq library sequencing were first filtered using the clone_filter program from the Stacks pipeline (Catchen et al., 2011, 2013), which excludes PCR replicates using the UMIs, which were subsequently trimmed away using the FASTX Toolkit trimmer v. 0.0.13 (Hannon, 2015). Trimmed reads were processed using the process_radtags function with the “rescue” feature activated in Stacks, which parses reads by barcode, confirms the presence of restriction digest cut sites, and discards reads lacking these features. Parsed reads were quality trimmed using Trimmomatic v. 0.32 (Bolger et al., 2014) and were aligned to the reference B. constrictor genome (Assemblethon2 team SGA assembly; Bradnam et al., 2013) using BWA v. 0.7.9 (Li and Durbin, 2009) with default settings (see Supplementary Table S5 for information on the number of quality-filtered and mapped reads). We identified single nucleotide polymorphisms (SNPs) using SAMtools and BCFtools v. 1.2 (Li, 2011; Li et al., 2009). We used default parameters for SNP calling (ignoring indels) and used VCFtools v. 0.1.14 (Danecek et al., 2011) to construct a stringently filtered dataset where sites were excluded that did not have a minimum Phred score of 20, that had >2 alleles per individual, that possessed a minor allele frequency <5%, or that contained >25% missing data across individuals after low confidence genotypes (Phred score < 20) were coded as missing data. This dataset was further filtered such that only the first SNP within a 50 kb window was used, to adhere to model assumptions in downstream analyses regarding independence of SNPs. This stringently filtered SNP dataset contained 1,686 SNPs and we used custom Python and R scripts to format datasets for several downstream analyses.
2.5 Estimating phylogenetic relationships and divergence times across Boa
We used the cyt-b alignment to estimate phylogenetic relationships and infer divergence times among Boa lineages using a fossilized birth-death model. This model removes the need for a priori node constraints and infers divergence times by integrating fossil dates into the lineage diversification and extinction model (Heath et al., 2014; Stadler, 2010). This model was implemented in BEAST v. 2.2.1 (Bouckaert et al., 2014) using the Sampled Ancestors add-on package (Gavryushkina et al., 2014). Fossils and associated dates (the average of the minimum and maximum dates in the age range) were acquired from the Paleobiology Database (http://paleobiodb.org), PaleoDB (http://paleodb.org), and from previous estimates of Boid divergence dates (Colston et al., 2013; Noonan and Chippindale, 2006a; Suárez-Atilano et al., 2014; see Supplementary Table S6 for full details). We specified a strict molecular clock and an HKY nucleotide substitution model with no codon partitioning to ensure proper mixing and convergence after experimenting with more complex models that showed signs of poor mixing and convergence. We performed the analysis using a total of 2.5 x 108 MCMC generations, sampling every 5000 generations, and discarded the first 20% as burn-in, based on likelihood stationarity visualized using Tracer v. 1.6 (Drummond and Rambaut, 2007). Phylogenetic trees were visualized and manipulated in R v. 3.2.0 (R Development Core Team, 2015) using the ape v. 3.3 (Paradis et al., 2004) and strap v. 1.4 (Bell and Lloyd, 2015) packages.
To further characterize the relationships among mitochondrial haplotypes and their frequencies within our dataset, we constructed a median-joining haplotype network using Network v. 4.613 (Bandelt et al., 2015; Bandelt et al., 1999). For this analysis, the mitochondrial alignment was further trimmed to eliminate any missing data located at the alignment ends (total alignment length was 878 bp). We used a recommended weighted transition:transversion ratio of 2:1 (per the Network manual) and used the maximum parsimony network method to minimize the number connections among haplotypes.
2.6 Mitochondrial estimates of landscape diversity and inter-clade gene flow among Boa populations
We assessed landscape-level patterns of genetic differentiation across the collective geographic range covered by our sampling, and individually on ranges occupied by the three major resolved population clusters (see Results section 3.1 for details). For this analysis we used only mitochondrial samples associated with precisely known collection localities (i.e., localities with geographic coordinate data or reliable descriptions for which coordinates could be well estimated; see Supplementary Table S4 for assignments) and applied a previously described methodology (Jezkova et al., 2015; Schield et al., 2015) that interpolates mitochondrial genetic distances across a geographic landscape and colors geographic regions based on the interpolated level of interpopulation genetic distance.
We used IMa2 (Hey and Nielsen, 2007) to estimate parameters of the isolation-migration model (Hey and Nielsen, 2004) between multiple island and mainland population pairs, and between populations east and west of the Isthmus of Tehuantepec (see Supplementary Table S4 for population assignments). We estimated burn-in to occur prior to 3.75 x 106 generations based on trial runs, and our full analyses included a total of 1.5 x 107 post burn-in MCMC generations, with sampling every 100 generations, and four independent runs per population comparison. We found these run times to be sufficient based on chain mixing and convergence, and parameter effective sample sizes >1000 for all parameters in each run. We rescaled parameter estimates into demographic units using generation time of three years (Lindemann, 2009) and a mitochondrial mutation rate estimate from Castoe et al. (2007).
2.7 Population genetic analyses of nuclear SNP data
We estimated the phylogenetic relationships among samples by inferring a maximum likelihood (ML) phylogeny using RAxML v. 8.1.20 (Stamatakis, 2014) with a GTR + Γ nucleotide substitution model with estimated base frequencies and 1000 bootstrap replicates (sensu Cariou et al., 2013). We visualized the resulting phylogeny and assessed bootstrap support using FigTree v. 1.4.2 (Rambaut, 2015).
We used NGSadmix (Skotte et al., 2013) and Entropy (Gompert et al., 2014), which are both similar to Structure (Pritchard et al., 2000), but leverage genotype likelihoods to infer admixture proportions across all samples and to investigate how ancestry may be partitioned under different numbers of assumed source populations (i.e., values of K population clusters). We conducted 10 independent runs for each value of K ranging from 1 to 11 and used the ΔK method (Evanno et al., 2005) to estimate the highest supported K value (i.e., the most likely number of source populations). Parallel runs were summarized using CLUMPP v. 1.1.2 (Jakobsson and Rosenberg, 2007) with the ‘greedy’ algorithm. Based on these results, we ran Entropy on a more targeted range of K from 1 to 8. We ran two MCMC chains for each value of K with 15,000 iterations per chain, with sampling every 5 iterations. We eliminated the first 20% of samples as burn-in and confirmed proper mixing and convergence before using Deviance Information Criteria (DIC) to determine the best-supported K value.
Based on the inferred genetic clustering of populations provided by NGSadmix and Entropy, we inferred population summary statistics for Central and North America populations. We used Stacks v. 1.34 (Catchen et al., 2011, 2013) to estimate nucleotide diversity (π), heterozygosity (H), and the inbreeding coefficient (FIS) at each locus, and determined the total number of private alleles per population. We also compared pairwise allelic differentiation (FST) between populations. This analysis was performed on a single Stacks-derived dataset (distinct from above-described SNP datasets) that we constructed from mapped RADseq data using the ref_map.pl tool and a minimum stack depth of 3. This dataset was filtered to allow for up to 50% missing data and retained loci with a minimum per-individual stack (i.e., read) depth of 10, resulting in 44,041 RAD loci.
We also tested for nuclear evidence of gene flow between major Boa lineages using TreeMix v. 1.12 (Pickrell and Pritchard, 2012). This analysis was conducted using population delineations informed from the results of several inferences (see Results and Supplementary Table S4). We allowed from zero to 12 migration events between lineages and calculated the fraction of the variance in relatedness between populations that is explained by each migration model.
2.8 Genome-wide Bayesian species delimitation of Boa
We used a subset of the total RADseq sampling to perform coalescent Bayesian species delimitation analysis (n = 33 samples; Supplementary Table S7). This subset was chosen to exclude individuals that contained higher levels of missing data (e.g., from low numbers of mapped reads), that when excluded did not result in major geographic/phylogenetic sampling gaps. We perform Bayes factor species delimitation using the BFD* method (Leaché et al., 2014) implemented using the SNAPP (Bryant et al., 2012) plugin for BEAST2. Overall, we tested three competing species models, including two “two species” models that lump either Central and North American populations (Model A) or Central and South American populations (Model B) into a single monophyletic species, and a third three species model that designates North, Central, and South American populations each as distinct species (Model C; Fig. 6 & Supplementary Table S7). These three models were informed by recent work (Hynková et al., 2009; Reynolds et al., 2014; Suárez-Atilano et al., 2014), and by our mitochondrial and nuclear analyses (see Results sections 3.1 and 3.3). For all three species models, we conducted path sampling for a total of 14 steps (100,000 MCMC steps, 10,000 burn-in steps each) to estimate marginal likelihoods for each competing model. Bayes factor support was compared between models to identify the best-supported species model. We visualized the best-supported species tree posterior from the final path sampling step (minus a 10% burn-in) using DensiTree v. 2.2.1 (Bouckaert, 2010).
Figure 6. Results from Bayes Factor comparisons of alternative species models.
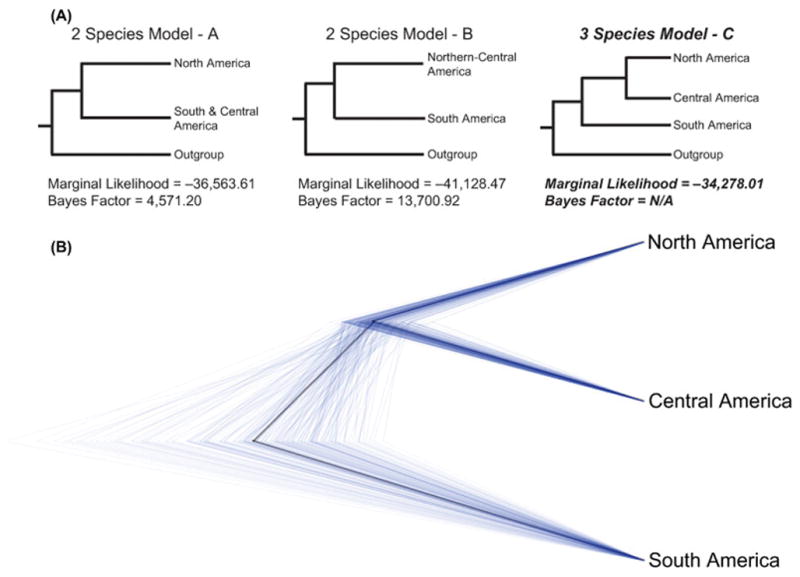
(A) Simplified trees showing the species model hypotheses tested using the BFD* framework and the Bayes Factor support obtained under each model. Outgroups are displayed only to aid comprehension and were not incorporated into any of the models. The best-supported species model and associated support values are bolded and italicized. (B) DensiTree of posterior estimates of the highest-supported species tree.
3. Results
3.1 Mitochondrial patterns of population structure, relationships, and divergence timing
The mitochondrial cyt-b alignment contained 305 total in-group samples and 1059 aligned bases. There were a total of 301 polymorphic sites and 250 total informative sites across the alignment. Phylogenetic inference in BEAST 2 resolved deeper relationships among Boa samples with high support (defined as >95% posterior support hereafter), but recent nodes received far less posterior support (Fig. 1). There was high posterior support for a sister relationship between a clade comprising Boa samples from Colombia and the remaining populations of Boa. Following this basal split, the core Boa radiation contains a highly supported split between South and Northern-Middle America (Fig. 1–2). Within the South American clade, there is also high support for two Ecuadorian samples being sister to the rest of the clade. A clade of Argentinian samples is resolved as the sister group to all other remaining samples, which includes individuals from Peru, Brazil, Guyana, and Surinam.
Figure 1. Phylogenetic patterns of population division within the genus Boa.
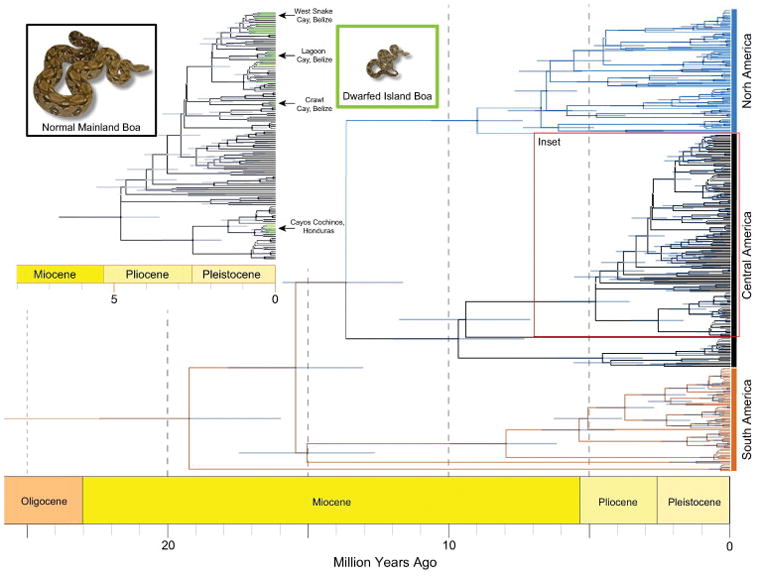
BEAST2 cladogram inferred using the Fossilized Birth-Death model with node bars reflecting the 95% HPD. Branches have been colored and annotated to reflect the broad geographic assignments of the major BCSC clades. The inset figure provides a high resolution view of Central American populations that contain island dwarf populations, with branches to these samples highlighted in bright green. Node symbols are colored according to posterior support: black = >95%, grey = 75% – 95%, white = 50% – 75%, and no symbols = <50%.
Figure 2. Geographic delimitation of major clades within the genus Boa.
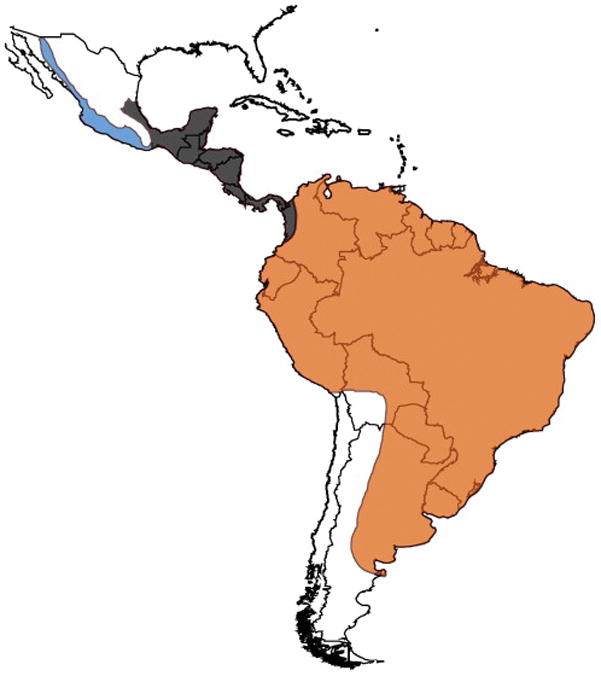
The three major of Boa snakes are localized roughly to the three major New World landmasses: South America, Central America (including parts of Colombia and the Gulf Coast of Mexico), and North America (the Pacific Coast of Mexico to the west of the Isthmus of Tehuantepec). Geographic ranges are colored to correspond to major clades outlined in Fig. 1 and 3.
Among Northern-Central American sampling, we found strong support for two mitochondrial clades. One clade includes samples from nuclear Central America, including localities that extend from northern South America through the Isthmus of Panama to the Isthmus of Tehuantepec, and along the Gulf coast of Mexico. The second clade includes samples west of the Isthmus of Tehuantepec, along the Pacific coast of Mexico (Fig. 1–2). Samples from Oaxaca, Mexico, located at the boundary between these two clades, fall into both of these two large clades, indicating a potential zone of introgression between these lineages in this region. Among island populations sampled, individuals from the Cay islands of Belize fall within one subclade of the Central American clade, while samples from Cayos Cochinos Menor in Honduras clustered with mainland samples from another subclade within the Central American clade. The split between these two Central American subclades is highly supported (see inset of Fig. 1).
We estimated the oldest split between the Boa clade containing Colombian samples and the rest of the Boa complex to have occurred almost 20 million years ago (Mya; 95% highest posterior density [HPD] = ca. 16 to 22.4 Mya) with a subsequent split between the North American and Northern-Central American clades occurring approximately 16 Mya (95% HPD = ca. 13.0 to 17.8 Mya). Within the well-resolved South American clade, we estimated the split between the Argentinian clade and its sister lineage to have occurred ca. 8 Mya (95% HPD = ca. 6.2 to 9.9 Mya). Other well-resolved divergences (i.e., > 95% posterior support) within the South American clade ranged from ca. 6 to 2 Mya. The split between the two Northern-Central American clades is estimated to have occurred 14 Mya (95% HPD = ca. 11.6 to 15.9 Mya), with subsequent splits in both lineages ranging from 5 to 10 Mya. The well-supported divergence between the two clades containing dwarfed island populations are estimated to have occurred 5 Mya (95% HPD = ca. 3.6 to 6.1 Mya), and 95% HPD ranges indicate that individual island divergences occurred within the past 1 My (Fig. 1).
3.2 Landscape patterns of mitochondrial diversity and admixture across populations
Pairwise mitochondrial genetic distance interpolations highlight several regions across the distribution of the genus Boa that contain particularly high genetic diversity. In South America, there is a region of high genetic diversity in Colombia, which coincides with the distribution of a deeply divergent lineage of Colombian Boa mitochondrial haplotypes that are sister to all Boa lineages in our mitochondrial tree (Fig. 3A). In Central America, regions of northern Honduras contain high average pairwise genetic distances (> 0.02). In North America, areas along the Pacific coast of Mexico also show average pairwise genetic distances higher than 0.02 (Fig. 3A). These results are corroborated by our haplotype network analysis, which indicated high levels of haplotype diversity in the North American and Central American clades overall, including these populations specifically (Fig. 3B). We also found high haplotype diversity within the South American clade. North American populations along the Pacific coast of Mexico show haplotype diversity patterns similar to South American populations, which coincide with the high levels of landscape genetic distances observed in the region (Fig. 3B).
Figure 3. Landscape patterns of mitochondrial genetic diversity and estimates of interpopulation gene flow.
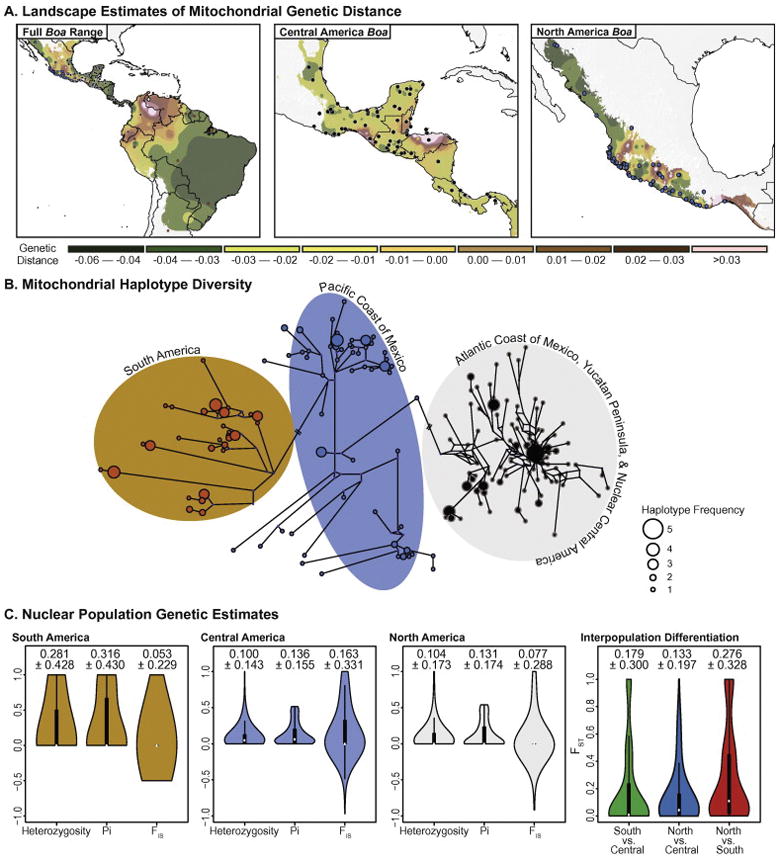
(A) Residual pairwise mitochondrial genetic distances interpolated across landscape for all Boa clades, the Central American clade, and the North American clade. (B) Median-joining haplotype network inferred using cyt-b haplotypes, with major geographic assignments indicated. (C) Violin plots of genome-wide estimates of interpopulation genetic statistics (Pi, Heterozygosity, and FIS) for South America, Central America, and North America, and of interpopulation genetic differentiation (FST) between each pairwise clade. For each violin plot, the white point indicates the median value and the black box indicates the interquartile range. The mean and standard deviations are reported above each respective violin plot.
Estimates of gene flow inferred using mitochondrial data and the Isolation-Migration model show evidence of gene flow from mainland populations to islands (approximately 1 – 20 migrants per generation; Supplementary Fig. S1A–C). In contrast, all three mainland-island comparisons provided no evidence of migration from any island to its respective mainland population. We also found no evidence of migrants shared between populations east and west of the Isthmus of Tehuantepec (Supplementary Fig. S1D), which contrasts with the phylogenetic findings that indicate admixture across the isthmus.
3.3. Patterns of population structure and relationships from nuclear SNP data
We recovered an average of 1.96 million quality-filtered (1.74 million mapped) Illumina reads per sample (Supplemental Table S5). Overall, three separate analyses – phylogenetic reconstruction using RAxML, admixture analyses from NGSadmix and Entropy, and inferences of population splits and mixtures using TreeMix – provide strong nuclear support for three distinct clades of Boa. The maximum likelihood analysis of concatenated SNPs inferred strong support for three major continental clades, mirroring the results from mitochondrial analyses (Fig. 4A), but also revealed considerable intra-clade lineage diversity, including two well-supported clades in Central America that each include island populations. In the Northern-Central American clade, analyses largely confirmed observations from the mitochondrial data. Populations along the Pacific coast of Mexico and Guatemala are distinct from those in the rest of Central America based on phylogenetic results (Fig. 4A). One major discordance between our mitochondrial and nuclear phylogenies, however, was that samples from the Pacific coast of Guatemala phylogenetically clustered with North American samples in Mexico (Fig. 4A), which conflicts with the Central American assignment evident in the mitochondrial phylogeny (Fig. 1).
Figure 4. Population structuring and relationships inferred from nuclear RADseq data.
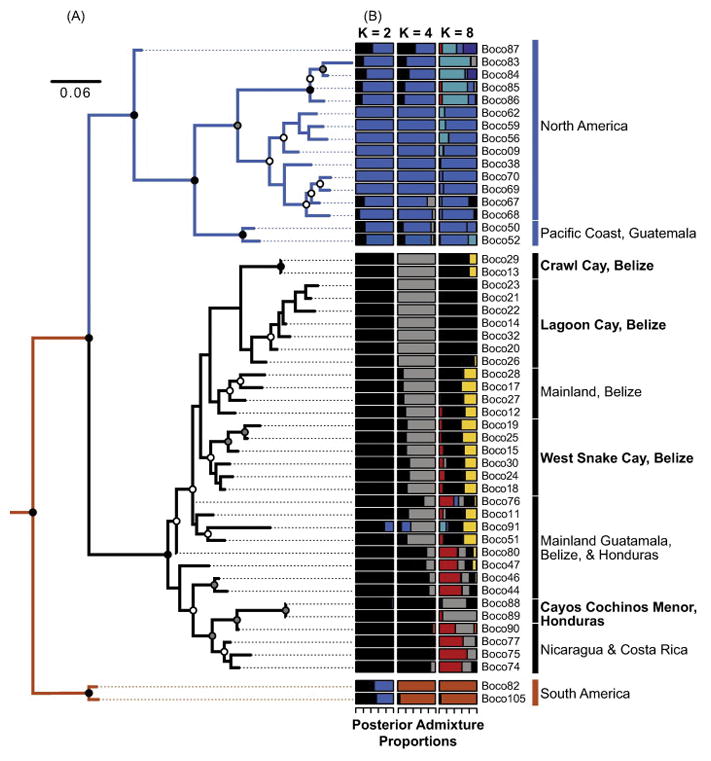
(A) Maximum likelihood phylogeny inferred from RAxML analysis of the nuclear SNP alignment with a topology, and color annotations, mirroring that of the mitochondrial phylogenies. Nodes symbols are colored according to bootstrap support: black = >95%, grey = 75% – 95%, white = 50% – 75%, and no symbols = <50%. (B) Admixture graphs K = 2, K = 4, and K = 8 allowed source populations inferred in Entropy.
The ΔK test of NGSadmix results supported an optimal model with two source populations, which divides the two Northern-Central American clades, with samples from South America clustering more closely with North America samples (Supplementary Fig. S2; Supplementary Table S8). Similar patterns of population assignment and ancestry proportions were obtained from the results of population clustering using the Bayesian framework implemented in Entropy, though these analyses favor an optimal model of K = 8 source populations based on comparisons of DIC values (Fig. 4B; Supplementary Fig. S3; Supplementary Table S8). Results from our population clustering analyses largely agree with phylogenetic results, even as additional source populations are allowed, and assignments to additional population clusters are intuitive given sampling geography (Supplementary Figs. S2–S3).
Population allelic differentiation inferred from nuclear SNPs is high between both the South America to Central America, and North America to Central America pairwise population comparisons (mean FST = 0.179 ± 0.300 standard deviation [SD] and FST = 0.133 ± 0.197 SD, respectively; Fig. 3C). We examined broad intra-clade genetic diversity in Central America using genome-wide SNP data and found modest levels of nucleotide diversity (mean = 0.136 ± 0.155 SD) and heterozygosity (mean = 0.100 ± 0.143 SD; Fig. 3C). Similar measures were observed in the North American Boa clade, as mean (± SD) nuclear nucleotide diversity and heterozygosity were 0.131 (± 0.174) and 0.104 (± 0.173), respectively (Fig. 3C). We found greater levels of nucleotide diversity and heterozygosity in the South American clade (mean = 0.316 ± 0.430 SD and 0.281 ± 0.428, respectively; Fig. 3C) than in either of the northern clades. Inbreeding coefficients were relatively high in Central America (mean = 0.163 ± 0.331 SD) compared to both South America (mean = 0.053 ± 0.229 SD) and North America (mean = 0.077 ± 0.228 SD). We observed 2,059 private alleles in the South American clade, 7,210 private alleles in the Central American clade, and 1,683 private alleles in the North American clade.
We found strong additional support for independent island population establishment (from the mainland) beyond the evidence already presented from phylogenetic and population clustering analyses (see above and Figs. 1 and 3). Moderately high population allelic differentiation (FST) is evident between pairwise comparisons of island and mainland populations, and varied from an average of 0.045 to 0.058 (Supplementary Fig. S4). FST estimates are lower between islands in Belize than between any pairwise comparison between islands in Belize and Cayos Cochinos Menor in Honduras (Supplementary Fig. S4), which is consistent with the large geographic distance between these two distinct island systems (ca. 200 km straight-line distance across ocean). Substantially different levels of heterozygosity, nucleotide diversity, and inbreeding coefficients were observed between island and mainland populations, but these intra-population statistics are consistent across island populations (Supplementary Fig. S4). We found that the mainland populations across Central America collectively contained the highest number of private alleles (6,403), while Crawl Cay, Belize and Cayos Cochinos Menor, Honduras contained modest numbers of private alleles (1,847 and 1,248, respectively), and Lagoon and West Snake Cays in Belize contained relatively few private alleles (489 and 270, respectively).
Overall, TreeMix produced a phylogeny which was similar to that based on nuclear phylogenetic analysis (Fig. 5). The amount of variance explained by the model plateaued at M=2 migration events, which explained about 99% of the variance in the dataset. The analysis supported an admixture event from a Central American population to the Guatemalan population of the North American clade. The second supported migration event was from a population ancestral to the Guatemalan population in North America to the mainland population of the Belize clade in Central America (Fig. 5C). These admixture events comprise a high (48%) and low (6%) portion of the recipient population ancestry, respectively (Fig. 5C).
Figure 5. Nuclear patterns of population divergence and gene flow from TreeMix.
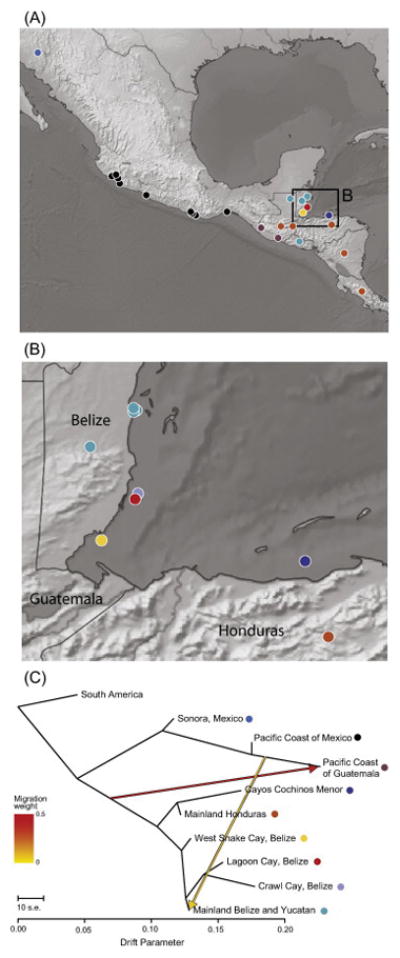
(A) Map of Northern-Central American nuclear sampling with samples color coded by population assignment (inferred from Fig. 4). (B) Fine-scale map of island and adjacent mainland sampling. (C) TreeMix population tree for the more stringent nuclear SNP dataset, which mirrors the topology observed in Figure 3A. The populations are color coded according to major population assignment in A and B. The drift parameter is ten times the average standard error of the estimated entries in the sample covariance matrix. Migration arrows are colored according to a weight that represents the fraction alleles in the descendent population that originated in the parental population. A model with two migration edges received the highest support – one from the Pacific Coast of Mexico and Guatemala to the Yucatan region and mainland Belize, and one from Central America to the Pacific Coast of Guatemala.
3.4. Results of Bayesian species delimitation
Marginal likelihood estimation and Bayes factor comparison of three competing species models found strong statistical support for a three species model that delineated Boa samples into a North American, Central American, and South American species (Model C, ln(Marginal Likelihood) = −34,278.01; Fig. 6). These three species designations largely coincide with our phylogenetic and population genetic analyses that show substantial lineage independence and divergence of these clades.
4. Discussion
4.1. Evidence for extensive lineage diversity and three species of Boa
Our results provide evidence from both mitochondrial and nuclear data that there are at least three well-differentiated species within the genus Boa. These three lineages correspond approximately to the three major landmasses of the Western Hemisphere inhabited by boas: North America (the Pacific coast of Mexico), Central America (including the Gulf coast of Mexico), and South America (Fig. 1–2). Mitochondrial data indicate a sharp division between individuals in the South American and Central American clades that appears to occur at the junction of lower Central America and South America. The transition from the Central American to the North American clade appears to be more diffuse, as mitochondrial haplotypes near the Isthmus of Tehuantepec in Oaxaca, Mexico fall in both the Central American and North American clades, suggesting potential gene flow between clades in this region. These same general patterns have been observed by Hynková et al. (2009) and, with much greater resolution, by Suárez-Atilano et al. (2014), whose mitochondrial datasets have been included in our own analyses. With our additional sampling of this region, we observed similar patterns and find additional evidence of mitochondrial admixture localized to areas surrounding the Isthmus of Tehuantepec.
Our nuclear SNP sampling, although geographically focused on Central America and Mexico, provides further support for three distinct species-level lineages of Boa. Our maximum likelihood analysis of the concatenated SNP alignment yielded a similar topology to that obtained from the more geographically well-sampled mitochondrial data (except for the deep divergence of some Colombian lineages from the mitochondrial data, discussed below). Multiple genetic clustering analyses indicate that at least three major genetic clusters exist within Boa, including a strong distinction between central-northern Mexican and Central American populations consistent with our North American and Central American mitochondrial clades. Based on our nuclear SNP data and mitochondrial IMa2 results, we found minimal evidence of admixture between lineages on either side of the Isthmus of Tehuantepec, which was somewhat surprising given indications of admixture from the mitochondrial data and previous results from microsatellites presented by Suárez-Atilano et al. (2014). Landscape diversity estimates based on mitochondrial data also indicate a pattern of high diversity in this region, highlighting the confluence of two highly distinct lineages there. Additional investigation with greater sampling from this region would help to establish the extent to which these populations are introgressing and the precise geographic boundaries of this apparent admixture zone.
A recent formal taxonomic revision of Boa constrictor (sensu lato) was conducted by Reynolds et al. (2014) in the context of a broad scale analysis of all Boid and Pythonid snakes. In their study they used two pet trade individuals from Puerto Rico, which had previously been examined in a continental context (Reynolds et al., 2013), to split the genus Boa into B. constrictor and B. imperator. Our sampling encompassed these two samples, and interestingly, we find that one individual clusters with the enigmatic Boa mitochondrial lineage containing samples from Colombia, which is sister to all other populations of Boa in our mitochondrial trees. The second sample, however, clusters with samples from Mexico. The fact that these samples, and many from Hynková et al. (2009), were from the pet trade is problematic because true sample provenance may be unclear or possibly erroneous. Nonetheless, the finding some Colombian samples form a lineage sister to all other boa in the mitochondrial phylogeny populations requires further investigation to determine if these are indeed mitochondrial sequences (versus nuclear inserts of mitochondrial genes; NUMTs; see Hazkani-Covo et al. (2010) for a review), deep coalescence of ancient mitochondrial haplotypes, or if these populations do indeed represent a fourth divergent lineage of Boa. These questions, however, fall outside the scope of the present study due to a lack of high-quality samples with known locality data from Colombia. Future studies that incorporate nuclear SNP sampling for Colombian and other South American samples would be valuable for further investigating patterns of Boa diversity.
Given both our nuclear and mitochondrial results, as well as previous work indicating the likelihood of multiple species-level lineages of Boa (Hynková et al., 2009; Reynolds et al., 2014; Suárez-Atilano et al., 2014), we were interested in explicitly testing three alternative models of species recognition for Boa lineages. Bayes Factor delimitation of the genome-wide SNP dataset rejected both of the alternative two species hypotheses that lumped either Central and North American clades (Model A) or Central and South American clades (Model B) into single species. Instead, Bayes factor comparisons overwhelmingly supported a three species model for the genus Boa in which North, Central and South American clades each represent distinct species (Model C). These results are highly consistent with our analyses of mitochondrial and nuclear variation and provide yet another level of support for the recognition of at least three species within the genus Boa.
Both our mitochondrial and nuclear analyses indicate that taxonomic revisions are necessary within the genus Boa. This genus has previously been recognized as monotypic, Boa constrictor, with 7 recognized subspecies (Uetz and Etzold, 1996; Uetz and Hošek, 2015). Based on mitochondrial data, Reynolds et al. (2014) elevated the subspecies B. c. imperator, comprising populations in Central and North America, to B. imperator. This change was previously suggested by Hynková et al. (2009). Suárez-Atilano et al. (2014) described greater population diversity and divergence across North and Central American populations, and concluded that the two major lineages in this region comprise evolutionary significant units, though did not make taxonomic recommendations. Our population clustering analyses, phylogenetic inference, and coalescent-based species delimitation methods spanning both mitochondrial and nuclear datasets provide multiple lines of evidence for three major lineages within the genus Boa. We recognize the South American lineage as B. constrictor and the Central American lineage (including South American populations in the Choco of Colombia and Ecuador [and probably Peru], and North American populations along the Gulf coast of Mexico [west of the Isthmus of Tehuantepec]) as B. imperator, in line with previous taxonomic discussions (Hynková et al., 2009; Reynolds et al., 2014; Suárez-Atilano et al., 2014). We recognize the North American lineage, comprising Mexican populations along the Pacific coast west of the Isthmus of Tehuantepec, as B. sigma (Smith, 1943). The taxon Constrictor c. sigma was described based on three specimens from María Madre Island, Tres Marías Islands, Nayarit, Mexico by Smith (1943); types: CAS 58681, USNM 24672 46484 [holotype]). The description notes that this population has the highest ventral counts of any other Boa population in Mexico, this character difference serving as diagnostic for the new taxon. Smith apparently was unaware that Slevin (1926) had mentioned the presence of the same taxon for María Magdalena Island. Zweifel (1960) reported on an American Museum expedition to the Tres Marías Islands and found seven more individuals, including specimens from María Madre, María Magdalena, and María Cleofas. In this publication Zweifel argues for the recognition of B. c. sigma as a junior synonym of Boa c. imperator based on expanded variation of ventral scale counts in the Tres Marías populations, which overlaps that found on the mainland (253 – 260 vs. 225 – 253 in the mainland of Mexico [including Pacific and Atlantic populations]). The Tres Marías population barely overlaps with the mainland in ventral counts, by one in nine specimens versus 41 from the mainland (given by Smith 1943). Although, we lack genetic sampling from the Tres Marías Islands, given our finding of a distinct species found in Western Mexico, B. sigma is the only available name we can unambiguously apply to a population within this North American lineage. Our taxonomic recommendation is to recognize B. sigma (Smith, 1943) as full species, encompassing the Western Mexico lineage. Finally, we acknowledge that further population-level investigations and analyses of morphology should be conducted to reinforce this recommendation.
4.2. Divergence time estimates and historical biogeography
Boid snakes in general, and the genus Boa in particular, are considered to be South American in origin, based on Gondwanan vicariance models of boine biogeography (e.g., (Noonan and Chippindale, 2006a, b), which are also consistent with early boid fossils from Colombia (Head et al., 2009) and a highly diverse boid radiation in South America (Burbrink, 2005; Noonan and Chippindale, 2006a). Using the newly-developed FBD model of divergence time estimation, we estimated the divergence between South American and Northern-Central American lineages at approximately 16 Mya (95% HPD = ca. 13.0 to 17.8 Mya), well earlier than findings from Suárez-Atilano et al. (2014), which place the split at 7.4 Mya (95% HPD = ca. 6.2 to 9.9 Mya). This divergence time substantially predates the historically recognized date of the closure of the Isthmus of Panama (estimated to occur ca. 5 Mya; Haug and Tiedemann, 1998; Haug et al., 2001; Keigwin, 1982; Ravelo et al., 2004), but also falls prior to a newly articulated date for the closure of the Isthmus of Panama (13 – 15 Mya; Montes et al., 2015). This suggests that boas may have successfully colonized Central America before the Panamanian land bridge was formed, an inference that is consistent with a Miocene Boa fossil known from Panama that was dated at 19.3 Mya (Head et al., 2012). Similarly, divergence times between other major Boa clades are also older than in previous estimates, as the split between the two major Northern-Central American clades is estimated to have occurred shortly after boas presumably colonized this landmass, at approximately 14 Mya (95% HPD = ca. 11.6 to 15.9 Mya). It is notable that this split may represent two coastal expansion fronts that moved northward through Central America, which were isolated by transcontinental mountain ranges. Even within the Central American clade, we find relatively deep divergences (ca. 5 – 10 Mya) among subclades, and thus significant population diversity that may warrant further investigation and taxonomic recognition, that indicates a long history of in situ Boa evolution in Central America. Lastly, mito-nuclear discordances in phylogenetic (including divergence timing) estimates have been recognized (see Toews and Brelsford (2012) for a review) and divergence estimates from a single gene is known to be difficult (Arbogast et al., 2002; Graur and Martin, 2004), facts that we acknowledge. However, given the concordance between our divergence estimates and limited fossil evidence, we believe our estimates are reasonable and may be even more realistic than the much younger divergence estimates from previous studies of Boid snakes (Noonan and Chippindale, 2006a, b; Suárez-Atilano et al., 2014).
4.3. Support for independent insular dwarfism in Central American Boa
Our results provide evidence that dwarf forms of boas that occur on multiple islands off the coast of Central America – from coastal islands in Belize and on Cayos Cochinos Menor in Honduras – have independent evolutionary origins. With regard to community assembly, this is not surprising, as it has been established that offshore islands are usually populated by the most common mainland species (Burbrink et al., 2015). However, it is particularly exciting that the dwarfed phenotype appears to be a product of convergent evolution, whereby similar insular ecosystems have independently selected for similar dwarf phenotypes. Mitochondrial haplotypes of individuals from these two separate island groups cluster within distinct highly-supported clades that are estimated to have diverged from one another approximately 5 Mya. A similar pattern is observed in our nuclear SNP-based phylogeny, where we find strong nodal support for the split between these two larger Central American clades, each of which includes one of the two groups of islands. Patterns observed from our SNP-based population cluster analyses also resolve these two population groups into separate distinct clusters, though there is some evidence of admixture across islands and Central American mainland source populations under various population models that we speculate represents standing genetic variation from the adjacent mainland populations more than recent gene flow (especially between islands).
Isolation-Migration analyses indicate that gene flow between the island and adjacent mainland populations is essentially unidirectional, from mainland to island in each of the two island systems. The broad posterior estimate on gene flow indicates a great deal of uncertainty in the degree of gene flow between island and mainland populations and is likely a product of small sample sizes, data from a single mitochondrial gene, and the confounding effects of multiple historical periods of gene flow and isolated with sea level change. Patterns of diversity in nuclear SNPs also indicate small effective population sizes on these islands that have likely allowed drift to substantially alter allele frequencies to the extent that pairwise allelic divergence (FST) is quite high between each island and mainland pair. This pattern is consistent with small empirical estimates of population sizes on the Belize islands (Boback, 2005) and on Cayos Cochinos Menor (Reed et al., 2007). Collectively our results support the hypothesis that evolutionary processes, including the evolution of dwarf phenotypes, have occurred in parallel between the two independent island population groups.
While drift is likely driving the majority of genetic differentiation in these island populations, it is likely that a subset of genetic differentiation observed between island and mainland populations may also be due to selection associated with these unique island ecosystems, which includes selection driving the evolution of dwarfism and other specialized phenotypes on these islands (Boback, 2003; Boback, 2005, 2006). Indeed, common garden experiments using dwarfed snakes from several Belize islands indicates that selection has favored genetic changes that are apparently causing dwarfism (Boback and Carpenter, 2007), a scenario also supported by the maintenance and breeding of dwarfed Boa from Cayos Cochinos and elsewhere in the pet trade. Beyond these two island systems, Boa populations exist on at least 50 near offshore islands (Henderson et al., 1995), and other known (but unsampled) populations of island dwarf populations exist from islands that are more widely geographically separated from those in our study. Collectively, this suggests that there is very likely to be more than two independent instances where island dwarfism evolved, though the proportion explained by genetic underpinnings versus phenotypic plasticity remains to be explored.
Conclusion
Our genome-wide nuclear and single-locus mitochondrial datasets both identified extensive population structure across the range of the genus Boa. Multiple lines of evidence indicate that there are (at least) three widely distributed clades, and each clade roughly corresponds to three major landmasses of the Western Hemisphere – North, Central, and South America. Our data also confirm results and taxonomic suggestions from previous studies, and further warranted the recognition of a third species in the genus Boa, B. sigma, corresponding to the North American clade. Additional studies using molecular data would be desirable to further test the hypothesis that the Mexican island populations from which the type specimens of B. sigma originate (Tres Marías) represent the same taxon as adjacent mainland Boa populations. Expanded sampling for South American Boa populations, especially those in Colombia where mitochondrial lineage diversity is high, would also be important for addressing outstanding questions about lineage diversity in Boa. Lastly, our data suggest two apparently independent instances of the evolution of dwarfism in Boa populations inhabiting offshore islands (in Belize and Cayos Cochinos Menor, Honduras) implicating substantial morphological convergence among these populations.
Supplementary Material
Supplementary Table S1. Voucher or collector identifiers for all new samples included as part of this work. Captive or pet animals lacking vouchers are encoded by ‘--’. UTA-ARDRC = University of Texas at Arlington Amphibian and Reptile Diversity Research Center.
Supplementary Table S2. All samples used for this study, along with the source of the data and the corresponding locality information. Missing or unsampled data is encoded by ‘--’. In some cases, coordinates were approximated for analyses by using coordinates at approximately the middle of the country or state/province (indicated with * following the coordinates) or using coordinates inferred from recorded locality data (indicated with ** following the coordinates).
Supplementary Table S3. Outgroup species used in the mitochondrial phylogenetic analysis. NCBI Genbank accession for the cyt-b sequence and the citation where the data was originally used are also included.
Supplementary Table S4. A summary of which ingroup samples were used for individual mitochondrial and nuclear genetic analyses. Data not included in an analysis – due either to not being collected or being excluded (see Materials and Methods) – is encoded by ‘--’.
Supplementary Table S5. Statistics on the number of high-quality and mapped Illumina reads for each sample with nuclear data.
Supplementary Table S6. Fossil data, including Paleobiology database collection number, date used, clade constrained, and relevant citation, used for divergence dating under the Fossilized Birth-Death model.
Supplementary Table S7. Sample assignments used for the three models compared using Bayesian Species Delimitation.
Supplementary Table S8. Summary table of highest supported number of source populations (K) deduced using the ΔK framework for NGSadmix and a DIC framework for Entropy. The values indicated the highest support for particular analyses are bolded.
Posterior density estimates of reciprocal migration parameters from the Isolation-Migration model for (A) Lagoon Cay island and mainland Belize, (B) West Snake Cay island and mainland Belize, (C) Cayos Cochinos Menor island and mainland Honduras, and (D) populations to the east and west of the Isthmus of Tehuantepec in Mexico.
Admixture graphs for all K = 2 – 8 allowed source populations inferred using the stringently filtered SNP dataset and NGSadmix. Samples are labeled based on the sample ID and the coarse locality information for the sample. In most cases, the province of the sample is included and “Crawl”, “Lagoon”, “WSnake” and “Cochinos” refer to Crawl, Lagoon, and West Snake Cays in Belize and to Cayos Cochinos Menor in Honduras, respectively. Nation names are abbreviated according to the ISO three-letter country codes.
Admixture graphs for all K = 2 – 8 allowed source populations inferred using the stringently filtered SNP dataset and Entropy. Samples are labeled based on the sample ID and the coarse locality information for the sample. In most cases, the province of the sample is included and “Crawl”, “Lagoon”, “WSnake” and “Cochinos” refer to Crawl, Lagoon, and West Snake Cays in Belize and to Cayos Cochinos Menor in Honduras, respectively. Nation names are abbreviated according to the ISO three-letter country codes.
Violin plots of genome-wide estimates of Pi (A), Heterozygosity (B), and FIS (C) for island and mainland populations in Central America and of FST (D) between each pairwise clade. For each violin plot, the white point indicates the median value and the black box indicates the interquartile range. The mean and standard deviations for each plot are evident above respective violin plots.
Acknowledgments
We thank Timothy Colston and Dr. Chad Montgomery for tissue samples and the numerous researchers at the University of Texas at Arlington who collected and deposited Boa tissue samples at the Amphibian and Reptile Diversity Research Center, which formed the basis of this work. Funding for fieldwork was provided in part by NSF grants DEB-0613802 and 0102383 to J. A. Campbell (whom we thank for making tissues available), and NSF DEB-0416160 and Bioclon grants to ENS. Collecting permits in Mexico were issued by the Secretaría de Medio Ambiente y Recursos Naturales (SEMARNAT) to V. León-Regañon and O. Flores-Villela, and we thank them for their help while working in Mexico. We thank Kyle Shaney, Nicole Hales, and Nicole Proctor for their help in the laboratory, and Chris Nice for assistance with SNP calling and filtering. We thank Mark Liu for the photographs used in Figure 1. Support was provided from startup funds from the University of Texas at Arlington to TAC and from a National Science Foundation Doctoral Dissertation Improvement Grant (DEB 1401747) to TAC and DCC.
References
- Arbogast BS, Edwards SV, Wakeley J, Beerli P, Slowinski JB. Estimating Divergence Times from Molecular Data on Phylogenetic and Population Genetic Timescales. Annual Review of Ecology and Systematics. 2002;33:707–740. [Google Scholar]
- Bandelt H-J, Forster P, Röhl A. Network. 2015. [Google Scholar]
- Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology & Evolution. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Barbour T. Vertebrata from the savanna of Panama. IV. Reptilia and Amphibia. Bulletin of the Museum of Comparative Zoology at Harvard College. 1906;46:224–229. [Google Scholar]
- Bauer AM. African-South American relationships: a perspective from the Reptilia. In: Goldblatt P, editor. Biological Relationships between African and South America. Yale University Press; New Haven, CT: 1993. pp. 244–288. [Google Scholar]
- Bell MA, Lloyd GT. strap: an R package for plotting phylogenies against stratigraphy and assessing their stratigraphic congruence. Palaeontology. 2015;58:379–389. [Google Scholar]
- Boback S, Carpenter M. Body size and head shape of island Boa constrictor in Belize: environmental versus genetic contributions. In: Henderson RW, Powell R, editors. Biology of the Boas and Pythons. Eagle Mountain Publishing; Eagle Mountain, UT: 2007. pp. 102–117. [Google Scholar]
- Boback SM. Body Size Evolution in Snakes: Evidence from Island Populations. Copeia. 2003;2003:81–94. [Google Scholar]
- Boback SM. Natural history and conservation of island boas (Boa constrictor) in Belize. Copeia. 2005;2005:879–884. [Google Scholar]
- Boback SM. A morphometric comparison of island and mainland boas (Boa constrictor) in Belize. Copeia. 2006;2006:261–267. [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Computational Biology. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert RR. DensiTree: making sense of sets of phylogenetic trees. Bioinformatics. 2010;26:1372–1373. doi: 10.1093/bioinformatics/btq110. [DOI] [PubMed] [Google Scholar]
- Bradnam K, Fass J, Alexandrov A, Baranay P, Bechner M, Birol I, Boisvert S, Chapman J, Chapuis G, Chikhi R, Chitsaz H, Chou WC, Corbeil J, Del Fabbro C, Docking T, Durbin R, Earl D, Emrich S, Fedotov P, Fonseca N, Ganapathy G, Gibbs R, Gnerre S, Godzaridis E, Goldstein S, Haimel M, Hall G, Haussler D, Hiatt J, Ho I. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. GigaScience. 2013;2:10. doi: 10.1186/2047-217X-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouat C, Chevallier H, Meusnier S, Noblecourt T, Rasplus JY. Specialization and habitat: spatial and environmental effects on abundance and genetic diversity of forest generalist and specialist Carabus species. Molecular Ecology. 2004;13:1815–1826. doi: 10.1111/j.1365-294X.2004.02206.x. [DOI] [PubMed] [Google Scholar]
- Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, RoyChoudhury A. Inferring Species Trees Directly from Biallelic Genetic Markers: Bypassing Gene Trees in a Full Coalescent Analysis. Molecular Biology & Evolution. 2012;29:1917–1932. doi: 10.1093/molbev/mss086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbrink FT. Inferring the phylogenetic position of Boa constrictor among the Boinae. Molecular Phylogenetics & Evolution. 2005;34:167–180. doi: 10.1016/j.ympev.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Burbrink FT, Lawson R, Slowinski JB. Mitochondrial DNA phylogeography of the polytypic North American rat snake (Elaphe obsoleta): A critique of the subspecies concept. Evolution. 2000;54:2107–2118. doi: 10.1554/0014-3820(2000)054[2107:MDPOTP]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Burbrink FT, McKelvy AD, Pyron RA, Myers EA. Predicting community structure in snakes on Eastern Nearctic islands using ecological neutral theory and phylogenetic methods. Proceedings of the Royal Society of London B: Biological Sciences. 2015;282:20151700. doi: 10.1098/rspb.2015.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariou M, Duret L, Charlat S. Is RAD-seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecology and Evolution. 2013;3:846–852. doi: 10.1002/ece3.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoe TA, Spencer CL, Parkinson CL. Phylogeographic structure and historical demography of the western diamondback rattlesnake (Crotalus atrox): A perspective on North American desert biogeography. Molecular Phylogenetics & Evolution. 2007;42:193–212. doi: 10.1016/j.ympev.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Molecular Ecology. 2013;22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3: Genes, Genomes, Genetics. 2011;1:171–182. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colston TJ, Grazziotin FG, Shepard DB, Vitt LJ, Colli GR, Henderson RW, Blair Hedges S, Bonatto S, Zaher H, Noonan BP, Burbrink FT. Molecular systematics and historical biogeography of tree boas (Corallus spp.) Molecular Phylogenetics & Evolution. 2013;66:953–959. doi: 10.1016/j.ympev.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Cope ED. Synopsis of the Cold Blooded Vertebrata, Procured by Prof. James Orton during His Exploration of Peru in 1876–77. Proceedings of the American Philosophical Society. 1877;17:33–49. [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R 100 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daudin FM, Sonnini CS. Histoire naturelle, générale et particulière des reptiles: ouvrage faisant suite à l’histoire naturelle générale et particulieère, composée par Leclerc de Buffon, et rédigée par CS Sonnini, membre de plusieurs sociétés savantes. Dufart: 1803. [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Eydoux F, Souleyet FA. Voyage autour du monde execute pendant les annees 1836 et 1837 sur la vorvette “La Bonite”, commandee par M. Vaillant 1842 [Google Scholar]
- Fields PD, Reisser C, Dukić M, Haag CR, Ebert D. Genes mirror geography in Daphnia magna. Molecular Ecology. 2015;24:4521–4536. doi: 10.1111/mec.13324. [DOI] [PubMed] [Google Scholar]
- Gavryushkina A, Welch D, Stadler T, Drummond AJ. Bayesian Inference of Sampled Ancestor Trees for Epidemiology and Fossil Calibration. PLoS Computational Biology. 2014;10:e1003919. doi: 10.1371/journal.pcbi.1003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompert Z, Lucas LK, Buerkle CA, Forister ML, Fordyce JA, Nice CC. Admixture and the organization of genetic diversity in a butterfly species complex revealed through common and rare genetic variants. Molecular Ecology. 2014;23:4555–4573. doi: 10.1111/mec.12811. [DOI] [PubMed] [Google Scholar]
- Graur D, Martin W. Reading the entrails of chickens: molecular timescales of evolution and the illusion of precision. Trends in Genetics. 2004;20:80–86. doi: 10.1016/j.tig.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Hannon GJ. FASTX-Toolkit. 2015. [Google Scholar]
- Haug GH, Tiedemann R. Effect of the formation of the Isthmus of Panama on Atlantic Ocean thermohaline circulation. Nature. 1998;393:673–676. [Google Scholar]
- Haug GH, Tiedemann R, Zahn R, Ravelo AC. Role of Panama uplift on oceanic freshwater balance. Geology. 2001;29:207–210. [Google Scholar]
- Hazkani-Covo E, Zeller RM, Martin W. Molecular Poltergeists: Mitochondrial DNA Copies (numts) in Sequenced Nuclear Genomes. PLoS Genetics. 2010;6:e1000834. doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head JJ, Bloch JI, Hastings AK, Bourque JR, Cadena EA, Herrera FA, Polly PD, Jaramillo CA. Giant boid snake from the Palaeocene neotropics reveals hotter past equatorial temperatures. Nature. 2009;457:715–717. doi: 10.1038/nature07671. [DOI] [PubMed] [Google Scholar]
- Head JJ, Rincon AF, Suarez C, Montes C, Jaramillo C. Fossil evidence for earliest Neogene American faunal interchange: Boa (Serpentes, Boinae) from the early Miocene of Panama. Journal of Vertebrate Paleontology. 2012;32:1328–1334. [Google Scholar]
- Heath TA, Huelsenbeck JP, Stadler T. The fossilized birth-death process for coherent calibration of divergence-time estimates. Proceedings of the National Academy of Sciences USA. 2014;111:E2957–E2966. doi: 10.1073/pnas.1319091111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson RW, Waller T, Micucci P, Puorto G, Bourgeois RW. Ecological correlates and patterns in the distribution of neotropical boines (Serpentes: Boidae): A preliminary assessment. Herpetological Natural History. 1995;3:15–27. [Google Scholar]
- Hey J, Nielsen R. Multilocus Methods for Estimating Population Sizes, Migration Rates and Divergence Time, With Applications to the Divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences USA. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull JM, Hull AC, Sacks BN, Smith JP, Ernest HB. Landscape characteristics influence morphological and genetic differentiation in a widespread raptor (Buteo jamaicensis) Molecular Ecology. 2008;17:810–824. doi: 10.1111/j.1365-294X.2007.03632.x. [DOI] [PubMed] [Google Scholar]
- Hynková I, Starostova Z, Frynta D. Mitochondrial DNA variation reveals recent evolutionary history of main Boa constrictor clades. Zoological Science. 2009;26:623–631. doi: 10.2108/zsj.26.623. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Jezkova T, Riddle BR, Card DC, Schield DR, Eckstut ME, Castoe TA. Genetic consequences of postglacial range expansion in two codistributed rodents (genus Dipodomys) depend on ecology and genetic locus. Molecular Ecology. 2015;24:83–97. doi: 10.1111/mec.13012. [DOI] [PubMed] [Google Scholar]
- Keigwin L. Isotopic Paleoceanography of the Caribbean and East Pacific: Role of Panama Uplift in Late Neogene Time. Science. 1982;217:350–353. doi: 10.1126/science.217.4557.350. [DOI] [PubMed] [Google Scholar]
- Langhammer J. A new subspecies of Boa constrictor, Boa constrictor melanogaster, from Ecuador (Serpentes: Boidae) Tropical Fish Hobbyist. 1985;32:70–79. [Google Scholar]
- Laurent RF. Herpetofaunal relationships between Africa and South America. In: Duellman WE, editor. The South American Herpetofauna: Its Origin, Evolution and Dispersal. Museum of Natural History, University of Kansas; Lawrence, KS: 1979. pp. 55–71. [Google Scholar]
- Lazell JD. The Lesser Antillean Representative of Bothrops and Constrictor. Bulletin of the Museum of Comparative Zoology. 1964;132:245–273. [Google Scholar]
- Leaché AD, Fujita MK, Minin VN, Bouckaert RR. Species Delimitation using Genome-Wide SNP Data. Systematic Biology. 2014;63:534–542. doi: 10.1093/sysbio/syu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann L. Boa constrictor. Animal Diversity Web; 2009. [Google Scholar]
- Linnaeus C. Systema naturae per regna tria naturae secundum classes, ordines, genera, species cum characteribus, differentiis, synonymis, locis. Holmiae (Salvius) 1758 [Google Scholar]
- Montes C, Cardona A, Jaramillo C, Pardo A, Silva JC, Valencia V, Ayala C, Pérez-Angel LC, Rodriguez-Parra LA, Ramirez V, Niño H. Middle Miocene closure of the Central American Seaway. Science. 2015;348:226–229. doi: 10.1126/science.aaa2815. [DOI] [PubMed] [Google Scholar]
- Noonan BP, Chippindale PT. Dispersal and vicariance: The complex evolutionary history of boid snakes. Molecular Phylogenetics & Evolution. 2006a;40:347–358. doi: 10.1016/j.ympev.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Noonan Brice P, Chippindale Paul T. Vicariant Origin of Malagasy Reptiles Supports Late Cretaceous Antarctic Land Bridge. The American Naturalist. 2006b;168:730–741. doi: 10.1086/509052. [DOI] [PubMed] [Google Scholar]
- O’Shea M. Boas and Pythons of the World. Princeton University Press; Princeton, NJ: 2007. [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippi R. Über die Boa der westlichen Provinzen der Argentinischen Republik. Zeitschrift für Gesammten Naturwissenschaften, Berlin. 1873;41:127–130. [Google Scholar]
- Pickrell JK, Pritchard JK. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genetics. 2012;8:e1002967. doi: 10.1371/journal.pgen.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price R, Russo P. Revisionary comments on the genus Boa with the description of a new subspecies of Boa constrictor from Peru. The Snake. 1991;23:29–35. [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of Population Structure Using Multilocus Genotype Data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. [Google Scholar]
- Rage JC. Gondwana, Tethys, and terrestrial vertebrates during the Mesozoic and Cainozoic. Geological Society, London, Special Publications. 1988;37:255–273. [Google Scholar]
- Rage JC. Fossil snakes from the Palaeocene of São José de Itaboraí, Brazil. Part II. Boidae. Palaeovertebrata. 2001;30:111–150. [Google Scholar]
- Rambaut A. FigTree v.1.4.2. 2015. [Google Scholar]
- Ravelo AC, Andreasen DH, Lyle M, Olivarez Lyle A, Wara MW. Regional climate shifts caused by gradual global cooling in the Pliocene epoch. Nature. 2004;429:263–267. doi: 10.1038/nature02567. [DOI] [PubMed] [Google Scholar]
- Reed R, Boback S, Montgomery C, Green S, Stevens Z, Watson D. Ecology and conservation of an exploited insular population of Boa constrictor (Squamata: Boidae) on the Cayos Cochinos, Honduras. In: Henderson RW, Powell R, editors. Biology of the Boas and Pythons. Eagle Mountain Publishing; Eagle Mountain, UT: 2007. pp. 389–403. [Google Scholar]
- Reynolds RG, Niemiller ML, Revell LJ. Toward a Tree-of-Life for the boas and pythons: Multilocus species-level phylogeny with unprecedented taxon sampling. Molecular Phylogenetics & Evolution. 2014;71:201–213. doi: 10.1016/j.ympev.2013.11.011. [DOI] [PubMed] [Google Scholar]
- Reynolds RG, Puente-Rolón A, Reed R, Revell L. Genetic analysis of a novel invasion of Puerto Rico by an exotic constricting snake. Biological Invasions. 2013;15:953–959. [Google Scholar]
- Schield DR, Card DC, Adams RH, Jezkova T, Reyes-Velasco J, Proctor FN, Spencer CL, Herrmann HW, Mackessy SP, Castoe TA. Incipient speciation with biased gene flow between two lineages of the Western Diamondback Rattlesnake (Crotalus atrox) Molecular Phylogenetics & Evolution. 2015;83:213–223. doi: 10.1016/j.ympev.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Skotte L, Korneliussen TS, Albrechtsen A. Estimating Individual Admixture Proportions from Next Generation Sequencing Data. Genetics. 2013:195. doi: 10.1534/genetics.113.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slevin JR. Expedition to the Revillagigedo Islands, Mexico, in 1925, III. Notes on a collection of reptiles and amphibians from the Tres Marias and Revillagigedo Islands, and the West Coast of Mexico, with description of a new species of Tantilla. Proceedings of the California Academy of Sciences. 1926:15. [Google Scholar]
- Smith HM. Summary of the collections of snakes and crocodilians made in Mexico under the Walter Rathbone Bacon Traveling Scholarship. US Government Printing Office; 1943. [Google Scholar]
- Stadler T. Sampling-through-time in birth-death trees. Journal of Theoretical Biology. 2010;267:396–404. doi: 10.1016/j.jtbi.2010.09.010. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull OG. Five new subspecies of the family Boidae. Occasional Papers of the Boston Society of Natural History. 1932;8:27. [Google Scholar]
- Suárez-Atilano M, Burbrink F, Vázquez-Domínguez E. Phylogeographical structure within Boa constrictor imperator across the lowlands and mountains of Central America and Mexico. Journal of Biogeography. 2014;41:2371–2384. [Google Scholar]
- Toews DPL, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology. 2012;21:3907–3930. doi: 10.1111/j.1365-294X.2012.05664.x. [DOI] [PubMed] [Google Scholar]
- Uetz P, Etzold T. The EMBL/EBI reptile database. Herpetological Review. 1996;27:175. [Google Scholar]
- Uetz P, Hošek J. The Reptile Database. 2015. [Google Scholar]
- Zweifel RG. Results of the Puritan-American Museum of Natural History Expedition to Western Mexico. 9, Herpetology of the Tres Marías Islands. Bulletin of the AMNH; v. 119. 1960 article 2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Voucher or collector identifiers for all new samples included as part of this work. Captive or pet animals lacking vouchers are encoded by ‘--’. UTA-ARDRC = University of Texas at Arlington Amphibian and Reptile Diversity Research Center.
Supplementary Table S2. All samples used for this study, along with the source of the data and the corresponding locality information. Missing or unsampled data is encoded by ‘--’. In some cases, coordinates were approximated for analyses by using coordinates at approximately the middle of the country or state/province (indicated with * following the coordinates) or using coordinates inferred from recorded locality data (indicated with ** following the coordinates).
Supplementary Table S3. Outgroup species used in the mitochondrial phylogenetic analysis. NCBI Genbank accession for the cyt-b sequence and the citation where the data was originally used are also included.
Supplementary Table S4. A summary of which ingroup samples were used for individual mitochondrial and nuclear genetic analyses. Data not included in an analysis – due either to not being collected or being excluded (see Materials and Methods) – is encoded by ‘--’.
Supplementary Table S5. Statistics on the number of high-quality and mapped Illumina reads for each sample with nuclear data.
Supplementary Table S6. Fossil data, including Paleobiology database collection number, date used, clade constrained, and relevant citation, used for divergence dating under the Fossilized Birth-Death model.
Supplementary Table S7. Sample assignments used for the three models compared using Bayesian Species Delimitation.
Supplementary Table S8. Summary table of highest supported number of source populations (K) deduced using the ΔK framework for NGSadmix and a DIC framework for Entropy. The values indicated the highest support for particular analyses are bolded.
Posterior density estimates of reciprocal migration parameters from the Isolation-Migration model for (A) Lagoon Cay island and mainland Belize, (B) West Snake Cay island and mainland Belize, (C) Cayos Cochinos Menor island and mainland Honduras, and (D) populations to the east and west of the Isthmus of Tehuantepec in Mexico.
Admixture graphs for all K = 2 – 8 allowed source populations inferred using the stringently filtered SNP dataset and NGSadmix. Samples are labeled based on the sample ID and the coarse locality information for the sample. In most cases, the province of the sample is included and “Crawl”, “Lagoon”, “WSnake” and “Cochinos” refer to Crawl, Lagoon, and West Snake Cays in Belize and to Cayos Cochinos Menor in Honduras, respectively. Nation names are abbreviated according to the ISO three-letter country codes.
Admixture graphs for all K = 2 – 8 allowed source populations inferred using the stringently filtered SNP dataset and Entropy. Samples are labeled based on the sample ID and the coarse locality information for the sample. In most cases, the province of the sample is included and “Crawl”, “Lagoon”, “WSnake” and “Cochinos” refer to Crawl, Lagoon, and West Snake Cays in Belize and to Cayos Cochinos Menor in Honduras, respectively. Nation names are abbreviated according to the ISO three-letter country codes.
Violin plots of genome-wide estimates of Pi (A), Heterozygosity (B), and FIS (C) for island and mainland populations in Central America and of FST (D) between each pairwise clade. For each violin plot, the white point indicates the median value and the black box indicates the interquartile range. The mean and standard deviations for each plot are evident above respective violin plots.