

Abstract
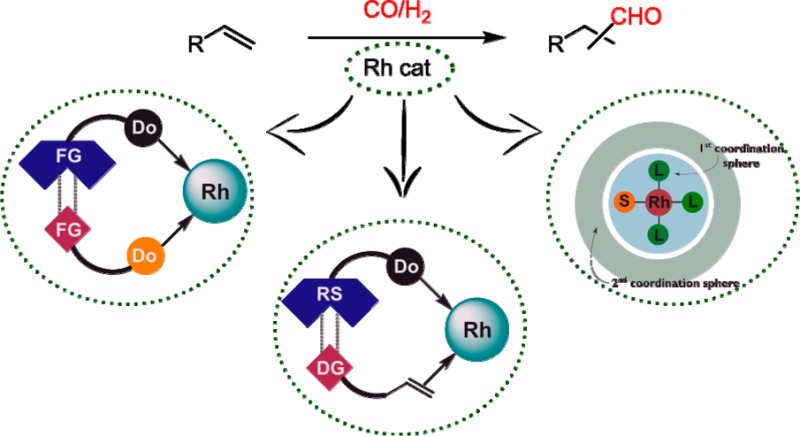
The hydroformylation reaction is one of the most intensively explored reactions in the field of homogeneous transition metal catalysis, and many industrial applications are known. However, this atom economical reaction has not been used to its full potential, as many selectivity issues have not been solved. Traditionally, the selectivity is controlled by the ligand that is coordinated to the active metal center. Recently, supramolecular strategies have been demonstrated to provide powerful complementary tools to control activity and selectivity in hydroformylation reactions. In this review, we will highlight these supramolecular strategies. We have organized this paper in sections in which we describe the use of supramolecular bidentate ligands, substrate preorganization by interactions between the substrate and functional groups of the ligands, and hydroformylation catalysis in molecular cages.
Keywords: hydroformylation, supramolecular chemistry, homogeneous catalysis, rhodium, phosphorus ligands, selectivity
1. Introduction
The hydroformylation reaction is the formal addition of a formyl group to an alkene functional group, which in practice involves a metal-catalyzed addition of CO and H2 to the alkene. The reaction was serendipitously found by Roelen during work on a Fischer–Tropsch process in 1938.1 In the 20 years after this discovery, the hydroformylation reaction did not receive much attention, which changed in the mid-1950s. In the 60 years that followed, the hydroformylation reaction was subject to intensive investigations, leading to many industrial applications. The first catalyst was a cobalt complex, and as a consequence, the first bulk processes also applied cobalt complexes. Later it was found that rhodium forms far more active catalysts for this reaction, and as such, the next generation of industrial processes applied rhodium catalysts, despite the fact that rhodium is more expensive. Other metals that can catalyze this reaction include palladium, platinum, ruthenium, and iridium, but no commercial applications based on these metals are known. So far, rhodium complexes remain a favorite because of their superior activity, allowing processes at lower temperatures and pressures, and the ability to control the selectivity of the reaction to a large extent by ligand variation. In volume, hydroformylation is still the largest industrial process in homogeneous catalysis, and several large companies have plants, including SHELL, BASF, Eastman, BP, DOW, and Evonik. The aldehyde products generally also have a characteristic smell, and as such, the hydroformylation reaction is also popular in the fragrance industry. Asymmetric hydroformylation leads to chiral compounds with an interesting aldehyde function that can be used for further functionalization, and as such, it is also a potentially interesting reaction for the fine chemical and pharmaceutical industry; however, it is so far a bit underexplored. The application of hydroformylation has recently been summarized by Börner in a review on applied hydroformylation2 and a beautiful review on hydroformylation of the flavor, fragrance, and food industry.3
The hydroformylation reaction is also an important reaction for educational purposes, not only because it has a rich history and plethora of applications but also because it is well understood despite its complexity.4 Next to the aldehyde products that are generally desired, there are several byproducts that can form (Scheme 1). Under hydroformylation reaction conditions, hydrogen is always present, and as such, both the substrate and the products can be hydrogenated, leading to alkanes and alcohols. In fact, alkene hydrogenation is thermodynamically more favorable, and therefore, the reaction should proceed under kinetic control. Next to this, the substrate can also isomerize under catalytic conditions, leading to a mixture of terminal and internal alkenes, which in turn can be hydroformylated. This isomerization can also be used as an advantage in cases in which isomerization–hydroformylation tandem reactions lead directly to the desired product, for example, the production of linear aldehydes from internal alkenes.5 All these side reactions depend on the catalyst properties and the conditions that are applied and can be qualitatively understood.
Scheme 1. General Reaction Scheme of the Hydroformylation Reaction, Converting Alkenes into Aldehydes, and Potential Side Products.

The mechanism originally proposed by Heck and Breslow6 is generally still valid, although more detailed insight has been obtained ever since.7 The general mechanism, displayed in Scheme 2, starts with rhodium(I)hydrido complex 1, and for this example, we use the bisphosphine biscarbonyl analogue. After decoordination of a CO ligand to create a vacant site, the alkene can coordinate to give common intermediate 3. In the next step, migration of the hydride to the carbon atom leads to the rhodium alkyl species. Depending on the orientation of the alkene, the migration is to either C2 or C1, leading to the linear (4) or the branched alkyl species (8), respectively. These two enter separate but identical catalytic cycles. After CO coordination and subsequent migration of the alkyl group to the CO, the rhodium acyl species (6 and 10) are formed. These four-coordinate species can directly coordinate CO to form 7 or 11 or directly react with molecular hydrogen to give the product and intermediate 2. Species 1, 7, and 11 are most often drawn as a part of the catalytic cycle, but recently, Landis showed that part of the reaction can go directly from acyl intermediate 6 to 2 without forming 7.8 As such, it is more accurate to draw these intermediates as off-cycle species. It is important to realize that all the steps except for the final hydrogenolysis are equilibria. As a consequence, the regio- and enantioselectivity could be determined by the hydride migration step from intermediate 3, but under certain conditions, it could also be that intermediates 6 and 10 are in fast equilibrium and the relative rate of the final hydrogenolysis of these species is determining the selectivity.
Scheme 2. Mechanism of Rhodium-Catalyzed Hydroformylation.

The reversibility of the hydride migration is the basis for the isomerization reaction, which can result in the formation of unwanted side products. For some applications, for example, when a mixture of alkenes is offered as the feed, an isomerization–hydroformylation sequence is required.5
Over the years, many different ligands have been explored for rhodium-catalyzed hydroformylation, of which some are displayed in Figure 1. Triphenylphosphine (TPP) has without a doubt been studied the most, as it is a cheap and readily available ligand, and several large-scale processes started in the 1970s using TPP. Also, water-soluble analogues have been developed, including monosulfonated TPP (TPPMS), and these have been used for aqueous phase hydroformylation. For these types of phosphine ligands, the rate-limiting step is early in the catalytic cycle (1–4), often termed type I kinetics,4a and the use of more electron-donating ligands such as alkyl phosphines results in very low activity. The use of more electron poor (or π-accepting) ligands such as phosphites results in much higher activities as CO dissociation is generally fast. As a consequence, the rate-determining step can be later in the cycle, typically the hydrogenolysis step, in which case one generally refers to type II kinetics.4a As the CO ligands are much more weakly coordinated on these electron poor complexes, the hydride migration reaction becomes more reversible, resulting in the formation of more isomerization side products. If the phosphite ligands are very bulky, there is only space for one ligand to coordinate to rhodium, and this leads to the most active catalysts.9
Figure 1.

Some typical ligands that have been used in the rhodium-catalyzed hydroformylation.
A lot of attention has been devoted to control the regioselectivity of the reaction, with the main target being the linear aldehyde. It was found that the use of a large excess of TPP can already provide reasonably high selectivity for the linear product, albeit at the expense of the activity of the catalyst.7b Many bidentate ligands have been explored, and it was found that ligands with a large bite angle, i.e., P–Rh–P angle around 110–120°, give rhodium complexes that produce the linear aldehyde in very high selectivity.10 BISBI and Xantphos are the most illustrative examples of such ligands (Figure 1). Catalysts that give very high selectivity for the branched aldehyde for aliphatic alkenes have not yet been found. For substrates such as styrene and vinyl acetate, the formation of a stabilizing allyl intermediate directs the hydroformylation toward the branched product.2
The field of enantioselective hydroformylation is far from mature, which is strange if one considers the potential impact for the fine chemical and pharmaceutical industry. The main reason must be the challenges associated with asymmetric hydroformylation processes. Whereas many asymmetric hydrogenation processes were reported between the 1970s and 1990s, the contributions in that period to asymmetric hydroformylation reported modest to good enantiomeric excess (ee) at best. This changed with the seminal work of Nozaki11 and later Landis,12 who reported BINAPHOS and diazaphospholanes, respectively, as novel bidentate ligands that gave high enantioselectivity in asymmetric hydroformylation. Substrates that have been mostly studied are those that typically give high branched selectivity, such as styrene derivatives and vinyl acetate. More recently, BOBphos was reported as a bidentate ligand that also gave relatively high branched selectivity and enantioselectivity in the hydroformylation of unfunctionalized 1-alkenes (Figure 2).13
Figure 2.

Some typical ligands that have successfully been used in asymmetric hydroformylation, typically of styrene derivatives.
It is clear that the hydroformylation reaction is a very powerful transformation for both the bulk, the fragrance, fine chemical, and pharmaceutical industry. Although many selectivity and activity issues have been solved, there are still many challenges left that would, when solved, really expand the scope of possibilities of this reaction. These challenges mainly involve selectivity issues, including the branched selective hydroformylation, the selective hydroformylation of internal alkenes, and the selective hydroformylation of tri- and tetrasubstituted alkenes. Also, the asymmetric hydroformylation of terminal disubstituted alkenes is a largely unsolved issue. In this light, the development of novel concepts to control selectivity in hydroformylation catalysis is of utmost importance. In this review, we will elaborate on the use of novel supramolecular strategies in hydroformylation. In the first part, we will discuss the use of supramolecular bidentate ligands in hydroformylation; then we highlight the supramolecular substrate orientation strategy, and we end with hydroformylation catalysis in cages.
2. Supramolecular Bidentate Ligands in Hydroformylation
Supramolecular bidentate ligands make up a recently introduced class of ligands that has the advantage of the synthetic accessibility of monodentate ligands but behaves as chelating bidentate. In such a supramolecular approach, two monodentate ligand building blocks are brought together by a self-assembly process using noncovalent interactions such as hydrogen bonds, ionic interactions, or dynamic metal–ligand coordination. As the number of supramolecular bidentate ligands that become accessible grows exponentially with the number of ligand building blocks available, this approach is well suited to the generation of large libraries of ligands. In addition, supramolecular bidentate ligands show clear chelating behavior. The application of such supramolecular ligands in catalysis has been a subject of earlier reviews.14 Two strategies to arrive at self-assembled ligands that have been successfully employed for hydroformylation are (I) metal-templated assembly and (II) direct interaction between functionalized ligand building blocks (Figure 3), and these will be discussed in this section.
Figure 3.
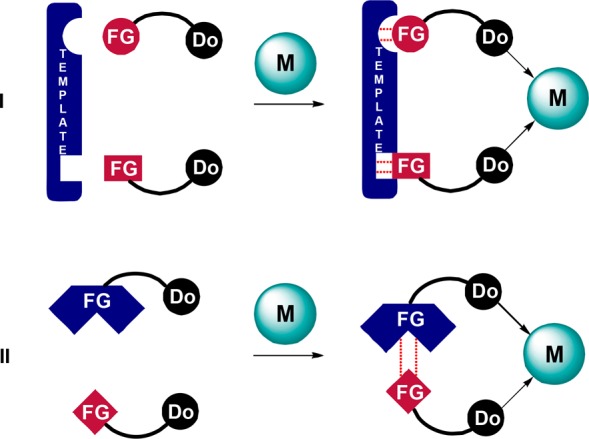
Schematic representation of supramolecular bidentates formed via a template (I) or via direct interactions between functionalized ligand building blocks (II). M = metal center. FG = functional group. Do = donor center.
Metal-Templated Assembly (I)
This strategy consists of the construction of chelating bidentate ligands using a template that contains binding sites for the selective assembly of two monodentate ligands (Figure 3, I). The first example of this strategy was based on the use of a bis-zinc(II)-porphyrin template (Scheme 3).15 The assembly process is based on selective coordination of the nitrogen-donor atoms of pyridylphosphorus building blocks a–c to the zinc atoms of the porphyrin template 13. The composition of the supramolecular ligand 13(a)2 in solution is confirmed by various spectroscopic techniques. In the presence of Rh(acac)(CO)2, high-pressure nuclear magnetic resonance (NMR) spectroscopy under 20 bar of syngas reveals the formation of [HRh(13(a)2(CO)2)], the active species for hydroformylation catalysis (Scheme 3, bottom).
Scheme 3. Bis-Zinc(II) and Bis-Tin(IV) Porphyrin Templates (top) and Phosphorus Monodentate Ligands (middle) Used for Supramolecular Assemblies.
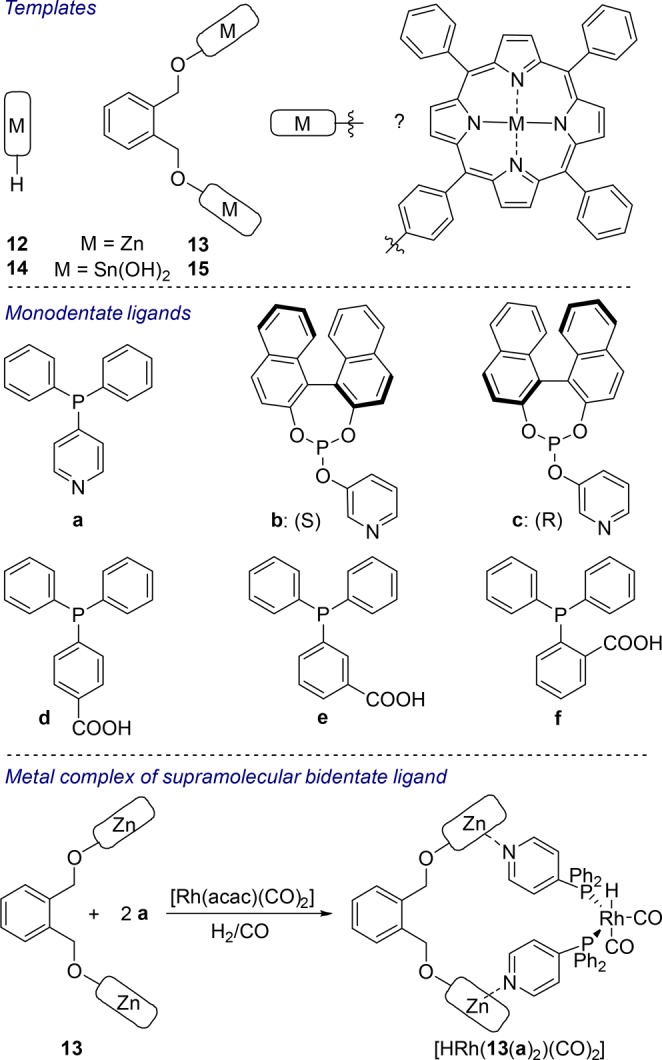
Synthesis of the rhodium hydroformylation catalyst based on the self-assembly of template 13 and ligand a.
The assemblies 12(a), 13(a)2, 12(b), and 13(b)2 are evaluated as catalysts in the rhodium-catalyzed hydroformylation of 1-octene. Although the chelating bidentate assemblies 13(a)2 and 13(b)2 exhibit activity that is lower than that of the corresponding monodentate analogues, they display slightly higher selectivity for the linear aldehyde [77:23 (13(a)2) vs 74:26 (12(a)) and 96:4 (13(b)2) vs 83:17 (12(b)) at 25 °C]. The chiral supramolecular catalysts containing 13(b)2 and 13(c)2 are also applied in the asymmetric rhodium-catalyzed hydroformylation of styrene, showing enantioselectivity (33%) that is higher than that of monodentate analogues (∼7%) and an increase in activity.15,16
After the initial results based on zinc(II), also bis-tin(IV) porphyrin-templated assemblies have been studied. The tin–oxygen interaction (Scheme 3) is stronger and gives rise to less dynamic bonds. The mixture of bis-tin(IV) porphyrin template 15 and carboxylic phosphorus ligands d–f results in novel supramolecular assemblies upon loss of water. The assemblies are active in the rhodium-catalyzed hydroformylation of 1-octene. While the rhodium complexes in which carboxylate-phosphines are coordinated to dihydroxotin(IV) porphyrin 15 lead to enhanced catalytic activity (≤40-fold) compared to that of complexes of 14 with d–f, the assemblies based on bis-zinc porphyrin templates are still more active in hydroformylation catalysis.16
The rigid bis-zinc(II) salphen building block has also been explored as a template for the assembly of ligands. The coordination of two identical monodentate pyridylphosphorus ligands on the zinc(II) salphen results in self-assembled bidentate ligands that in the rhodium-catalyzed hydroformylation of styrene and 1-octene outperform their nontemplated analogues. However, in comparison with those of the bis-zinc(II) porphyrin template, the regio- and enantioselectivities are not improved by using the more rigid template.17 Interestingly, the coordination of two different monodentate pyridylphosphorus ligands on the bis-zinc(II) salphen template provides an efficient supramolecular approach to form heterobidentate ligands (e.g., Figure 4). Remarkably, templated heterobidentate ligands are selectively formed, which is attributed to steric effects; two bulky ligands just do not fit on the rigid template. In the asymmetric hydroformylation of styrene, these supramolecular heterobidentate ligands are characterized by higher enantioselectivities (≤72% ee) compared to those of the nontemplated mixed ligand combinations (≤13% ee).18 The bis-zinc(II) salphen template has been also utilized in combination with a chiral 3-pyridyl-substituted monodentate phosphoramidite to prepare a supramolecular “box” 78,19 which will be discussed in more detail in section 4.
Figure 4.
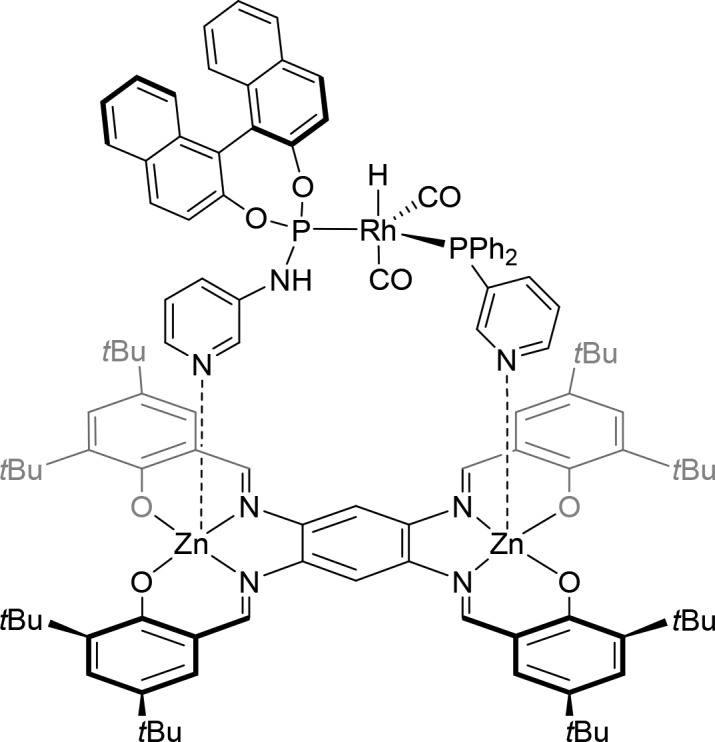
Bis-zinc(II) salphen templated heterobidentate complex 16.
The self-assembly of bidentate ligands can be extended to multicomponent structures. Employing an N-donor ditopic ligand, such as 1,4-diazabicyclo[2.2.2]octate (DABCO), in combination with tris(zinc(II) porphyrin)phosphite 17 leads to a five-component self-assembled bidentate ligand (Scheme 4). The bidentate ligand system is assembled by three bridging ditopically coordinated DABCO molecules, if precisely 1.5 equiv of DABCO is used, and in the presence of Rh(I), the chelating rhodium complex 18 is formed. Complex 18 displays high selectivity in the rhodium-catalyzed hydroformylation of 1-octene for the linear aldehyde (96:4) and a reduced activity in comparison to that of the rhodium catalyst based on monomeric ligand 17. This demonstrates that the multicomponent self-assembled ligand shows bidentate coordination behavior under catalytic conditions. Importantly, in the presence of a small excess of DABCO, the selectivity drops, suggesting that all ditopic ligands must be present to achieve sufficient stability to induce the high selectivity.20
Scheme 4. Synthesis of a Multicomponent Assembly from Ligand 17 and DABCO (as templates) and the Rhodium(I) Complex.

Self-Assembly of Functionalized Ligand Building Blocks To Form Bidentate Ligands (II)
In the previous part, we showed that one can form bidentate ligands by preorganization of ligand building blocks on a template, and in this part, we discuss the examples in which two functionalized ligand building blocks self-assemble to form a bidentate ligand (Figure 3). The assembly process can be based on selective metal–ligand interactions or hydrogen bonding.
Reek and co-workers explored the construction of bidentate ligands using coordination of axial nitrogen to zinc(II) porphyrins. A set of six phosphite-functionalized porphyrins (19–24) and eight monodentate phosphorus-donor ligands (a–h) equipped with N-donor functions generates a library of 48 chelating bidentates, coined SUPRAPhos (Figure 5).21 This library of bidentate ligand assemblies is evaluated in the rhodium-catalyzed hydroformylation of styrene. An increase in selectivity for the branched product (b:l of 10.4 for 19a and 9.3 for 20a) and a decrease in activity compared to those of monodentate phosphorus ligands 19 and 20 are observed when bidentate 19a and 20a are used, which is in agreement with the formation of chelating bidentate ligand assemblies.22
Figure 5.

Building blocks used to generate bidentate SUPRAPhos ligands via self-assembly.
In 2008, the library was extended to 450 SUPRAPhos bidentate ligands, based on 15 different porphyrin phosphite and phosphoramidite and 30 pyridyl-appended phosphoramidite, phosphite, and chiral phosphine ligands. The introduction of the building blocks with stereogenic centers at the phosphorus atom allowed the application of these assemblies in the asymmetric hydroformylation of styrene.23 Generally, high activity is observed for the catalysts formed from phosphine-containing or bulky phosphite ligands, which unfortunately displayed low enantioselectivities. In contrast, ligands based on phosphoramidite ligands show relatively high enantioselectivities (≤76% ee) but at low conversion.24 Also, the regioselectivity was strongly influenced by the supramolecular bidentate ligand used, giving rise to high selectivity for either the branched or the linear aldehyde (b:l ratio between 0.5 and 8.9).
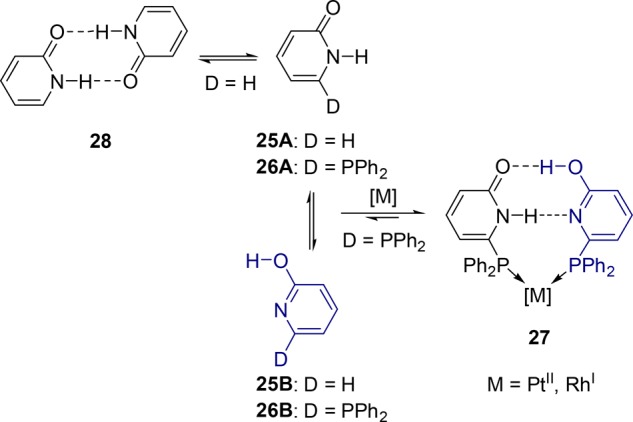
The first self-assembled bidentate ligands based on hydrogen bonding used in hydroformylation were reported by Breit and Seiche.25 The 2-pyridone 25A/2-hydroxypyridine 25B tautomer system is used as a self-complementary hydrogen bonding motif. Introduction of donor groups (PPh2) capable of binding to a metal center is straightforward. Importantly, the equilibrium shifts toward the mixed hydroxypyridine/pyridone dimer 27 in the presence of a coordinating metal, compared to symmetrical pyridone dimer 28 formed in apolar solvents in the absence of the metal or the donor groups (Scheme 5). The formation of the hydrogen-bond pattern in 27 is evidenced from the X-ray structure of the cis-[PtCl2(26)2] complex, and the structure also reveals the presence of the ligand 26 (6-DPPon = 6-diphenylphosphanylpyridone) in two different tautomeric forms.26
Scheme 5. Self-Assembly of the 2-Pyridone/2-Hydroxypyridine System via Hydrogen Bonding.
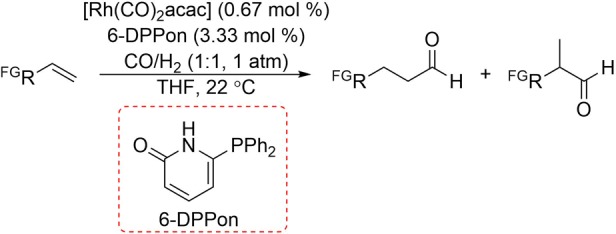
The chelating behavior of the hydrogen-bonded ligand in rhodium complex 27 is clear from the results in the hydroformylation of 1-octene as a high selectivity for the linear aldehyde is obtained (l:b ratio of 32), comparable to the well-established bidentate ligand Xantphos (l:b ratio of 49). The Rh-based catalyst also hydroformylates amide-, alcohol-, and ester-functionalized terminal alkenes and produces the corresponding linear aldehydes in high yields and regioselectivities. The Rh-based catalyst 27 even converts terminal alkenes at room temperature and ambient pressure (RTAP), with low catalyst loading (Scheme 6).27 Addition of a surfactant such as tocopherol derivative polyoxyethanyl α-tocopheryl sebacate (PTS) also makes it possible to conduct the RTAP hydroformylation experiments in aqueous media, while excellent regioselectivity for the linear aldehyde confirms the stability of the hydrogen bonding motif.28
Scheme 6. Room-Temperature, Ambient-Pressure Hydroformylation of Functionalized Terminal Alkenes with the Rhodium/6-DPPon Catalyst (FG = functional group).
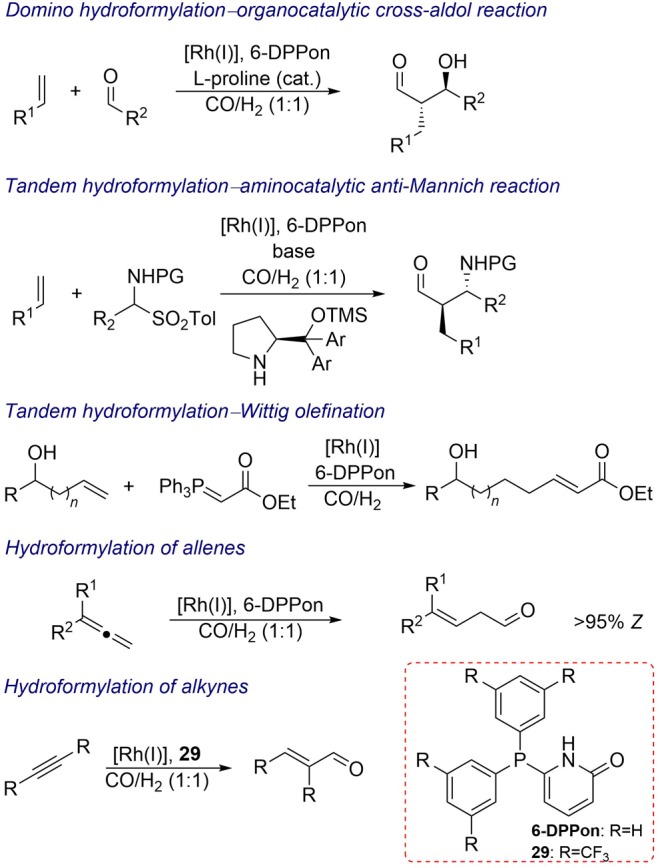
The efficiency of the rhodium/6-DPPon catalyst at room temperature and ambient pressure has enabled the development of tandem processes such as a domino hydroformylation/enantioselective organocatalytic cross-aldol reaction sequence.29 The application of ligand 26 also allowed the development of highly selective tandem hydroformylation/hydrogenation processes,30 and tandem regio-, diastereo-, and enantioselective hydroformylation–organocatalytic anti-Mannich reaction.31 The 6-DPPon ligand and its derivatives are also utilized in the tandem rhodium-catalyzed hydroformylation–Wittig olefination reaction of homoallylic alcohols.32 Moreover, the 6-DPPon ligand and its derivative (29) form excellent catalysts in rhodium-catalyzed hydroformylation of 1,1-disubstituted allenes,33 as well as dialkyl- and diaryl-substituted alkynes,34 demonstrating excellent chemo-, regio-, and stereoselectivities for the formation of β,γ-unsaturated aldehydes and enals, respectively (Scheme 7).
Scheme 7. Application of Rhodium/6-DPPon and Rhodium/29 Catalysts in Tandem Hydroformylation Reactions and Hydroformylation of Allenes and Alkynes.
To gain a deeper insight into the mechanism of the rhodium-catalyzed hydroformylation of terminal alkenes using self-assembled 6-DPPon ligands, detailed experimental (including kinetic studies35 as well as in situ ESI-MS,36 IR, and NMR investigations) and computational studies37,38 have been performed. The existence of the hydrogen bonding motif during catalysis is verified by in situ IR and NMR studies of a catalytically competent intermediate (acyl complex [(COR)Rh(26)2(CO)2]) and confirmed by density functional theory (DFT) calculations. DFT calculations also show that hydrogen bonding is present not only in the resting state but also throughout the catalytic cycle. By calculation of the free energy surface of the prolinear and probranched catalytic cycle (propene is used as a model substrate for DFT calculations), hydrometalation was shown to be a selectivity-determining step. The rate-determining transition state is stabilized by hydrogen bonding by at least 7 kcal mol–1. In line with this, O- and N-methylated derivatives of the 6-DPPon catalyst, possessing no hydrogen bonding, are synthesized and applied in the hydroformylation under identical conditions. These complexes lead to selectivities (l:b ratios of 76:24 and 74:26) and activities typically observed for complexes based on monodentate PPh3, once again showing the importance of the hydrogen bonds between the ligands for catalyst activity and selectivity.37
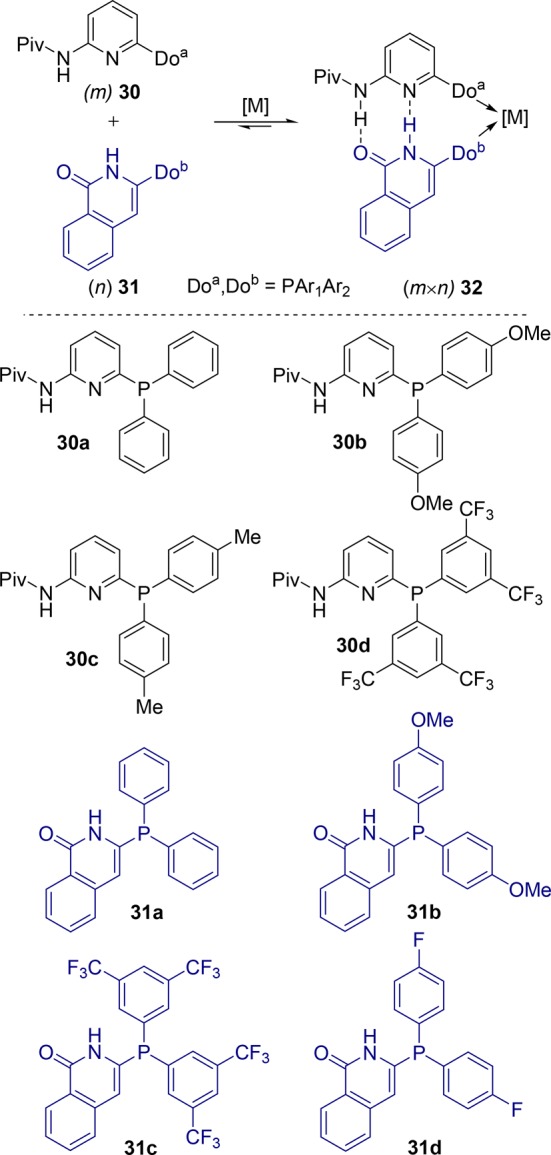
The 6-DPPon ligand can form only homobidentate ligands, and to generate a heterobidentate ligand by self-assembly, the Breit group extended their chemistry to the aminopyridine 30/isoquinolone 31 hydrogen bond motif. These building blocks consist of a donor/acceptor and acceptor/donor combination, akin to A-T base pairs in DNA, and therefore, these units bind selectively to each other; on the other hand, self-aggregation leads to systems in which the donor atoms are pointing in the opposite directions (Scheme 8).39,40 When the ligand building blocks 30a and 31a are mixed with [PtCl2(1,5-cod)], the heterodimeric 32aa–PtCl2 complex is formed exclusively. The hydrogen bonding between the building blocks is confirmed by X-ray crystal structure determination as well as by NMR studies. From eight phosphine ligand building blocks, a set of 16 (4 × 4) heterobidentate ligand assemblies can be formed. Evaluation of this library in rhodium-catalyzed hydroformylation of terminal alkenes shows a variation in activity and selectivity. Generally, phosphine-substituent modification of the aminopyridines 30a–d has the strongest influence on the catalyst; in particular, electron-donating groups decrease the catalyst activity, while electron-withdrawing groups lead to its increase. The best catalyst is competitive with the best traditional bidentate ligands in terms of activity and selectivity for the linear aldehydes (≤96:4).
Scheme 8. Self-Assembly of the Aminopyridine/Isoquinolone System To Generate Heterodimeric Bidentate Ligands (Do = donor center).
Other acceptor/donor donor/acceptor hydrogen bond pairs based on heterocyclic scaffolds have been explored for the purpose of generating self-assembled ligands. A new 5 × 2 self-assembled ligand library is generated and explored in rhodium-catalyzed hydroformylation of 1-octene (Figure 6).41 All the ligand combinations demonstrate high regioselectivities (89:11 to >99:1) consistent with bidentate ligand catalysts that are operative in the catalytic cycle. Interestingly, the highest selectivities for the linear aldehyde (>99:1) are obtained for the catalysts formed by thiazole/isoquinolone (37/38) and thiazole/7-azaindole (37/39) combinations. This is explained by formation of stronger hydrogen bonds between the self-assembled building blocks and, hence, a more rigid or stable ligand system. Remarkably, the strength of hydrogen bonding is such that it also allows the hydroformylation reactions in protic solvents such as methanol. While the aminopyridine/isoquinolone system (33/38) shows a significant drop in regioselectivity with a change in the solvent from toluene (94:6) to methanol (82:18), the thiazole/isoquinolone combinations (36/38 and 37/38) keep their high selectivities observed in toluene (98:2 and 99:1) when the solvent is changed to methanol (97:3 and 96:4, respectively).41 In the search for selective asymmetric hydrogenation catalysts, such a ligand library was evaluated using a combinatorial iterative deconvolution strategy, which allowed identification of the best catalyst in a limited number of experiments.42 For finding the most selective hydroformylation catalysts, similar strategies can be envisioned; however, reports that describe these are lacking at this stage.
Figure 6.
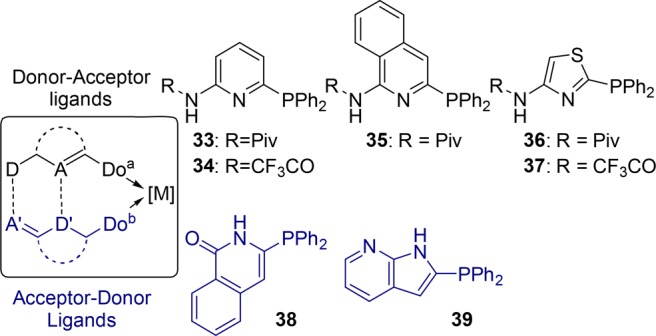
Self-assembly of heterocyclic systems to generate methanol-stable heterodimeric bidentate ligands. D = hydrogen-bond donor. A = hydrogen-bond acceptor. Do = donor center.
Subsequent development of the self-assembly approach by hydrogen bonding afforded new bidentate β-sheet-like P-ligands based on peptidic structures (Figure 7). Rhodium catalysts, generated in situ upon mixing Rh(acac)(CO)2 with C-linked phosphine-functionalized peptidyl ligands (LC) and complementary N-linked peptidic systems (LN), are applied in the asymmetric hydroformylation of styrene. While homocombinations of the ligands display low enantioselectivities (5–8% ee), the heterobidentate ligand combinations give significant levels of enantioselectivities (≤38% ee) despite a remote position of the stereocenters in relation to the catalytically active centers.43
Figure 7.

Metal-templated self-assembly of peptide-based P-ligands.
Finally, Reek and co-workers have developed a novel type of phosphinourea ligand that forms supramolecular homo- and heterobidentate ligands via self-assembly in the presence of rhodium(I) precursor Rh(acac)(CO)2 (Scheme 9).44 The rhodium complexes based on six phosphinoureas (40–45) are evaluated in the asymmetric hydroformylation of styrene. All the catalysts exhibit good conversions (64–100%) and high selectivities (86–97%) for the branched product and moderate enantioselectivities (≤46% ee for the 43/43 homocombination). It has been proposed that these functionalized ligands operate via a slightly different mechanism, involving ligand cooperativity leading to an intermediate unknown for traditional ligands (see Scheme 9).44
Scheme 9. Phosphinourea Ligands and Synthesis of Supramolecular Rhodium Complexes Thereof.

The Rh complex is also suggested as one of the intermediates of the hydroformylation cycle.
3. Selective Hydroformylation by Substrate Preorganization
Another recently introduced strategy for achieving selectivity in transition metal catalysis is through preorganization of the substrate with respect to the metal center. A commonly explored way to achieve preorganization is through ditopic binding of the substrate to the metal center, in which a second functional group controls the orientation of the substrate (Figure 8, I).45,46 Such a directing group can already be present on the substrate or alternatively needs to be introduced temporarily to the substrate before it is subjected to the catalytic conversion. These strategies have been demonstrated to be very powerful in C–H activation and asymmetric hydrogenation; however, there are some limitations and drawbacks. For example, the strategies are limited to substrates that have a specific directing group close to the reactive bond, or additional synthesis steps are required for the introduction and removal of the directing group. Moreover, an additional vacant coordination site has to be present on the metal center for the coordination of the directing group. Using supramolecular chemistry, some of these issues can be resolved as substrate orientation no longer has to be established via the catalytically active metal. Recently, several groups have focused on ditopic binding of the substrate as a way to preorganize the substrate with respect to the metal center via a recognition site installed on the ligand (Figure 8, II). The structure of the bifunctional ligand can be adjusted to the structure of the substrate. This can allow for remote selectivity control as well as catalyst design in a predictable fashion when combined with in-depth mechanistic knowledge. This is far more challenging using traditional approaches. Through supramolecular substrate preorganization, highly selective reactions have been engineered, and this strategy was highlighted specifically in recent perspectives by Phipps et al.47 and Reek et al.48
Figure 8.
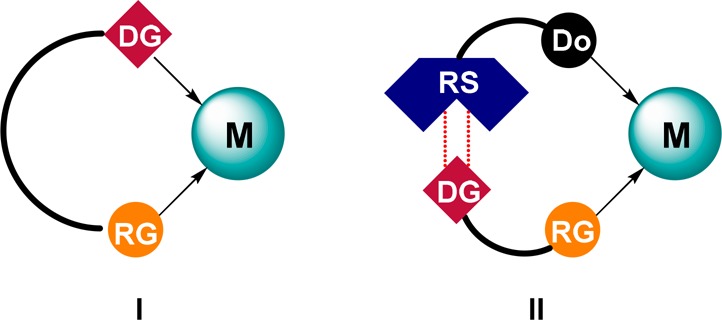
Schematic representation of substrate preorganization via traditional approaches (I) or via supramolecular strategies (II). M = metal center. DG = directing group. RG = reactive group. RS = recognition site. Do = donor center.
In rhodium-catalyzed hydroformylation, the selectivity is typically determined during the hydride migration step. Generally, the alkene substrate can coordinate in various manners, and upon migration of the hydride, the alkene rotates. As such, substrate orientation via supramolecular interactions should be a viable strategy for controlling the selectivity, and indeed, this approach has been shown to be very powerful; in this section, we will discuss the relevant examples reported so far.
Hydrogen-bonded systems have been exceptionally effective in the context of hydroformylation combined with supramolecular substrate preorganization. The neutral Rh(I) complex often observed as a resting state has a strong affinity for CO and the phosphorus ligands, and as such, the functional groups required for substrate orientation generally do not coordinate. Breit et al. reported a guanidinium-functionalized phosphine ligand that acts as a receptor for unsaturated carboxylic acids (Figure 9).49
Figure 9.

Guanidinium-functionalized phosphine ligands.
Hydrogen bonds between the guanidinium group of the ligand and the carboxylic acid moiety of the substrate preorganize the alkene with respect to the metal center. When 3-butenoic acid is converted by a rhodium catalyst based on this ligand, a very high selectivity is achieved for the linear product (l:b ratio of 41) (Scheme 10, 49a). This catalyst is also active for internal alkenes, which are generally less reactive. When 3-pentenoic acid is hydroformylated with this catalyst system, the product is formed in which the aldehyde is introduced at the unsaturated carbon atom farthest from the carboxylic acid [o:i ratio of 18:1; o = outermost, and i = innermost (Scheme 10, 49b)]. The selectivity was found to be highly dependent on the distance between the acid moiety and the alkene function. 4-Pentenoic acid hydroformylation with the supramolecular system gave selectivity to levels typically found for triphenyl phosphine-based catalysts, indicating that substrate preorganization does not play a role. This clearly shows that for this catalyst system the alkene–acid distance has to be precise to control the selectivity by supramolecular preorganization. This can be exploited for substrates containing two alkenes at different distances from the carboxylate (Scheme 11, 50). The alkene with the proper carboxylic acid–olefin distance is converted at a higher rate (8.8:1) and with a higher selectivity for the linear aldehyde (l:b ratio of 32), compared to the alkene moiety that is farther from the carboxylic acid (l:b ratio of 3).50
Scheme 10. Regioselective Hydroformylation of Unsaturated Carboxylic Acids (o = outermost; i = innermost).

Scheme 11. Hydroformylation of a Substrate Containing Multiple Olefinic Sites.

DFT calculations show that the lowest energy is obtained when two ligands are coordinated to the metal center and the carboxylic acid moiety of the substrate forms four hydrogen bonds with the two guanidine groups of the ligands (Figure 10). No substrate–ligand interaction can be observed when only one ligand coordinates to the metal center, and as such, the biscoordinated species is proposed to be the most likely intermediate responsible for the high selectivity.50 Analysis of the calculated structures indicates that preceding the hydride migration step the alkene is rotated toward the hydride through the hydrogen bonds between the guanidinium moieties and the carboxylic acid moiety of the substrate. In experiments in which competitive guests (with carboxylic acid functional groups) are present, the substrates are converted with lower selectivity and activity in line with the substrate preorganization model.
Figure 10.
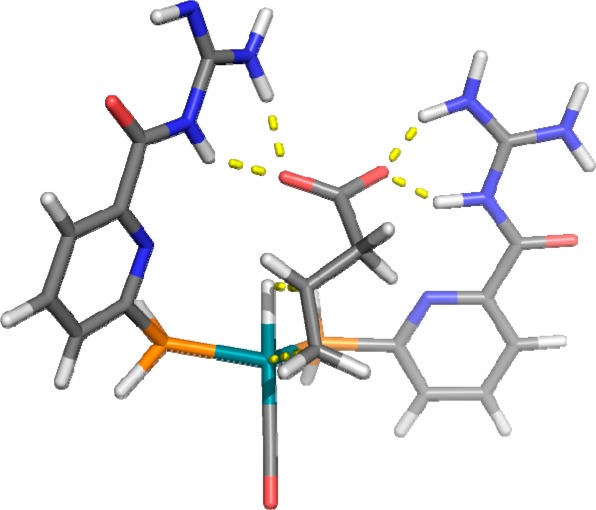
Substrate orientation in the selectivity-determining hydride migration step (DFT study).
46 is also effective in a decarboxylative hydroformylation of α,β-unsaturated carboxylic acids (Scheme 12).51 In this cascade reaction, the formyl group is introduced on the substrate, after which the carboxylate leaves the substrate as CO2. Under similar conditions, but using triphenylphosphine as the ligand, the double bond is reduced instead of hydroformylated, which exemplifies the need for supramolecular interactions between the substrate and the catalyst to yield the terminal aldehyde product.
Scheme 12. Decarboxylative Hydroformylation of α,β-Unsaturated Carboxylic Acids.

When the pyridine moiety of the previously discussed ligand (Figure 9, 46) is replaced with a benzene moiety or a pyrrole moiety, aldehyde hydrogenation is observed (Figure 9, 47 and 48).52 As such, these ligands can be used in the context of a tandem hydroformylation–hydrogenation sequence converting 1-octene into 1-nonanol. The selectivity for the linear alcohol can be enhanced by combining the pyrrole (48) analogue of the guanidinium catalyst with the 2-pyridone/2-hydroxypyridine supramolecular bidentate (6-DPPon) to yield a highly selective hydroformylation–hydrogenation reaction of 1-octene to 1-nonanol (Scheme 13).30
Scheme 13. Tandem Processes Using Supramolecular Substrate Preorganization Ligands.
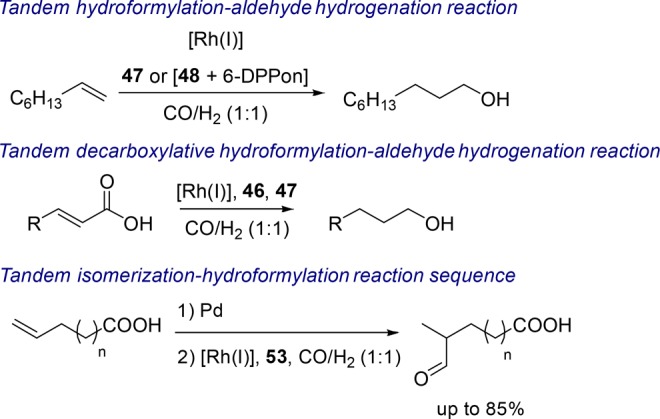
Combining the decarboxylative hydroformylation approach of α,β-unsaturated acids with a supramolecular aldehyde hydrogenation catalyst yields a tandem decarboxylative hydroformylation–hydrogenation catalytic system (Scheme 13).53 The system works most effectively when a mixture of the most active ligand in decarboxylative (46) hydroformylation is used in combination with an analogue effective in the hydrogenation of aldehydes (47).
Regioselective hydroformylation of unsaturated acids can also be achieved with a series of bidentate phosphines and phosphite ligands, coined DIMPhos, functionalized with a highly selective anion receptor, 7,7-diamido-2,2-diindolylmethane (Figure 11).54
Figure 11.

Anion receptor-functionalized bisphosphines (DIMPhos).
Terminal unsaturated carboxylates can be hydroformylated with a phosphine analogue (Figure 11, 52) of the ligand. 4-Pentenoate to 10-undecenoate are converted to the aldehyde with high selectivities for the linear product (Scheme 14, 54). 3-Butenoate is not converted selectively because the substrate is too short to bind to the receptor moiety and the metal center simultaneously. Unsaturated phosphate analogues are also converted with high selectivities. Upon protonation or methylation of the substrate, the selectivity is lost and the conversion is significantly lower. It is interesting to note that, contrary to the monodentate guanidinium ligands (Figure 9, 46) reported by Breit et al., the high selectivity for the linear product is obtained for a variety of substrates with different distances between the alkene and the carboxylate group.49
Scheme 14. Regioselective Hydroformylation of ω-Unsaturated Carboxylic Acids (DIPEA = N,N-diisopropylethylamine).

In situ spectroscopy, kinetic data, and DFT calculations show that the hydride migration step is selectivity-determining. As seen for the guanidinium phosphine systems, DFT data show that due to the binding of the substrate in the DIM pocket, the alkene is properly preorganized with respect to the Rh–H bond for the hydride migration step leading to the linear rhodium alkyl species (Figure 12). The hydride migration to form the branched alkyl species cannot proceed without the carboxylate leaving the pocket, and also other competitive pathways leading to the branched product are significantly higher in energy.54
Figure 12.
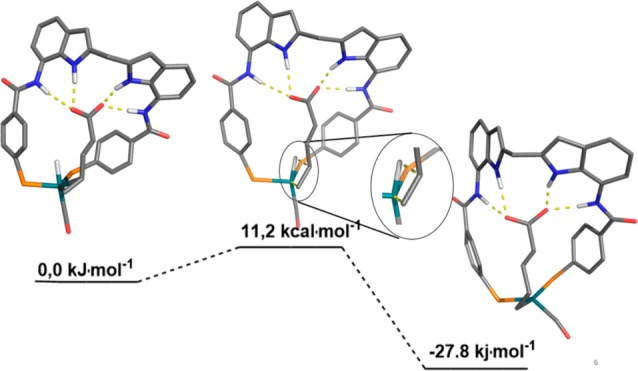
Substrate preorganization in the selectivity-determining hydride migration step (DFT study).
Phosphite analogues of the DIMPhos ligands (Figure 11, 53) give rhodium catalysts that are sufficiently active to hydroformylate internal aliphatic alkenes under mild conditions, which is not possible with the phosphine-based systems.55 The CO inserts farthest from the carboxylate, and exceptionally high selectivities are observed for internal alkenes (i.e., o:i ratio of ≤78) using the substrate orientation strategy with 53 as the ligand (Scheme 15). Again, a series of substrates with different distances between the alkene and the carboxylate were selectively converted with the highest selectivity obtained for the internal alkene at position 4.
Scheme 15. Regioselective Hydroformylation of Internal Unsaturated Carboxylic Acids.

Also, carboxy-vinylarenes are hydroformylated with the same system to form the linear product with the highest selectivities reported to date (>98%) (Scheme 16, 56a). The branched aldehyde is not detected, whereas this is usually the dominant product. Electronic factors dictate that these aromatic substrates mainly form branched aldehydes (Scheme 16, 56b).56 Remarkably, the methyl-substituted and cyclic analogues were also converted with very high selectivity (Scheme 16, 57).
Scheme 16. Regioselective Hydroformylation of 2-Carboxyvinylarenes and Cyclic Analogues.

When the phosphite-based DIMPhos hydroformylation system is combined with a palladium isomerization catalyst, terminal alkenes are converted into α-branched methyl aldehydes (Scheme 13).57 The advantage is that branched aldehydes can be synthesized from inexpensive terminal alkenes with selectivities surpassing those of direct branched selective hydroformylation catalysts.
A related strategy for selective hydroformylation catalysis relies on dynamic covalent chemistry. Instead of supramolecular interactions, the substrate is temporarily bound to a scaffolding ligand by reversible bond formation between the substrate and such a ligand.58 Via the scaffolding ligand, the substrate binds in a ditopic fashion to the metal complex. The exchange of the substrate with the scaffolding ligand should be compatible with the hydroformylation reaction.59,60
One scaffolding ligand that can be used in the context of regioselective hydroformylation is 58 that has a hemilabile C–O bond (Figure 13). Through reversible cleavage of the C–O bond, the hydroxy group of the substrate can bind to the ligand.59 Preorganization reverses the regioselectivity of several substituted homoallylic alcohols to form the branched/innermost product in excess. After hydroformylation, an oxidation reaction yields five-membered ring lactones in good selectivities of ≤98:2 (Scheme 17, 62). The strategy is feasible for both internal and terminal alkenes. A control reaction using PPh3 yields the six-membered ring lactone in excess (six-membered:five-membered ratio of 3:1). The same ligand is also successfully applied in the hydroformylation of substrates containing a sulfonamide as a directing group instead of an alcohol group.61
Figure 13.
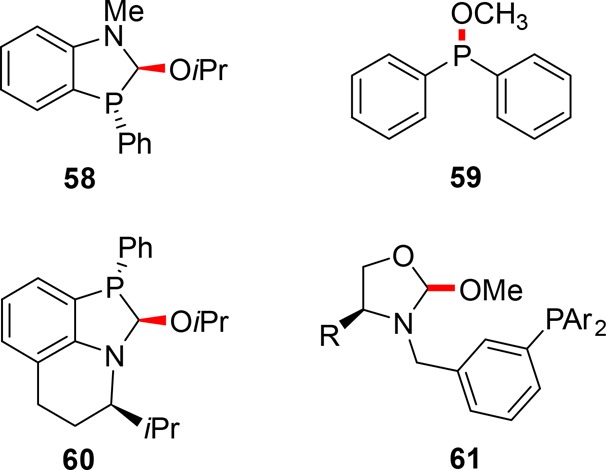
Catalytic scaffolding ligands (reversible bond colored red).
Scheme 17. Regioselective Hydroformylation Employing Catalytic Scaffolding Ligands (PCC = pyridinium chlorochromate).

In a similar approach, methyl diphenylphosphinite has been used as a scaffolding ligand (Figure 13, 59).60 This ligand has a labile P–O bond, and the methoxy moiety can exchange with hydroxy groups on the substrate. A catalytic amount of the ligand can be combined with homoallylic alcohols to yield the branched product (innermost for internal alkenes) in near perfect selectivities of <99%. A lactone is formed after oxidation of the formed lactol in this reaction (Scheme 18, 63). This ligand can also be applied in the regioselective hydroformylation of bishomoallylic alcohols to selectively yield six-membered lactones as a product (Scheme 18, 64).62
Scheme 18. Regioselective Hydroformylation of Homoallylic and Bishomoallylic Alcohols Employing Catalytic Scaffolding Catalysts.

The same strategy can also be applied to form quaternary carbon centers via a hydroformylation reaction, which is considered as one of the most challenging reactions in hydroformylation.63,64 Hydroformylation to form quaternary carbon centers was achieved when α,α-disubstituted olefins were used with the previously discussed scaffolding ligands 58 and 59. As the aldehyde inserts on the carbon center closest to the alcohol group, the “Keulemans’ rule”, which dictates that addition of a formyl group never occurs at the tertiary position of the olefin in hydroformylation, is overruled (Scheme 19, 65 and 66).65
Scheme 19. Hydroformylation of α,α-Disubstituted Alcohols To Form Quaternary Carbon Centers.

When amine-based substrates are hydroformylated in combination with an enantioenriched version (60) of the previously discussed scaffolding ligand (58), high enantioselectivities of ≤92% are obtained (Scheme 20, 67).66 Directed hydroformylation of 2,5-cyclohexadienyl-1-carbinols with diphenylphosphite as a ligand allowed excellent regio- and diastereocontrol (Scheme 21, 68).67
Scheme 20. Enantioselective Hydroformylation of Amine-Based Substrates.

Scheme 21. Directed Hydroformylation of 2,5-Cyclohexadienyl-1-carbinols.

Remarkably, placement of the binding moiety of the scaffolding ligand farther from the phosphorus atom (Figure 13, 61) reverses the selectivity completely. This leads to the insertion at the outermost carbon providing the product with selectivities of o:i ≤ 19:1 (Scheme 22, 69).68
Scheme 22. Divergent Selectivity upon Variation of the Scaffolding Ligand.
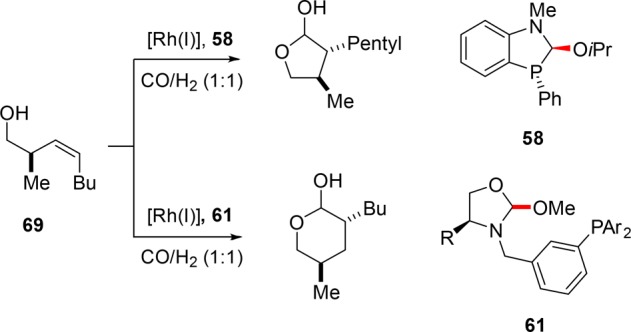
Altering the distance between the alcohol and the olefinic moiety reveals that the homoallylic alcohols reacted with the highest selectivity using this system to form six-membered lactols and lactones. The reaction proceeds most selectively when the R-based enantiomer of the substrate is reacted with the enantiopure l-valine-based catalyst.
4. Hydroformylation Catalysis in Confined Spaces
In the previous section, selective hydroformylation by controlling the substrate orientation was discussed. For this approach, the substrate needs to have functional groups to establish the proper interaction with the functional groups of the catalyst system. Many substrates of interest for hydroformylation do not have functional groups. To achieve selective hydroformylation for this class of substrates, the hydride migration step must be controlled in an alternative manner. One strategy that has been explored is the application of catalysts in confined spaces, and this will be discussed in this section.
For the sake of brevity, in this section solely the modulation of the activity and/or selectivity of rhodium-based hydroformylation catalysts as a result of confinement in a synthetic, molecular cage-like structure will be discussed. Organic transformations and other metal-catalyzed reactions have recently been described elsewhere; excellent reviews on using molecular containers in multistep reaction cascades and tandem enzymatic reactions,69 in reactivity modulation,70 and in the context of enzyme mimics based upon supramolecular coordination chemistry,71 along with transition metal and organocatalysis in functional molecular flasks,72 have already been published.
In pioneering work by Monflier and co-workers, cyclodextrins have been applied in combination with water-soluble rhodium phosphine-based catalysts for biphasic hydroformylation of higher olefins.73 The cyclodextrin cavity is essentially hydrophobic and can therefore host various organic molecules in water (Figure 14). Generally, the cyclodextrin host forms a water-soluble host–guest complex with the substrate, thereby increasing the water solubility of the alkene. As such, it brings it into closer contact with the catalyst residing in the aqueous layer, typically resulting in increased catalytic rates (Scheme 23). In this regard, the cyclodextrin acts as a phase-transfer catalyst by facilitating the migration of a reactant from one phase into the phase where the reaction occurs. Furthermore, in most examples, the regioselectivity is also affected as a direct consequence of confinement. Examples of cyclodextrin acting exclusively as a phase-transfer catalyst will not be discussed in this review.
Figure 14.
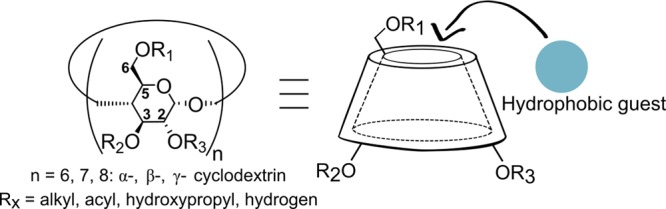
Schematic representation of the α-, β-, and γ-cyclodextrins that have a hydrophobic cavity that can host organic guest molecules.
Scheme 23. Phase-Transfer Catalysis Mediated by Cyclodextrins.
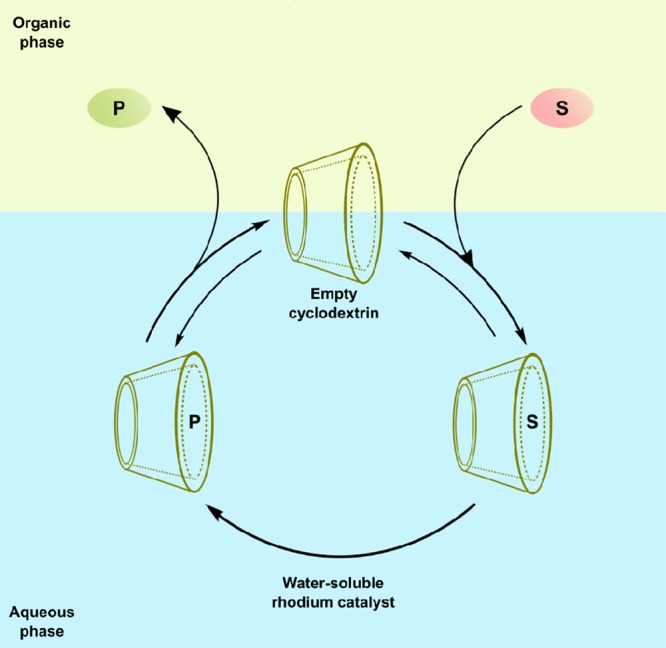
The first example reported along these lines was the application of partially methylated β-cyclodextrins in the biphasic rhodium 3,3′,3″-phosphinetriyltris(benzenesulfonate) (TPPTS)-catalyzed hydroformylation of various water-insoluble terminal and internal alkenes, enabling efficient biphasic hydroformylation of higher alkenes.73a In the absence of cyclodextrins, only substrates with sufficient water solubility, and hence shorter carbon chain lengths, are efficiently converted under biphasic hydroformylation conditions. Different substrates ranging from terminal and internal aliphatic alkenes to aromatic styrene derivatives can be converted with the novel catalytic system. A significant increase in the catalytic rate was observed in the presence of the methylated β-cyclodextrins, especially for the insoluble alkenes. For example, for 1-decene the conversion after 6 h was 10-fold, and the increase in reaction rate is estimated to be 25-fold. The effect of confinement is also clear in the hydroformylation of internal alkenes, although the overall conversions are rather low. This low reactivity is attributed to the lack of accessibility of the double bond of the substrate, which most likely resides too deep inside the hydrophobic cavity of the cyclodextrin host. The presence of the cyclodextrin also has an influence on the regioselectivity in the hydroformylation of 1-alkenes. For all substrates, the selectivity for the linear aldehyde decreases in the presence of the cyclodextrin. This is explained by the fact that not only the alkene but also the water-soluble phosphine ligand interacts with the cavity.73b By encapsulation of the phosphine ligand, the equilibria between different rhodium species are shifted to low-coordinate complexes that are generally more active and less selective, explaining the decrease in selectivity.
The group of Monflier extended the cyclodextrin-based catalytic system to fully solvent free conditions.74 Substrate molecules and a rhodium phosphine-based catalyst were dispersed in a mixture of acyclic saccharides and cyclodextrins, resulting in a heterogeneous mixture. The saccharides ensure complete dispersion of the substrates in the solid mixture, leading to increased catalytic rates. Exposure of this mixture to syngas in a mixing planetary ball mill results in the hydroformylation of a variety of styrene derivatives, always with 100% chemoselectivity to provide the corresponding aldehydes. In the absence of cyclodextrins, the branched aldehyde is predominantly formed, which is typical for styrene derivatives. However, the presence of cyclodextrins results in a 5-fold increase in the selectivity for the linear aldehyde product. This altered selectivity is due to the binding of the aromatic alkene within the hydrophobic cavity of the cyclodextrin, favoring hydroformylation at the less-hindered position of the vinyl function (Scheme 24). The substrate displays a stronger inclusion in the larger cyclodextrin (n = 7), which results in steric hindrance between the alkene functionality and the host. As a consequence, the formyl group will be transferred to the least hindered carbon atom that yields the linear aldehyde as the main product. For the smaller cyclodextrin (n = 6), the binding does not result in shielding as it is shallower in the pocket, and as a result, the selectivity is solely determined by the electronics of the substrate itself.
Scheme 24. Representation of the Effect of the Depth of Inclusion of the Substrate in the Cyclodextrin Cavity on the Observed Aldehyde Product Selectivity.

To prevent the earlier discussed inclusion of the phosphine ligand within the cavity of the cyclodextrin, bulkier mono- and bidentate ligands were subsequently applied in the search for catalytic systems that display a higher selectivity for the linear aldehyde product.73c Indeed, bulky water-soluble phosphine ligands such as 1,3,5-triaza-7-phosphaadamantane-based ligands73d (71) (Figure 15, middle) and 2-naphthylphosphines73e (72a–72c) (Figure 15, right) do not form strong inclusion complexes with cyclodextrins compared to those formed with TPPTS; thus, a smaller decrease in regioselectivity is observed in the hydroformylation of 1-decene, and at the same time, the conversions are much higher. The bidentate sulfonated 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene ligand (70) is of particular interest as it forms strong complexes with rhodium, preventing the formation of monoligated complexes, and because of its large bite angle, it already favors the formation of the linear product (Figure 15, left). Furthermore, because it is larger than the previously employed TPPTS ligand, it does not form a strong inclusion complex with the cyclodextrin host, allowing more efficient substrate transportation. In the presence of β-cyclodextrin, the catalytic system displays a 5-fold increase in the conversion of 1-octene to nonanal under standard hydroformylation conditions. Interestingly, the selectivity for the linear aldehyde increased (l:b ratio from 14 to 33) when cyclodextrin was present in the catalytic mixture. This enhanced selectivity is proposed to be due to the steric bulk around the coordination sphere of rhodium, forcing the hydride to preferentially migrate to C2 over C1.
Figure 15.

Structures of the water-soluble ligands 70, 71, and 72a–72c.
As an alternative to the approach described above, systems relying on the direct covalent functionalization of the β-cyclodextrin with phosphine ligands have been reported.75,76 The group of Reetz reported on the application of rhodium complexes of various β-cyclodextrin-modified diphosphine ligands in the biphasic hydroformylation of 1-octene. Interestingly, this catalytic system converts not only terminal alkenes but also relatively unreactive internal olefins. A proposed mode of action for the diphosphine-based catalyst is displayed in Scheme 25. The modified cyclodextrin-based catalysts display a 150-fold increase in activity at 80 °C compared to systems without cyclodextrin at 120 °C in the conversion of 1-octene to nonanal. The catalyst converts the substrate with a selectivity for the linear product of 76%, which is typical for rhodium complexes based on small bite angle ligands.
Scheme 25. Proposed Mode of Action of the β-Cyclodextrin-Modified Diphosphine-Based Catalytic System.
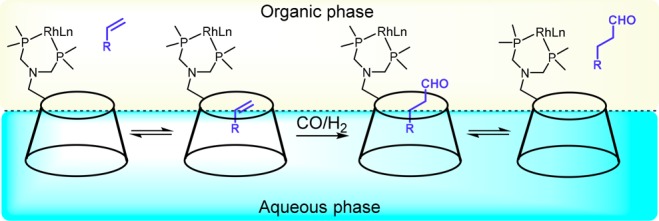
A similar approach to the cyclodextrin system relies on the use of hemispherical rhodium calixarene-based phosphite and phosphine complexes. These systems have been shown to promote the formation of linear aldehyde products over branched ones in hydroformylation catalysis. While it has been postulated that the confined space around the catalytically active site plays a role in the product selectivity, more detailed mechanistic studies are needed to confirm this.77,78
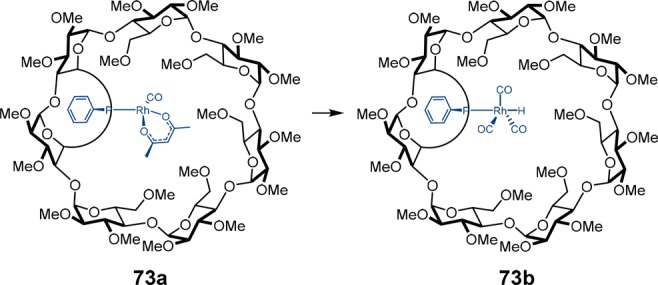
Recently, Matt and co-workers described the use of rhodium monophosphine complexes for the hydroformylation of styrene.76 By covalent confinement of the phosphine ligand inside a chiral cyclodextrin cavity, exclusive formation of rhodium monophosphine complex 73b is observed, even in the presence of excess ligand (Scheme 26). This is a clear example in which the second coordination sphere around the catalytically active site controls the coordination mode of the metal. Remarkably, the catalyst converts styrene with both a high branched selectivity (98%) and enantioselectivity (≤95%). It is interesting to note that the cyclodextrin is the only source of chirality, and as such, the enantioselectivity is controlled by the chiral second coordination sphere around the metal complex.
Scheme 26. Formation of the Active Species 73b from the Monophosphine Rhodium Complex 73a.
Reek and co-workers introduced a general strategy for encapsulating transition metal complexes in an efficient way that involves a ligand-template approach.79 In this strategy, the ligand serves a dual role as it operates as a template for the self-assembly of the capsule and coordinates to the catalytically active metal center; hence, it was coined ligand-template approach. The first example reported using this strategy was structure 74 (Figure 16), formed by the self-assembly of three zinc meso-tetraphenylporphyrin (ZnTPP) units around the ligand-template tris(m-pyridyl)phosphine [P(m-py)3], relying on the selective N–Zn coordination. The phosphine atom of the ligand-template is coordinated to rhodium and forms the active species [HRhP(CO)3] under syngas. The overall formed encapsulated complex is an efficient hydroformylation catalyst that converts various terminal alkene substrates with enhanced activity and selectivity for the branched aldehyde. Importantly, because of the steric bulk imposed by the porphyrin moieties, monoligation of the formed rhodium complexes is enforced. Via the variation of the structure of the template and the surrounding zinc building blocks, the effect of variations in the second coordination sphere on the activity and selectivity of the encapsulated rhodium catalyst has been further explored.
Figure 16.
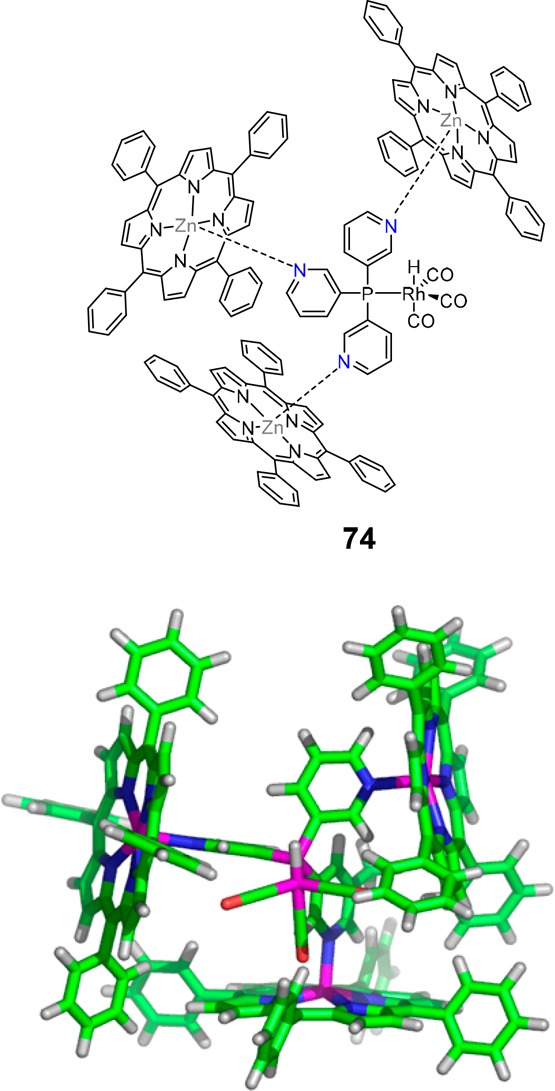
Structure of HRh(CO)3 coordinated to the central phosphine of the first-generation assembly. Molecular structure (top) and modeled structure (bottom) of the encapsulated catalyst.
Application of 74 in 1-octene hydroformylation at room temperature leads to a 10-fold increase in the catalytic activity as compared to that with the rhodium complex in the absence of porphyrin under otherwise identical conditions.79a,79b This rate enhancement can, at least partly, be explained by the higher reactivity of monophosphine complexes. Indeed, DFT calculations show that the catalytic pathway of the rhodium monophosphine catalyst has a free energy barrier that is lower than that of the bisphosphine analogue, leading to an inherently more active catalyst.80 Remarkably, the encapsulated catalyst dominantly forms the branched aldehyde product (l:b ratio of 0.6), which is highly unusual for aliphatic, terminal alkenes. Upon generation of the encapsulated catalysts based on ruthenium porphyrin building blocks instead of zinc, less dynamic capsules that display even higher regioselectivity are formed (l:b ratio of 0.4), but at the cost of the catalytic activity. The orientation of the porphyrin with respect to the phosphine is crucial; the use of tris(p-pyridyl)phosphine [P(p-py)3] ligand-templates instead of the meta analogue results in a catalyst that displays the selectivity typical for rhodium bisphosphine complexes.81 Indeed, a more open structure is formed, allowing the formation of a bisphosphine-coordinated rhodium complex that is encapsulated by six porphyrins. In the solid state, an unusual supramolecular structure is formed according to X-ray analysis, in which one zinc porphyrin unit acts as a bridging moiety between two capsules via an unusual hexacoordinate zinc (Figure 17).
Figure 17.
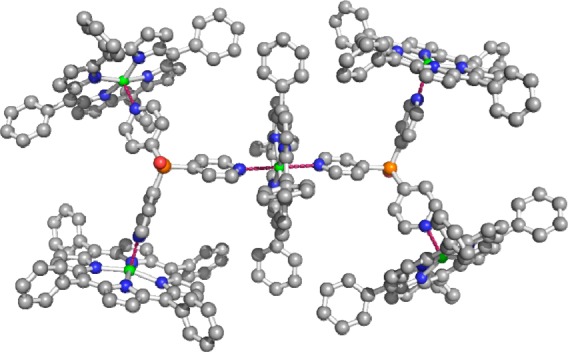
Highly unusual supramolecular structure containing a mixture of penta- and hexacoordinate zinc porphyrins.
The generality of the ligand-template approach is demonstrated by the application of a variety of building blocks. The ligand-template can be combined with zinc salphens (75a–75d) and zinc bis(thiosemicarbazonato) complexes (76) (Figure 18),82 which both display a supramolecular binding with pyridine that is stronger than that of zinc porphyrins. As these building blocks are significantly smaller, the exclusive formation of encapsulated catalysts cannot be enforced. The smaller size allows considerable conformational flexibility in the self-assembled structures, resulting in a mixture of mono- and bisligated complexes. The catalysts encapsulated by 75a–75d display some selectivity for the branched aldehyde product when applied in 1-octene hydroformylation, but the selectivity is much lower than for the first-generation capsule 74. Moreover, these systems also show an increase in undesired isomerization. Similar results have been obtained for the capsules based on 76.
Figure 18.
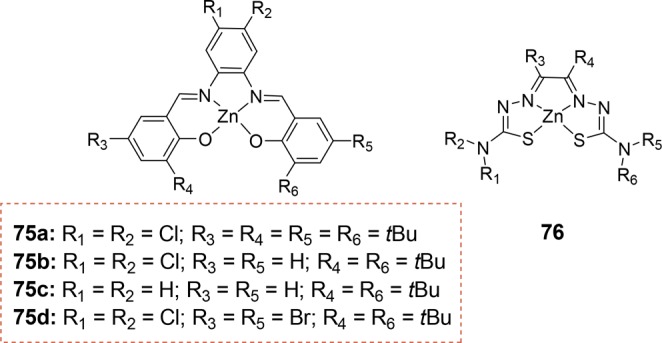
Molecular structures of the smaller building blocks 75a–75d and 76.
Following the publication of the promising results obtained with terminal alkene substrates, the more challenging internal alkenes were subjected to capsule-controlled hydroformylation.83,84 Regioselective hydroformylation of internal alkenes is challenging as the two carbon atoms of the double bond are electronically identical and sterically similar. In addition, the reactivity of internal alkenes is lower, and the application of harsher conditions leads in general to more side reactions as isomerization. The rhodium complex of the first-generation capsule 74 converts trans-2-octene and trans-3-octene with very high regioselectivity (o:i of 1:9 for 2 octene and 1:4 for 3-octene), where the formyl group is installed on the innermost carbon atom, and also the activity is higher compared to that displayed by the unencapsulated catalyst. This selectivity is in line with the results obtained for 1-octene where a preference for the branched aldehyde is observed. Interestingly, a rather high selectivity could still be retained at 40 °C, whereas at 80 °C, isomerization side products lead to a loss of regioselectivity. A combination of DFT calculations and detailed kinetic and mechanistic studies demonstrate that the selectivity is determined in the hydride migration step (Figure 19).84 The path to the 3-alkylrhodium intermediate has an energy considerably lower than that of the 2-alkylrhodium intermediate. Significant structural rearrangements of the capsule would be necessary to arrive at the 2-alkylrhodium intermediate, resulting in a high energy penalty. Consequently, the 3-alkylrhodium intermediate is favored leading to a high selectivity toward the C3-aldehyde. This is related to the mechanism in which the selectivity is controlled by substrate orientation, the difference being that the rotation of the alkene associated with the hydride migration step is not controlled by hydrogen bonds, but by the sterics of the cage.
Figure 19.
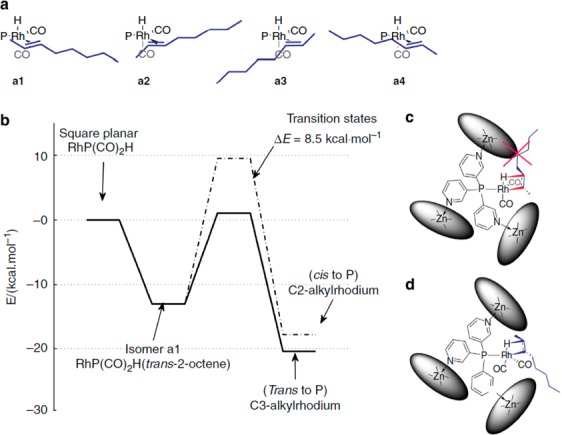
Energy profile for the hydride migration step leading to the two possible intermediates b and c. Reprinted with permission from ref (84).
All these hydroformylation reactions were performed at room temperature or at slightly elevated temperatures, as the supramolecular N–Zn coordinate bond was expected to weaken at elevated temperatures. To expand the application of the capsule to more industrially relevant conditions, catalytic experiments were conducted at 75 °C.85 To retain the unusual branched selectivity in the hydroformylation of 1-octene at this temperature, a higher partial pressure of CO is required. Remarkably, when the reaction is performed at 75 °C and 80 bar of CO/H2 (2:1), a selectivity (l:b ratio of 0.7) nearly identical to that at room temperature is obtained. At these elevated temperatures, the reaction is highly dependent on the partial CO pressure; at 20 bar, the selectivity for the branched aldehyde was lost. In line with that, high-pressure infrared measurements show the formation of bisphosphine-coordinated rhodium complexes under these low-pressure conditions.
Next to temperature, the solvent scope was also investigated. As the zinc–pyridine interaction is strongest in apolar and noncoordinating solvents, these solvents were preferably applied in the initial experiments. As expected, upon using more polar solvents, the zinc–pyridine binding constant decreases, and as a result, the equilibrium shifts to the non-encapsulated catalyst, resulting in a loss of activity and selectivity. Interestingly, the oxidized analogues of zinc TPP, zinc porpholactones, bind more strongly to pyridine by nearly an order of magnitude.86 Consequently, the application window of the ligand-template approach can be extended to industrially relevant more polar and coordinating solvents, such as dioctyl terephthalate, while retaining the typical branched selectivity in 1-octene hydroformylation. Furthermore, as the capsule 77 has a slightly smaller size and a different shape, it also allows the branched selective hydroformylation of propene, which is inherently challenging (l:b ratio of 0.84) (Figure 20).
Figure 20.
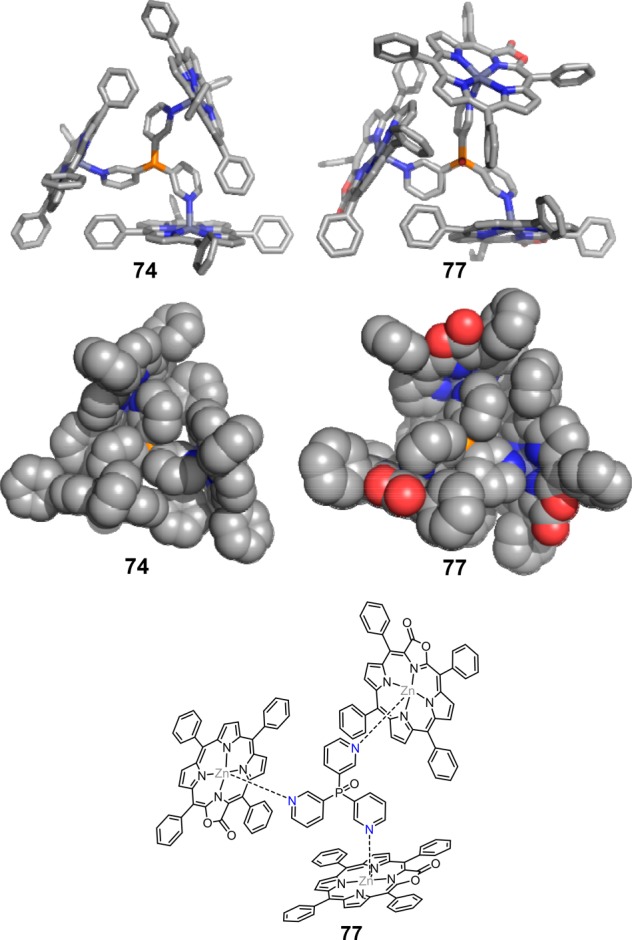
Comparison of the crystal structures of assemblies 74 and 77, formed by the self-assembly of 3 equiv of Zn(II) meso-tetraphenylporpholactone and tris(m-pyridyl)phosphine in toluene (letop) and a molecular structure of assembly 77 (bottom). Reprinted with permission from ref (86). Copyright 2017 Creative Commons.
While the regioselectivity is now controlled, these branched aldehydes are formed in racemic form as the catalyst is not chiral. Next, the ligand-template approach was extended to the enantioselective hydroformylation of internal alkenes and styrene derivatives.19,87 Replacing the ligand-template with a chiral pyridine-functionalized phosphoramidite ligand, while maintaining the original zinc porphyrin building blocks, results in novel chiral capsules. Interestingly, addition of the zinc porphyrin building blocks to the activated rhodium–phosphoramidite hydride complex enforces a change from an equatorial to axial coordination mode for the ligand. This change in the coordination mode also changes the properties of the catalyst in the hydroformylation of trans-2-octene. Both the conversion (56%) and the enantioselectivity (45%) increase in the presence of porphyrin, leading to the capsule, yet a preference for the addition of the formyl group to the innermost carbon atom was again seen. The modest increase in enantioselectivity can be attributed to the dynamic nature of the capsule, and therefore, a more rigid capsule with less rotational freedom was explored.19 The same chiral ligand was also encapsulated in a metal–organic coordination cage containing two zinc porphyrins.88 The supramolecular complex does not result in significant enantioselective induction in the asymmetric hydroformylation of 1-octene; however, it gives rise to a high chiral induction (ee of ≤74%) in the hydroformylation of styrene compared to that of the non-encapsulated catalyst (ee of <10%).
The mixing of two bis-[ZnII(salphen)] building blocks with two chiral pyridine-functionalized phosphoramidite templates results in the formation of a supramolecular “box” 78 with the ligands functioning as pillars (Figure 21).19 Modeling of the complex shows that the rhodium complex indeed is embedded in a cage defined by the bis-[ZnII(salphen)] building blocks. The application of this supramolecular box in the hydroformylation of internal alkenes such as trans- and cis-2-octene results in the formation of the aldehyde product with high regioselectivity and enantioselectivity (ee of ≤86%). This demonstrates that both the regioselectivity and the enantioselectivity can be controlled by spatially confining the catalytically active site in a tight pocket.
Figure 21.
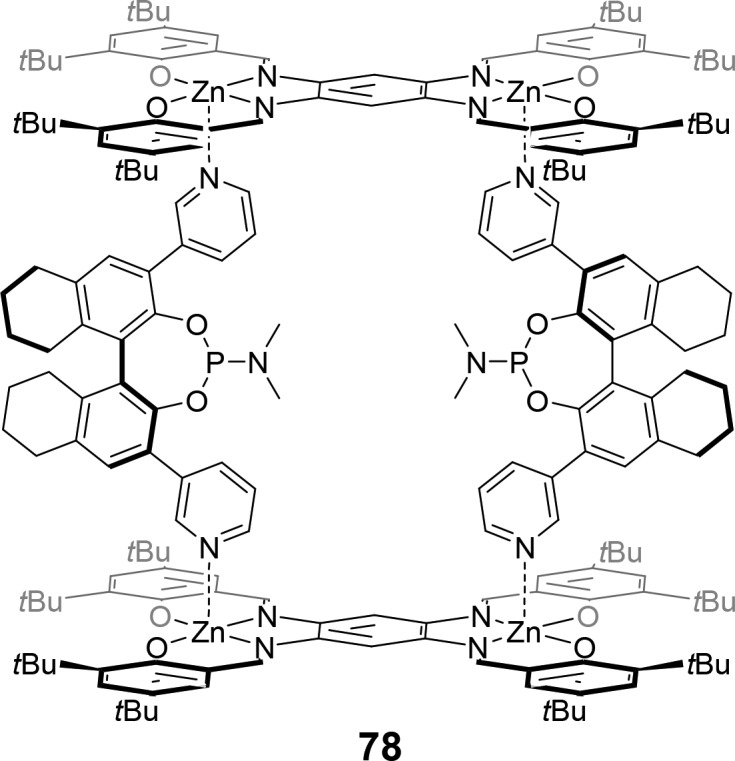
Bis-zinc(II) salphen-templated supramolecular “box” 78.
5. Conclusion and Outlook
In this review, we have discussed various supramolecular strategies for generating selective hydroformylation catalysts, which are in many examples far more selective than the traditional catalysts. In the first section, we show that you can generate supramolecular bidentate ligands by self-assembly of ligand building blocks. The main advantage of this strategy is that the ligand library you generate grows exponentially with the number of building blocks you can use. It has been demonstrated that rhodium catalysts that are very selective for the linear product can be generated. The strategy has also been applied for asymmetric hydroformylation, and although the proof of principle has been demonstrated, catalysts based on supramolecular bidentate ligands that are more enantioselective in styrene hydroformylation have yet to be discovered. The second approach that has been discussed involves substrate orientation via interactions between the functional groups of the substrate and those of the ligand. Typically, the hydride migration step of the catalytic cycle, often the selectivity-determining step, is controlled by this substrate preorganization. This has resulted in many examples of hydroformylation reaction in which the product is formed with an unusually high selectivity. As such, this is a very promising strategy for solving selectivity problems in hydroformylation catalysis that cannot be solved by simple ligand design. As a final approach, we discussed hydroformylation catalysis in molecular cages. Using this strategy, selectivities can also be achieved that are beyond those displayed by traditional catalysts, and importantly, it also works for unfunctionalized substrates.
We are just at the beginning of exploring these new supramolecular concepts in hydroformylation catalysis, and we are convinced that upon expanding the number of systems we will be able to solve many selectivity problems in hydroformylation catalysis.
Acknowledgments
The authors thank all the co-workers that contributed to our work over the past decades whose names appear in the references and all the institutes that financed our research programs, including support for the authors of the current paper [(NWO) Dutch National Science Foundation, ERC advanced grant (N° 339786-NAT-CAT), Eastman, and IncatT BV].
The authors declare no competing financial interest.
References
- Roelen O. (to Chemische Verwertungsgesellschaft Oberhausen m.b.H.) German Patent DE 849548, 1938/1952; U.S. Patent 2327066, 1943; Chem. Abstr.1944, 38, 3631.
- Franke R.; Selent D.; Börner A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. 10.1021/cr3001803. [DOI] [PubMed] [Google Scholar]
- Gusevskaya E. V.; Jiménez-Pinto J.; Börner A. Hydroformylation in the Realm of Scents. ChemCatChem 2014, 6, 382–411. 10.1002/cctc.201300474. [DOI] [Google Scholar]
- a van Leeuwen P. W. N. M. In Homogeneous catalysis: Understanding the art; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; pp 125–174. [Google Scholar]; b Cornils B.; Herrmann W. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Three Volumes; Wiley-VCH: Weinheim, Germany, 2002; Vol. 1, pp 31–194. [Google Scholar]
- Vilches-Herrera M.; Domke L.; Börner A. Isomerization–Hydroformylation Tandem Reactions. ACS Catal. 2014, 4, 1706–1724. 10.1021/cs500273d. [DOI] [Google Scholar]
- a Heck R. F.; Breslow D. S. The Reaction of Cobalt Hydrotetracarbonyl with Olefins. J. Am. Chem. Soc. 1961, 83, 4023–4027. 10.1021/ja01480a017. [DOI] [Google Scholar]; b Heck R. F. Addition Reactions of Transition Metal Compounds. Acc. Chem. Res. 1969, 2, 10–16. 10.1021/ar50013a002. [DOI] [Google Scholar]
- a Agbossou F.; Carpentier J.-F.; Mortreux A. Asymmetric Hydroformylation. Chem. Rev. 1995, 95, 2485–2506. 10.1021/cr00039a008. [DOI] [Google Scholar]; b van Leeuwen P. W. N. M.; Claver C. In Rhodium Catalyzed Hydroformylation; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000. [Google Scholar]; c Wiese K.-D.; Obst D. In Catalytic Carbonylation Reactions; Beller M., Ed.; Springer: Berlin, 2006; pp 1–33. [Google Scholar]; d Kubis C.; Baumann W.; Barsch E.; Selent D.; Sawall M.; Ludwig R.; Neymeyr K.; Hess D.; Franke R.; Börner A. Investigation into the Equilibrium of Iridium Catalysts for the Hydroformylation of Olefins by Combining In Situ High-Pressure FTIR and NMR Spectroscopy. ACS Catal. 2014, 4, 2097–2108. 10.1021/cs500368z. [DOI] [Google Scholar]; e Kubis C.; Sawall M.; Block A.; Neymeyr K.; Ludwig R.; Börner A.; Selent D. An Operando FTIR Spectroscopic and Kinetic Study of Carbon Monoxide Pressure Influence on Rhodium-Catalyzed Olefin Hydroformylation. Chem. - Eur. J. 2014, 20, 11921–11931. 10.1002/chem.201402515. [DOI] [PubMed] [Google Scholar]; f Kubis C.; Ludwig R.; Sawall M.; Neymeyr K.; Börner A.; Wiese K.-D.; Hess D.; Franke R.; Selent D. A Comparative In Situ HP-FTIR Spectroscopic Study of Bi- and Monodentate Phosphite-Modified Hydroformylation. ChemCatChem 2010, 2, 287–295. 10.1002/cctc.200900292. [DOI] [Google Scholar]; g Kubis C.; Selent D.; Sawall M.; Ludwig R.; Neymeyr K.; Baumann W.; Franke R.; Börner A. Exploring Between the Extremes: Conversion-Dependent Kinetics of Phosphite-Modified Hydroformylation Catalysis. Chem. - Eur. J. 2012, 18, 8780–8794. 10.1002/chem.201200603. [DOI] [PubMed] [Google Scholar]; h Casey C. P.; Petrovich L. M. (Chelating diphosphine)rhodium-Catalyzed Deuterioformylation of 1-Hexene: Control of Regiochemistry by the Kinetic Ratio of Alkylrhodium Species Formed by Hydride Addition to Complexed Alkene. J. Am. Chem. Soc. 1995, 117, 6007–6014. 10.1021/ja00127a014. [DOI] [Google Scholar]; i Lazzaroni R.; Uccello-Barretta G.; Scamuzzi S.; Settambolo R.; Caiazzo A. 2H NMR Investigation of the Rhodium-Catalyzed Deuterioformylation of 1,1-Diphenylethene: Evidence for the Formation of a Tertiary Alkyl–Metal Intermediate. Organometallics 1996, 15, 4657–4659. 10.1021/om960462e. [DOI] [Google Scholar]; j Horiuchi T.; Shirakawa E.; Nozaki K.; Takaya H. Mechanistic Aspects of Asymmetric Hydroformylation of Olefins Catalyzed by Chiral Phosphine–Phosphite–Rhodium(I) Complexes. Organometallics 1997, 16, 2981–2986. 10.1021/om970134u. [DOI] [PubMed] [Google Scholar]; k Lazzaroni R.; Settambolo R.; Uccello-Barretta G.; Caiazzo A.; Scamuzzi S. Rhodium-Catalyzed Hydroformylation of Vinylidenic Olefins: the Different Behaviors of the Isomeric Alkyl–Metal Intermediates as the Origin of the β-Regioselectivity. J. Mol. Catal. A: Chem. 1999, 143, 123–130. 10.1016/S1381-1169(98)00374-4. [DOI] [Google Scholar]; l van der Slot S. C.; Duran J.; Luten J.; Kamer P. C. J.; van Leeuwen P. W. N. M. Rhodium-Catalyzed Hydroformylation and Deuterioformylation with Pyrrolyl-Based Phosphorus Amidite Ligands: Influence of Electronic Ligand Properties. Organometallics 2002, 21, 3873–3883. 10.1021/om010760y. [DOI] [Google Scholar]; m Watkins A. L.; Landis C. R. Origin of Pressure Effects on Regioselectivity and Enantioselectivity in the Rhodium-Catalyzed Hydroformylation of Styrene with (S, S, S)-BisDiazaphos. J. Am. Chem. Soc. 2010, 132, 10306–10317. 10.1021/ja909619a. [DOI] [PubMed] [Google Scholar]
- Brezny A. C.; Landis C. R. Unexpected CO Dependencies, Catalyst Speciation, and Single Turnover Hydrogenolysis Studies of Hydroformylation via High Pressure NMR Spectroscopy. J. Am. Chem. Soc. 2017, 139, 2778–2785. 10.1021/jacs.6b12533. [DOI] [PubMed] [Google Scholar]
- a van Rooy A.; de Bruijn J. N. H.; Roobeek K. F.; Kamer P. C. J.; van Leeuwen P. W. N. M. Rhodium-Catalysed Hydroformylation of Branched 1-Alkenes; Bulky Phosphite vs. Triphenylphosphine as Modifying Ligand. J. Organomet. Chem. 1996, 507, 69–73. 10.1016/0022-328X(95)05748-E. [DOI] [Google Scholar]; b Kamer P. C. J.; van Rooy A.; Schoemaker G. C.; van Leeuwen P. W. N. M. In Situ Mechanistic Studies in Rhodium Catalyzed Hydroformylation of Alkenes. Coord. Chem. Rev. 2004, 248, 2409–2424. 10.1016/j.ccr.2004.06.006. [DOI] [Google Scholar]
- a Casey C. P.; Whiteker G. T.; Melville M. G.; Petrovich L. M.; Gavney J. A.; Powell D. R. Diphosphines with Natural Bite Angles near 120° Increase Selectivity for n-Aldehyde Formation in Rhodium-Catalyzed Hydroformylation. J. Am. Chem. Soc. 1992, 114, 5535–5543. 10.1021/ja00040a008. [DOI] [Google Scholar]; b Kranenburg M.; van der Burgt Y. E. M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Goubitz K.; Fraanje J. New Diphosphine Ligands Based on Heterocyclic Aromatics Inducing Very High Regioselectivity in Rhodium-Catalyzed Hydroformylation: Effect of the Bite Angle. Organometallics 1995, 14, 3081–3089. 10.1021/om00006a057. [DOI] [Google Scholar]; c van der Veen L. A.; Boele M. D. K.; Bregman F. R.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Goubitz K.; Fraanje J.; Schenk H.; Bo C. Electronic Effect on Rhodium Diphosphine Catalyzed Hydroformylation: The Bite Angle Effect Reconsidered. J. Am. Chem. Soc. 1998, 120, 11616–11626. 10.1021/ja981969e. [DOI] [Google Scholar]; d van der Veen L. A.; Keeven P. H.; Schoemaker G. C.; Reek J. N. H.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Lutz M.; Spek A. L. Origin of the Bite Angle Effect on Rhodium Diphosphine Catalyzed Hydroformylation. Organometallics 2000, 19, 872–883. 10.1021/om990734o. [DOI] [Google Scholar]; e Carbó J. J.; Maseras F.; Bo C.; van Leeuwen P. W. N. M. Unraveling the Origin of Regioselectivity in Rhodium Diphosphine Catalyzed Hydroformylation. A DFT QM/MM Study. J. Am. Chem. Soc. 2001, 123, 7630–7637. 10.1021/ja0101678. [DOI] [PubMed] [Google Scholar]; f Kumar M.; Chaudhari R. V.; Subramaniam B.; Jackson T. A. Importance of Long-Range Noncovalent Interactions in the Regioselectivity of Rhodium-Xantphos-Catalyzed Hydroformylation. Organometallics 2015, 34, 1062–1073. 10.1021/om5012775. [DOI] [Google Scholar]
- a Sakai N.; Mano S.; Nozaki K.; Takaya H. Highly Enantioselective Hydroformylation of Olefins Catalyzed by New Phosphine Phosphite-Rhodium(I) Complexes. J. Am. Chem. Soc. 1993, 115, 7033–7034. 10.1021/ja00068a095. [DOI] [Google Scholar]; b Nozaki K.; Sakai N.; Nanno T.; Higashijima T.; Mano S.; Horiuchi T.; Takaya H. Highly Enantioselective Hydroformylation of Olefins Catalyzed by Rhodium(I) Complexes of New Chiral Phosphine–Phosphite Ligands. J. Am. Chem. Soc. 1997, 119, 4413–4423. 10.1021/ja970049d. [DOI] [Google Scholar]; c Horiuchi T.; Shirakawa E.; Nozaki K.; Takaya H. Mechanistic Aspects of Asymmetric Hydroformylation of Olefins Catalyzed by Chiral Phosphine–Phosphite–Rhodium(I) Complexes. Organometallics 1997, 16, 2981–2986. 10.1021/om970134u. [DOI] [PubMed] [Google Scholar]; For a related ligand, see:; d Yan Y.; Zhang X. A Hybrid Phosphorus Ligand for Highly Enantioselective Asymmetric Hydroformylation. J. Am. Chem. Soc. 2006, 128, 7198–7202. 10.1021/ja057065s. [DOI] [PubMed] [Google Scholar]
- a Landis C. R.; Jin W.; Owen J. S.; Clark T. P. Rapid Access to Diverse Arrays of Chiral 3,4-Diazaphospholanes. Angew. Chem., Int. Ed. 2001, 40, 3432–3434. . [DOI] [PubMed] [Google Scholar]; b Clark T. P.; Landis C. R.; Freed S. L.; Klosin J.; Abboud K. A. Highly Active, Regioselective, and Enantioselective Hydroformylation with Rh Catalysts Ligated by Bis-3,4-diazaphospholanes. J. Am. Chem. Soc. 2005, 127, 5040–5042. 10.1021/ja050148o. [DOI] [PubMed] [Google Scholar]; c Klosin J.; Landis C. R. Ligands for Practical Rhodium-Catalyzed Asymmetric Hydroformylation. Acc. Chem. Res. 2007, 40, 1251–1259. 10.1021/ar7001039. [DOI] [PubMed] [Google Scholar]
- a Noonan G. M.; Fuentes J. A.; Cobley C. J.; Clarke M. L. An Asymmetric Hydroformylation Catalyst that Delivers Branched Aldehydes from Alkyl Alkenes. Angew. Chem., Int. Ed. 2012, 51, 2477–2480. 10.1002/anie.201108203. [DOI] [PubMed] [Google Scholar]; b Dingwall P.; Fuentes J. A.; Crawford L.; Slawin A. M. Z.; Bühl M.; Clarke M. L. Understanding a Hydroformylation Catalyst that Produces Branched Aldehydes from Alkyl Alkenes. J. Am. Chem. Soc. 2017, 139, 15921–15932. 10.1021/jacs.7b09164. [DOI] [PubMed] [Google Scholar]
- A Wilkinson M. J.; van Leeuwen P. W. N. M.; Reek J. N. H. New Directions in Supramolecular Transition Metal Catalysis. Org. Biomol. Chem. 2005, 3, 2371–2383. 10.1039/b503407h. [DOI] [PubMed] [Google Scholar]; b Breit B. Supramolecular Approaches to Generate Libraries of Chelating Bidentate Ligands for Homogeneous Catalysis. Angew. Chem., Int. Ed. 2005, 44, 6816–6825. 10.1002/anie.200501798. [DOI] [PubMed] [Google Scholar]; c Sandee A. J.; Reek J. N. H. Bidentate Ligands by Supramolecular Chemistry—the Future for Catalysis?. Dalton Trans. 2006, 3385–3391. 10.1039/B606045P. [DOI] [PubMed] [Google Scholar]; d Bellini R.; van der Vlugt J. I.; Reek J. N. H. Supramolecular Self-Assembled Ligands in Asymmetric Transition Metal Catalysis. Isr. J. Chem. 2012, 52, 613–629. 10.1002/ijch.201200002. [DOI] [Google Scholar]
- Slagt V. F.; van Leeuwen P. W. N. M.; Reek J. N. H. Bidentate Ligands Formed by Self-Assembly. Chem. Commun. 2003, 2, 2474–2475. 10.1039/b306683e. [DOI] [PubMed] [Google Scholar]
- Slagt V. F.; van Leeuwen P. W. N. M.; Reek J. N. H. Supramolecular Bidentate Phosphorus Ligands Based on Bis-zinc(II) and Bis-tin(IV) Porphyrin Building Blocks. Dalton Trans. 2007, 2302–2310. 10.1039/b702462m. [DOI] [PubMed] [Google Scholar]
- Kuil M.; Goudriaan P. E.; Kleij A. W.; Tooke D. M.; Spek A. L.; van Leeuwen P. W. N. M.; Reek J. N. H. Rigid Bis-zinc(II) Salphen Building Blocks for the Formation of Template-Assisted Bidentate Ligands and Their Application in Catalysis. Dalton Trans. 2007, 2311–2320. 10.1039/b702375h. [DOI] [PubMed] [Google Scholar]
- Kuil M.; Goudriaan P. E.; van Leeuwen P. W. N. M.; Reek J. N. H. Template-Induced Formation of Heterobidentate Ligands and Their Application in the Asymmetric Hydroformylation of Styrene. Chem. Commun. 2006, 4679–4681. 10.1039/b609321c. [DOI] [PubMed] [Google Scholar]
- Gadzikwa T.; Bellini R.; Dekker H. L.; Reek J. N. H. Self-Assembly of a Confined Rhodium Catalyst for Asymmetric Hydroformylation of Unfunctionalized Internal Alkenes. J. Am. Chem. Soc. 2012, 134, 2860–2863. 10.1021/ja211455j. [DOI] [PubMed] [Google Scholar]
- Slagt V. F.; van Leeuwen P. W. N. M.; Reek J. N. H. Multicomponent Porphyrin Assemblies as Functional Bidentate Phosphite Ligands for Regioselective Rhodium-Catalyzed Hydroformylation. Angew. Chem., Int. Ed. 2003, 42, 5619–5623. 10.1002/anie.200352525. [DOI] [PubMed] [Google Scholar]
- Slagt V. F.; Röder M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Reek J. N. H. Supraphos: A Supramolecular Strategy to Prepare Bidentate Ligands. J. Am. Chem. Soc. 2004, 126, 4056–4057. 10.1021/ja038955f. [DOI] [PubMed] [Google Scholar]
- Reek J. N. H.; Röder M.; Goudriaan P. E.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Slagt V. F. Supraphos: A Supramolecular strategy to Prepare Bidentate Ligands. J. Organomet. Chem. 2005, 690, 4505–4516. 10.1016/j.jorganchem.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Goudriaan P. E.; Jang X.-B.; Kuil M.; Lemmens R.; van Leeuwen P. W. N. M.; Reek J. N. H. Synthesis of Building Blocks for the Development of the SUPRAPhos Ligand Library and Examples of Their Application in Catalysis. Eur. J. Org. Chem. 2008, 2008, 6079–6092. 10.1002/ejoc.200800499. [DOI] [Google Scholar]
- Goudriaan P. E.; Kuil M.; Jiang X.-B.; van Leeuwen P. W. N. M.; Reek J. N. H. SUPRAPhos Ligands for the Regioselective Rhodium Catalyzed Hydroformylation of Styrene Forming the Linear Aldehyde. Dalton Trans. 2009, 1801–1805. 10.1039/b818665k. [DOI] [PubMed] [Google Scholar]
- Breit B.; Seiche W. Hydrogen Bonding as a Construction Element for Bidentate Donor Ligands in Homogeneous Catalysis: Regioselective Hydroformylation of Terminal Alkenes. J. Am. Chem. Soc. 2003, 125, 6608–6609. 10.1021/ja0348997. [DOI] [PubMed] [Google Scholar]
- Gellrich U.; Huang J.; Seiche W.; Keller M.; Meuwly M.; Breit B. Ligand Self-Assembling through Complementary Hydrogen-Bonding in the Coordination Sphere of a Transition Metal Center: The 6-Diphenylphosphanylpyridin-2(1H)-one System. J. Am. Chem. Soc. 2011, 133, 964–975. 10.1021/ja108639e. [DOI] [PubMed] [Google Scholar]
- Seiche W.; Schuschkowski A.; Breit B. Bidentate Ligands by Self-Assembly through Hydrogen Bonding: A General Room Temperature/Ambient Pressure Regioselective Hydroformylation of Terminal Alkenes. Adv. Synth. Catal. 2005, 347, 1488–1494. 10.1002/adsc.200505174. [DOI] [Google Scholar]
- Straub A. T.; Otto M.; Usui I.; Breit B. Room Temperature Ambient Pressure (RTAP)-Hydroformylation in Water Using a Self-Assembling Ligand. Adv. Synth. Catal. 2013, 355, 2071–2075. 10.1002/adsc.201300258. [DOI] [Google Scholar]
- Abillard O.; Breit B. Domino Hydroformylation/Enantioselective Cross-Aldol Addition. Adv. Synth. Catal. 2007, 349, 1891–1895. 10.1002/adsc.200700216. [DOI] [Google Scholar]
- Fuchs D.; Rousseau G.; Diab L.; Gellrich U.; Breit B. Tandem Rhodium-Catalyzed Hydroformylation-Hydrogenation of Alkenes by Employing a Cooperative Ligand System. Angew. Chem., Int. Ed. 2012, 51, 2178–2182. 10.1002/anie.201108946. [DOI] [PubMed] [Google Scholar]
- Diezel S.; Breit B. Tandem Regioselective Rhodium-Catalyzed Hydroformylation–Enantioselective Aminocatalytic anti-Mannich Reaction. Synthesis 2014, 46, 1311–1320. 10.1055/s-0033-1338602. [DOI] [Google Scholar]
- Ruan Q.; Zhou L.; Breit B. New Phosphorus Self-Assembling Ligands for the Tandem Hydroformylation–Wittig Olefination Reaction of Homoallylic Alcohols — A Key Step for Stereoselective Pyran Synthesis. Catal. Commun. 2014, 53, 87–90. 10.1016/j.catcom.2014.04.022. [DOI] [Google Scholar]
- Köpfer A.; Breit B. Rhodium-Catalyzed Hydroformylation of 1,1-Disubstituted Allenes Employing the Self-Assembling 6-DPPon System. Angew. Chem., Int. Ed. 2015, 54, 6913–6917. 10.1002/anie.201502086. [DOI] [PubMed] [Google Scholar]
- Agabekov V.; Seiche W.; Breit B. Rhodium-Catalyzed Hydroformylation of Alkynes Employing a Self-Assembling Ligand System. Chem. Sci. 2013, 4, 2418–2422. 10.1039/c3sc50725d. [DOI] [Google Scholar]
- Gellrich U.; Koslowski T.; Breit B. Full Kinetic Analysis of a Rhodium-Catalyzed Hydroformylation: Beyond the Rate-Limiting Step Picture. Catal. Sci. Technol. 2015, 5, 129–133. 10.1039/C4CY01209G. [DOI] [Google Scholar]
- Beierlein C. H.; Breit B.; Paz Schmidt R. A.; Plattner D. A. Online Monitoring of Hydroformylation Intermediates by ESI-MS. Organometallics 2010, 29, 2521–2532. 10.1021/om100131t. [DOI] [Google Scholar]
- Gellrich U.; Seiche W.; Keller M.; Breit B. Mechanistic Insights into a Supramolecular Self-Assembling Catalyst System: Evidence for Hydrogen Bonding during Rhodium-Catalyzed Hydroformylation. Angew. Chem., Int. Ed. 2012, 51, 11033–11038. 10.1002/anie.201203768. [DOI] [PubMed] [Google Scholar]
- Gellrich U.; Himmel D.; Meuwly M.; Breit B. Realistic Energy Surfaces for Real-World Systems: An IMOMO CCSD(T):DFT Scheme for Rhodium-Catalyzed Hydroformylation with the 6-DPPon Ligand. Chem. - Eur. J. 2013, 19, 16272–16281. 10.1002/chem.201302132. [DOI] [PubMed] [Google Scholar]
- Breit B.; Seiche W. Self-Assembly of Bidentate Ligands for Combinatorial Homogeneous Catalysis Based on an A-T Base-Pair Model. Angew. Chem., Int. Ed. 2005, 44, 1640–1643. 10.1002/anie.200462499. [DOI] [PubMed] [Google Scholar]
- Breit B.; Seiche W. Self-Assembly of Bidentate Ligands for Combinatorial Homogeneous Catalysis Based on an A-T Base Pair Model. Pure Appl. Chem. 2006, 78, 249–256. 10.1351/pac200678020249. [DOI] [PubMed] [Google Scholar]
- Waloch C.; Wieland J.; Keller M.; Breit B. Self-Assembly of Bidentate Ligands for Combinatorial Homogeneous Catalysis: Methanol-Stable Platforms Analogous to the Adenine–Thymine Base Pair. Angew. Chem., Int. Ed. 2007, 46, 3037–3039. 10.1002/anie.200605202. [DOI] [PubMed] [Google Scholar]
- Wieland J.; Breit B. A Combinatorial Approach to the Identification of Self-Assembled Ligands for Rhodium-Catalysed Asymmetric Hydrogenation. Nat. Chem. 2010, 2, 832–837. 10.1038/nchem.800. [DOI] [PubMed] [Google Scholar]
- Laungani A. C.; Slattery J. M.; Krossing I.; Breit B. Supramolecular Bidentate Ligands by Metal-Directed in situ Formation of Antiparallel β-Sheet Structures and Application in Asymmetric Catalysis. Chem. - Eur. J. 2008, 14, 4488–4502. 10.1002/chem.200800359. [DOI] [PubMed] [Google Scholar]
- Meeuwissen J.; Sandee A. J.; de Bruin B.; Siegler M. A.; Spek A. L.; Reek J. N. H. Phosphinoureas: Cooperative Ligands in Rhodium-Catalyzed Hydroformylation? On the Possibility of a Ligand-Assisted Reductive Elimination of the Aldehyde. Organometallics 2010, 29, 2413–2421. 10.1021/om901028z. [DOI] [Google Scholar]
- Hoveyda A. H.; Evans D. A.; Fu G. C. Substrate-Directable Chemical Reactions. Chem. Rev. 1993, 93, 1307–1370. 10.1021/cr00020a002. [DOI] [Google Scholar]
- Rousseau G.; Breit B. Removable Directing Groups in Organic Synthesis and Catalysis. Angew. Chem., Int. Ed. 2011, 50, 2450–2494. 10.1002/anie.201006139. [DOI] [PubMed] [Google Scholar]
- Davis H.; Phipps R. J. Harnessing Non-Covalent Interactions to Exert Control over Regioselectivity and Site-Selectivity in Catalytic Reactions. Chem. Sci. 2017, 8, 864–877. 10.1039/C6SC04157D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dydio P.; Reek J. N. H. Supramolecular Control of Selectivity in Transition-Metal Catalysis through Substrate Preorganization. Chem. Sci. 2014, 5, 2135–2145. 10.1039/C3SC53505C. [DOI] [Google Scholar]
- Šmejkal T.; Breit B. A Supramolecular Catalyst for Regioselective Hydroformylation of Unsaturated Carboxylic Acids. Angew. Chem., Int. Ed. 2008, 47, 311–315. 10.1002/anie.200703192. [DOI] [PubMed] [Google Scholar]
- Šmejkal T.; Gribkov D.; Geier J.; Keller M.; Breit B. Transition-State Stabilization by a Secondary Substrate-Ligand Interaction: A New Design Principle for Highly Efficient Transition-Metal Catalysis. Chem. - Eur. J. 2010, 16, 2470–2478. 10.1002/chem.200902553. [DOI] [PubMed] [Google Scholar]
- Šmejkal T.; Breit B. A Supramolecular Catalyst for the Decarboxylative Hydroformylation of α,β-Unsaturated Carboxylic Acids. Angew. Chem., Int. Ed. 2008, 47, 3946–3949. 10.1002/anie.200800956. [DOI] [PubMed] [Google Scholar]
- Diab L.; Šmejkal T.; Geier J.; Breit B. Supramolecular Catalyst for Aldehyde hydrogenation and Tandem Hydroformylation-Hydrogenation. Angew. Chem., Int. Ed. 2009, 48, 8022–8026. 10.1002/anie.200903620. [DOI] [PubMed] [Google Scholar]
- Diab L.; Gellrich U.; Breit B. Tandem Decarboxylative Hydroformylation–Hydrogenation Reaction of α,β-Unsaturated Carboxylic Acids toward Aliphatic Alcohols under Mild Conditions Employing a Supramolecular Catalyst System. Chem. Commun. 2013, 49, 9737–9739. 10.1039/c3cc45547e. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Dzik W. I.; Lutz M.; de Bruin B.; Reek J. N. H. Remote Supramolecular Control of Catalyst Selectivity in the Hydroformylation of Alkenes. Angew. Chem., Int. Ed. 2011, 50, 396–400. 10.1002/anie.201005173. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Detz R. J.; Reek J. N. H. Precise Supramolecular Control of Selectivity in the Rh-Catalyzed Hydroformylation of Terminal and Internal Alkenes. J. Am. Chem. Soc. 2013, 135, 10817–10828. 10.1021/ja4046235. [DOI] [PubMed] [Google Scholar]
- a Dydio P.; Reek J. N. H. Supramolecular Control of Selectivity in Hydroformylation of Vinyl Arenes: Easy Access to Valuable β-Aldehyde Intermediates. Angew. Chem., Int. Ed. 2013, 52, 3878–3882. 10.1002/anie.201209582. [DOI] [PubMed] [Google Scholar]; b Dydio P.; Detz R. J.; de Bruin B.; Reek J. N. H. Beyond Classical Reactivity Patterns: Hydroformylation of Vinyl and Allyl Arenes to Valuable β- and γ-Aldehyde Intermediates using Supramolecular Catalysis. J. Am. Chem. Soc. 2014, 136, 8418–8429. 10.1021/ja503033q. [DOI] [PubMed] [Google Scholar]; c Dydio P.; Reek J. N. H. Scalable and Chromatography-Free Synthesis of 2-(2-Formylalkyl)Arenecarboxylic Acid Derivatives through the Supramolecularly Controlled Hydroformylation of Vinylarene-2-Carboxylic Acids. Nat. Protoc. 2014, 9, 1183–1191. 10.1038/nprot.2014.077. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Ploeger M.; Reek J. N. H. Selective Isomerization-Hydroformylation Sequence: A Strategy to Valuable α-Methyl-Branched Aldehydes from Terminal Olefins. ACS Catal. 2013, 3, 2939–2942. 10.1021/cs400872a. [DOI] [Google Scholar]
- a Tan K. L. Induced Intramolecularity: An Effective Strategy in Catalysis. ACS Catal. 2011, 1, 877–886. 10.1021/cs2002302. [DOI] [Google Scholar]; b Tan K. L.; Sun X.; Worthy A. D. Scaffolding Catalysis: Expanding the Repertoire of Bifunctional Catalysts. Synlett 2012, 23, 321–325. 10.1055/s-0031-1290321. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yeung C. S.; Dong V. M. A new Direction in Enantioselective Catalysis: Scaffolding Ligands in Olefin Hydroformylation. Angew. Chem., Int. Ed. 2011, 50, 809–812. 10.1002/anie.201006489. [DOI] [PubMed] [Google Scholar]
- Lightburn T. E.; Dombrowski M. T.; Tan K. L. Catalytic Scaffolding Ligands: An Efficient Strategy for Directing Reactions. J. Am. Chem. Soc. 2008, 130, 9210–9211. 10.1021/ja803011d. [DOI] [PubMed] [Google Scholar]
- Grünanger C. U.; Breit B. Branched-Regioselective Hydroformylation with Catalytic Amounts of a Reversibly Bound Directing Group. Angew. Chem. 2008, 120, 7456–7459. 10.1002/ange.200802296. [DOI] [PubMed] [Google Scholar]
- Worthy A. D.; Gagnon M. M.; Dombrowski M. T.; Tan K. L. Regioselective Hydroformylation of Sulfonamides Using a Scaffolding Ligand. Org. Lett. 2009, 11, 2764–2767. 10.1021/ol900921e. [DOI] [PubMed] [Google Scholar]
- Grünanger C. U.; Breit B. Remote Control of Regio- and Diastereoselectivity in the Hydroformylation of Bishomoallylic Alcohols with Catalytic Amounts of a Reversibly Bound Directing Group. Angew. Chem., Int. Ed. 2010, 49, 967–970. 10.1002/anie.200905949. [DOI] [PubMed] [Google Scholar]
- Sun X.; Frimpong K.; Tan K. L. Synthesis of Quaternary Carbon Centers via Hydroformylation. J. Am. Chem. Soc. 2010, 132, 11841–11843. 10.1021/ja1036226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki Y.; Ito H.; Usui I.; Breit B. Formation of Quaternary Carbon Centers by Highly Regioselective Hydroformylation with Catalytic Amounts of a Reversibly Bound Directing Group. Chem. - Eur. J. 2011, 17, 8555–8558. 10.1002/chem.201101186. [DOI] [PubMed] [Google Scholar]
- Keulemans A. I. M.; Kwantes A.; van Bavel T. The Structure of the Formylation (OXO) Products Obtained from Olefines and Watergas. Recl. des Trav. Chim. Pays-Bas 1948, 67, 298–308. 10.1002/recl.19480670406. [DOI] [Google Scholar]
- Worthy A. D.; Joe C. L.; Lightburn T. E.; Tan K. L. Application of a Chiral Scaffolding Ligand in Catalytic Enantioselective Hydroformylation. J. Am. Chem. Soc. 2010, 132, 14757–14759. 10.1021/ja107433h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui I.; Nomura K.; Breit B. Diastereoselective Hydroformylation of 2,5-Cyclohexadienyl-1-Carbinols with Catalytic Amounts of a Reversibly Bound Directing Group. Org. Lett. 2011, 13, 612–615. 10.1021/ol1028546. [DOI] [PubMed] [Google Scholar]
- Joe C. L.; Blaisdell T. P.; Geoghan A. F.; Tan K. L. Distal-Selective Hydroformylation Using Scaffolding Catalysis. J. Am. Chem. Soc. 2014, 136, 8556–8559. 10.1021/ja504247g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarra S.; Wood D. M.; Roberts D. A.; Nitschke J. R. Molecular Containers in Complex Chemical systems. Chem. Soc. Rev. 2015, 44, 419–432. 10.1039/C4CS00165F. [DOI] [PubMed] [Google Scholar]
- Breiner B.; Clegg J. K.; Nitschke J. R. Reactivity Modulation in Container Molecules. Chem. Sci. 2011, 2, 51–56. 10.1039/C0SC00329H. [DOI] [Google Scholar]
- Wiester M. J.; Ulmann P. A.; Mirkin C. A. Enzyme Mimics Based upon Supramolecular Coordination Chemistry. Angew. Chem., Int. Ed. 2011, 50, 114–137. 10.1002/anie.201000380. [DOI] [PubMed] [Google Scholar]
- a Leenders S. H. A. M.; Gramage-Doria R.; de Bruin B.; Reek J. N. H. Transition Metal Catalysis in Confined Spaces. Chem. Soc. Rev. 2015, 44, 433–448. 10.1039/C4CS00192C. [DOI] [PubMed] [Google Scholar]; b Brown C. J.; Toste F. D.; Bergman R. G.; Raymond K. N. Supramolecular Catalysis in Metal–Ligand Cluster Hosts. Chem. Rev. 2015, 115, 3012–3035. 10.1021/cr4001226. [DOI] [PubMed] [Google Scholar]; c Vriezema D. M.; Comellas Aragonès M.; Elemans J. A. A. W.; Cornelissen J. J. L. M.; Rowan A. E.; Nolte R. J. M. Self-Assembled Nanoreactors. Chem. Rev. 2005, 105, 1445–1490. 10.1021/cr0300688. [DOI] [PubMed] [Google Scholar]; d Yoshizawa M.; Klosterman J. K.; Fujita M. Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]; e Hooley R. J.; Rebek J. Chemistry and Catalysis in Functional Cavitands. Chem. Biol. 2009, 16, 255–264. 10.1016/j.chembiol.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Monflier E.; Tilloy S.; Fremy G.; Castanet Y.; Mortreux A. A Further Breakthrough in Biphasic, Rhodium-Catalyzed Hydroformylation: the Use of Per(2,6-di-O-methyl)-β-Cyclodextrin as Inverse Phase Transfer Catalyst. Tetrahedron Lett. 1995, 36, 9481–9484. 10.1016/0040-4039(95)02073-X. [DOI] [Google Scholar]; b Mathivet T.; Méliet C.; Castanet Y.; Mortreux A.; Caron L.; Tilloy S.; Monflier E. Rhodium Catalyzed Hydroformylation of Water Insoluble Olefins in the Presence of Chemically Modified β-Cyclodextrins: Evidence for Ligand-Cyclodextrin Interactions and Effect of Various Parameters on the Activity and the Aldehydes Selectivity. J. Mol. Catal. A: Chem. 2001, 176, 105–116. 10.1016/S1381-1169(01)00229-1. [DOI] [Google Scholar]; c Leclercq L.; Hapiot F.; Tilloy S.; Ramkisoensing K.; Reek J. N. H.; Van Leeuwen P. W. N. M.; Monflier E. Sulfonated Xantphos Ligand and Methylated Cyclodextrin: A Winning Combination for Rhodium-Catalyzed Hydroformylation of Higher Olefins in Aqueous Medium. Organometallics 2005, 24, 2070–2075. 10.1021/om048994f. [DOI] [Google Scholar]; d Legrand F.-X.; Hapiot F.; Tilloy S.; Guerriero A.; Peruzzini M.; Gonsalvi L.; Monflier E. Aqueous Rhodium-Catalyzed Hydroformylation of 1-Decene in the Presence of Randomly Methylated β-cyclodextrin and 1,3,5-Triaza-7-phosphaadamantane Derivatives. Appl. Catal., A 2009, 362, 62–66. 10.1016/j.apcata.2009.04.017. [DOI] [Google Scholar]; e Elard M.; Denis J.; Ferreira M.; Bricout H.; Landy D.; Tilloy S.; Monflier E. Rhodium Catalyzed Hydroformylation Assisted by Cyclodextrins in Biphasic Medium: Can Sulfonated Naphthylphosphanes Lead to Active, Selective and Recyclable Catalytic Species?. Catal. Today 2015, 247, 47–54. 10.1016/j.cattod.2014.06.002. [DOI] [Google Scholar]; f Vanbésien T.; Sayede A.; Monflier E.; Hapiot F. A Self-Emulsifying Catalytic System for the Aqueous Biphasic Hydroformylation of Triglycerides. Catal. Sci. Technol. 2016, 6, 3064–3073. 10.1039/C5CY01758K. [DOI] [Google Scholar]
- Cousin K.; Menuel S.; Monflier E.; Hapiot F. Hydroformylation of Alkenes in a Planetary Ball Mill: From Additive-Controlled Reactivity to Supramolecular Control of Regioselectivity. Angew. Chem., Int. Ed. 2017, 56, 10564–10568. 10.1002/anie.201705467. [DOI] [PubMed] [Google Scholar]
- Reetz M. T.; Waldvogel S. R. β-Cyclodextrin-Modified Diphosphanes as Ligands for Supramolecular Rhodium Catalysts. Angew. Chem., Int. Ed. Engl. 1997, 36, 865–867. 10.1002/anie.199708651. [DOI] [Google Scholar]
- Jouffroy M.; Gramage-Doria R.; Armspach D.; Sémeril D.; Oberhauser W.; Matt D.; Toupet L. Confining Phosphanes Derived from Cyclodextrins for Efficient Regio- and Enantioselective Hydroformylation. Angew. Chem., Int. Ed. 2014, 53, 3937–3940. 10.1002/anie.201311291. [DOI] [PubMed] [Google Scholar]
- van Leeuwen P. W. N. M.; Kamer P. C. J.. Phosphorus(III) Ligands in Homogeneous Catalysis Design and Synthesis; John Wiley & Sons, Ltd.: New York, 2012; pp 405–426. [Google Scholar]
- Sémeril D.; Matt D. Synthesis and Catalytic Relevance of P(III) and P(V)-Functionalised Calixarenes and Resorcinarenes. Coord. Chem. Rev. 2014, 279, 58–95. 10.1016/j.ccr.2014.06.019. [DOI] [Google Scholar]
- a Slagt V. F.; Reek J. N. H.; Kamer P. C. J.; van Leeuwen P. W. N. M. Assembly of Encapsulated Transition Metal Catalysts. Angew. Chem. 2001, 113, 4401–4404. . [DOI] [PubMed] [Google Scholar]; b Slagt V. F.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Reek J. N. H. Encapsulation of Transition Metal Catalysts by Ligand-Template Directed Assembly. J. Am. Chem. Soc. 2004, 126, 1526–1536. 10.1021/ja0386795. [DOI] [PubMed] [Google Scholar]; c Kleij A. W.; Reek J. N. H. Ligand-Template Directed Assembly: An Efficient Approach for the Supramolecular Encapsulation of Transition-Metal Catalysts. Chem. - Eur. J. 2006, 12, 4218–4227. 10.1002/chem.200500875. [DOI] [PubMed] [Google Scholar]
- Jacobs I.; de Bruin B.; Reek J. N. H. Comparison of the Full Catalytic Cycle of Hydroformylation Mediated by Mono- and Bis-Ligated Triphenylphosphine-Rhodium Complexes by Using DFT Calculations. ChemCatChem 2015, 7, 1708–1718. 10.1002/cctc.201500087. [DOI] [Google Scholar]
- Kleij A. W.; Kuil M.; Tooke D. M.; Spek A. L.; Reek J. N. H. Template-Assisted Ligand Encapsulation; the Impact of an Unusual Coordination Geometry on a Supramolecular Pyridylphosphine–Zn(II)porphyrin Assembly. Inorg. Chem. 2005, 44, 7696–7698. 10.1021/ic050858v. [DOI] [PubMed] [Google Scholar]
- a Kleij A. W.; Lutz M.; Spek A. L.; van Leeuwen P. W. N. M.; Reek J. N. H. Encapsulated Transition Metal Catalysts Comprising Peripheral Zn(ii)salen Building Blocks: Template-Controlled Reactivity and Selectivity in Hydroformylation Catalysis. Chem. Commun. 2005, 3661–3663. 10.1039/b503708e. [DOI] [PubMed] [Google Scholar]; b Jacobs I.; van Duin A. C. T.; Kleij A. W.; Kuil M.; Tooke D. M.; Spek A. L.; Reek J. N. H. Conformational Studies of Ligand-Template Assemblies and the Consequences for Encapsulation of Rhodium Complexes and Hydroformylation Catalysis. Catal. Sci. Technol. 2013, 3, 1955. 10.1039/c3cy20665c. [DOI] [Google Scholar]; c Bocokić V.; Lutz M.; Spek A. L.; Reek J. N. H. Bis-(thiosemicarbazonato) Zn(II) Complexes as Building Blocks for Construction of Supramolecular Catalysts. Dalton Trans. 2012, 41, 3740–3750. 10.1039/c2dt12096h. [DOI] [PubMed] [Google Scholar]
- Kuil M.; Soltner T.; van Leeuwen P. W. N. M.; Reek J. N. H. High-Precision Catalysts: Regioselective Hydroformylation of Internal Alkenes by Encapsulated Rhodium Complexes. J. Am. Chem. Soc. 2006, 128, 11344–11345. 10.1021/ja063294i. [DOI] [PubMed] [Google Scholar]
- Bocokić V.; Kalkan A.; Lutz M.; Spek A. L.; Gryko D. T.; Reek J. N. H. Capsule-Controlled Selectivity of a Rhodium Hydroformylation Catalyst. Nat. Commun. 2013, 4, 2670. 10.1038/ncomms3670. [DOI] [PubMed] [Google Scholar]
- Besset T.; Norman D. W.; Reek J. N. H. Supramolecular Encapsulated Rhodium Catalysts for Branched Selective Hydroformylation of Alkenes at High Temperature. Adv. Synth. Catal. 2013, 355, 348–352. 10.1002/adsc.201200790. [DOI] [Google Scholar]
- Wang X.; Nurttila S. S.; Dzik W. I.; Becker R.; Rodgers J.; Reek J. N. H. Tuning the Porphyrin Building Block in Self-Assembled Cages for Branched-Selective Hydroformylation of Propene. Chem. - Eur. J. 2017, 23, 14769–14777. 10.1002/chem.201702113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bellini R.; Chikkali S. H.; Berthon-Gelloz G.; Reek J. N. H. Supramolecular Control of Ligand Coordination and Implications in Hydroformylation Reactions. Angew. Chem., Int. Ed. 2011, 50, 7342–7345. 10.1002/anie.201101653. [DOI] [PubMed] [Google Scholar]; b Bellini R.; Reek J. N. H. Coordination Studies on Supramolecular Chiral Ligands and Application in Asymmetric Hydroformylation. Chem. - Eur. J. 2012, 18, 7091–7099. 10.1002/chem.201200225. [DOI] [PubMed] [Google Scholar]; c Bellini R.; Reek J. N. H. Application of Supramolecular Bidentate Hybrid Ligands in Asymmetric Hydroformylation. Chem. - Eur. J. 2012, 18, 13510–13519. 10.1002/chem.201202044. [DOI] [PubMed] [Google Scholar]
- García-Simón C.; Gramage-Doria R.; Raoufmoghaddam S.; Parella T.; Costas M.; Ribas X.; Reek J. N. H. Enantioselective Hydroformylation by a Rh-Catalyst Entrapped in a Supramolecular Metallocage. J. Am. Chem. Soc. 2015, 137, 2680–2687. 10.1021/ja512637k. [DOI] [PubMed] [Google Scholar]