

Abstract
Brain malformations involving the corpus callosum are common in children with developmental disabilities. We identified DCC mutations in four families and five sporadic individuals with isolated agenesis of the corpus callosum (ACC) without intellectual disability. DCC mutations result in variable dominant phenotypes with decreased penetrance, including mirror movements and ACC associated with a favorable developmental prognosis. Possible phenotypic modifiers include the type and location of mutation and the sex of the individual.
The corpus callosum, the main cerebral commissure in placental mammals, has a key role in communication between the brain hemispheres1. Formation of the corpus callosum is a complex process involving ligands, such as those in the Netrin, Ephrin, Semaphorin and Slit families, and their receptors2. ACC is the complete or partial absence of the corpus callosum. This frequent brain malformation affects approximately 1 in 4,000 newborns and 3–5% of children with intellectual disability (ID)3,4 and is a common cause of late-pregnancy termination5. Mutations in many genes cause syndromes with ID and ACC, whereas the genetics of isolated ACC remain poorly understood3,6,7. The Netrin receptor Dcc plays a critical role in corpus callosum development in mice by guiding callosal axons at the midline8. Whereas mutations in DCC have been associated with congenital mirror movements (MMs) in humans9, they have not been described in individuals with ACC.
We investigated four multigenerational families with individuals presenting with ACC, MMs or both phenotypes segregating as autosomal-dominant traits (Fig. 1a). Neuroimaging and clinical studies confirmed that complete or partial ACC was isolated in most cases (Fig. 1b and Supplementary Fig. 1) and was associated with a range of intellectual abilities (normal to borderline impaired); additionally, specific cognitive impairments, including language delay or visuospatial deficits, were documented (Supplementary Table 1). Diffusion MRI tractography-based probabilistic constrained spherical deconvolution identified decreased crossing of descending corticospinal tract projections at the pyramidal decussation in all affected individuals in families 2 and 4 with either ACC and MMs (n = 5) or MMs only (n = 2) (Fig. 1c and Supplementary Fig. 2). The tractography results for other commissural fibers, including the decussation of the superior cerebellar peduncles, anterior commissure, posterior commissure and optic chiasm, were comparable between the affected individuals and controls.
Figure 1.
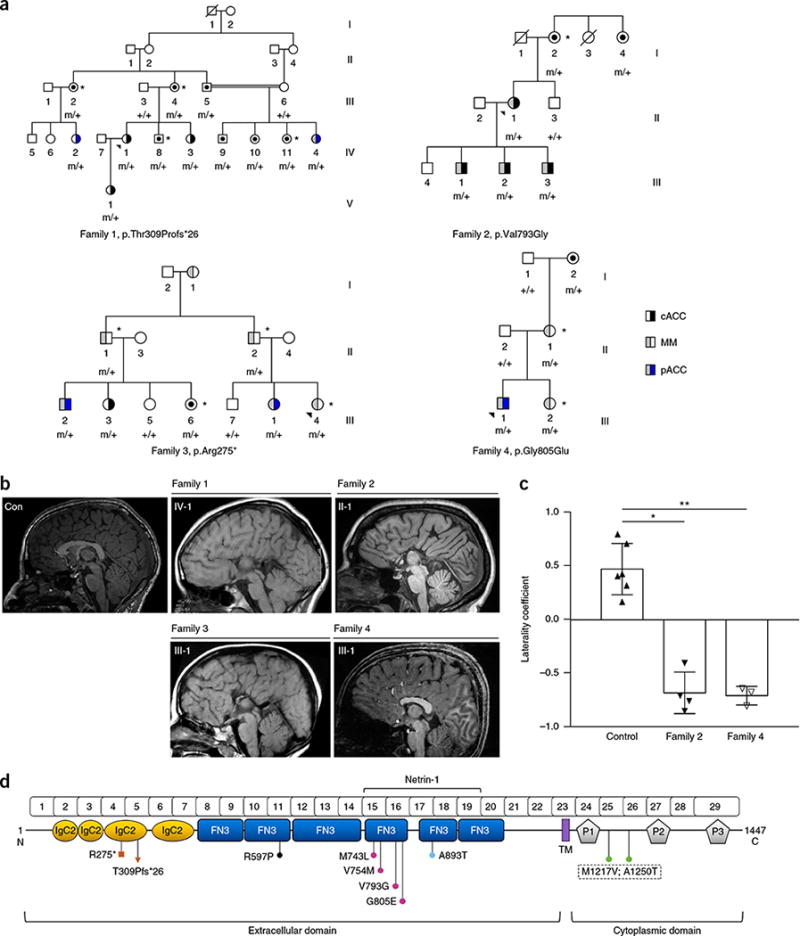
DCC mutations cause isolated ACC and/or MMs associated with significantly decreased crossing of descending corticospinal tract projections at the pyramidal decussation. (a) All available family samples were analyzed; m, mutation; black dot, mutation carrier; blue, partial ACC (pACC); black, complete ACC (cACC); gray, MMs. Asterisk indicates neuroimaging data for mutation carrier or individual with MMs. (b) Midsagittal MRI of control and family proband/representative individual (1 or 2, complete ACC; 3, near-complete ACC with thin rostrum and genu remaining; 4, partial ACC with absence of the rostrum and genu). Con, control. (c) Groupwise comparison of laterality coefficients in both families (family 2, n = 4; family 4, n = 3) were compared with controls (n = 6). For each individual, a laterality coefficient for the corticospinal tract was calculated as the ratio of the difference between the numbers of crossed and uncrossed streamlines to the total number of streamlines. Right and left coefficients were averaged to determine the laterality coefficient of each individual. Greater positive values indicate more crossed streamlines, and negative values indicate more uncrossed streamlines (mean ± s.d, *P = 0.0238; **P = 0.0095 by two-tailed Mann–Whitney U test). (d) Protein domain structure depicting the location of the DCC truncation (red square and triangle) and missense mutations (colored dots). The Netrin-1-binding region is indicated. IgC2, immunoglobulin-like type C2 domain; FN3, fibronectin type III–like domain; TM, transmembrane domain; P1–3, proline-rich conserved motifs.
Linkage analysis and exome sequencing of three affected individuals in family 1 identified two shared candidate variants in the 16 linkage regions (Supplementary Fig. 3 and Supplementary Table 2), including a truncating mutation in DCC (NM_005215.3: c.925delA, p.Thr309ProfsTer26). For family 2, 48 candidate variants were identified in 28 linkage regions (Supplementary Fig. 4 and Supplementary Table 3), including a missense variant (c.2378T>G, p.Val793Gly) in DCC. The previously reported nonsense mutation (c.823C>T, p.Arg275Ter) in DCC segregated with MMs in five individuals of family 3 (ref. 10). Further investigation identified two additional female mutation carriers with ACC and a male carrier with MMs who had a thin rostrum. Direct screening in family 4 identified a heterozygous DCC missense variant (c.2414G>A, p.Gly805Glu). All four DCC mutations were absent from public databases, including the 1000 Genomes and Exome Aggregation Consortium (ExAC) databases, and segregated with ACC and/or MMs in all available individuals tested (Fig. 1a). In addition, we sequenced DCC in 70 unrelated individuals with ACC, including 46 with normal cognitive development. Five individuals, all with isolated complete ACC, had at least one heterozygous missense variant altering a conserved amino acid of DCC (Fig. 1d, Table 1 and Supplementary Figs. 1 and 5). Analysis of all available imaging in mutation-positive individuals with complete ACC also showed absence of the hippocampal commissure and cingulate gyri, and dysmorphic lateral ventricles (usually colpocephaly), as would be expected (details in Online Methods). Apart from these observations, no consistent additional brain malformations were seen.
Table 1.
Summary of DCC variants identified in individuals with ACC (6MMs) in this study
| Family number | Phenotype | cDNA | Protein | Protein domain | SIFT | PolyPhen-2 | ExAC | dbSNP |
|---|---|---|---|---|---|---|---|---|
| 1 | cACC or pACC ± MMs | c.925delA | p.Thr309ProfsTer26 | IgC2-3 | – | – | No | – |
| 2 | cACC and MMs | c.2378T>G | p.Val793Gly | FN3-4 | Deleterious | Probably damaging | No | – |
| 3 | cACC or MMs ± pACC | c.823C>T | p.Arg275Ter | IgC2-3 | No | – | ||
| 4 | pACC and/or MMs | c.2414G>A | p.Gly805Glu | FN3-4 | Deleterious | Probably damaging | No | – |
| 5 | cACC | c.1790G>C | p.Arg597Pro | FN3-2 | Deleterious | Probably damaging | No | – |
| 6 | cACC | c.2227A>T | p.Met743Leu | FN3-4 | Deleterious | Benign | No | rs199651452 |
| 7 | cACC | c.2260G>A | p.Val754Met | FN3-4 | Deleterious | Possibly damaging | Yes (×19) | – |
| 8 | cACC | c.2677G>A | p.Ala893Thr | FN3-5 | Deleterious | Benign | No | – |
| 9 | cACC | c.3649A>G; c.3748G>A | p.Met1217Val; p.Ala1250Thr | Cytoplasmic | Tolerated; tolerated | Benign; probably damaging | No; Yes (×2) | – |
cACC, complete isolated agenesis of the corpus callosum; pACC, partial isolated agenesis of the corpus callosum; MMs, mirror movements; IgC2, immunoglobulin-like type C2 domain; FN3, fibronectin type III–like domain; PolyPhen-2, Polymorphism Phenotyping v2; ExAC, Exome Aggregation Consortium; dbSNP, dbSNP reference SNP identification number. Reference sequences used are NM_005215.3 and NP_005206.2.
We analyzed the phenotypes of individuals with DCC mutations reported in the literature and in this study (Supplementary Table 4) to assess the penetrance of MMs and ACC. Of the 88 individuals with DCC mutations identified to date, 50 had MMs; among the 39 who had brain imaging, 19 exhibited ACC. After exclusion of the index individuals from the analysis, the penetrance of MMs was estimated to be 42%, and the penetrance of ACC was estimated to be 26% (Supplementary Table 5). Overall, males (n = 31) exhibited MMs more frequently than females (n = 19, male/female ratio = 1.8, P = 0.0027, two-tailed Fisher’s exact test; Supplementary Table 5), whereas in individuals with truncating DCC variants, ACC was more often present in females (n = 7) than males (n = 1, male/female ratio = 0.2). Sex differences in corpus callosum anatomy have been associated with testosterone levels during prenatal brain development11–13; therefore, we tested the effect of androgens on DCC expression. Independent analysis by RNA-seq and RT–qPCR demonstrated a significant dose-dependent increase in DCC expression in human neural stem cells treated with 10 nM or 100 nM testosterone (Supplementary Fig. 6). Because variants introducing a premature stop codon generally result in haploinsufficiency, owing to nonsense-mediated decay of the mutant mRNA, it is possible that ACC may occur when the amount of DCC mRNA (and DCC protein) falls below a threshold level during corpus callosum development, as would occur more frequently in females. However, given the incomplete penetrance observed in both sexes, the phenotypic outcome must also be influenced by additional genetic, epigenetic and/or environmental factors. Interestingly, families 1 and 3, in which a majority of females displayed ACC, are both of North African background, thus supporting the hypothesis of genetic modifiers.
In contrast to truncating variants, missense-mutant proteins are usually present in the cell and can interfere with the function of the wild-type protein, thus potentially resulting in differing phenotypes compared with those associated with haploinsufficiency of the same protein. Binding of Netrin-1 to DCC results in intracellular homodimerization or heterodimerization with UNC5, another axon-guidance receptor, and is critical for both the chemoattractive and chemorepulsive properties of the signaling complexes14,15. The Netrin-1-binding region involves the fourth, fifth and sixth fibronectin type III–like domains of DCC14,16; therefore, amino acid substitutions in this binding region may compromise DCC function. Five of the eight DCC missense variants identified in individuals with ACC are located in the Netrin-1-binding region (Fig. 1d), thus representing a considerable enrichment compared with missense variants located in this domain in ExAC (5/74, 6.7% versus 519/~60,000, 0.86%; P = 5 × 10−4 (all rare variants) or 284/~60,000, 0.47%; P = 3 × 10−5 (rare variants predicted to be damaging by the SIFT (sorting intolerant from tolerant) program, two-tailed Fisher’s exact test; Supplementary Table 6). Given the decreased penetrance and mild phenotype of DCC-related ACC, it is possible that some individuals described in ExAC have pathogenic DCC mutations and undiagnosed ACC.
Modeling of DCC missense variants showed that the amino acid substitutions in families 2 and 4, both located within the DCC/Netrin-1 binding interface, are predicted to be most disruptive. The p.Val793Gly substitution abolishes a hydrophobic interaction with Thr147 of Netrin-1, whereas p.Gly805Glu introduces a highly unfavorable charged moiety within a hydrophobic pocket, thus disrupting interaction with Leu113 of Netrin-1 (Supplementary Figs. 7 and 8). The predicted effects of the three substitutions within the Netrin-1-binding region but outside the binding interface (Supplementary Figs. 9–12) are consistent with results from in vitro studies demonstrating that even conservative mutations to residues in this binding region can disrupt DCC dimerization, Netrin-1 binding and axon guidance16.
In addition to the effects of sex hormones and the type and location of DCC mutations, developmental differences between the corpus callosum and corticospinal tract may also contribute to the variable ACC and MM phenotypes. Callosal and subcerebrally projecting pyramidal neurons of the cortex are specified at early stages of development, and the molecular identity of each population directly affects its axonal connectivity15. Whereas corticospinal axons use DCC and Netrin-1 signaling to reach the midline, callosal axons use DCC and Netrin-1 chemoattraction to attenuate ROBO1- and SLIT2-mediated chemorepulsion to approach and cross the midline17. Therefore, a DCC mutation may differentially affect commissural versus subcerebral axon trajectories, thus leading to ACC, MMs or both. MMs were consistently associated with decreased crossing of descending corticospinal tract projections at the pyramidal decussation in this study as well as in individuals with RAD51-related MMs18, thus suggesting that DCC-mediated MMs are primarily the result of corticospinal tract decussation abnormalities.
In conclusion, our results provide compelling evidence that DCC mutations cause isolated ACC in humans, in addition to the previously reported MM phenotype. The factors determining the phenotypic variability are complex and probably include the hormonal context during development, the type and location of DCC mutation and the genetic background of the individual. Although the spectrum of phenotypes associated with DCC mutations remains to be fully characterized, individuals described in this study have an intellectual quotient within the normal-to-borderline range. Heterozygous mutations in DCC therefore appear to result in isolated ACC with a mild phenotype and favorable cognitive outcomes, in contrast with the unfavorable developmental outcomes associated with syndromic ACC. Given the high frequency of DCC mutations detected in our cohorts, this observation has prenatal diagnostic and parental counseling implications for fetuses with ACC, because the condition currently has unclear prognosis. Our data suggest that the prenatal detection of isolated ACC related to a pathogenic DCC mutation indicates a lower risk of an abnormal neurodevelopmental outcome.
URLs
1000 Genomes Project, http://www.internationalgenome.org/; Exome Variant Server, http://exac.broadinstitute.org/; SIFT, http://sift.jcvi.org/; PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/; dbSNP, https://www.ncbi.nlm.nih.gov/SNP/; PyMOL, https://www.pymol.org/; Novoalign, http://www.novocraft.com/; UniProt, http://www.uniprot.org/; Clustal Omega, http://www.ebi.ac.uk/Tools/msa/clustalo/.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Patients and genetic material
Informed written consent was obtained from each individual or the individual’s parents or legal representatives before blood sampling. The study received approval from the relevant ethical standards committees on human experimentation (France, Pitié-Salpêtrière; Australia, Royal Children’s Hospital and University of Queensland; Canada, Montreal Neurological Institute; US, UCSF Benioff Children’s Hospital). No statistical method was used to predetermine sample size. The analyses were performed on anonymized samples with no further randomization or blinding. Clinical information was obtained by review of medical records and examination of affected individuals. All available imaging and associated imaging reports for DCC-positive individuals were reviewed by a neuroradiologist (S.A.M.) and neurologist (R.J.L.), both of whom have extensive experience in imaging features of human brain malformations. The brain MRI scans were reviewed in a single session during which consensus was reached. The scans of 18 mutation-positive individuals from families 1, 2, 3, 4 and 9 were reviewed, including 12 with complete or partial ACC, and six with an intact corpus callosum. The imaging was from a mix of studies, including 1.5T and 3T MRI, and one individual had a CT only. All available sequences were reviewed, which, for the majority, included axial, coronal and sagittal T1 and T2 sequences at a minimum. The scans were reviewed systematically to include an assessment of the corpus callosum, commissures (anterior, posterior and hippocampal), cerebral cortex, white matter, ventricles, hippocampi, cortex, basal ganglia, cerebral and cerebellar peduncles, optic chiasm, brainstem, cerebellar vermis and hemispheres, and pituitary gland. All patients with complete ACC also had absence of the hippocampal commissure, cingulate gyri and dysmorphic lateral ventricles (usually colpocephaly), as expected. Apart from these observations, no consistent additional brain malformations were seen.
A systematic neurological exam was performed to assess MMs in these individuals. Specific neuropsychological data or a clinical diagnosis regarding the presence or absence of intellectual disability were available in all affected individuals from families 1 to 4 and probands 5 to 9. Genomic DNA was extracted from blood cells through standard procedures. The reference sequences for DCC used were NM_005215.3 (human) and protein sequences NP_005206.2 (human), P43146 (human) and P70211 (mouse).
Family 1 originated from North Africa and comprises five female individuals, three with complete ACC and two with partial ACC. One of the females with partial ACC also has MMs. The proband and her sister have complete ACC, which was fortuitously detected by brain MRI after workup for headaches. Isolated complete or partial ACC was later detected in the female fetuses by ultrasound during pregnancy. Individuals with ACC in this family had mild learning difficulties in school, especially in language and mathematics, but their intelligence is within normal range.
Family 2 is an Australian family of European ancestry, including four male children from a nonconsanguineous marriage. The mother and three of the four offspring have isolated complete ACC and mild clinical sequelae, including MMs, and, according to test results, the three children have global behavioral/academic impairments and low-average-to-borderline intellectual functioning. In their early years, they attended mainstream schooling.
The clinical features of family 3, also originating from North Africa and comprising five individuals presenting with MMs, have previously been reported10. Reassessment of this family included a neurological examination and brain imaging in additional family members and identified partial ACC in the female index subject, who also had mild MMs, complete ACC in a female without MMs, and a thin rostrum in a male carrier with MMs (pedigree in Fig. 1a). ACC in the two female individuals was detected prenatally but was previously considered to be a phenotype independent of MMs before this study.
Family 4 is a three-generation nonconsanguineous Australian kindred of European ancestry. The mother and both offspring have MMs, and the son has additional features of isolated partial ACC (absence of rostrum, genu and anterior body), language disorder, poor memory, ataxic-type tremors and borderline intellectual impairment.
Notably, ACC appeared to be more frequent in recent generations, possibly because of an ascertainment bias.
We collected the DNA of 46 individuals with complete or partial ACC but no intellectual disability, who were recruited in the Trousseau and La Pitié-Salpêtrière hospitals (Paris, France) or different French centers. Twenty-six of these individuals had a normal array-CGH analysis, and six were screened through next-generation sequencing for mutations in 423 selected genes involved in ACC in humans or mice (Callosome target enrichment panel, performed at the Necker Hospital), but no obvious genetic abnormalities were identified. The remaining 20 patients had no genetic analysis before DCC screening. An additional 24 probands with ACC were recruited from the Royal Children’s Hospital (Melbourne, Australia), McGill University and UCSF; the majority had not had genetic analysis performed before this study.
Probands 5, 7 and 8 have isolated complete ACC that was detected prenatally by ultrasound and was further confirmed by brain MRI; proband 6 has isolated complete ACC that was fortuitously detected by brain MRI after a workup for migraine (Supplementary Fig. 1). All probands as well as the affected sister of proband 7 have normal intellectual and social abilities, although probands 5 and 7, respectively, showed language and learning difficulties in school. Proband 9 has ADHD plus social and executive-function deficits. Brain MRI confirmed complete ACC. Additional features, which have occasionally been described in association with callosal agenesis, included an interhemispheric cyst, complex gray-matter heterotopia in the periventricular regions, nodular periventricular gray-matter heterotopia in both temporal horns and malrotated, dysmorphic hippocampi (Supplementary Fig. 1). The mother is an unaffected carrier who was confirmed to have a normal corpus callosum via brain MRI and who has no learning, cognitive or intellectual deficits.
Diffusion tensor imaging and tractography
Diffusion MRI data for families 2 and 4 were collected on Siemens Magnetom Trio 3T scanners at the Royal Children’s Hospital (Melbourne, Australia) and the Queensland Brain Institute (Brisbane, Australia), respectively, by using identical sequence parameters (b value, 3,000 s/mm2; repetition time (TR), 8,200 ms; echo time (TE), 112 ms; voxel size, 2.3 × 2.3 × 2.3 mm3, with 60 diffusion-encoding directions and seven b = 0 s/mm2 volumes). High-resolution 3D T1-weighted MR images were also acquired for family 2 (Royal Children’s Hospital, Magnetom Trio 3T, voxel size, 0.9 × 0.9 × 0.9 mm3; TR, 2,530 m; multiecho TE, 7.2, 5.32, 3.51 and 1.77 ms) and family 4 (Queensland Brain Institute, Magnetom 7T, voxel size, 0.75 × 0.75 × 0.75 mm3; TR, 5,000 ms; TE, 3.4 ms). Motion and eddy current correction was performed with the FMRIB Software Library (FSL v5)19. Reversed phase-encode blips were acquired for individuals scanned in Melbourne, and inhomogeneity distortion correction was performed20,21. Diffusion-weighted images were upsampled by a factor of 2, the brain was extracted, and fiber orientation distribution was estimated with MRtrix3 through constrained spherical deconvolution22–24 with default processing parameters (maximum harmonic order lmax = 8). Fractional anisotropy and color fractional anisotropy maps were generated with FSL v5. For medullary pyramid tractography, four bilateral ROIs were specified for each individual at the base of the pons, upper medulla, lower medulla, and lateral funiculus (Supplementary Fig. 2); in addition, a midline ROI was specified at the pyramidal decussation. Ipsilateral projections were found by specifying the ipsilateral funiculus and midline decussation ROIs as inclusion and exclusion ROIs, respectively. Contralateral projections were found by specifying the contralateral funiculus and ipislateral medullary ROIs as inclusion ROIs. Streamlines were seeded from the ROIs for the ipsilateral and contralateral base of pons (relative to the lateral funiculus) for uncrossed and crossed projections, respectively. All tractography was performed with the iFOD2 algorithm in Mrtrix3 with the following parameters: step size, 0.115 mm; curvature, 90°; maximum number of streamlines, 100,000; FOD cutoff, 0.2. A laterality coefficient was generated for the corticospinal tract of each individual by calculating the ratio of the difference between the number of crossed and uncrossed streamlines to the total number of streamlines. That is, laterality coefficient = (contralateral streamlines − ipsilateral streamlines)/(contralateral streamlines + ipsilateral streamlines). Right and left coefficients were averaged to determine the laterality coefficient of each individual. Group analysis between each family and controls (n = 6, three individuals scanned at each site) was based on two-tailed Mann–Whitney U tests, and P <0.05 was considered statistically significant.
Linkage analysis and whole-exome sequencing
Genome-wide linkage analysis was performed on 13 members (five symptomatic individuals, three obligate carriers, three at-risk individuals and two spouses) of family 1 by using OmniExpress-24 microarrays (Illumina). Illumina microarray experiments were performed according to the automated manufacturer’s protocol. Multipoint LOD scores on autosomes were calculated with Merlin 1.0 (affected-only analysis, autosomal-dominant trait; disease-allele frequency, 0.0001; phenocopy rate, 0.0001). Multipoint LOD scores on chromosome X were calculated with the same parameters with minx (Supplementary Fig. 3).
Exome sequencing was performed for three members of family 1 (IV-1, IV-2 and IV-3) at the genotyping and sequencing facility of ICM. Exons were captured from fragmented genomic-DNA samples with the SeqCap EZ Exome v3 enrichment protocol (Roche), and paired-end 75-base massively parallel sequencing was carried out on an Illumina NextSeq500 platform, according to the manufacturers’ protocols. Bioinformatics analyses were performed with an in-house pipeline developed by the bioinformatics and biostatistics core facility of ICM (iCONICS), as follows: sequencing reads passing quality filtering were aligned to the human reference genome (hg19) with Burrows–Wheeler Aligner (BWA)25, and GATK26 was used to recalibrate base quality scores, realign around indels and mark duplicate reads. Variant filtering was performed with Polyweb (University Paris-Descartes) with the following criteria: (i) variant present in the heterozygous state in the three members of family 1; (ii) variant present in a positive linkage region; (iii) variant altering the coding sequence (nonsense, missense, frameshift or essential splice-site variants); (iv) variant with a minor-allele frequency (MAF) <1% in the 1000 Genomes, HapMap and ExAC databases; (v) variant present in fewer than two individuals in a local database. Two variants met these criteria (Supplementary Table 2). Sanger sequencing confirmed the presence of the DCC variant in all affected family members and several nonmanifesting carriers.
For family 2, WES and linkage analysis was performed on all four affected individuals (II-1, III-1, III-2 and III-3), essentially as previously described27. Exons were captured from fragmented genomic-DNA samples with the SureSelect Human All Exon, V5+UTRs (Agilent), and sequencing was performed by the Australian Genome Research Facility with 100-base paired-end reads on an Illumina HiSeq 2000 platform. Reads were aligned to the reference human genome (GRch37/hg19) with Novoalign. Variants were called with SAMtools 0.1.17 and annotated with ANNOVAR. Genotypes were extracted and processed with LINKDATAGEN. Parametric multipoint linkage analysis was subsequently performed with MERLIN, specifying a rare dominant disease model (Supplementary Fig. 4). The candidate list was filtered to retain rare variants predicted to alter the coding sequence or mRNA splicing (minor-allele frequency <0.01) within the linkage regions with public databases and an in-house database of 132 unrelated exomes (Supplementary Table 3). Sanger sequencing confirmed the presence of the DCC variant in all affected family members. Familial segregation testing also identified the DCC variant in two unaffected family members (both in DNA derived from peripheral blood and buccal cells), one of whom (I-2) was confirmed through MRI to have a normal corpus callosum.
Amplification and sequencing of the DCC coding region
The entire coding region of the DCC gene (NM_005215.3) was amplified by PCR, as previously described28. Purified PCR products were sequenced in both directions with a Big Dye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems). Sanger sequencing was performed on an ABI 3730×l sequencer by GATC Biotech (Constance, Germany), and the sequences were analyzed with SeqScape 2.1 (Applied Biosystems).
Brain malformations gene panel
A custom in-house next-generation-sequencing gene panel targeting 287 genes (including DCC) associated with brain malformations was developed with the HaloPlexHS target enrichment system (Agilent Technology). We used 50 ng of gDNA to prepare the sequencing library according to the manufacturer’s instructions and performed 2 × 150-bp paired-end sequencing to a median depth of 400× on a MiSeq instrument (Illumina). The raw data files (fastq.gz) were analyzed with SureCall software (Agilent Technology). Sanger sequencing confirmed the presence of the mutation and segregation.
Interpretation of genetic variants
Missense variants were assessed in silico for possible pathogenicity with Alamut 2.3 (Biointeractive Software), PolyPhen-2 and SIFT. Orthologous sequences of human DCC (NP_005206) were retrieved from UniProt and aligned with Clustal Omega (see URLs).
Effect of testosterone treatment on DCC expression
Human neural stem cells (NSCs) derived from the embryonic stem-cell line (ESC, SA001, 46XY, Cellartis) were obtained from I-Stem. Stability of the hNSCs was monitored by karyotype analysis at I-Stem. Morphology, SOX2 expression and the ability to differentiate into neurons was verified before the experiments were performed. hNSCs were cultured in depleted medium (phenol red–free medium without N2B27) for 48 h before androgen treatments. The cells were then treated with DMSO, 10 nM testosterone or 100 nM testosterone for 24 h. Total RNA was extracted from treated cells with TRI reagent (Molecular Research Center), treated with 1 U/μg DNase I (Roche) for 30 min at 37 °C and then purified with an RNeasy Mini kit (Qiagen). Biological triplicates of treated versus control (DMSO) conditions were compared in RNA-seq experiments. RNA sequencing was performed at the IGBMC Microarray and Sequencing platform (Strasbourg, France). DNA libraries were generated with a TruSeq RNA Sample Preparation v2 kit (Illumina). Sequencing was performed on an Illumina HiSeq 2500 instrument as single-end 50-base reads. Image analysis and base calling were performed with RTA 1.18.61 and CASAVA 1.8.2. DCC mRNA levels were further quantified in three independent experiments, each composed of at least four well replicates (first experiment, DMSO, n = 6; all other conditions, n = 4), by RT–qPCR with two different primer pairs in DCC and two different control genes: PPIA29 and GAPDH (Supplementary Table 7). Reverse transcription was performed from 500 ng of total RNA with random hexamers and SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer’s recommendations. RT–qPCR was performed on a LightCycler 480 II instrument (Roche) with QuantiTect SYBR Green PCR Master Mix (Qiagen), in triplicates. The relative expression of DCC versus GAPDH or PPIA was calculated with the 2−(ΔΔ Ct) method.
Protein modeling analyses
Proteins were modeled with the web-based flexible backbone modeling program RosettaBackrub30. Substitutions within the DCC FN4 and FN5 domains were modeled with the previously determined crystal structures of the DCC–netrin-1 complex (PDB 4PLO and 4URT)14,16. The p.Arg597Pro substitution was modeled with the solution structure for the DCC FN2 domain (PDB 2ED8). Models were visualized with PyMOL and Chimera31. Electrostatic surface potentials were calculated with PDB2PQR32 and Adaptive Poisson-Boltzmann Solver (APBS)33. Interacting residues within the binding interface of various DCC–netrin-1 complexes were identified with the program LIGPLOT+ (v1.4)34. The evolutionary conservation of amino acid residues on the basis of phylogenetic relations between homologous sequences was determined with the bioinformatics tool ConSurf35.
Statistical analyses
Frequencies of rare variants in patients were compared with two-tailed Fisher’s exact tests. The relative expression of DCC versus GAPDH or PPIA was compared with parametric two-tailed Student’s t-tests. Laterality coefficients were compared between affected families (four individuals from family 2 and three individuals from family 4) and controls (six individuals) with two-tailed Mann–Whitney U tests.
Data availability
Families included in this study have not consented to have next-generation sequence data publicly released. Variants identified in this study have been deposited in ClinVar under accession numbers SCV000485052–SCV000485060.
Supplementary Material
Acknowledgments
We thank the studied families and the Lefroy family for their participation in and support of this study. We thank the DNA and cell bank of the ICM (Paris, France) for DNA extraction, Sinead Eyre (QBI) for study coordination and M. Kean (RCH) and M. Seal (MCRI) for assistance with MRI protocols and scanning. This work was funded in part by National Health and Medical Research Council (NHMRC) Australia Project grants (GNT1059666, GNT631466, GNT1064174, GNT1048849, GNT1104455 and GNT1064174), the Agence Nationale de la Recherche (ANR Blanc CILAXCAL, ANR Blanc HARTaGeNe), Assistance Publique des Hôpitaux de Paris (APHP), the ‘Programme Hospitalier de Recherche Clinique’ (PHRC) ACCREM, and the ‘Investissements d’Avenir’ programs ANR-10-IAIHU-06 (IHU-A-ICM), ANR-10-LABX-0030-INRT and ANR-10-IDEX-0002-02. A.P.L.M. and L.R.M. are supported by an Australian Postgraduate Award, T.J.E. is supported by a University of Queensland Research Scholarship, and A.P. is supported by a QBI PhD scholarship. S.H. and A.Q. are respectively supported by a master’s and a doctoral grant from the Fondation pour la Recherche Médicale (FRM). M.B. is supported by an NHMRC Senior Research Fellowship and an NHMRC Program Grant (GNT1054618). E.H.S. is supported by a grant from the NIH (2R01NS058721), and R.J.L. is supported by a Melbourne Children’s Clinician Scientist Fellowship. L.J.R. is supported by an NMHRC Principal Research Fellowship, and P.J.L. is supported by an NHMRC Career Development Fellowship (GNT1032364). C.D. and C.N. are supported as members of the Bio-Psy Labex. This work was supported in part by the Victorian Government’s Operational Infrastructure Support Program and Australian Government NHMRC IRIISS.
Footnotes
A full list of authors affiliations appears at the end of the paper.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
J.-L.M., D.J.A., E.R., A. Piton, M.B., T.B.d.V., E.H.S., R.J.L., L.J.R., P.J.L. and C.D. contributed to critically revising the article for important intellectual content.J.-L.M., D.J.A., E.R., A. Piton, M.B., T.B.d.V., E.H.S., R.J.L., L.J.R., P.J.L. and C.D. contributed to critically revising the article for important intellectual content.A.P.L.M., D.H., T.J.E., C. Galea, E.H.S., R.J.L., L.J.R., P.J.L. and C.D. contributed to formulation of theory and prediction. A.P.L.M., D.H., T.J.E., C.N., S.E.M.S., J.-L.M., A. Piton, L.J.R., P.J.L. and C.D. contributed to experimental conception and design. A.P.L.M., D.H., T.J.E., C. Galea, A.Q., C.N., A.R., M.-L.M., V.A., P.B., J.B., A.F., C. Garel, G.G., I.G., J.G., S.H., B.K., F.L., V.L., S.A.M., G.M., A. McIlroy, A. Meneret, C.M., L.R.M., S.O., A. Paolino, K.P., F.R., G.A.R., M.S.-S., M.S., S.E.M.S., R.T., O.T., Q.W., A.W., E.R., A. Piton, M.B., T.B.d.V., E.H.S., R.J.L., L.J.R., P.J.L. and C.D. contributed to acquisition, analysis and/or interpretation of data. A.P.L.M., T.J.E., P.J.L. and C.D. contributed to drafting the article. A.P.L.M., D.H., T.J.E., A.Q., C. Galea, C.N., M.-L.M., V.A., S.A.M., G.M., A.M., G.A.R., A.B., G.R., T.A.-B., M.B.D., J.-L.M., D.J.A., E.R., A. Piton, M.B., T.B.d.V., E.H.S., R.J.L., L.J.R., P.J.L. and C.D. contributed to critically revising the article for important intellectual content.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Lindwall C, Fothergill T, Richards LJ. Curr Opin Neurobiol. 2007;17:3–14. doi: 10.1016/j.conb.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Chédotal A. Curr Opin Neurobiol. 2011;21:68–75. doi: 10.1016/j.conb.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Paul LK, et al. Nat Rev Neurosci. 2007;8:287–299. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- 4.Glass HC, Shaw GM, Ma C, Sherr EH. Am J Med Genet A. 2008;146A:2495–2500. doi: 10.1002/ajmg.a.32418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rouleau C, et al. Arch Dis Child Fetal Neonatal Ed. 2011;96:F360–F364. doi: 10.1136/adc.2010.201483. [DOI] [PubMed] [Google Scholar]
- 6.Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Brain. 2014;137:1579–1613. doi: 10.1093/brain/awt358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sotiriadis A, Makrydimas G. Am J Obstet Gynecol. 2012;206:337.e1–337.e5. doi: 10.1016/j.ajog.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 8.Fazeli A, et al. Nature. 1997;386:796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- 9.Srour M, et al. Science. 2010;328:592. doi: 10.1126/science.1186463. [DOI] [PubMed] [Google Scholar]
- 10.Méneret A, et al. Neurology. 2014;82:1999–2002. doi: 10.1212/WNL.0000000000000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ardekani BA, Figarsky K, Sidtis JJ. Cereb Cortex. 2013;23:2514–2520. doi: 10.1093/cercor/bhs253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moffat SD, Hampson E, Wickett JC, Vernon PA, Lee DH. Brain Res. 1997;767:297–304. doi: 10.1016/s0006-8993(97)00614-8. [DOI] [PubMed] [Google Scholar]
- 13.Chura LR, et al. Psychoneuroendocrinology. 2010;35:122–132. doi: 10.1016/j.psyneuen.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 14.Xu K, et al. Science. 2014;344:1275–1279. doi: 10.1126/science.1255149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Nat Rev Neurosci. 2013;14:755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finci LI, et al. Neuron. 2014;83:839–849. doi: 10.1016/j.neuron.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fothergill T, et al. Cereb Cortex. 2014;24:1138–1151. doi: 10.1093/cercor/bhs395. [DOI] [PubMed] [Google Scholar]
- 18.Gallea C, et al. Brain. 2013;136:3333–3346. doi: 10.1093/brain/awt258. [DOI] [PubMed] [Google Scholar]
- 19.Andersson JL, Sotiropoulos SN. Neuroimage. 2016;125:1063–1078. doi: 10.1016/j.neuroimage.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersson JL, Skare S, Ashburner J. Neuroimage. 2003;20:870–888. doi: 10.1016/S1053-8119(03)00336-7. [DOI] [PubMed] [Google Scholar]
- 21.Smith SM, et al. Neuroimage. 2004;23(Suppl. 1):S208–S219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 22.Tournier JD, Calamante F, Gadian DG, Connelly A. Neuroimage. 2004;23:1176–1185. doi: 10.1016/j.neuroimage.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 23.Tournier JD, Calamante F, Connelly A. Neuroimage. 2007;35:1459–1472. doi: 10.1016/j.neuroimage.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 24.Tournier JD, Calamante F, Connelly A. Int J Imaging Syst Technol. 2012;22:53–66. [Google Scholar]
- 25.Li H, Durbin R. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna A, et al. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith KR, et al. Genome Biol. 2011;12:R85. doi: 10.1186/gb-2011-12-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Depienne C, et al. Neurology. 2011;76:260–264. doi: 10.1212/WNL.0b013e318207b1e0. [DOI] [PubMed] [Google Scholar]
- 29.Dupasquier S, et al. BMC Mol Biol. 2014;15:9. doi: 10.1186/1471-2199-15-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lauck F, Smith CA, Friedland GF, Humphris EL, Kortemme T. Nucleic Acids Res. 2010;38:W569–W575. doi: 10.1093/nar/gkq369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pettersen EF, et al. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laskowski RA, Swindells MB. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 35.Landau M, et al. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Families included in this study have not consented to have next-generation sequence data publicly released. Variants identified in this study have been deposited in ClinVar under accession numbers SCV000485052–SCV000485060.