

Summary
Enterovirus D68 (EV-D68) is a medically important respiratory plus-strand RNA virus of children that has been linked to acute flaccid myelitis. We have determined that EV-D68 induces autophagic signaling and membrane formation. Autophagy, a homeostatic degradative process that breaks down protein aggregates and damaged organelles, promotes replication of multiple plus-strand viruses. Induction of autophagic signals promotes EV-D68 replication, but the virus inhibits the downstream degradative steps of autophagy in multiple ways. EV-D68 proteases cleave a major autophagic cargo adaptor and the autophagic SNARE SNAP29, which reportedly regulates fusion between autophago-some to amphisome/autolysosome. Although the virus inhibits autophagic degradation, SNAP29 promotes virus replication early in infection. An orphan SNARE, SNAP47, is shown to have a previously unknown role in autophagy, and SNAP47 promotes the replication of EV-D68. Our study illuminates a mechanism for subversion of autophagic flux and redirection of the autophagic membranes to benefit EV-D68 replication.
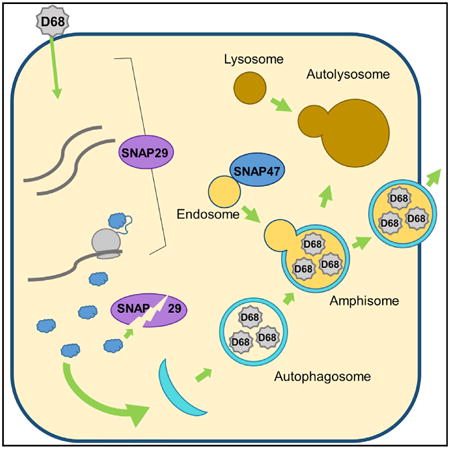
Graphical abstract
Enterovirus D68, a medically important respiratory virus, benefits from signaling to the host degradation pathway of autophagy. Corona et al. show that EV-D68 disrupts autophagic degradation in multiple ways, including manipulation of cellular SNAREs, to promote replication and dissemination. This suggests that redirected autophagy promotes exit of virus from cells.
Introduction
Enterovirus D68 (EV-D68) is a positive-sense, single-stranded RNA virus of the Picornaviridae family, first isolated in 1962, which causes a mild, upper respiratory illness (Esposito et al., 2015; Schieble et al., 1967). EV-D68 cases have been increasing in frequency and severity over the last decade, with outbreaks in the US in 2014 and 2016 (Midgley et al., 2015; Wang et al., 2017). It is likely that the number of cases is severely under-reported, as only the most severe clinical presentations are tested for EV-D68, allowing many infections to go undetected as asymptomatic or mild flu-like cases (Oberste et al., 2004). Recently, infections are often correlated with cases of acute flaccid myelitis (AFM) (Aliabadi et al., 2016; Messacar et al., 2015). AFM is a rare condition that causes weakening in the muscles and is seen most often in children (Andersen et al., 2017; Oster et al., 2017, Soc. Neuroped., abstract). Though the link between EV-D68 and AFM is still under investigation, a mouse model fulfills Koch's postulates for virus as a cause of paralysis, suggesting that EV-D68 may be a cause of AFM cases (Hixon et al., 2017). EV-D68 remains relatively understudied, and much about the interactions between the cell and the virus has yet to be elucidated.
Many pathogens interact positively or negatively with the host autophagic pathway. Autophagy is a process of homeostatic degradation in a cell, used to create nutrients in times of stress and asa mechanism to recycle damaged organelles or microbes in the cytosol (Karanasios and Ktistakis, 2016). Autophagy initiation is regulated by a tightly controlled web of post-translational modifications and is monitored by the conversion of microtu-bule-associated protein 1A/1B-light chain 3 (LC3) from its cytosolic resting state (LC3-I) to the lipidated form LC3-II, with the addition of a phosphatidylethanolamine by E3-like conjugation enzymes (Nakatogawa, 2013). The process is characterized by the formation of a crescent membrane that engulfs cytosolic cargo and forms a double-membraned vesicle, termed an autophagosome (Landajuela et al., 2016). The autophagosome fuses with an acidifying endosome, which delivers vacuolar ATPases, and the resultant vesicle is termed the amphisome (Seglen, 2008). This acidification is required for lysosomal fusion. The amphisome fuses with the lysosome to form an autolysosome, in which autophagic cargo is degraded, including cargo adaptor molecules, such as SQSTM1 (Ni et al., 2011). SQSTM1 levels indicate whether or not autophagosomes are successfully delivering their cargo to lysosomes for degradation.
The fusion events in autophagy are coordinated by a class of proteins called soluble N-ethylmaleimide-sensitive factor attachment receptors (SNAREs) (Wang et al., 2016). SNARE fusion bundles require 4 a helices to function: Qa; Qb; Qc; and R (Rizo, 2003). STX17 is the Qa SNARE on the autophagosome that coordinates its fusion with other vesicles (Itakura et al., 2012). SNAP29 is a cytosolic Qbc SNARE that binds to STX17, donating its two helices to the forming fusion bundle (Morelli et al., 2014). VAMP8 is an R SNARE, found on lysosomal and endosomal membranes, that binds to STX17-SNAP29, tethering the membranes together for fusion (Diao et al., 2015). The family of known Qbc SNARES is small, consisting of SNAP25 exclusively in neurons, SNAP23 at plasma membranes, SNAP29, and the larger SNAP47, which is thought to be associated with endosomes (Holt et al., 2006; Kloepper et al., 2007; Kuster et al., 2015). It has been suggested that picornavirus proteins can interact with autophagosomal SNAREs to help form the autolyso-some (Lai et al., 2017).
Many pathogens are degraded by autophagy, but some sub-vert the process for their own benefit, including several picorna-viruses (Jackson, 2014; Shi and Luo, 2012). Previous work by our group has demonstrated that poliovirus 1 (PV) uses acidic amphisomes to promote virus replication and maturation (Richards and Jackson, 2012). Coxsackievirus B3 induces the autophagic pathway but may inhibit degradation of autophagic cargo (Alirezaei et al., 2015; Shi et al., 2014; Wong et al., 2008). We report here that EV-D68 induces autophagy signaling to benefit its replication and manipulates the autophagosomal SNAREs. SNAP29 has important roles early in EV-D68 replication and late in cellular exit, when SNAP29 is cleaved by EV-D68 viral protease 3C. We show that SNAP47 is required for autophagic flux and benefits virus replication. Our data suggest that the manipulation of SNAREs can impact the growth and trafficking of picornaviruses.
Results
EV-D68 Is Affected by Autophagy-Altering Treatments
To characterize the effect of autophagy on EV-D68, we treated cells with an autophagy inhibitor, bafilomycin A1 (BafA1). BafA1 inhibits acidification of cellular compartments, including amphisomes, blocking autolysosome formation. H1HeLa cells were treated with BafA1 for 18 hr and then infected with EV-D68. Viral titers were analyzed by plaque assay for cell-associated (Figure 1A) and extracellular (Figure 1B) virus. We found a significant drop in cell-associated virus in BafA1-treated cells. Parallel uninfected cells were collected for western blotting for LC3 and SQSTM1 to assess the effect of the treatment on autophagy (Figure 1C) and found buildup of these markers, consistent with autophagic inhibition.
Figure 1. EV-D68 Is Affected by Autophagy-Altering Treatments.
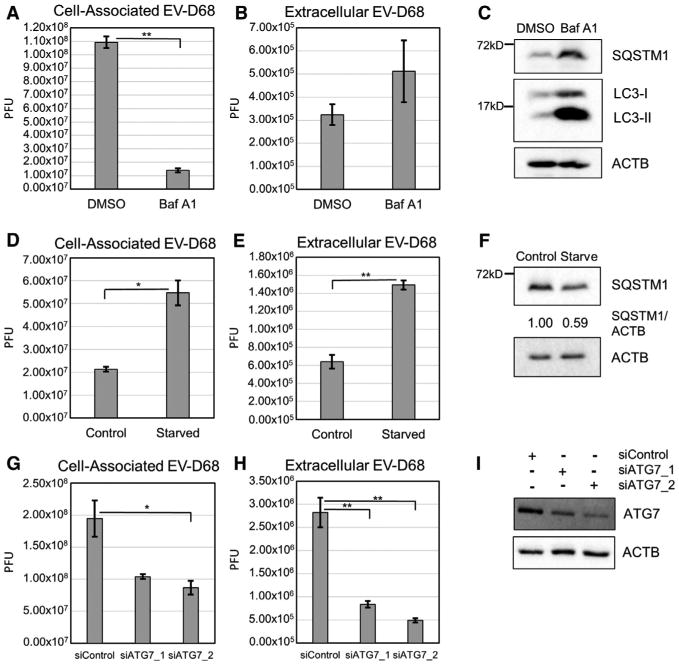
(A and B) H1HeLa cells were treated with 0.1 mM BafA1 for 18 hr prior to infection and then infected at an MOI of 0.1 with EV-D68 for 6 hr. Viral titers were analyzed by plaque assay for both cell-associated virus (A; p = 0.0017) and extracellular virus (B).
(C) Autophagy markers SQSTM1 and LC3 were analyzed by western blot of parallel samples from (A) and (B).
(D and E) H1HeLa cells were treated with starvation medium for 4 hr prior to infection and then infected at an MOI of 0.1 with EV-D68 for 5 hr. See also Figure S1. Viral titers were analyzed byplaque assay for both cell-associated virus (D; p = 0.046) and extracellular virus (E; p = 0.0019).
(F) Parallel samples from (D) and (E) were analyzed by western blot. (G and H) H1HeLa cells were transfected with a control siRNA or one of two different siRNAs against ATG7 for 40 hr. Cells were infected at an MOI of 0.1 with EV-D68 for 5 hr.
(G and H) Viral titers were analyzed by plaque assay for both cell-associated virus (G; p = 0.044) and extracellular virus (H; p = 0.0078 and p = 0.0042, respectively). (I) Parallel samples from (G) and (H) were analyzed by western blot for knockdown efficiency. Western blots are representative from one of the 3 independent experiments. Viral titers are represented as the mean ± SEM. Statistical tests were done using an unpaired Student's t test with statistical significance set at **p < 0.01 and *p ≤ 0.05.
Because autophagic inhibition was detrimental to virus replication, we wanted to test autophagic activation. Cells were incubated in starvation medium and induction of autophagy in H1HeLa cells validated by western blot (Figure S1). Cells were starved for 4 hr, infected with EV-D68, and placed in complete media. Starvation prior to infection caused a significant increase in cell-associated (Figure 1D) and extracellular (Figure 1E) virus. SQSTM1 levels were analyzed by western blot at the end of the starvation period, confirming autophagy induction (Figure 1F). The increase in cell-associated virus supports that autophagy benefits viral replication. The increase in extracellular virus may suggest that autophagy also promotes exit of viral particles from the cell.
To confirm these data, we used a small-molecule autophagy activator, trifluoperazine, which induces a buildup of LC3-II (Zhang et al., 2007). Cells were treated for 18 hr with 8.3 mM trifluoperazine and assayed for autophagy markers. LC3-II levels did increase post-treatment (Figure S2A). SQSTM1 levels, however, also increased upon treatment, suggesting trifluoperazine may block autophagic flux, similar to BafA1 treatment. Trifluo-perazine treatment induced a significant drop in both cell-associated and extracellular virus titers (Figures S2B and S2C). These data suggest, again, that autophagy is beneficial for the virus and open new questions to the mechanism of action of trifluoperazine.
To avoid problems associated with pharmaceutical autophagy regulators, we targeted the specific autophagy protein ATG7, a critical player in the LC3 conjugation process required for autophagosome formation (Nakatogawa, 2013). We used two different small interfering RNAs (siRNAs) to successfully target ATG7, and knockdown caused a significant drop in cell-associated and extracellular EV-D68 titers (Figures 1G and 1H). This supports the model that EV-D68 benefits from the presence of autophagy in host cells.
EV-D68 Alters Intracellular Morphology during Infection
Picornaviruses manipulate the membrane landscape in a cell during an infection. Membrane rearrangements seen in the cell include single-membraned vesicles, tubular structures, and autophagic membranes (Belov et al., 2012). Because, as shown in Figure 1, EV-D68 infection benefits from autophagy, it would be expected that EV-D68 would also generate double-membraned vesicles (DMVs) characteristic of induction of the autophagy pathway. Cells were infected with an MOI of 25 and prepared for transmission electron microscopy (TEM). Figure 2 shows representative images of mock and infected cells. In the infected cells, there are increased groupings of vesicles when compared to mock cells, some of which were double membraned, as seen on the more magnified image. These DMVs are suggestive of autophagic vesicles and demonstrate that EV-D68 induces autophagosome-like vesicles during its replicative cycle.
Figure 2. EV-D68 Changes Lipid Morphology upon Infection in H1HeLa Cells.

(A–D) H1HeLa cells were infected with EV-D68 at an MOI of 25 for 5 hr. Cells were collected, fixed, and subjected to transmission electron microscopy preparations and imaging. Mock (A and B) and EV-D68-infected (C and D) samples were imaged. Images shown are representative of the dataset.
Traditional Autophagy Markers Respond to EV-D68 Infection
We investigated the subcellular localization of LC3 during EV-D68 infection. Previous reports have shown that LC3, normally diffuse in the cytosol, aggregates to visible puncta when autophagy is stimulated. H1HeLa cells were transfected with a GFP-LC3 plasmid for 24 hr and then infected with EV-D68 or placed in starvation medium. We show in Figure 3A that fed uninfected cells display diffuse cytosolic localization of GFP-LC3 as expected, whereas starvation or infection induces formation of numerous puncta throughout the cell. This suggests that autophagosomes increase in both infected and starved cells.
Figure 3. Traditional Autophagy Markers Respond to EV-D68 Infection.
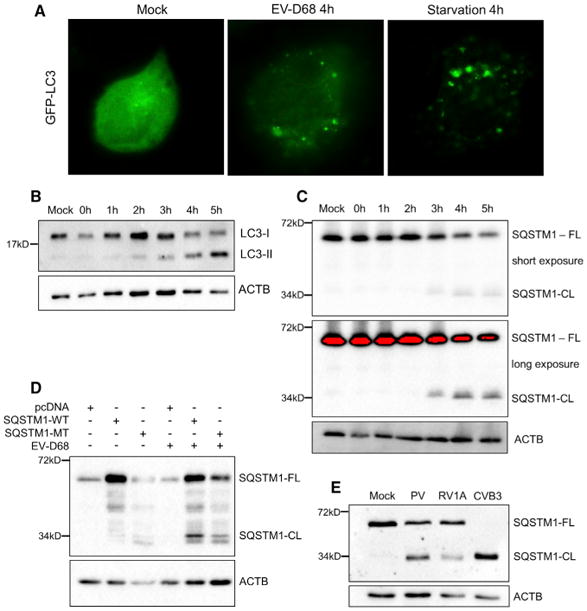
(A) H1HeLa cells were transfected with GFP-LC3 for 24 hr prior to experiment. Cells were either untreated/mock, EV-D68 infected at an MOI of 25 for 4 hr, or treated with starvation medium for 4 hr. Cells were imaged for GFP fluorescence. Images shown are representative of 3 experiments.
(B and C) H1HeLa cells were infected with EV-D68 for 5 hr and samples collected every hour during infection. Samples were subjected to western blot analysis and immunoblotted for LC3 (B) or SQSTM1-FL and SQSTM1-CL (C).
(D) H1HeLa cells were transfected with pcDNA,SQSTM1-WT, or SQSTM1-MT 24 hr prior toinfection. Cells were infected at an MOI of 25 with EV-D68 for 5 hr. Parallel samples were immuno-blotted for SQSTM1.
(E) H1HeLa cells were infected with PV, RV1A, or CVB3 for 6 hr. Samples were subjected to western blot analysis for SQSTM1. CL, cleavage fragment;FL, full length.
We tested for the presence of LAMP1 and cathepsin B (CTSB) on GFP-LC3-labeled vesicles in Figure S3. LAMP1 is a marker of endosomes and target vesicles that have fused with endosomes, whereas CTSB is a marker of lysosomes and vesicles that have fused with lysosomes. GFP-LC3-transfected H1HeLa cells were infected or starved and then stained for LAMP1 or CTSB. Normally, LAMP1 is observed in punctate structures, consistent with endosomes, which do not colocalize with GFP-LC3 puncta. During infection, LAMP1 partially colocalizes with GFP-LC3 puncta, suggesting that some autophagosomes have fused with endosomes. This is similar to the result seen during starvation (Figure S3A).
The mock condition for CTSB staining showed a few, punctate, presumably lysosomal structures. During infection, GFP-LC3 puncta and CTSB puncta do not significantly overlap, in contrast to starvation conditions, in which autophagy progresses to the maturation of the autophagosome or amphisome to an autolysosome. An autolysosome is expected to be LC3 positive on the outer membrane but CSTB positive in the center of the enlarged vesicle. We have denoted structures consistent with this (solid white arrowheads) in the starved cell image. However, similar structures do not appear in the EV-D68 condition. These data suggest that the autophagosome or amphisome is unable to form autolysosomes in virus-infected cells.
LC3-I and LC3-II levels over the course of an EV-D68 infection were examined by western blot (Figure 3B). LC3-II levels start increasing at 2 hr post-infection. Another set of samples was immunoblotted for SQSTM1 (Figure 3C). Full-length SQSTM1 levels decrease late in infection; longer exposures reveal the appearance of a minor, approximately 34-kDa band beginning at 3 hr post-infection. Similar data from studies of coxsackievirus B3 were demonstrated to represent cleavage of SQSTM1 by the 2A viral protease (Shi et al., 2013).
We investigated the similarity between this cleavage of SQSTM1 by EV-D68 and the cleavage seen by Shi et al. (2014) and obtained SQSTM1-expressing plasmids from the authors. The first plasmid was a SQSTM1 wild-type plasmid, originally sourced from Yu et al. (2009). The second plasmid was SQSTM1-MT, which was mutated at G241 (G241E), the site found in the prior publication for the protease-mediated cleavage of SQSTM1. Either pcDNA vector, SQSTM1-WT, or SQSTM1-MT were transfected into H1HeLa cells for 40 hr and then infected at an MOI of 25 with EV-D68 for 5 hr (Figure 3D). Cell samples were collected and immunoblotted for full-length and cleaved forms of SQSTM1. Our data suggest a common mechanism of SQSTM1 cleavage between these two viruses.
To determine whether SQSTM1 cleavage is common among picornaviruses, other family members were tested for their ability to induce this cleavage of SQSTM1. Three viruses were tested: poliovirus 1 (PV); rhinovirus 1A (RV1A); and coxsackievirus B3-Woodruff (CVB3). Our group and others have reported that PV subverts autophagy to benefit its own replication, and reduction of full-length SQSTM1 was previously interpreted as an indication of active autophagic degradation (Richards and Jackson, 2012). RV1A does not require the autophagy pathway to replicate (Quiner and Jackson, 2010). Finally, CVB3 was tested as a positive control (Shi et al., 2013, 2014). At 6 hr post-infection with PV, a strong 34-kDa band appears cross-reacting with SQSTM1 (Figure 3E). RV1A infection generated a minor 34-kDa band, whereas during CVB3-Woodruff infection, essentially all detectable SQSTM1 was 34 kDa, indicating that a high percentage of the host protein is cleaved (Figure 3E). These data strongly suggest that monitoring full-length SQSTM1 is not a valid measurement of degradative autophagy during picornavirus infection.
EV-D68 Infection Affects Autophagosomal SNARE Protein Levels
Because the standard assay for degradation by autophagy pathway cannot be interpreted during EV-D68 infection, due to cleavage of SQSTM1, we decided to monitor the SNARE complex responsible for fusion of the autophagosome with the lysosome, facilitating the delivery of degradative enzymes into the lumen of the autophagosome: STX17; SNAP29; and VAMP8. We were interested in autophagy-specific SNAREs, so we focused on SNAP29 and STX17. H1HeLa cells were infected for 5 hr and lysates immunoblotted for SQSTM1, LC3, STX17, and SNAP29 (Figure 4). We observe a surprising increase in STX17 levels; because EV-D68 shuts down both RNA Pol II transcription and cap-dependent mRNA translation, new production of host proteins is unexpected. An internal ribosomal entry site (IRES) would allow more protein translation from existing RNAs during the course of the infection, but to our knowledge, STX17 has not been identified in screens for host mRNAs that contain an IRES (http://www.iresite.org). It is possible that the actual abundance of the protein is not increasing, but that there are changes in its cellular localization or membrane association, and that one of these factors makes STX17 more easily detected by western blot over the course of the infection.
Figure 4. EV-D68 Infection Affects Autophagosomal Fusion SNARE Protein Levels.
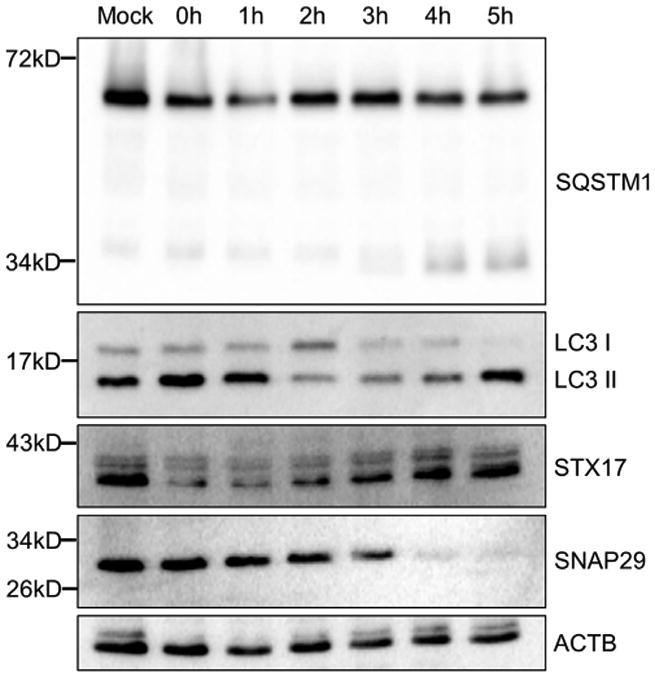
H1HeLa cells were infected at an MOI of 25 with EV-D68. Cells were collected every hour for 5 hr. Samples were prepared and subjected to western blotting analysis and then immunoblotted for SQSTM1, LC3, STX17, and SNAP29.
We detect lower levels of the cytosolic SNARE SNAP29 beginning at 3 hr post-infection. SNAP29 is a necessary component of the amphisome-lysosome fusion complex. The timing of this loss of full-length SNAP29, at a similar time post-infection as SQSTM1 cleavage and about 1 hr after LC3-II protein levels increase, suggests that EV-D68 is inducing autophagosomes, but both inhibiting delivery of cargo to the autophagosomes and preventing delivery of any surviving cargo to the lysosome. This suggests that EV-D68 interrupts autophagy at the autophagosome or amphisome stage.
SNAP29 Reduction Is a Result of Cleavage by EV-D68 3C Protease
To understand the mechanism by which SNAP29 protein levels are reduced during infection, we infected H1HeLa cells and then treated with pepstatin A and E64D (to inhibit lysosomal proteases), MG132 (to inhibit the proteasome), cycloheximide (to inhibit translation), or DMSO (as a vehicle control; Figure S4A). SNAP29 levels remained consistent throughout the treatments in uninfected cells. SNAP29 levels also remained consistent during cycloheximide treatment, suggesting that the reduction in protein is not due to turnover in the absence of new host protein synthesis during EV-D68 infection. In cells which were infected and then DMSO, pepstatin A/E64D, or MG132 treated, SNAP29 levels were reduced. Data in Figures S3B–S3D confirm that treatment of cells with pepstatin A/E64D and MG132 did not inhibit virus replication.
Because the inhibitors failed to rescue SNAP29 levels, we investigated a role for viral proteases in eliminating full-length SNAP29. We used two different SNAP29 antibodies to probe for SNAP29 in infected cell lysates: one that detects the C-terminal region of SNAP29 (ab138500) and one that detects the N-terminal domain of SNAP29 (ab181151; Figure 5A). We were able to detect an N-terminal fragment of SNAP29 beginning 3 hr into infection at approximately 18 kDa. We detected the C-terminal fragment of SNAP29 beginning at 3 hr into infection at approximately 11 kDa.
Figure 5. SNAP29 Reduction Is a Result of Cleavage by EV-D68's 3C Protease.
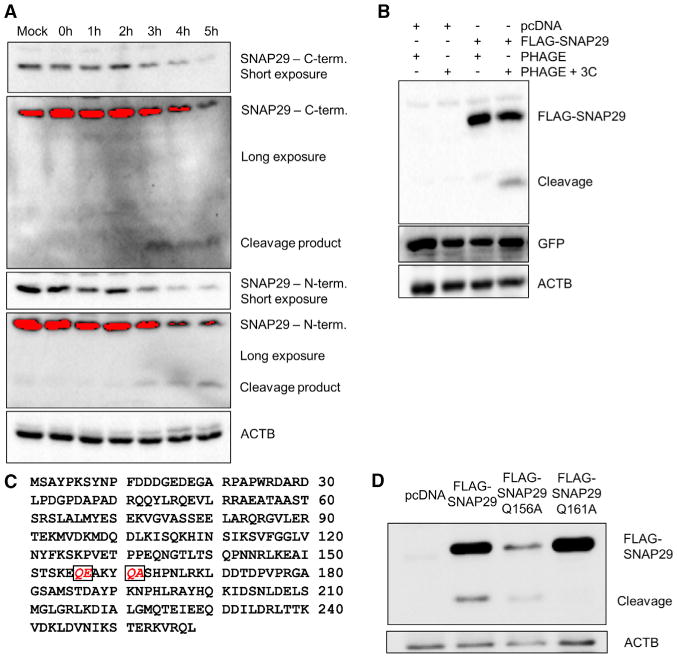
(A) H1HeLa cells were infected at an MOI of 25 for 5 hr, and cells were collected every hour. Samples were subjected to western blot analysis and probed for SNAP29 using antibodies recognizing the C terminus and N terminus of the protein. Both short and long exposures are provided.
(B) H1HeLa cells were transfected with pcDNA, FLAG-SNAP29, PHAGE, or PHAGE + 3C for 48 hr. Samples immunoblotted for FLAG and GFP are shown.
(C) Amino acid alignment of SNAP29, with putative cleavage sites italicized and boxed.
(D) 293T cells were transfected with pcDNA, FLAG-SNAP29, FLAG-SNAP29 Q156A, or FLAG-SNAP29 Q161A for 24 hr. Cells were then infected with EV-D68 for 5 hr, and samples were collected. Samples were then immunoblotted for FLAG; full-length and cleavage bands are marked.
We constructed a 3C-expressing plasmid that independently expresses GFP to confirm transfection. We transfected the construct, the FLAG-SNAP29 plasmid, and/or corresponding empty vectors into H1HeLa cells for 48 hr. Lysates were immunoblotted for FLAG and GFP (Figure 5B). Upon cotransfection with FLAG-SNAP29 and EV-D68 3C protease, a FLAG-reactive band is observed at approximately 25 kDa, the expected molecular weight of the N-terminal cleavage fragment with the 3×-FLAG tag. Based on this size information and the SNAP29 protein sequence, two putative cleavage sites for the viral 3C protease were identified, which we would expect to produce fragments of approximately 11 kDa and 18 kDa (Figure 5C). We generated point mutant-containing versions of the FLAG-SNAP29 plasmid for both of the putative cleavage sites, Q156A and Q161A. These constructs were transfected into 293T cells for 24 hr and then cells were infected with EV-D68. We show in Figure 5D that SNAP29 mutant Q156A, although not expressed at high levels, was still cleaved during EV-D68 infection. Mutant Q161A, however, was not cleaved. Subsequent experiments co-expressing both mutants with the EV-D68 2A and 3C protease-expressing plasmids indicate that the 3C protease can cleave FLAG-SNAP29-Q156A, but not FLAG-SNAP29-Q161A (Figure S5). These data together demonstrate that reduction in full-length SNAP29 during EV-D68 infection is the result of cleavage by the viral 3C protease at Q161.
SNAP29 Affects EV-D68 Replication in H1HeLa Cells
To further examine the role that SNAP29 plays during EV-D68 infection, siRNA-mediated knockdown of SNAP29 was carried out (Figure 6C). After a 48-hr knockdown and subsequent low-MOI infection, cell-associated virus (Figure 6A) and extracellular virus (Figure 6B) were analyzed by plaque assay. There was an approximately 10-fold reduction in cell-associated virus and an approximate 8-fold reduction in extracellular virus. We had hypothesized that loss of SNAP29 would have a positive effect on viral replication, because SNAP29 levels decrease late in infection (Figure 4); however, as shown here, a reduced level of SNAP29 prior to infection results in a significant drop in titer. These data may suggest an important role for SNAP29 early in the replication cycle of EVD68.
Figure 6. SNAP29 Affects EV-D68 Infection in H1HeLa Cells.
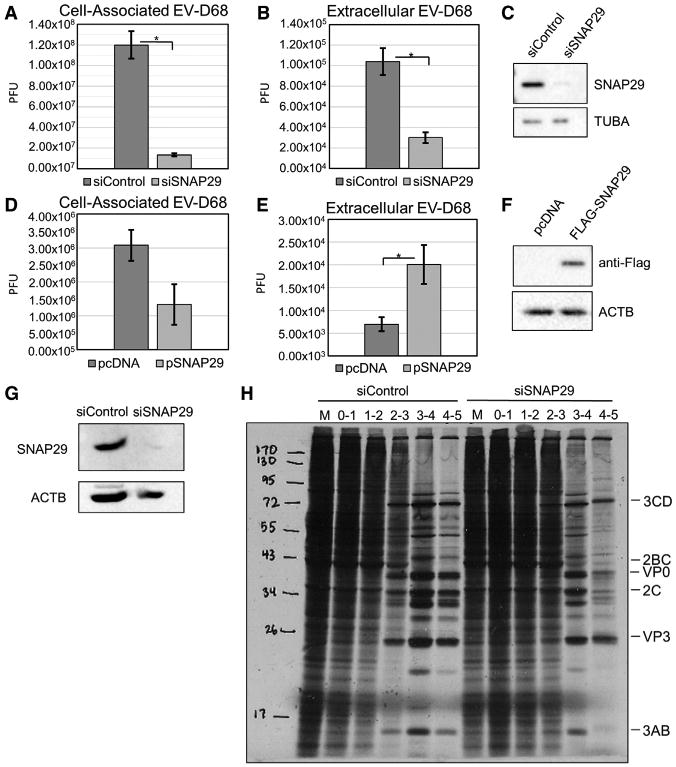
(A and B) Cells were transfected with a siRNA targeting SNAP29 40 hr prior to infection and then infected with EV-D68 at an MOI of 0.1 for 5 hr. Viral titers were analyzed by plaque assay for both cell-associated virus (A; p = 0.018) and extracellular virus (B; p = 0.012).
(C) Parallel samples were analyzed by western blot for SNAP29. H1HeLa cells were transfected with a plasmid containing FLAG-SNAP29 40 hr prior to infection.
(D and E)Transfected H1HeLa cells were infected at an MOI of 0.1 with EV-D68 for 5 hr. Viral titers were analyzed by plaque assay for both cell-associated virus (D) and extracellular virus (E; p = 0.017).
(F) Western blot to examine FLAG-SNAP29 ectopic expression. Western blots are representative of 3 independent experiments.
(G and H) H1HeLa cells were transfected with siSNAP29 40 hr prior to infection, infected at an MOI of 25 with EV-D68, and metabolically labeled each hour with 35-S methionine.
(G) Samples were analyzed for SNAP29 in knockdown cells.
(H) Samples were subjected to SDS-PAGE and exposed to X-ray film.
Viral titers are represented as the mean ± SEM. Statistical tests were done using Student's t test with statistical significance set at *p ≤ 0.05.
We next used a FLAG-tagged expression construct for SNAP29 and transfected the plasmid into H1HeLa cells 40 hr prior to infection. By overexpressing SNAP29, we hoped to maintain a population of full-length SNAP29 throughout the entire EV-D68 infection. Uninfected transfected H1HeLa cells were collected to confirm the overexpression by western blot (Figure 6F). We found that there was a slight reduction in the cell-associated virus but a significant 4-fold increase in extracellular virus in cells still retaining SNAP29 (Figure 6DE). This suggests that any remaining full-length SNAP29 may play a role in the release of the virus. It is clear that SNAP29 plays very different roles in the EV-D68 life cycle early and late in infection.
Because SNAP29 requires STX17 and VAMP8 to carry out its membrane fusion function, we next co-expressed the proteins in multiple combinations (Figure S4C). Overexpression of SNAP29-VAMP8 and the combination of SNAP29-STX17-VAMP8 both significantly decreased cell-associated EV-D68 (Figures S4A and S4B). We hypothesize that co-expression creates more fusion complex availability within the cell, decreasing the ability of the virus to escape degradative autophagy. Consistent with this hypothesis, overexpression of SNAP29 significantly increased extracellular virus levels (Figure 6E). We next analyzed the effect on autophagy. SQSTM1 levels were increased slightly in the SNAP29 and VAMP8 dual cotransfection lanes and then dropped below control levels in the triple SNARE overexpression lanes (Figure S4C). LC3-II levels were increased in the single-transfection lanes of SNAP29, STX17, or VAMP8 but dropped in co-transfection conditions. This suggests that autophagic flux is only enhanced in the presence of the autophagic SNARE complex and transfection of a single autophagic SNARE is not sufficient for enhanced flux.
Because our data suggested a role for SNAP29 in early steps of the EV-D68 life cycle, we analyzed virus entry, host shutoff, and polyprotein production and processing using metabolic labeling of infected cells. Post-infection, cells were either kept in complete media or placed in methionine-, glutamine-, and cysteine-free minimum essential medium (MEM) supplemented with 100 μCi/mL of 35S-methionine and collected immediately following a 1-hr treatment. Lysates were subjected to SDS-PAGE and exposed to film (Figure 6H). As expected, EV-D68 shuts down host translation beginning 2 or 3 hr into the infection. In SNAP29 siRNA-treated samples, there is a reproducible 1-hr delay in host translation shutoff, although from these data, we cannot identify the specific step in early virus events that SNAP29 affects.
Role of SNAP47 in EV-D68 Replication
We have demonstrated that SNAP29 has a complex role in EV-D68 replication. There are two other SNAP homologs in non-neuronal cells, SNAP23 and SNAP47 (Holt et al., 2006; Kloepper et al., 2007; Kuster et al., 2015). Because SNAP23 localizes to the plasma membrane, we decided to analyze SNAP47, which, due to its putative endosomal association, could play a role in autophagosome acidification and maturation (Kuster et al., 2015). No role for SNAP47 in autophagy or RNA virus infection has been described to date, but we have previously demonstrated that autophagosome acidification is crucial for picornavirus particle maturation (Richards and Jackson, 2012). We began with siRNA knockdown of SNAP47 alone and in combination with SNAP29. Due to the high level of homology between these two proteins, we confirmed siRNA specificity (Figure 7A). Autophagic activity was assessed by LC3-II formation and SQSTM1 steady-state levels (Figure 7B). Surprisingly, the data show that loss of SNAP47 inhibits autophagic degradation to a greater degree than loss of SNAP29, as monitored by both LC3-II formation and SQSTM1 degradation. We find that loss of SNAP47 inhibits virus production by more than two logs (Figure 7C) and extracellular virus by more than one log (Figure 7D). We conclude that SNAP47 plays a major role in producing infectious virions but inhibits release of virus from cells.
Figure 7. SNAP47 Is Essential for Degrada-tive Autophagy and Negatively Affects Viral Exit.

H1HeLa cells were transfected with siControl, siSNAP29, or siSNAP47 40 hr prior to infection.
(A) Immunoblot for SNAP29 and SNAP47.
(B) Transfected cells were either starved for 5 hr, infected at an MOI of 25 with EV-D68, or untreated as control. Samples were immunoblotted for LC3 and SQSTM1. Densitometry is taken from 3 independent experiments.
(C and D) Transfected cells were infected at an MOI of 0.1 for 5 hr and collected for plaque assay analysis. Cell-associated (C; p = 0.00021) and extracellular virus (D; p = 0.0051) were analyzed via plaque assay. Viral titers are represented as the mean ± SEM. Statistical tests were done using an unpaired Student's t test with statistical significance set at ***p < 0.001, **p < 0.01, and *p % 0.05.
(E) H1HeLa cells were infected with EV-D68 at an MOI of 25 for 5 hr. Samples were collected each hour and a mock sample collected at 5 hr. Samples were immunoblotted for SNAP47.
During a time course of infection of H1HeLa cells, there is a modest reduction in SNAP47 protein levels by 5 hr post-infection (Figure 7E). Whereas not a complete loss of detectable protein, as observed for SNAP29, it is reproducible, and we investigated the mechanism of protein loss. Co-expression of EV-D68 proteases and SNAP47 provided no detection of apparent cleavage products (Figure S7A). We also tested whether it was a classical, cell-mediated method of degradation, utilizing the lysosome or proteasome, or if the cap-dependent translation shutoff was enough to affect SNAP47 protein levels. There is a statistically significant drop in SNAP47 levels when cells are treated with cycloheximide. This implicates virus-mediated translation inhibition, although little is known about the half-life of SNAP47 protein and more investigation is yet required to support this conclusion (Figures S7B and S7C). We conclude SNAP47 plays a significant role in both autophagy and EV-D68 replication and speculate that these roles are connected.
Discussion
In general, positive-strand RNA viruses have been shown to either benefit from, or be indifferent to, the autophagy pathway (Shi and Luo, 2012). Here, we demonstrate, through use of pharmacological agents and genetic knockdown, that activating the autophagy pathway promotes replication of EV-D68. Inhibition of autophagosome acidification and cargo delivery to lysosomes using BafA1 reduced cell-associated EV-D68 titers by more than one log. Silencing of ATG7 to disrupt proper autophagic membrane formation also significantly decreased viral titers. Induction of autophagy through cell starvation increased cell-associated virus by more than two-fold but reduced extracellular titers to approximately the same degree (Figure 1). Autophagic signaling is, therefore, a positive force for virus replication.
We therefore expected that the virus induces the autophagy pathway during infection, and we have identified multiple hallmarks of autophagic activation. Double-membraned vesicular structures resembling autophagosomes are generated during infection (Figure 2). These vesicles have apparent cytosolic contents and are approximately 100–300 nm in diameter, which is consistent with autophagic structures. The LC3 protein islipidated during infection, a hallmark of autophagy signaling, and GFP-LC3 puncta are observed, consistent with autophagosome formation (Figure 3). We conclude that infection promotes signaling to the autophagy pathway and generation of autophagic structures.
Autophagy is regulated at multiple steps, and induction of the pathway does not necessarily mean degradation will be induced. Delivery of cargo to autophagosomes, autophagosome acidification into amphisomes, and fusion of amphisomes with lysosomes can all be regulated. Autophagic degradation is commonly assayed by monitoring the steady-state levels of SQSTM1, a primary cargo adaptor for contents destined for the autophagosome. As seen in Figure 3, SQSTM1 is cleaved during infection by multiple picornaviruses, including PV, RV1A, and EV-D68. This was previously shown for CVB3 by the Luo lab, but here, we demonstrate that this is a common viral mechanism (Shi et al., 2014).
This significance of SQSTM1 cleavage is two-fold. First, because SQSTM1 is the most common assay for autophagic degradation, loss of the full-length protein can be misinterpreted if a relatively minor 34-kDa species is missed. In our previous work on PV, we misinterpreted loss of SQSTM1 as induction of autophagic degradation (Richards and Jackson, 2012). At the time, we were puzzled: if non-infectious particles were maturing into infectious virions inside amphisomes, how is autophagic degradation occurring without a loss of titer? Here, this issue is resolved with the realization that loss of full-length SQSTM1 during infection does not indicate autophagic degradation. Second, cleavage of SQSTM1 indicates that cargo is not being delivered to autophagosomes. Not only is this a false positive for degradation, it actually means the opposite is taking place: an inhibition of degradation.
To further investigate EV-D68 inhibition of the degradative step of autophagy, examination of a SNARE complex, which mediates fusion events of autophagic vesicles, was carried out. The three members of the complex, STX17, SNAP29, and VAMP8, each contribute coiled-coil domains to form a tight complex mediating the membrane fusion steps between autophagosomes, amphisomes, and autolysosomes (Wang et al., 2016). SNAP29 interacts with enterovirus A71 2BC protein, making it a known target for picornavirus regulation (Lai et al., 2017). As seen in Figure 4, we observe a distinct loss of full-length SNAP29 during infection and an apparent increase in detectable STX17. A loss of SNAP29 should result in inhibition of fusion, halting degradative autophagy.
Given our data regarding SQSTM1, we wanted to investigate the mechanism of SNAP29 loss. We demonstrate an EV-D68 3C-mediated proteolytic cleavage of SNAP29 at Q161 (Figure 5). Based on size and potential cleavage sites, the data predict that the N-terminal coiled-coil domain of SNAP29 (amino acids [aas] 50–132) would be separated from the C-terminal coiled-coil domain (aas 197–258) by this cleavage event (Scales et al., 2000). The two halves should independently interact with STX17 and VAMP8, preventing formation of the membrane-fusion complex. It has been reported that, in non-starvation conditions, SNAP29 is O-GlcNac modified on residues Ser2, Ser61, Thr130, and Ser153, and these modifications inhibit the autophago-some-lysosome fusion process (Guo et al., 2014). These modifications are all N-terminal to the two putative cleavage sites, creating an N-terminal cleavage fragment that may inhibit fusion. It is possible, as with SQSTM1, that relatively minor cleaved species could act as dominant negatives, preventing autophagic cargo delivery to lysosomes. Our data support this model. The endosomal marker LAMP1 partially colocalizes with LC3 during infection, whereas CTSB does not colocalize with LC3 (Figure S3). This indicates that endosome-autophagosome fusion can occur, delivering LAMP1 and acidifying vacuolar ATPases, but the subsequent lysosome fusion step is blocked. We hypothesize this block is due to SNAP29 cleavage.
SNAP29 promotes both virus replication and virion exit from cells (Figure 6). There is a slight delay in an early step, possibly virus entry, translation, or host-shutoff, in the absence of SNAP29, as seen in Figure 6H. This difference is reproducible, and we plan to investigate the role of SNAP29 early in EV-D68 infection. Overexpression of SNAP29 only slightly decreased cell-associated virus. Surprisingly, overexpression increased virus release significantly. It is possible that SNAP29 may be able to bind to target SNAREs on the plasma membrane to promote exit in a non-physiological situation, such as overexpression, where full-length SNAP29 persists through viral replication. SNAP29 has been shown to interact with enterovirus A71 capsid protein (Lai et al., 2017).
The family of Snap Qbc SNAREs is relatively small: the neuron-specific SNAP25; the plasma-membrane localized SNAP23; cytosolic SNAP29; and the less-characterized membrane-associated SNAP47. We were interested in determining whether SNAP47, which is largely cytosolic but associates with endosome- and Golgi-resident VAMP proteins, plays a role in EV-D68 replication (Holt et al., 2006; Kuster et al., 2015). We found that knockdown of SNAP47 had a larger inhibitory effect on EV-D68 growth than SNAP29 knockdown (Figure 7). In addition, there was a significant increase in released virus, despite the loss of cell-associated virus. We analyzed autophagy in cells knocked down for SNAP47 and found a major reduction in degradation, with little change in signaling, as would be expected for a SNARE protein with a role in autophagy. In fact, our data indicate that knockdown of SNAP47 has a more significant effect on autophagy than knockdown of SNAP29, paralleling the relative effects of knockdown of these proteins on EV-D68 replication. Our data indicate that SNAP47 is a major SNARE in the autophagic pathway, and the specific role of SNAP47 in autophagy is being investigated.
We hypothesize that, whereas both SNAP29 and SNAP47 play roles during EV-D68's viral cycle, they participate at different stages. In Figure 4, we see that SNAP29 protein levels are reduced late in infection, which is, as shown in Figure 5, likely to be the result of cleavage by a viral protease, such as 3C. However, when SNAP29 protein levels are reduced by siRNA knockdown prior to infection, resulting viral titers significantly dropped (Figures 6A and 6B). We conclude that SNAP29 plays a positive role early in infection but is not required late in infection. How SNAP29 promotes EV-D68 replication early in the virus life cycle is still being determined.
The importance of acidification to EV-D68 made us question how the endosome and lysosome were able to fuse with the autophagosome in the viral-mediated absence of SNAP29. The potential role of SNAP47 in endosome trafficking led us to investigate SNAP47 in autophagy and EV-D68's life cycle. After we observed the inhibition of SQSTM1 degradation in the absence of SNAP47, we concluded that there may be more than one SNAP involved with the autophagic process. A previous study on SNAP47 showed subcellular localization to be throughout the cytosol yet often punctate. The same study also showed the ability for SNAP47 to substitute for another SNARE, SNAP25, forming an active fusion complex (Holt et al., 2006). SNAP47 itself interacts with VAMP7 and VAMP4, which are associated with the endosome and trans-Golgi, respectively (Kuster et al., 2015). During an infection with EV-D68, a modest decrease of SNAP47 occurs (Figure S7), which may be due to the relatively short half-life of the protein and the presence of a translational shutoff during infection, common among picornavi-ruses, but which we demonstrate here for EV-D68 (Figure 6H). We hypothesize SNAP47 is able promote autophagosome acidification, which benefits EV-D68.
In summary, we have identified a major role for autophagic signaling and autophagic membrane rearrangements in EV-D68 replication. However, the virus inhibits the downstream, degradative aspects of autophagy at multiple levels by cleaving a major cargo adaptor and a SNARE protein, SNAP29, reported to be important for delivering amphisome cargo to the lysosome. We have demonstrated that a second SNARE, SNAP47, has major effects on autophagic degradation and EV-D68 growth. We suggest that EV-D68 reshapes the autophagic pathway to prevent degradation of virus and promote virus replication and exit from the cell. In the last several years, it has been demonstrated that multiple picornaviruses exit cells in membraned form, and in some cases, these membranes are derived from the autophagy pathway (Chen et al., 2015; Feng et al., 2013; Robinson et al., 2014). We suggest that EV-D68 may be reshaping the autophagy pathway to allow autophagosome-like vesicles filled with infectious virus to exit the cell instead of being targeted to the lysosome and are currently testing this model. We believe that this may be a general model for picornavirus egress from cells, and another paper in this issue of Cell Reports by Mohamud et al. (2018), showing similar data for coxsackievirus B3, demonstrates the wide potential for understanding the roles of autophagic SNAREs in replication of positive-strand RNA viruses.
Experimental Procedures
Viral Infections
Viral infections were carried out in H1HeLa cells, except where noted. Adsorptions were carried out in PBS with added calcium chloride and magnesium chloride (PBS+) for 30 min at 37°C and then rinsed with PBS and covered in complete media for the incubation period.
Drug Treatments
Bafilomycin A1 was prepared in ethanol and used at 0.1 mM for an 18-hr pre-treatment before and during viral infection. Starvation media as described in Axe et al. (2008) was used prior to viral infection. Pepstatin A (10 mg/mL) and E64D (100 mM) were prepared in DMSO. MG132 (20 mM) and cycloheximide (100 mM) were purchased ready made in DMSO.
Plaque Assays
Extracellular virus was collected by taking a 1-mL sample of the supernatant. Cell-associated virus was collected by aspirating supernatant off the cell monolayer, adding 1 mL PBS+, and scraping the cells into a microcentrifuge tube. Samples were freeze thawed, and viral titers were then determined by plaque assay. Samples were serially diluted in PBS+ and added to H1HeLa cells. After adsorption, plates were overlaid with a 23 MEM/2% agar mixture and incubated for 40 hr. Agar was removed, and cells were fixed and stained.
Western Blotting
Cell samples were lysed and loaded onto polyacrylamide tris-glycine gels for SDS-PAGE. Gels were run at 90 V and transferred to polyvinylidene fluoride (PVDF) via iBlot 2 (Invitrogen). Blots were blocked in 5% milk/TBS-T for 1 hr and then incubated with primary antibody overnight at 4°C. Blots were washed three times and incubated with secondary antibody for 1 hr at room temperature. Blots were developed using Western Lightning ECL on a Bio-Rad ChemiDoc.
siRNA Knockdowns
siRNAs against ATG7, SNAP29, and SNAP47 were manufactured by Sigma and sequences provided in Supplemental Experimental Procedures. Either Mission siRNA Universal Negative Control 1 or gene-of-interest siRNAs were transfected using Lipofectamine 2000 into H1HeLa cells 40 hr prior to experiment. Knockdown efficiency was analyzed by immunoblotting each experiment.
Plasmid Overexpression
FLAG-SNAP29, FLAG-STX17, and FLAG-VAMP8 plasmid constructs were obtained through Addgene as a gift from Noboru Mizushima (Addgene plasmids 45915, 45911, and 45912, respectively). EVD68-2A and 3C, FLAG-SNAP29-Q156A and -Q161A plasmids were generated by PCR cloning. SQSTM1-WT and SQSTM1-MT plasmids were a gift from Honglin Luo. GFP-SNAP47 was purchased from OriGene Technologies. Plasmid-of-interest or pcDNA empty vector was transfected into H1HeLa cells or 293T cells for 24 hr prior to experiment, except where noted. Expression of protein was analyzed by western blot.
Fluorescence Microscopy
Coverslips were fixed in cold methanol for 15 min and blocked in PBS/5% normal goat serum and either 0.1% saponin or 0.3% Triton X-100 where noted. Coverslips were stained as described in Supplemental Experimental Procedures and mounted using Vectashield (H-1200). Images were obtained on a Zeiss LSM 710 using a 1003 oil-immersion objective.
Electron Microscopy
Samples were fixed in 2% paraformaldehyde/2.5% glutaraldehyde 5 hr postinfection. Samples were prepared for imaging as described in Supplemental Experimental Procedures and examined on a FEI Tecnai T12 electron microscope.
Autoradiography
Cells were incubated in a methionine and cysteine-free MEM that contained 100 mCi/mL 35S methionine. Cells were treated for an hour at a time throughout the course of infection and collected after for SDS-PAGE. Gel dried was exposed to X-ray film for 24 hr.
Statistical Analysis
Statistical testing was done using Student's unpaired t tests, with significance defined as ***p < 0.001, **p < 0.01, and *p ≤ 0.05. p > 0.05 was deemed not significant. “N” represents sum total of technical and biological replicates. All experiments were performed 2–4 times.
Additional details for all methods can be found in the Supplemental Expermental Procedures.
Supplementary Material
Highlights.
Enterovirus D68 (EV-D68) induces and benefits from autophagy
The autophagic SNARE SNAP29 promotes EV-D68 in early infection but is later cleaved
The orphan SNARE SNAP47 promotes EV-D68 infection
SNAP47 plays a major role in autophagy and EV-D68 replication
Acknowledgments
This work was funded by NIAID grant AI104928 to William T.Jackson. We thank Joseph Mauban and University of Maryland School of Medicine Center for Innovative Biomedical Resources, Confocal Microscopy Core – Baltimore, Maryland for help with the fluorescence work and Core resources. EM work utilized a sample preparation instrument that was purchased with funding from an NIH SIG grant (1S10RR26870-1) awarded to the University of Maryland-Baltimore. We thank Dr. J. Lindsay Whitton for the infectious clone for Coxsackievirus Woodruff. We thank Dr. Ru-ching Hsia for electron microscopy guidanceand support.Wealso thank Dr. Honglin Luo and Yasir Mohamud for enlightening discussions and communication of results prior to publication.
Footnotes
Author Contributions: Conceptualization, A.K.C., A.F.C.V., and W.T.J.; Methodology, A.K.C. and W.T.J.; Investigation, A.K.C., A.F.C.V., and W.T.J.; Resources, H.M.S.; Writing – Original Draft, A.K.C. and W.T.J.; Writing – Review & Editing, A.K.C. and W.T.J.; Supervision, W.T.J.; Funding Acquisition, W.T.J.
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.003.
Declaration Of Interests: The authors declare no competing interests.
References
- Aliabadi N, Messacar K, Pastula DM, Robinson CC, Leshem E, Sejvar JJ, Nix WA, Oberste MS, Feikin DR, Dominguez SR. Enterovirus D68 infection in children with acute flaccid myelitis, Colorado, USA, 2014. Emerg Infect Dis. 2016;22:1387–1394. doi: 10.3201/eid2208.151949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Flynn CT, Wood MR, Harkins S, Whitton JL. Coxsackievirus can exploit LC3 in both autophagy-dependent and -independent manners in vivo. Autophagy. 2015;11:1389–1407. doi: 10.1080/15548627.2015.1063769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EW, Kornberg AJ, Freeman JL, Leventer RJ, Ryan MM. Acute flaccid myelitis in childhood: a retrospective cohort study. Eur J Neurol. 2017;24:1077–1083. doi: 10.1111/ene.13345. [DOI] [PubMed] [Google Scholar]
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Haber-mann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. Complex dynamic development of poliovirus membranous replication complexes. J Virol. 2012;86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Du W, Hagemeijer MC, Takvorian PM, Pau C, Cali A, Brantner CA, Stempinski ES, Connelly PS, Ma HC, et al. Phos-phatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160:619–630. doi: 10.1016/j.cell.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520:563–566. doi: 10.1038/nature14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito S, Bosis S, Niesters H, Principi N. Enterovirus D68 infection. Viruses. 2015;7:6043–6050. doi: 10.3390/v7112925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong SH, Walker C, Lanford RE, Lemon SM. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013;496:367–371. doi: 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovács AL, Zhang Z, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014;16:1215–1226. doi: 10.1038/ncb3066. [DOI] [PubMed] [Google Scholar]
- Hixon AM, Yu G, Leser JS, Yagi S, Clarke P, Chiu CY, Tyler KL. A mouse model of paralytic myelitis caused by enterovirus D68. PLoS Pathog. 2017;13:e1006199. doi: 10.1371/journal.ppat.1006199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, Varoqueaux F, Wiederhold K, Takamori S, Urlaub H, Fasshauer D, Jahn R. Identification of SNAP-47, a novel Qbc-SNARE with ubiquitous expression. J Biol Chem. 2006;281:17076–17083. doi: 10.1074/jbc.M513838200. [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Jackson WT. Poliovirus-induced changes in cellular membranes throughout infection. Curr Opin Virol. 2014;9:67–73. doi: 10.1016/j.coviro.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanasios E, Ktistakis NT. Autophagy at the Cell, Tissue and Organismal Level (Springer) 2016. [Google Scholar]
- Kloepper TH, Kienle CN, Fasshauer D. Anelaborate classification of SNARE proteins sheds light on the conservation of the eukaryotic endo-membrane system. Mol Biol Cell. 2007;18:3463–3471. doi: 10.1091/mbc.E07-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuster A, Nola S, Dingli F, Vacca B, Gauchy C, Beaujouan JC, Nunez M, Moncion T, Loew D, Formstecher E, et al. The Q-soluble N-ethylmaleimide-sensitive factor attachment protein receptor (Q-SNARE) SNAP-47 regulates trafficking of selected vesicle-associated membrane proteins (VAMPs) J Biol Chem. 2015;290:28056–28069. doi: 10.1074/jbc.M115.666362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JKF, Sam IC, Verlhac P, Baguet J, Eskelinen EL, Faure M, Chan YF. 2BC non-structural potein of enterovirus A71 interacts with SNARE proteins to trigger autolysosome formation. Viruses. 2017;9:E169. doi: 10.3390/v9070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landajuela A, Hervás JH, Antón Z, Montes LR, Gil D, Valle M, Rodriguez JF, Goñi FM, Alonso A. Lipid geometry and bilayer curvature modulate LC3/GABARAP-mediated model autophagosomal elongation. Biophys J. 2016;110:411–422. doi: 10.1016/j.bpj.2015.11.3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messacar K, Schreiner TL, Maloney JA, Wallace A, Ludke J, Oberste MS, Nix WA, Robinson CC, Glodé MP, Abzug MJ, Dominguez SR. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet. 2015;385:1662–1671. doi: 10.1016/S0140-6736(14)62457-0. [DOI] [PubMed] [Google Scholar]
- Midgley CM, Watson JT, Nix WA, Curns AT, Rogers SL, Brown BA, Conover C, Dominguez SR, Feikin DR, Gray S, et al. EV-D68 Working Group. Severe respiratory illness associated with a nationwide outbreak of enterovirus D68 in the USA (2014): a descriptive epidemiological investigation. Lancet Respir Med. 2015;3:879–887. doi: 10.1016/S2213-2600(15)00335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamud Y, Shi J, Qu J, Poon T, Xue YC, Deng H, Zhang J, Luo H. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication Cell Rep. 2018;22:3292–3303. doi: 10.1016/j.celrep.2018.02.090. this issue. [DOI] [PubMed] [Google Scholar]
- Morelli E, Ginefra P, Mastrodonato V, Beznoussenko GV, Rusten TE, Bilder D, Stenmark H, Mironov AA, Vaccari T. Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy. 2014;10:2251–2268. doi: 10.4161/15548627.2014.981913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013;55:39–50. doi: 10.1042/bse0550039. [DOI] [PubMed] [Google Scholar]
- Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autoly-sosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste MS, Maher K, Schnurr D, Flemister MR, Lovchik JC, Peters H, Sessions W, Kirk C, Chatterjee N, Fuller S, et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J Gen Virol. 2004;85:2577–2584. doi: 10.1099/vir.0.79925-0. [DOI] [PubMed] [Google Scholar]
- Quiner CA, Jackson WT. Fragmentation of the Golgi apparatus provides replication membranes for human rhinovirus 1A. Virology. 2010;407:185–195. doi: 10.1016/j.virol.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AL, Jackson WT. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 2012;8:e1003046. doi: 10.1371/journal.ppat.1003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J. SNARE function revisited. Nat Struct Biol. 2003;10:417–419. doi: 10.1038/nsb0603-417. [DOI] [PubMed] [Google Scholar]
- Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014;10:e1004045. doi: 10.1371/journal.ppat.1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales SJ, Chen YA, Yoo BY, Patel SM, Doung YC, Scheller RH. SNAREs contribute to the specificity of membrane fusion. Neuron. 2000;26:457–464. doi: 10.1016/s0896-6273(00)81177-0. [DOI] [PubMed] [Google Scholar]
- Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol. 1967;85:297–310. doi: 10.1093/oxfordjournals.aje.a120693. [DOI] [PubMed] [Google Scholar]
- Seglen PO. Hepatic Endocytosis. Trivandrum, India: Transworld Research Network); 2008. Where Endocytosis and Autophagy Meet: The Amphisome; pp. 61–100. [Google Scholar]
- Shi J, Luo H. Interplay between the cellular autophagy machinery and positive-stranded RNA viruses. Acta Biochim Biophys Sin (Shanghai) 2012;44:375–384. doi: 10.1093/abbs/gms010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wong J, Piesik P, Fung G, Zhang J, Jagdeo J, Li X, Jan E, Luo H. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy. 2013;9:1591–1603. doi: 10.4161/auto.26059. [DOI] [PubMed] [Google Scholar]
- Shi J, Fung G, Piesik P, Zhang J, Luo H. Dominant-negative function of the C-terminal fragments of NBR1 and SQSTM1 generated during enteroviral infection. Cell Death Differ. 2014;21:1432–1441. doi: 10.1038/cdd.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li L, Hou C, Lai Y, Long J, Liu J, Zhong Q, Diao J. SNARE-mediated membrane fusion in autophagy. Semin Cell Dev Biol. 2016;60:97–104. doi: 10.1016/j.semcdb.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhuge J, Huang W, Nolan SM, Gilrane VL, Yin C, Dimitrova N, Fallon JT. Enterovirus D68 subclade B3 strain circulating and causing an outbreak in the United States in 2016. Sci Rep. 2017;7:1242. doi: 10.1038/s41598-017-01349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J Virol. 2008;82:9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HB, Kielczewska A, Rozek A, Takenaka S, Li Y, Thorson L, Hancock REW, Guarna MM, North JR, Foster LJ, et al. Sequesto-some-1/p62 is the key intracellular target of innate defense regulator peptide. J Biol Chem. 2009;284:36007–36011. doi: 10.1074/jbc.C109.073627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, Yuan J. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci USA. 2007;104:19023–19028. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.