

Abstract
C-reactive protein (CRP) is an acute-phase protein with therapeutic activity in mouse models of systemic lupus erythematosus and other inflammatory and autoimmune diseases. To determine the mechanism by which CRP suppresses immune complex disease, an adoptive transfer system was developed in a model of immune thrombocytopenic purpura (ITP). Injection of 200 μg of CRP 24 h before induction of ITP markedly decreased thrombocytopenia induced by anti-CD41. CRP-treated splenocytes also provided protection from ITP in adoptive transfer. Splenocytes from C57BL/6 mice were treated with 200 μg/ml CRP for 30 min, washed, and injected into mice 24 h before induction of ITP. Injection of 106 CRP-treated splenocytes protected mice from thrombocytopenia, as did i.v. Ig-treated but not BSA-treated splenocytes. The suppressive cell induced by CRP was found to be a macrophage by depletion, enrichment, and the use of purified bone marrow-derived macrophages. The induction of protection by CRP-treated cells was dependent on FcRγ-chain and Syk activation, indicating an activating effect of CRP on the donor cell. Suppression of ITP by CRP-treated splenocytes required FcγRI on the donor cell and FcγRIIb in the recipient mice. These findings suggest that CRP generates suppressive macrophages through FcγRI, which then act through an FcγRIIb-dependent pathway in the recipient to decrease platelet clearance. These results provide insight into the mechanism of CRP regulatory activity in autoimmunity and suggest a potential new therapeutic approach to ITP.
Introduction
C-reactive protein (CRP) 3 is an acute-phase serum protein and a classical pentraxin (1, 2). Serum levels of CRP may increase up to 1000-fold in response to inflammation, infection or trauma. Pentraxins are pattern recognition molecules composed of five identical subunits arranged in cyclic pentameric configuration (3). CRP interacts with the immune system through interaction with FcR and activation of the classical complement pathway. The best described functions of CRP include protection from bacterial infection, clearance of damaged tissue, and regulation of the inflammatory response (1, 4). We have been particularly interested in the role of CRP in protection from autoimmunity, which was suggested by its ability to bind to nuclear Ags that are targets of autoantibodies in connective tissue diseases, including chromatin, histones, and small nuclear ribonucleoproteins (5–7).
Studies in our laboratory first showed that CRP prevented accelerated autoimmune disease in female (NZB x NZW)F1 mice injected with chromatin (8). Subsequent studies by our laboratory and others have confirmed that CRP treatment suppresses glomerulonephritis and delays mortality in both the (NZB x NZW) F1 and the MRL/Fas lpr mouse models of systemic lupus erythematosus (9–11). CRP-transgenic mice are also protected from experimental autoimmune encephalomyelitis, a predominantly T cell-mediated autoimmune disease (12). Others established potent anti-inflammatory activity of CRP in endotoxin- and cytokine-induced shock (13), and we showed that this activity of CRP was dependent on FcγR (14). Our results in the systemic lupus erythematosus models suggested that the anti-inflammatory activity of CRP was also central to its effectiveness in treating ongoing lupus nephritis (9, 11). Using a nephrotoxic nephritis model, we established that CRP-mediated suppression of IC-induced renal disease required FcγRI and macrophages (15). However, identification of the mechanisms involved in CRP-mediated regulation of inflammation has been limited by the requirements for direct administration of CRP into the animal model or the expression of CRP as a transgene product. Therefore, we sought to develop a model in which CRP would induce a suppressive cell in vitro with activity after adoptive transfer. This approach would allow for the identification of the active cell, the receptors required for its generation and the mechanism by which it induces regulation of the inflammatory response.
Immune thrombocytopenic purpura (ITP) is a common human autoimmune disease, which is caused by the production of antiplatelet Abs (16, 17). These autoantibodies opsonize platelets for splenic clearance, resulting in low levels of circulating platelets. The disease is characterized by recurring episodes of severe thrombocytopenia and is associated with a bleeding diathesis. Treatments for ITP include splenectomy, high-dose corticosteroids and i.v. Ig (IVIg). During the last decade, the mechanism by which IVIg modulates ITP has been under intense investigation, but no consensus exists for a single protective mechanism (18, 19). The development of an adoptive transfer approach to study the mechanism of IVIg-mediated protection from ITP was recently reported (20).
FcγR on splenic macrophages are the primary mediators of ITP in humans and mice (16, 21). FcγRI, FcγRIII, and FcγRIV are activating receptors in mice that signal through ITAMs on the associated γ-chain (22). FcγRIIb is the only inhibitory FcγR, and it contains an ITIM in its intracellular domain. The balance between activating and inhibitory FcγR is critical in the ability of cells not only to respond but also to regulate the intensity of the response (23). For example, mice that are normally resistant to collagen-induced arthritis become susceptible when FcγRIIb is genetically eliminated (24). CRP binding to FcγR was first demonstrated for FcγRI (25). Additional reactivity of CRP with FcRII was later established in both humans and mice (26–28). Recently, we defined the crystal structure of the related pentraxin serum amyloid P component in complex with FcγRIIa as a model for CRP-FcγR interaction (29). These studies confirmed the interaction of CRP with FcγR and defined the binding characteristics of the human pentraxins for all three classes of human FcγR.
In these experiments, we examine the effect of CRP treatment in vitro and in vivo in a mouse model of ITP. Adoptive transfer of CRP-treated cells was used in this model to examine the role of activating and inhibitory FcγR in CRP-mediated suppression. A primary role for macrophages in transfer of suppression was established using depletion and enrichment strategies. The results demonstrate that activation of macrophages by CRP through FcγRI produces a cell capable of suppressing immune complex (IC) disease in the absence of the continued presence of CRP. We propose that induction of this suppressive macrophage is the initial event in CRP suppression of a range of inflammatory and autoimmune diseases.
Materials and Methods
Reagents
CRP was purified from human pleural fluids as described previously (30). All preparations were examined on overloaded SDS-PAGE gels stained with SYPRO Ruby protein gel stain (Invitrogen) to ensure purity. No additional bands other than the major band at 23 kDa were seen. All preparations used contained <3 endotoxin U (0.3 ng) of endotoxin per mg of protein determined by a quantitative chromogenic Limulus amebocyte lysate assay (Lonza). IVIg purchased from Bayer was used for most experiments. In some experiments, IVIg (Baxter), a gift from Dr. Ralph C. Williams (University of New Mexico, Albuquerque, NM), was used with equivalent results. Chicken OVA, piceatannol, egg phosphatidylcholine, BSA, and cholesterol were purchased from Sigma-Aldrich. Dichloromethylene bisphosphonate (Clodronate) was provided by Boehringer Mannheim. rIFN-γ was purchased from R&D Systems. Abs were obtained as follows: rabbit IgG anti-OVA from MP Biomedical; rat mAb specific for mouse CD41 (IgG1k, clone MWReg 30), FcγRI (CD64), and CD11b from BD Biosciences; rat mAb specific for F4/80 and FcγRIIb (clone MM7301) from Caltag; rat mAb specific for CD11c from Miltenyi Biotec. Rat mAb 2.4G2 was used as an FcγR-blocking agent. Corresponding isotype control Abs were used in all flow cytometry experiments. Magnetic microbeads along with separation columns were purchased from Miltenyi Biotec. ICs were prepared by mixing chicken OVA with a 10-fold molar excess of anti-OVA for 30 min at room temperature.
Mice
C57BL/6 (B6) mice were obtained from the National Cancer Institute. FcγRIIB−/− mice and IL-10 −/− mice were obtained from The Jackson Laboratory and backcrossed to 10 generations on B6 mice. FcγRI −/− mice were obtained from Dr. J. S. Verbeek (Leiden University Medical Center, Leiden, The Netherlands) and further backcrossed onto B6 mice (31). Female mice at 6–12 wk of age were used in all experiments. All procedures involving animals were approved by the Institutional Review Board of the Department of Veterans Affairs Medical Center.
Induction of ITP
Mice were injected i.v. with 4 μg of anti-CD41 in 0.2 ml of PBS, pH 7.2. Blood was taken from the retro-orbital plexus and diluted in Unopettes (BD Bioscience). Platelets were counted using a hemocytometer. Baseline platelet counts were obtained from mice before treatment. In the adoptive transfer experiments, the average baseline platelet counts for each mouse strain were determined and are shown as normal platelet counts on the graphs.
Treatment of ITP in vivo
Mice were injected i.p. with 50 mg of IVIg 24 h before the injection of anti-CD41 (20). For CRP, mice were injected i.p. with 200 μg of CRP 24 h before treatment with anti-CD41 or i.v. with 200 μg of CRP 4 h before treatment with anti-CD41.
Treatment of spleen cells in vitro and adoptive transfer
Splenocytes were prepared from donor mice, and erythrocytes were lysed with ammonium chloride (ACK) lysis buffer. In the standard protocol, splenocytes were suspended in serum-free RPMI 1640 at a concentration of 1.4 × 106 cells/ml and treated with 200 μg/ml CRP, 200 μg/ml BSA, or 18 mg/ml IVIg for 0.5 h at 37°C. After treatment, cells were washed twice in RPMI 1640 and resuspended at a concentration of 5 × 106 cells/ml, and 0.2 ml of the suspension was injected i.v. into recipient mice 24 h before induction of ITP.
Culture of bone marrow macrophages (BMM)
Bone marrow cells were cultured for 1 wk in DMEM (Sigma-Aldrich; high glucose) with 20% FBS and 30% L cell-conditioned medium, as a source of M-CSF. BMMs were >95% positive for the F4/80 macrophage marker by flow cytometry. BMMs were treated with 100 U/ml IFN-γ for 24 h before treatment with CRP or IVIg. Cells were harvested using 10 mM EDTA for 5 min at 4°C, washed, and suspended to desired concentration in RPMI 1640 for treatment, as described for splenocytes.
Macrophage depletion and enrichment
Liposomes containing Clodronate were prepared as previously described (32). The liposomes were washed by ultracentrifugation, and mice were injected i.v. with 0.2 ml of liposomes containing Clodronate or control liposomes prepared with PBS. Spleens were taken from mice 48 h after injection of liposomes. We previously found that treatment with Clodronate liposomes depletes 99% of Kupffer cells and 95% of splenic macrophages (33). Clodronate liposome treatment of mice resulted in an 80% decrease in F4/80+ spleen cells by flow cytometry.
F4/80+ cells were enriched from adherent splenocytes using biotin antiF4/80 and streptavidin beads (Miltenyi Biotec). The target cell population was selected, following the manufacturer’s instructions, and was verified by analysis using flow cytometry.
Immunoprecipitation and Western blot analysis of Syk phosphorylation
BMMs (10 × 106) in 1 ml were stimulated with 200 μg/ml CRP for the desired time, then immediately put on ice, and subsequently washed twice with PBS. Cells were lysed with lysis buffer (1% Nonidet P-40, 0.15 M NaCl, 0.05 M Tris, pH 7.2, with Halt Protease Inhibitor 2x (Thermo Scientific), and 100 μM activated NaVO4). The protein concentration was determined by a bicinchoninic acid assay (Thermo Scientific). Syk was immunoprecipitated with anti-Syk (N-19) Ab overnight with protein A/G PLUS-agarose beads (Santa Cruz Biotechnology). Beads were washed five times with lysis buffer and then boiled for 5 min in 2x loading buffer.
Samples were loaded on a 4–20% SDS-polyacrylamide precast gel (Invitrogen); then SDS-PAGE was conducted for 3 h at 120 V. Proteins were transferred to a polyvinylidene difluoride membrane with a wet transfer system (NOVEX) at 25 V for 90 min. The membrane was blocked with 5% BSA in TBST for 1 h and then probed for Phospho-Syk (Cell Signaling Technology) with polyclonal Phospho-Syk Ab (1/1000) in TBST plus 5% BSA overnight at 4°C. The membrane was washed three times for 5 min in TBST. HRP-conjugated secondary anti-rabbit IgG (Cell Signaling Technologies; 1/2000) was incubated with membrane for 1 h at room temperature. The blot was washed three times with TBST before detection with ECL (SuperSignal West Dura Extended Duration Substrate; Thermo Scientific). The blot was imaged using a charge-coupled device camera (Gene Gnome; Syngene Bio Imaging System) and analyzed with ImageJ (National Institutes of Health). OD was determined and normalized for each sample using the IgG H chain as an internal loading control.
Data analysis
Graphic and statistical analyses were performed using GraphPad Prism software (GraphPad). Data are presented as the means SEM for the number of mice indicated in the figure legends. Comparisons between means were done using Student’s t test or one-way ANOVA. p values are indicated in the figure legends as: *, p < 0.05; **, p < 0.01; ***, p > 0.001.
Results
Injection of CRP protects mice from ITP
An experimental model of ITP was established based on the previous studies of Siragam et al. (20). B6 mice and FcγRIIB−/− mice were injected with a mAb to CD41, and platelet counts were performed over time. At 24 h after injection, platelet levels were found to be at their lowest (Fig. 1A). In all subsequent experiments, platelets were counted 24 h after injection of the anti-platelet Ab. To determine whether CRP would suppress ITP, mice were injected i.p. with 200 μg of CRP 24 h before injection of anti-CD41. An additional set of mice was injected with 50 mg of IVIg i.p. as described (20). IVIg was administered i.p. to avoid fluid overload due to rapidly increasing the intravascular volume. As shown in Fig. 1B, i.p. injection of 200 g of CRP protected mice from developing ITP to a degree similar to that of administration of 50 mg of IVIg (Fig. 1C). In data not shown, injection of 200 μg of CRP i.v. 4 h before induction of ITP was as effective as injection of CRP 24 h before induction of ITP.
FIGURE 1.
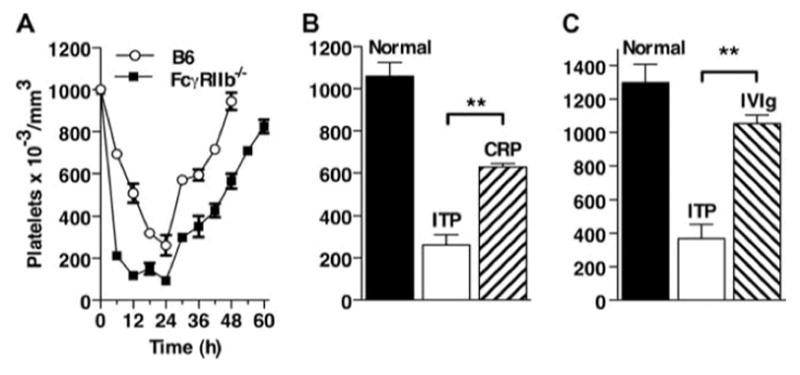
Injection of CRP or IVIg protects mice from ITP. A, ITP model. B6 and FcγRIIB −/− mice were injected with mAb to CD41, and platelets were counted every 6 h. n = 3. B, B6 mice were injected i.p. with 200 μg of CRP 24 h before injection with anti-CD41. After 24 h, platelets were counted. n = 3. C, B6 mice were injected i.p. with 50 mg of IVIg 24 h before injection with anti-CD41. After 24 h, platelets were counted. Normal indicates platelet counts in mice before treatment. n = 3.
CRP protects mice from ITP in the adoptive transfer system
CRP inhibited the induction ITP to a similar degree as, but at a much lower dose than, IVIg when injected directly. To determine whether CRP was acting to block FcR-mediated uptake or whether an effector cell was generated, adoptive transfer of suppression was tested. Previous studies indicated that treatment of splenocytes in vitro with 18 mg/ml IVIg rendered them capable of adoptive transfer of suppression of ITP (20). Spleen cells were treated with 0–500 μg/ml CRP or 18 mg/ml IVIg as a positive control and then transferred to mice, which were subsequently treated with mAb to CD41. As seen in Fig. 2, A and B, CRP-treated splenocytes were as effective as IVIg-treated splenocytes with maximum effectiveness at a concentration of ~200 μg/ml, a dose 90 times less than that of IVIg. Subsequent experiments used a CRP dose of 200 μg/ml for adoptive transfer studies. We next determined the number of splenocytes that were required to transfer suppression of ITP. Splenocytes treated with 200 μg/ml CRP were transferred at numbers ranging from 10 4 to 10 7 cells. The effect of cell transfer was seen with as few as 104 cells, but increasing numbers of cells gave progressively more suppression of thrombocytopenia (Fig. 2C).
FIGURE 2.
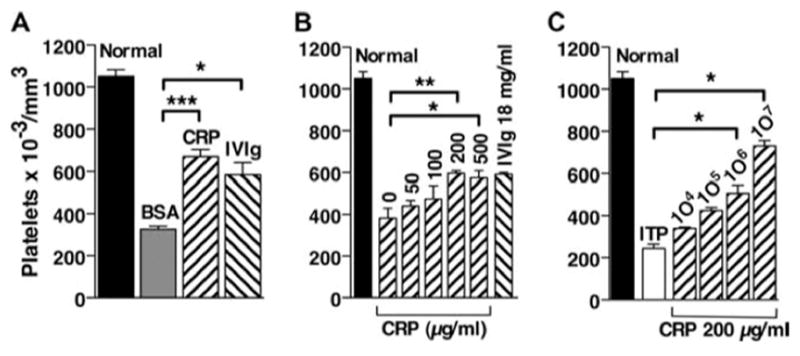
Transfer of CRP-treated spleen cells protects mice from ITP. Spleen cells from B6 mice were treated with CRP, BSA or IVIg for 30 min, washed, and injected i.v. into B6 mice. Twenty-four hours after cell transfer, mice were injected i.v. with anti-CD41 to induce ITP, and 24 h later platelets were counted. A, Spleen cells were treated with 200 μg of CRP or BSA or with 18 mg/ml IVIg and 106 cells were transferred. B, Spleen cells were treated with 0–500 μg/ml CRP or 18 mg/ml IVIg before transfer. C, Spleen cells were treated with 200 μg/ml CRP before transfer of the indicated number of cells. ITP indicates that no spleen cells were transferred. n = 3.
Induction of the suppressive phenotype requires signaling through the γ-chain and Syk activation
Our previous studies indicated that many of the effects of CRP on inflammation in vivo are due to interaction with FcγR (15). We first examined whether the effects of CRP were mediated by activating FcγR using γ-chain −/− mice. Mouse FcγRI, -III, and -IV are expressed in association with the γ-chain and use this chain to initiate signaling. FcγR signaling also requires Syk, which is recruited to phosphorylated ITAMs and activated. Syk then phosphorylates adaptor proteins, which are necessary for downstream signaling events (23). Splenocytes from γ-chain −/− mice were treated with CRP and transferred to B6 mice after which ITP was induced. Splenocytes from γ-chain −/− mice provide no protection from ITP (Fig. 3A). Therefore, to determine whether the γ-chain is needed simply for CRP to interact with the cell or if signaling downstream of the γ-chain is also required, inhibition of FcγR signaling was examined. Splenocytes from B6 mice were treated with CRP after incubation with the Syk inhibitor, piceatannol, or ethanol (vehicle) before transfer. Inhibition of Syk eliminated the suppressive effect of CRP-treated cells, whereas vehicle alone did not. Thus, generation of the suppressive phenotype required signaling through a γ-chain-associated receptor and Syk activation. To verify that signaling was initiated, BMMs from B6 mice were stimulated with CRP for various time points and then lysed and immunoprecipitated with anti-Syk. Proteins were transferred and probed for phosphorylated Syk. Fig. 3C demonstrates that CRP induced maximum phosphorylation at 5 min (Fig. 3B). Together, these results indicate that CRP induces intracellular signaling events through Syk to mediate protection in recipient mice.
FIGURE 3.
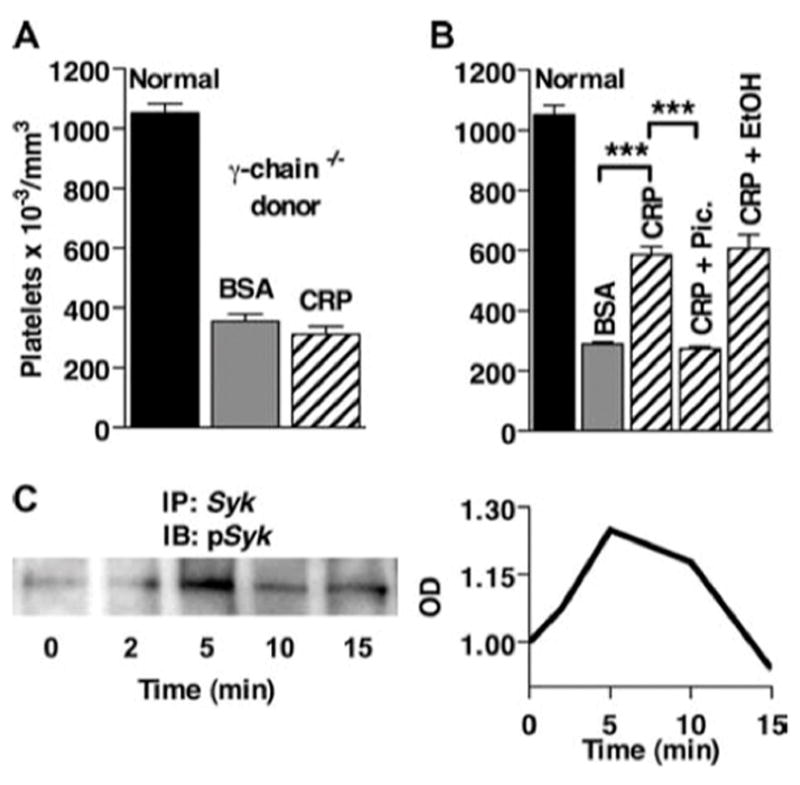
CRP-mediated protection from ITP requires the FcR γ-chain and Syk activation. Spleen cells were treated with 200 μg/ml CRP or BSA for 30 min and washed, and 10 6 cells were injected i.v. into B6 recipient mice. Twenty-four hours after spleen cell transfer, mice were injected with anti-CD41, and 24 h later platelets were counted. A, Spleen cells from FcR γ-chain −/− mice injected into B6 mice. n = 3. B, Spleen cells from B6 mice were treated with 10 μM piceatannol or ethanol (vehicle control) before CRP treatment and transfer into B6 mice. n = 3. C, BMMs from B6 mice were stimulated with 200 μg/ml CRP for 0, 2, 5, 10, and 15 min and then immunoprecipitated (IP) with anti-Syk. The blot (IB) was probed for phospho-Syk, and the bands were normalized to IgG H chain in each respective lane.
FcγRI is required for CRP-mediated protection
Our previous studies of CRP-mediated suppression of inflammation suggested a central role for FcγRI (15). FcγRI is the highaffinity receptor for IgG and binds CRP with moderate to high affinity. Spleen cells from FcγRI −/− mice were treated with CRP in vitro, washed, and then transferred to B6 mice before induction of ITP. No protective effect on the development of ITP was seen (Fig. 4A), indicating that FcγRI expression on the donor cell is required for CRP-mediated suppression. The requirement for FcγRI in the recipient mouse of adoptive transfer was also examined. In contrast to the results using FcγRI −/− donor cells, FcγRI was not required for development of ITP or for suppression by CRP-treated B6 splenocytes (Fig. 4B). These results suggest that FcγRI is an integral part of the protective effect of CRP in the donor cell but that its presence is not required in the recipient. These results indicate that the initial event in the suppressive cell is an activating event, which induces the suppressive phenotype.
FIGURE 4.
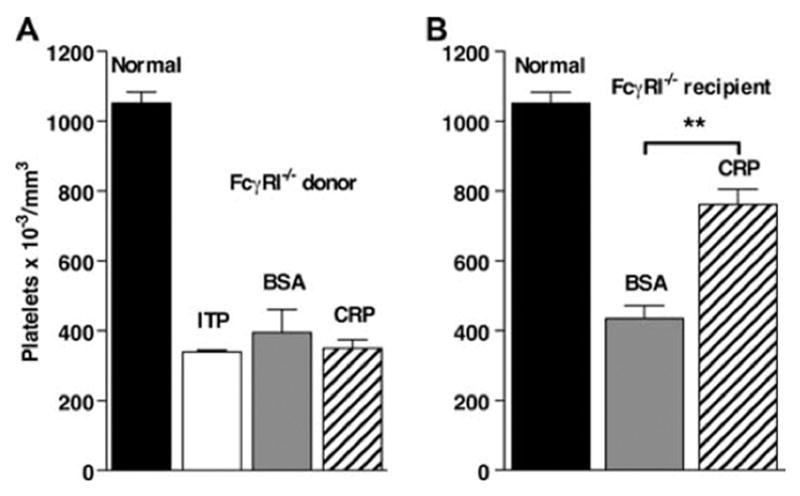
CRP-mediated protection from ITP requires FcγRI on donor splenocytes. Spleen cells were treated with 200 μg/ml CRP or BSA for 30 min and washed, and 10 6 cells were injected i.v. into recipient mice. Twenty-four h after spleen cell transfer, mice were injected with anti-CD41, and 24 h later platelets were counted. A, Spleen cells from FcγRI −/− mice injected into B6 mice. B, Spleen cells from B6 mice were injected into FcγRI −/− mice. ITP indicates that no spleen cells were transferred. n = 3.
CRP interaction with FcγRIIb is not required for its protective role
FcγRIIb is an inhibitory receptor and has been shown to regulate phagocytosis through activating FcγR (34). It has also been shown that FcγRIIb is required in recipients for the adoptive transfer of suppression of ITP by IVIg-treated spleen cells (20). To test the requirement for FcγRIIb expression in CRP-mediated suppression, FcγRIIB −/− mice were used either as a source of splenocyte suppressor cells or as recipients undergoing ITP. Spleen cells from FcγRIIB −/− mice were fully active in adoptive transfer of suppression (Fig. 5A). However, CRP-treated splenocytes from B6 mice were unable to suppress ITP in FcγRIIB −/− mice (Fig. 5B). These findings indicate a crucial role for FcγRIIb in the recipient mouse as previously found for IVIg (19, 20).
FIGURE 5.
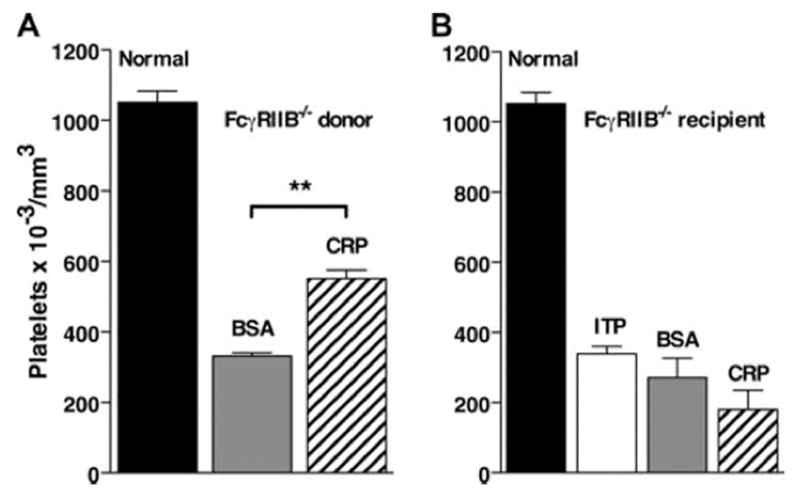
CRP-mediated protection from ITP requires FcγRIIb in recipient mice. Spleen cells were treated with 200 μg/ml CRP or BSA for 30 min and washed, and 10 6 cells were injected i.v. into recipient mice. Twenty-four hours after spleen cell transfer, mice were injected with anti-CD41 and 24 h later platelets were counted. A, Spleen cells from FcγRIIB −/− mice injected into B6 mice. B, Spleen cells from B6 mice injected into FcγRIIB −/− mice. n = 3.
IL-10 is not required for suppressor cell activity
Previous studies in our laboratory on the suppression of nephrotoxic nephritis indicated a requirement for IL-10 in CRP-mediated suppression. We therefore tested IL-10 −/− mice as donors and recipients in the ITP model. Unlike the case of nephrotoxic nephritis, IL-10 was not required in either the donor or the recipient for suppression of ITP by CRP-treated splenocytes (Fig. 6). CRP-treated splenocytes from IL-10 −/− donor mice showed a small decrease in the ability to transfer suppression of ITP compared with wild-type cells. Thus, suppression by CRP-treated splenocytes does not rely completely on IL-10.
FIGURE 6.
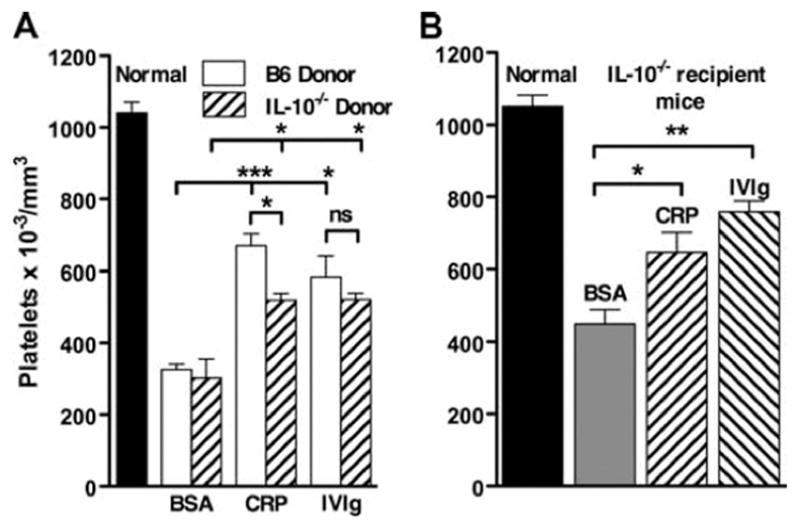
CRP-mediated protection from ITP does not require IL-10. Spleen cells were treated with 200 μg/ml CRP or BSA or 18 mg/ml IVIg for 30 min and washed, and 10 6 cells were injected i.v. into recipient mice. Twenty-four hours after spleen cell transfer, mice were injected with antiCD41, and 24 h later platelets were counted. A, Spleen cells from B6 or IL-10 −/− mice were injected into B6 mice. B, Spleen cells from B6 mice injected into IL-10 −/− mice. n = 3.
The adoptive transfer of CRP-mediated suppression requires macrophages
Previous adoptive transfer studies of IVIg-mediated suppression suggested that dendritic cells were the major cell population responsible for suppression (20). Preliminary studies were performed to determine whether dendritic cells were responsible for CRP-mediated suppression. CD11c+ cells were isolated using magnetic bead separation, treated with CRP, and tested in the adoptive transfer system. CRP-treated CD11c+ dendritic cells were less effective than CD11c-cells in transferring suppression (not shown). To determine whether macrophages were the cells in the splenocyte population that mediates the protective effect of CRP, mice were treated with Clodronate-containing liposomes 2 days before harvesting spleens. Mice treated with Clodronate-containing liposomes lack splenic macrophages for 5–6 days (32). Spleen cells from these mice were then treated with CRP in vitro and tested for adoptive transfer of suppression. CRP-treated splenocytes from Clodronate-treated mice were unable to transfer suppression of thrombocytopenia (Fig. 7A). However, CRP-treated spleen cells from control mice were effective (Fig. 7A) as were spleen cells from mice that were pretreated with PBS-liposomes (not shown). In contrast, IVIg-mediated protection was not eliminated by Clodronate pretreatment of mice (Fig. 7A). We next used magnetic bead separation with anti-F4/80 to enrich for splenic macrophages. F4/80-enriched spleen cells transferred suppression of ITP after CRP treatment but were less effective after IVIg treatment (Fig. 7B). These studies suggest that macrophages are the predominant cell type required for the transfer of CRP-mediated suppression of ITP and that different cells are essential for transfer of suppression after CRP and IVIg treatment.
FIGURE 7.
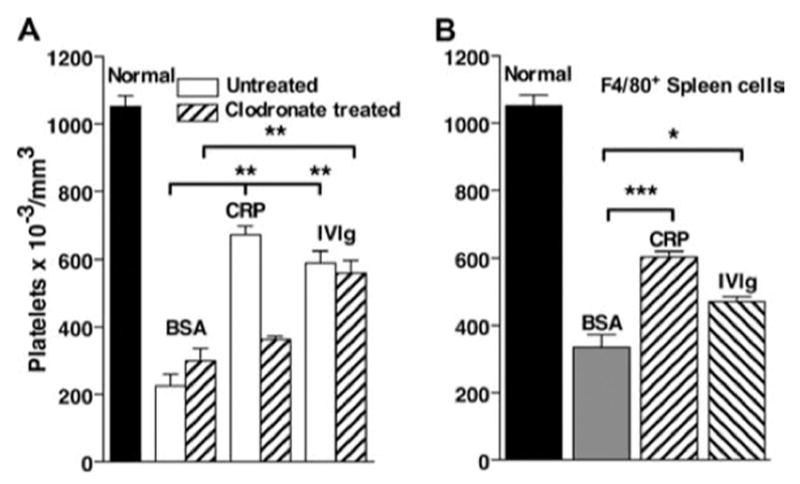
Splenic macrophages are required for the transfer of protection from ITP by CRP-treated splenocytes. A, B6 mice were untreated or injected with liposomes containing Clodronate to deplete macrophages. After 48 h, spleens were removed and cells were treated with 200 μg/ml CRP or BSA or with 18 mg/ml IVIg for 30 min and washed, and 106 cells were injected i.v. into recipient mice. Twenty-four hours after spleen cell transfer, mice were injected with anti-CD41, and 24 h later platelets were counted. n = 3. B, Spleen cells from B6 mice were enriched for macrophages by selection for F4/80+ cells. These cells were treated with CRP, BSA, or IVIg and washed, and then 1 × 106 cells were injected into B6 mice. Twenty-four hours after treatment, mice were injected with antiCD41 to induce ITP. After 24 h, blood was sampled and platelets were counted. n = 5.
BMMs transfer the protective effect from CRP
The previous data suggested that splenic macrophages are required to transfer the protective effect of CRP. To determine whether macrophages alone could mediate this effect, purified BMMs were used as the donor cell population. BMMs from B6 mice were primed with IFN-γ for 24 h, then treated with CRP, and transferred to B6 mice 24 h before induction of ITP (Fig. 8A). BMMs treated with 18 mg/ml IVIg or 200 μg/ml IC before transfer were able to protect mice from developing ITP. BMMs are ≥95% F4/80+, arguing against a requirement for other cell types. A dose-response experiment was done using BMMs treated with 200 μg/ml CRP (Fig. 8B). BMMs were maximally effective when 10 5 or 10 6 cells were transferred, and 10 5 BMMs were as protective as 10 7 spleen cells (Fig. 2C), further suggesting that macrophages are the active cell population in the spleen. To examine the role of FcγRI on the generation of the suppressive phenotype of macrophages by CRP, BMMs from FcγRI −/− or B6 mice were treated with 200 μg/ml CRP and then transferred to B6 mice. BMMs that lack expression of FcγRI −/− were not able to protect mice from developing ITP (Fig. 8C).
FIGURE 8.
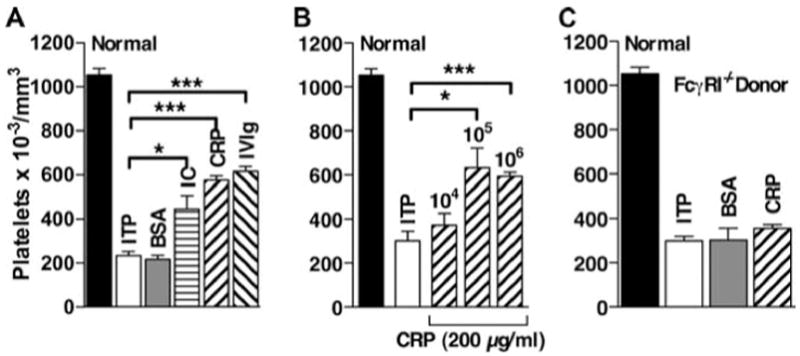
BMM reproduce the protective effect of CRP-treated splenocytes on ITP. BMMs from B6 mice were prepared as described and treated with IFN-γ for 24 h before treatment with 200 μg/ml CRP, BSA, or IC or with 18 mg/ml IVIg for 30 min. IC were prepared as described in Materials and Methods. Cells were then washed and injected into B6 mice. Twenty-four hours after transfer, mice were injected with anti-CD41 to induce ITP, and 24 h later platelets were counted. A, BMMs were treated 200 g/ml CRP, BSA, and IC or with 18 mg/ml IVIg, and 10 6 cells were injected per mouse. n = 3. B, BMMs were treated with 200 μg/ml CRP, and 104, 105, or 106 cells were injected per mouse. n = 5 combined from 2 experiments. C, BMMs from FcγRI −/− mice were treated with 200 μg/ml CRP, and 106 cells were injected per mouse. n = 3. ITP indicates that no BMMs were transferred.
Discussion
We and others have previously reported that CRP suppresses inflammation in mouse models (13–15). However, these activities were dependent on the endogenous expression of CRP from a transgene or the direct injection of CRP. These requirements made identification of the cell type responsible for immune regulation and the mediators involved difficult to determine. The major new finding presented here is that CRP can be used in an adoptive transfer system to induce immunomodulation in a nontreated host. This approach allowed us to determine that macrophages activated by CRP through FcγRI are responsible for this activity.
CRP injection markedly suppressed the induction of ITP by antiCD41. The degree of protection from ITP was similar to that provided by IVIg. However, CRP suppressed ITP at a much lower dose. IVIg is a heterogeneous preparation composed of multiple IgG isotypes with broad antigenic reactivity. The mechanism by which IVIg suppresses ITP remains unresolved. It is unknown whether the high dose of IVIg required is due to the low activity of the IgG or whether a minor fraction of the IgG is active based on, e.g., glycosylation status, aggregation, or antigenic reactivity (35). Most recently, it has been suggested that the interaction of IVIg occurs through a receptor for sialylated Fc and not through FcγR (35, 36). However, dendritic cells activated through FcR suppressed ITP in the adoptive transfer model studied here (20).
CRP binds to a limited group of determinants and is essentially homogeneous with respect to binding specificity. CRP is a nonglycosylated molecule with no sequence variation or isotypes; thus binding to FcγR is expected to be homogeneous. CRP binding to FcγR occurs with 1:1 stoichiometry, and monomeric CRP is therefore unlikely to induce receptor cross-linking or signaling (29). The preparations used did not contain aggregates before injection, as determined by analytical gel filtration, but it is possible that ligands for CRP exist in vivo, which could result in receptor cross-linking. CRP was effective up to 24 h before induction of ITP, suggesting that CRP need not be present throughout the course of ITP. Human CRP is known to have a short half-life (4 h) in mice (37).
CRP was also found to be active in a transfer model of suppression. This finding represents the first model in which one can transfer suppression using CRP-treated cells. The effect was very unlikely to be due to CRP carryover as splenocytes were washed before transfer to recipient mice. The dose-response curve would suggest that a much higher dose of CRP would be required than any amount carried over in the washed cells. In addition, cells lacking FcγRI or γ-chain or cells treated with a tyrosine kinase inhibitor were inactive. FcRI was not required in the recipient, suggesting that at least two different cells could be involved in the adoptive transfer of suppression of ITP. The suppressive cell generated in the donor by CRP treatments must act on the effector cell in the recipient undergoing ITP that is responsible for platelet clearance. However, it is likely that, in the case of direct injection of CRP into mice undergoing ITP, no second cell is involved and CRP acts directly on the effector cell. The spleen cell dose response in adoptive transfer suggested that the suppressive cell was active at relatively low numbers or represented activity in a broad population of cells.
The results of additional experiments indicate that the active cell is a macrophage. This was somewhat unexpected as previous studies using IVIg indicated a central role for dendritic cells in this adoptive transfer model of ITP (20). Our experiments, using enrichment and depletion of splenic macrophage and dendritic cell populations, are consistent with a requirement for macrophages for CRP-induced suppression, but not for IVIg-induced suppression. BMMs treated with IFN- were, however, sufficient to transfer suppression of ITP after treatment with CRP, IVIg, or IC. CRP-treated BMMs were active at 10-fold lower numbers than splenocytes, consistent with the numbers of macrophages in the spleen. Although the overall effect of CRP appears similar to that of IVIg, the receptor interactions undoubtedly differ between the two agents.
Suppressive macrophages with different phenotypes have been described in several systems (38). Mosser’s group (39) has characterized a macrophage that he has called type II activated macrophages. These macrophages produce IL-10 and down-regulate IL-12 production but are distinguished from alternatively activated macrophages by their expression of NO rather than arginase and other characteristics. Activation of this phenotype is induced by cross-linking activating FcγR together with TLR stimulation (40). Type II activated macrophages were active in the regulation of endotoxin-induced inflammation and altered T cell responses to Ag, but have not been studied in IC inflammation models.
Our previous studies have suggested an important role for the suppressive effect of CRP on inflammatory models in mice (15). The results indicated a central role for FcγRI, the high-affinity receptor for IgG and a receptor of intermediate affinity for CRP. The ability of CRP to induce the suppressive macrophage was strictly dependent on FcγRI. Ongoing studies indicate that CRP binds FcγRI with lower affinity than IgG (29). FγRI is an activating, γ-chain-associated receptor, suggesting that the initial event involved in generating the suppressive phenotype is also activating. FcRI has been implicated in the suppressive activities of IC (41), and FcγRI −/− mice have enhanced Ab production in response to immunization (42).
In the recipient mice undergoing ITP, the presence of FcγRIIb was required for suppression. A requirement for FcγRIIb was shown previously in both direct treatment and adoptive transfer models of IVIg suppression of ITP (20). However, it is possible that the somewhat greater efficacy of the mAb in induction of thrombocytopenia makes the FcγRIIB −/− mouse more resistant to suppression by CRP.
Because the protective effect of CRP in nephrotoxic nephritis required IL-10 and not FcγRIIb, we tested the requirement for IL-10 in the ITP transfer model. Unlike the case of nephrotoxic nephritis, no requirement for IL-10 was seen, given that IL-10 deficient mice were fully responsive to CRP treatment. The effect of the CRP-induced suppressive macrophage population was not limited to ITP, given that these cells were also effective in the Arthus model (data not shown). The Arthus reaction is a well-established and defined model of IC-mediated inflammation. Neutrophil recruitment to the peritoneal cavity of the mouse is mediated primarily by two chemokines that are produced by resident macrophages.
We cannot conclusively determine that the mechanism involved in the adoptive transfer corresponds to the in vivo protection from ITP shown by CRP. However, these studies establish an initial common pathway for anti-inflammatory effects of CRP in two distinctly different models. We have defined a population of macrophages induced by CRP acting through FcγRI that is active in an adoptive transfer model. The mechanisms by which these cells mediate suppressive effects are currently incompletely understood, but they are likely to play an initial role in many of the antiinflammatory activities of CRP.
CRP could be considered as an alternative therapeutic agent to IVIg for several reasons. CRP is a homogeneous, well-defined molecule of consistent properties. It was active at a much lower dose than IVIg in vitro and in vivo. IVIg can contain antigenic specificities that are potentially deleterious. The supply of IVIg is limited due to the current increasing range of therapeutic indications and the inherent requirement for very large numbers of donors.
Acknowledgments
The authors thank Dr. J. Sjef Verbeek for generously providing breeder pairs of FcRI −/− mice and Dr. Ralph C. Williams for IVIg.
Disclosures: The authors have no financial conflict of interest.
Abbreviations used in this paper
- CRP
C-reactive protein
- IC
immune complex
- BMM
bone marrow macrophage
- Clodronate
dichloromethylene bisphosphonate
- ITP
immune thrombocytopenic purpura
- IVIg
i.v. Ig
Footnotes
This work was supported by the Department of Veterans Affairs and the University of New Mexico Research Allocations Committee.
References
- 1.Marnell L, Mold C, Du Clos TW. C-reactive protein: ligands, receptors and role in inflammation. Clin Immunol. 2005;117:104–111. doi: 10.1016/j.clim.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol Immunol. 2001;38:189–197. doi: 10.1016/s0161-5890(01)00042-6. [DOI] [PubMed] [Google Scholar]
- 3.Shrive AK, Cheetham GM, Holden D, Myles DA, Turnell WG, Volanakis JE, Pepys MB, Bloomer AC, Greenhough TJ. Three dimensional structure of human C-reactive protein. Nat Struct Biol. 1996;3:346–354. doi: 10.1038/nsb0496-346. [DOI] [PubMed] [Google Scholar]
- 4.Szalai AJ. The antimicrobial activity of C-reactive protein. Microbes Infect. 2002;4:201–205. doi: 10.1016/s1286-4579(01)01528-3. [DOI] [PubMed] [Google Scholar]
- 5.Du Clos TW, Mold C. C-reactive protein as a regulator of autoimmune disease. In: Zouali M, editor. Molecular Autoimmunity: In Commemoration of the 100th Anniversary of the First Description of Human Autoimmune Disease. Springer Science and Business Media; New York: 2005. pp. 27–41. [Google Scholar]
- 6.Du Clos TW. C-reactive protein reacts with the U1 small nuclear ribonucleoprotein. J Immunol. 1989;143:2553–2559. [PubMed] [Google Scholar]
- 7.Du Clos TW. The interaction of C-reactive protein and serum amyloid P component with nuclear antigens. Mol Biol Rep. 1996;23:253–260. doi: 10.1007/BF00351177. [DOI] [PubMed] [Google Scholar]
- 8.Du Clos TW, Zlock LT, Hicks PS, Mold C. Decreased autoantibody levels and enhanced survival of (NZB x NZW) F1 mice treated with C-reactive protein. Clin Immunol Immunopathol. 1994;70:22–27. doi: 10.1006/clin.1994.1005. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez W, Mold C, Kataranovski M, Hutt J, Marnell LL, Du Clos TW. Reversal of ongoing proteinuria in autoimmune mice by treatment with C-reactive protein. Arthritis Rheum. 2005;52:642–650. doi: 10.1002/art.20846. [DOI] [PubMed] [Google Scholar]
- 10.Szalai AJ, Weaver CT, McCrory MA, van Ginkel FW, Reiman RM, Kearney JF, Marion TN, Volanakis JE. Delayed lupus onset in (NZB x NZW) F1 mice expressing a human C-reactive protein transgene. Arthritis Rheum. 2003;48:1602–1611. doi: 10.1002/art.11026. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez W, Mold C, Marnell LL, Hutt J, Silverman GJ, Tran D, Du Clos TW. Prevention and reversal of nephritis in MRL/lpr mice with a single injection of C-reactive protein. Arthritis Rheum. 2006;54:325–335. doi: 10.1002/art.21556. [DOI] [PubMed] [Google Scholar]
- 12.Szalai AJ, Nataf S, Hu XZ, Barnum SR. Experimental allergic encephalomyelitis is inhibited in transgenic mice expressing human C-reactive protein. J Immunol. 2002;168:5792–5797. doi: 10.4049/jimmunol.168.11.5792. [DOI] [PubMed] [Google Scholar]
- 13.Xia D, Samols D. Transgenic mice expressing rabbit C-reactive protein are resistant to endotoxemia. Proc Natl Acad Sci USA. 1997;94:2575–2580. doi: 10.1073/pnas.94.6.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mold C, Rodriguez W, Rodic-Polic B, Du Clos TW. C-reactive protein mediates protection from lipopolysaccharide through interactions with FcR. J Immunol. 2002;169:7019–7025. doi: 10.4049/jimmunol.169.12.7019. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez W, Mold C, Kataranovski M, Hutt JA, Marnell LL, Verbeek JS, Du Clos TW. C-reactive protein-mediated suppression of nephrotoxic nephritis: role of macrophages, complement, and Fcγ receptors. J Immunol. 2007;178:530–538. doi: 10.4049/jimmunol.178.1.530. [DOI] [PubMed] [Google Scholar]
- 16.Cines DB, V, Blanchette S. Immune thrombocytopenic purpura. N Engl J Med. 2002;346:995–1008. doi: 10.1056/NEJMra010501. [DOI] [PubMed] [Google Scholar]
- 17.Stasi R, Evangelista ML, Stipa E, Buccisano F, Venditti A, Amadori S. Idiopathic thrombocytopenic purpura: current concepts in pathophysiology and management. Thromb Haemost. 2008;99:4–13. doi: 10.1160/TH07-08-0513. [DOI] [PubMed] [Google Scholar]
- 18.Crow AR, Lazarus AH. The mechanisms of action of intravenous immunoglobulin and polyclonal anti-D immunoglobulin in the amelioration of immune thrombocytopenic purpura: what do we really know? Transfus Med Rev. 2008;22:103–116. doi: 10.1016/j.tmrv.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291:484–486. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 20.Siragam V, Crow AR, Brinc D, Song S, Freedman J, Lazarus AH. Intravenous immunoglobulin ameliorates ITP via activating Fc receptors on dendritic cells. Nat Med. 2006;12:688–692. doi: 10.1038/nm1416. [DOI] [PubMed] [Google Scholar]
- 21.Ravetch JV, Clynes RA. Divergent roles for Fc receptors and complement in vivo. Annu Rev Immunol. 1998;16:421–432. doi: 10.1146/annurev.immunol.16.1.421. [DOI] [PubMed] [Google Scholar]
- 22.Nimmerjahn F, Ravetch JV. Fcγ receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 24.Yuasa T, Kubo S, Yoshino T, Ujike A, Matsumura K, Ono M, Ravetch J, Takai T. Deletion of Fc receptor IIB renders H-2 b mice susceptible to collagen-induced arthritis. J Exp Med. 1999;189:187–194. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marnell LL, Mold C, Volzer MA, Burlingame RW, Du Clos TW. C-reactive protein binds to FcRI in transfected COS cells. J Immunol. 1995;155:2185–2193. [PubMed] [Google Scholar]
- 26.Stein MP, Mold C, Du Clos TW. C-reactive protein binding to murine leukocytes requires Fc receptors. J Immunol. 2000;164:1514–1520. doi: 10.4049/jimmunol.164.3.1514. [DOI] [PubMed] [Google Scholar]
- 27.Bharadwaj D, Stein MP, Volzer M, Mold C, Du Clos TW. The major receptor for C-reactive protein on leukocytes is Fc receptor II. J Exp Med. 1999;190:585–590. doi: 10.1084/jem.190.4.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein MP, Edberg JC, Kimberly RP, Mangan EK, Bharadwaj D, Mold C, Du Clos TW. C-reactive protein binding to FcγRIIa on human monocytes and neutrophils is allele-specific. J Clin Invest. 2000;105:369–376. doi: 10.1172/JCI7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcγR by innate pentraxins. Nature. 2008 doi: 10.1038/nature07468. In press. [DOI] [PMC free article] [PubMed]
- 30.Du Clos TW, Marnell L, Zlock LR, Burlingame RW. Analysis of the binding of C-reactive protein to chromatin subunits. J Immunol. 1991;146:1220–1225. [PubMed] [Google Scholar]
- 31.Ioan-Facsinay A, de Kimpe SJ, Hellwig SM, van Lent PL, Hofhuis FM, van Ojik HH, Sedlik C, da Silveira SA, Gerber J, de Jong YF, et al. FcγRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 2002;16:391–402. doi: 10.1016/s1074-7613(02)00294-7. [DOI] [PubMed] [Google Scholar]
- 32.van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 33.Du Clos TW, Volzer MA, Hahn FF, Xiao R, Mold C, Searles RP. Chromatin clearance in C57Bl/10 mice: interaction with heparan sulphate proteoglycans and receptors on Kupffer cells. Clin Exp Immunol. 1999;117:403–411. doi: 10.1046/j.1365-2249.1999.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunter S, Indik ZK, Kim MK, Cauley MD, Park JG, Schreiber AD. Inhibition of Fc receptor-mediated phagocytosis by a nonphagocytic Fcγ receptor. Blood. 1998;91:1762–1768. [PubMed] [Google Scholar]
- 35.Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. J Exp Med. 2007;204:11–15. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 37.Baltz ML, I, Rowe F, Pepys MB. In vivo turnover studies of C-reactive protein. Clin Exp Immunol. 1985;59:243–250. [PMC free article] [PubMed] [Google Scholar]
- 38.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 39.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukocyte Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutterwala FS, Noel GJ, Salgame P, Mosser DM. Reversal of proinflammatory responses by ligating the macrophage Fcγ receptor type I. J Exp Med. 1998;188:217–222. doi: 10.1084/jem.188.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boekhoudt GH, Frazier-Jessen MR, Feldman GM. Immune complexes suppress IFN- signaling by activation of the FcγRI pathway. J Leukocyte Biol. 2007;81:1086–1092. doi: 10.1189/jlb.0906543. [DOI] [PubMed] [Google Scholar]
- 42.Barnes N, Gavin AL, Tan PS, Mottram P, Koentgen F, Hogarth PM. FcγRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity. 2002;16:379–389. doi: 10.1016/s1074-7613(02)00287-x. [DOI] [PubMed] [Google Scholar]