

Abstract
Several S. cerevisiae deletion strains involving heat-shock response factors were among the most sensitive mutants identified in a previous genetic screen for doxorubicin hypersensitivity. These strains included ydj1Δ, ssz1Δ and zuo1Δ mutants. In addition, new1Δ, whose function was unknown, also displayed significant sensitivity to anthracyclines. We further investigated the basis for the sensitivity of these mutants. We determined that heat-shock could partially rescue the sensitivity of the strains to doxorubicin, including the homologous recombination mutant rad52Δ, which is sensitive to doxorubicin-mediated DNA double strand breaks (DSBs). However, none of the heat-shock response mutants were sensitive to DSBs, but were highly sensitive to reactive oxygen species (ROS) generated by quinone-ring-containing agents, such as anthracyclines and menadione. A fluorescent-based assay indicates that doxorubicin causes protein aggregation. Interestingly, the disaggregase mutant hsp104Δ is not sensitive to anthracyclines or menadione suggesting that Hsp104p does not play a role in disaggregating doxorubicin-induced protein aggregates. However New1p, which has been recently shown to be a novel disaggregase, is essential for cell viability after exposure to anthracyclines and menadione and it is not involved in thermotolerance. Our data suggest that in S. cerevisiae, doxorubicin produces protein aggregation through ROS and requires Ydj1p and New1p for resolution.
Keywords: Anthracyclines, Heat-Shock Response, Oxidative Stress, Molecular Chaperones, Cancer
Introduction
Anthracycline antibiotics are commonly used chemotherapeutic drugs, which are effective in a wide range of hematologic malignancies and solid tumors [1, 2]. In the absence of targets for biological therapy, such as in triple negative breast cancer, cytotoxic chemotherapy may provide the only therapeutic alternative [3]. The mechanism by which anthracyclines exert their therapeutic action has not been completely elucidated. The inhibition of DNA topoisomerase II by anthracyclines, which results in the perpetuation of DNA double-stranded breaks (DSBs) and leads to apoptosis, is a major contributor to their anticancer activity [2, 4]. The aglycone moiety of anthracyclines [2], which contains a ring which cycles between the quinone and the quinol [5], is responsible for the generation of reactive oxygen species (ROS) which damage cellular structures, and has also been proposed to mediate anthracyclines activity [2, 6]. The effectiveness of anthracyclines is dose-dependent, however the development of side effects from cumulative exposure to the drug can limit its use. Of particular concern is the development of chronic cardiomyopathy [7, 8]. Anthracycline toxicity results in apoptosis of the cardiomyocyte, leading to dilated cardiomyopathy which results in congestive heart failure. This condition is refractory to therapeutic treatment and can only be treated by heart transplantation [9]. It is proposed that anthracycline-induced ROS is responsible for damage to the cardiac mitochondria [10], and has been partially managed by the reduction of the cellular load of ROS using the iron chelator dexrazoxane [6]. In addition, the development of drug resistance is a significant concern, and commonly encountered during anthracycline-based chemotherapy. Primary resistance due to the overexpression of P-glycoproteins cannot be overcome by increasing the drug dose due to the increased risk of side effects [11] [12–14].
To investigate the mechanism that protect cells from anthracyclines we carried out a genome-wide genetic screening in S. cerevisiae to identify mutant strains that display hypersensitivity to doxorubicin. A total of 71 deletion strains displayed varying levels of sensitivity to anthracyclines [15]. Of these, several factors involved in the heat-shock response (HSR) were identified, including the heat shock protein 40 (HSP40) Ydj1p and Zuo1p, the HSP70 Ssz1p and New1p, until then of unknown function. The HSR is activated by proteotoxic stress which leads to the release of transcription factor Hsf1 from an inactive complex containing HSP40/HSP70/HSP90 and its translocation into the nucleus where it induces the expression of other HSPs, also referred to as chaperones [16, 17]. The HSR is a complex series of reactions, which restores the native structure of unfolded peptides. HSP40s play a crucial role by binding unfolded peptide, presenting them to the HSP70s and stimulating their ATPase activity to promote refolding [18, 19]. Failure to refold denatured peptides can lead to the formation of protein aggregates, which require the function of a disaggregase, such as Hsp104p, in S. cerevisiae [20, 21].
There is growing evidence on the significance of the HSR in cancer and currently, the potential of targeting cancer cells with HSP90 inhibitors is an extensively investigated field [22–24]. In addition, despite knowledge of anthracyclines producing oxidative stress, which can potentially damage proteins, little is known about the consequences of the oxidative damage and the factors involved in protecting the cell.
In this study we investigate the basis for the sensitivity of the HSR mutants. We determined that heat-shock is protective to yeast cells. Using a fluorescent-based assay we determine that doxorubicin causes protein aggregation. However, the activity of the HSP104p disaggregase is not essential for protection from doxorubicin-induced toxicity, and that function may be performed by New1p, which has been shown to possess disaggregase activity [25]. We also confirm a role for Ssz1p and Zuo1p in drug resistance, a function previously described [26]. All HSR mutants were sensitive to the ROS-generating agent menadione and not sensitive to DSBs. Our data suggest that, in S. cerevisiae, doxorubicin produces protein aggregation through ROS and requires Ydj1p and New1p for resolution. These results may provide novel leads for addressing both tumor hypersensitization and side-effects reduction in regimens including anthracyclines.
Materials and Methods
General Genetic Methods
YPD (1% yeast extract, 2% peptone, 2% dextrose, 2% agar) and synthetic complete (SC, 0.67% yeast nitrogen base without amino acid, 0.087% amino acid mixture, 2% dextrose, 2% agar) media or the corresponding drop-out media (SD), were as described [27] [28].
Strains
Homozygous haploid deletion strains library (Parental strain BY4741: MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was obtained from Dharmacon, Inc. (Pittsburgh, PA). Gene-disrupted strains were constructed using a 3-step PCR method using DNA cassettes containing exogenic regions of the gene of interest (i.e. SSA1, SSA2), flanking a selective marker (i.e. HIS3, TRP1, URA3). These disruption cassettes were integrated into the genomes by homologous recombination. Double knockout mutants were obtained by sequential gene disruption using different selective markers. All strains were confirmed by PCR.
Chemicals
Doxorubicin-HCl and daunorubicin were obtained from Bedford Laboratories (Eatontown, NJ, USA); menadione sodium bisulfite was purchased from Sigma-Aldrich (St Louis, MO, USA); and etoposide was obtained from Enzo Life Sciences (Farmingdale, NY).
Sensitivity of Strains to Chemotherapeutic Agents and Cytotoxic Stressors
The concentration of the drugs used for strain exposure was as previously described [15]. Briefly, single colonies were grown overnight in liquid YPD media, at 30°C with shaking. Cells were then washed and re-suspended in ultra-pure sterile water. Strains were then separated into control and treatment groups. Ten-fold serial dilutions were spotted onto YPD agar plates containing the drugs as indicated for each experiment. Doxorubicin plates contained 20 μM of the drug; daunorubicin contained 10 μM and etoposide contained 1 mM of the drug. Exposure to menadione (6.6 mM) and MNNG (3 μM) was done for 1 hour in cells resuspended in sterile water, followed by washing, serial dilutions and plating onto YPD agar plates. All plates were incubated at 30°C for the indicated time. Heat shock treatment was performed by plating serial dilutions of the strains and incubated at 37°C. Cell growth was monitored daily and colonies were counted at day 3. Survival was calculated relative to the corresponding untreated control and sensitivity was determined relative to the survival of the wild type strain.
Thermotolerance
Cells were grown to exponential phase in YPD at 30°C and switched to 39°C to induce the heat-shock response. Aliquots were transferred to 50°C and incubated for the indicated time, then placed on ice before spotting. Serial dilutions (5-fold) were spotted onto YPD plates and incubated at 30°C. Cell growth was monitored daily and colonies were counted at day 3.
Protein Aggregation and Fluorescent Microscopy: The assay is based on a firefly luciferase-GFP fusion. The GFP chromophore is resistant to denaturation while the luciferase component becomes denatured and aggregates, allowing the monitoring of GFP fluorescence in the cell. Aggregation will result in uneven distribution of the GFP. Plasmid pFFL-GFP (gift of Dr. John Glover, U. of Toronto) was transformed into wild type cells and selected in leu− drop-out plates. These cells were grown on liquid SD-leu− media, washed, resuspended in sterile water and exposed to doxorubicin (300 μM) for 30 min. Cells were washed and prepared as described in spot assay. Cells (5 μl) were immobilized on a glass slide pre-coated with 0.1% poly-L-lysine solutions (Sigma) covered and sealed with Cytoseal™ XYL mounting medium (Richard-Allan Scientific). Cells were viewed under a Zeiss Axioplan 2 imaging microscope (Carl Zeiss, Thornwood, NY) with a water-immersion Achroplan 63X/0.9W/DIC III objective. The illumination source was a 100-W mercury arc lamp. Cell images were taken using an AxioCam HRm digital camera operated via AxioVision 4.5 software. Confocal images were captured with a LSM 510 META system operated via META 3.2 software. The wavelengths of the filters used to visualize the FFL-GFP were: excitation 488 nm; emission 509 nm. Unless otherwise noted, all experiments were performed at room temperature.
Results
Heat-shock rescues the sensitivity of yeast strains to doxorubicin
To determine the effect of the heat-shock response in the sensitivity of yeast cells to doxorubicin, we determined the survival of selected strains when exposed to doxorubicin under heat-shock conditions. As observed in figure 1, at the concentration of doxorubicin tested and in the absence of heat-shock (30°C, Fig 1, top panels), wild type cells displayed approximately a 10-fold reduction of viability, while rad52Δ mutant was completely inviable. The co-chaperone defective ydj1Δ strain displayed a ~10,000-fold reduced viability. We also tested ssa1Δ, which is defective in an HSP70 normally associated to Ydj1p, displaying a 100-fold reduction in viability after exposure to doxorubicin. When the mutant strains were exposed to doxorubicin under heat-shock conditions (37°C, Fig. 1, bottom panels), a significant increase in viability was observed. The doxorubicin-exposed wild type strain showed no difference with untreated control (Fig 1), while the viability of rad52Δ, was increased from inviable to a 1,000-fold more sensitive than untreated (Fig 1). Interestingly, ssa1Δ was almost completely rescued, which contrasts with the inviability of the ydj1Δ strain (Fig 1). This result suggests that Ssa1p is not the exclusive HSP70 of Ydj1p, and other HSP70s may play a redundant role. We generated and tested the ssa1Δ ssa2Δ, ssa1Δ ssa3Δ and ssa2Δ ssa3Δ double mutants, as well as the ssa1Δ ssa2Δ ssa3Δ triple mutant, but failed to see a hypersensitive phenotype for doxorubicin and heat-shock sensitivity (data not shown).
Figure 1.

Heat-shock rescues strains sensitivity to doxorubicin. Serial dilutions of selected strains were plated onto YPD with or without doxorubicin, as described in the Materials and Methods. The plates were incubated at either 30°C (normal temperature) or 37°C (heat-shock). Growth was scored after 3 days of incubation. The result of a representative experiment is presented.
Doxorubicin exposure leads to protein aggregation in yeast
Since one of the functions of Ydj1p is to prevent protein aggregation, we tested if doxorubicin was capable of inducing protein aggregation. We transformed wild type cells with a luciferase-GFP fusion construct (pFFL-GFP) that has been used to monitor and study protein aggregation in vivo [29]. Under non-stressed conditions (− Doxo, Fig 2b) GFP is homogenously distributed throughout the cell. However, when wild type cells were exposed to doxorubicin, considerable aggregation was observed, as indicated by the uneven distribution of the GFP fluorescence, which appears punctuate rather than homogenous (+ Doxo, Fig 2b). This aggregation was not a consequence of cell dying from doxorubicin toxicity, since the concentration/exposure time used did not affect viability as indicated in the survival spot assay (Fig 2a, triplicate experiment shown).
Figure 2.

Doxorubicin exposure leads to protein aggregation. The evaluation of doxorubicin-induced protein aggregation was performed in wild type strain using a model substrate of firefly luciferase (FFL) fused to green fluorescent protein (GFP) as described in the Materials and Methods. a) Serial dilutions of the control (− Doxo) and doxorubicin-exposed cells (+ Doxo) were plated onto Leu-drop out plates and incubated at 30°C to determine growth. b) Protein aggregation as determined by aggregation of the FFL-GFP fusion reporter. GFP and DIC images are presented for doxorubicin treated (− Doxo) and control (− Doxo) wild type cells.
NEW1 is not required for thermotolerance
The observation that new1Δ mutants are sensitive to doxorubicin, and the description of its disaggregase activity, we tested if NEW1 was required for thermotolerance. While the hsp104Δ strain was sensitive to heat-shock, the new1Δ strain was not affected, displaying similar survival to the wild type strain (Fig 3). This indicates that New1p is not required for disaggregation of heat-shock aggregates.
Figure 3.

HSP104 is not essential for protection of cells to doxorubicin. Cells were grown to exponential phase in YPD at 30°C and switched to 39°C to induce the heat-shock response. Aliquots were transferred to 50°C and incubated for the indicated time, then placed on ice before spotting. Serial dilutions (5-fold) were spotted onto YPD plates and incubated at 30°C. Cell growth was monitored daily and colonies were counted at day 3.
Heat-shock defective strains are not sensitive to DNA double-strand breaks
Doxorubicin mechanism of action involves the inhibition of topoisomerase II, which leads to the stabilization of DNA double-strand breaks (DSBs), as well as the generation of ROS. To investigate if the heat-shock response defective strains are sensitive to DNA DSBs we exposed ydj1Δ, ssz1Δ, zuo1Δ, hsp104Δ and new1Δ mutants to the topo II inhibitor etoposide. Unlike doxorubicin, etoposide lacks the quinone ring responsible for the generation of ROS, thus only generates DSBs. The homologous recombination mutant strain rad52Δ was used as a positive control. As shown in figure 4, none of the heat-shock mutant strains were affected by etoposide, while rad52Δ was sensitive, as expected. This data suggest that the heat-shock strains are not sensitive to the DSBs-inducing activity of anthracyclines. We further tested the sensitivity of the strains to other DNA damaging agents. While new1Δ was unaffected by ionizing radiation, zuo1Δ and ydj1Δ mutants were partially sensitive and ssz1Δ was highly sensitive (Table 1). DNA alkylating agent MNNG reduced the viability of zuo1Δ by 5-fold and ssz1Δ by 10-fold, while it did not affect ydj1Δ or new1 mutants (Table 1). Similarly, topoisomerase I inhibitor reduced the viability of zuo1Δ by 5-fold and ydj1Δ and ssz1Δ mutants by 10-fold (Table 1). As expected, all strains were sensitive to the anthracycline daunorubicin (Table 1).
Figure 4.

Heat-shock defective strains are not sensitive to DNA double-strand breaks generated by doxorubicin. Serial dilutions of heat-shock response mutants were plated onto etoposide-containing plates as described in the Materials and Methods. The rad52Δ sensitive strain was used as the control. Growth was scored after 3 days of incubation at 30°C. Sensitivity was assessed by comparison to growth of the strains on plates without etoposide.
Table 1.
Sensitivity of the heat shock response mutants to cytotoxic agents.
| Strain | Growth | Sensitivity (fold)c | |||
|---|---|---|---|---|---|
| Irradiationa (50 Gy) |
Heatb (37°C) |
MNNG (3 μM) |
Camptothecin (10 μM) |
Daunorubicin (10 μM) |
|
| zuo1Δ | + | 5 | 5 | 1,000 | |
| ydj1Δ | + | S | 10 | 10,000 | |
| new1Δ | ++ | 1,000 | |||
| ssz1Δ | − | 10 | 10 | 1,000 | |
Growth after irradiation was determined relative to wild type cells. Strains were classified as: not sensitive: +++ (≥ 50% survival), slightly sensitive: ++ (10–50% survival), sensitive: + (1–10%) and hypersensitive: - (<1%). Survival of wild type cells was 50% after irradiation.
Heat sensitive strains did not grow when patched onto YPD plates and incubated at 37°C for 3 days (indicated by an S).
Sensitivity to MNNG, camptothecin and daunorubicin was calculated by determining cell survival and is expressed as fold reduction in viability. Survival of the wild type strain was >90% at the concentrations of drugs used.
Strains defective in the heat-shock response are sensitive to agents that generate oxidative stress
Doxorubicin is capable of generating ROS through its quinone ring. To determine if the heat-shock defective strains are sensitive to ROS, we determined their viability after exposure to menadione, a commonly used ROS generating agent, which shares rings C and D of the aglycone moiety of the anthracyclines [2], containing the quinone ring. As shown in figure 5, ydj1Δ, new1Δ, ssz1Δ, and zuo1Δ mutants were highly sensitive to menadione, with a reduction in survival equivalent to that resulting from doxorubicin treatment. The positive control sod1Δ, which is defective in superoxide dismutase, is also highly sensitive as expected. Under the conditions tested, wild type cells presented a 5-fold reduction in viability for doxorubicin and a 10-fold reduction in viability for menadione. The hsp104Δ mutant was not considerably affected displaying a 10-fold reduction in viability for both agents, similar to those of the wild type strain.
Figure 5.
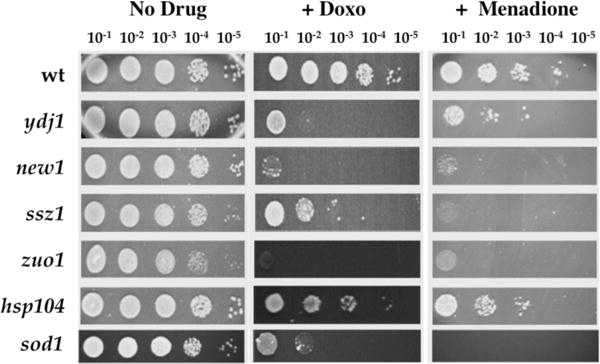
Strains defective in the heat-shock response are sensitive to agents that generate oxidative stress. Serial dilutions of heat-shock response mutants exposed to menadione were plated onto non-selective plates as described in the Materials and Methods. The sod1Δ sensitive strain was used as the control. Growth was scored after 3 days of incubation at 30°C, and sensitivity was assessed by comparison to the growth of the untreated cells (No Drug) and to cells grown on plates containing doxorubicin (+ Doxo).
Discussion
The results presented in this paper describe the basis for the sensitivity of the heat-shock defective strains to chemotherapeutic agent doxorubicin. It also provides evidence for doxorubicin-induced protein aggregation, as a consequence of the generation of reactive oxygen species.
We had previously identified ssz1Δ, zuo1Δ and ydj1Δ mutants in a genome-wide screen for strains that are hypersensitive to doxorubicin [15]. The role of the heat-shock response (HSR) in protecting cells from doxorubicin was confirmed by the rescue of the mutant strains sensitivity at high temperature. This effect is not limited to HSR factors, but it can also rescue homologous recombination mutant rad52Δ and the wild type strain. As expected, the ydj1Δ mutant which is sensitive to high temperatures was inviable, indicating a crucial role of Ydj1p in the survival response to doxorubicin.
Ydj1p has also been shown to participate in protein disaggregation, as co-chaperone of the Hsp104p disaggregase. In fact, we found that exposure of yeast cells to doxorubicin resulted in protein aggregation, as determined by a firefly luciferase-GFP fusion reporter. The absence of a requirement for HSP104 in the response to doxorubicin exposure was intriguing, considering the observation of doxorubicin-induced protein aggregation. However, a strain defective in NEW1, an ATP binding cassette protein proposed to be a prion in S. cerevisiae [30], presented high sensitivity to doxorubicin. New1p, which has been shown to be capable of dispersing Sup35 amyloid fibers, in fact acting as a disaggregase [25], may be a good candidate to perform this function in doxorubicin-induced protein aggregates. Interestingly, this observation suggests that cells can effectively distinguish between heat-induced protein aggregates and those generated by other types of stresses. In fact, oxidative damage to protein can result in various modifications, including carbonyl derivatives, oxidized groups and protein fragmentation [31] not occurring in heat-denatured proteins. While there is substantial evidence of a partnership between Ydj1p and HSP104p in the disaggregation of heat-denatured proteins, we do not know at this time, if YDJ1p and NEW1p work in conjunction in doxorubicin-induced protein aggregates. Similarly, the requirement for an HSP70 in this process is not clear. Single, double and triple deletions strains of ssa1Δ, ssa2Δ and ssa3Δ were constructed, tested and found to be not hypersensitive to anthracyclines.
Anthracyclines are complex drugs that exert different effects in the exposed cells. Two well-known effects are the generation of DSBs through the inhibition of DNA topoisomerase II and the generation of ROS, mediated by the anthracycline quinone ring. To investigate which of these injuries resulted in reduced viability of the HSR mutants, we exposed the strains to agents which exclusively caused DSBs by inhibition of topoisomerase II (etoposide), or generated ROS through a quinone ring identical to that present in anthracyclines (menadione). The HSR mutants did not display significant sensitivity to etoposide, or to other types of DNA damaging agents, such as alkylating agent MNNG and the topoisomerase I inhibitor camptothecin (a 5 to 10-fold increase). However, ssz1Δ, ydj1Δ and zuo1Δ mutants did display sensitivity to ionizing radiation. Considering that the majority of the damage to cellular components caused by ionizing radiation is due to the generation of ROS [32] [33], is possible that, upon irradiation, cellular proteins suffer oxidative damage similar to that generated by anthracyclines. The requirement for these HSR factors in the response to oxidative stress is observed when the deletion strains are exposed to menadione, a quinone ring-containing compound that closely resembles rings C and D of anthracyclines [2], responsible for the generation of ROS.
Interestingly, Ssz1p and Zuo1p have previously been shown to activate pleiotropic drug resistance [26]. This function is distinct from their role as endoplasmic reticulum (ER) chaperones, and is independent from ribosome binding [26], consistent with a response needed in different cellular compartments not limited to the ER. The ssz1Δ and zuo1Δ mutant strains were also sensitive to menadione, suggesting that they also participate in the response to ROS-induced protein denaturation.
Significant effort is currently devoted to reducing side effects of cytotoxic chemotherapy. For anthracyclines, chronic cardiomyopathy is the most serious undesired effect resulting from cumulative toxicity and limits their therapeutic use. Attempts to redesign the drug to analogs with lower toxicity results in compounds with lower therapeutic efficacy [2] [34]. In the search for alternatives to improve cytotoxic chemotherapy, the option of sensitizing tumors is an attractive one. In the case of anthracyclines, in addition to increasing efficacy, the use of lower doses would concomitantly result in reduced side effects to sensitive tissue such as the heart. Modulating the HSR for this purpose may be an option, as well. In fact, the HSR is already a therapeutic target in cancer chemotherapy [22–24]. This may be accomplished by activating the HSR to protect normal tissue from the toxicity of anthracyclines. Currently, the accepted option for reducing the risk of cardiotoxicity involves the use of dexrazoxane, an iron chelator intended to reduce the load of ROS by blocking Fe-dependent ROS formation [35].
We are currently devoting significant effort to identify the mammalian counterparts of the yeast genes. If found, they will provide potential new targets for tumor sensitization to chemotherapy as well as a potential alternative for modulating the response of normal tissue to the cytotoxicity of ROS-generating agents, such as anthracyclines.
Conclusion
Exposure to doxorubicin in the yeast S. cerevisiae results in increased reactive oxygen species that lead to protein aggregation. The heat-shock response proteins Ydj1p, Ssz1p and Zuo1p are critical for the survival to the anthracycline, while the disaggregase Hsp104p is not. This role appears to be performed by the novel disaggregase New1p.
Acknowledgments
The authors are grateful for the support provided by the Gene and Cell Manipulation Facility of Florida A&M University. This project was supported by the National Center for Research Resources and the National Institute of Minority Health and Health Disparities of the National Institutes of Health through Grant G12MD007582 and P20MD006738.
Footnotes
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Piekarski M, Jelinska A. Anthracyclines still prove effective in anticancer therapy. Mini Rev Med Chem. 2013;13(5):627–34. doi: 10.2174/1389557511313050001. [DOI] [PubMed] [Google Scholar]
- 2.Minotti G, et al. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 3.von Minckwitz G, Martin M. Neoadjuvant treatments for triple-negative breast cancer (TNBC) Ann Oncol. 2012;23(Suppl 6):vi35–9. doi: 10.1093/annonc/mds193. [DOI] [PubMed] [Google Scholar]
- 4.Dal Ben D, et al. DNA topoisomerase II structures and anthracycline activity: insights into ternary complex formation. Curr Pharm Des. 2007;13(27):2766–80. doi: 10.2174/138161207781757105. [DOI] [PubMed] [Google Scholar]
- 5.Cummings J, et al. The enzymology of doxorubicin quinone reduction in tumour tissue. Biochem Pharmacol. 1992;44(11):2175–83. doi: 10.1016/0006-2952(92)90344-i. [DOI] [PubMed] [Google Scholar]
- 6.Minotti G, et al. Doxorubicin cardiotoxicity and the control of iron metabolism: quinone-dependent and independent mechanisms. Methods Enzymol. 2004;378:340–61. doi: 10.1016/S0076-6879(04)78025-8. [DOI] [PubMed] [Google Scholar]
- 7.Toldo S, et al. Comparative cardiac toxicity of anthracyclines in vitro and in vivo in the mouse. PLoS One. 2013;8(3):e58421. doi: 10.1371/journal.pone.0058421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol. 2003;93(3):105–15. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 9.Lenneman AJ, et al. Heart transplant survival outcomes for adriamycin-dilated cardiomyopathy. Am J Cardiol. 2013;111(4):609–12. doi: 10.1016/j.amjcard.2012.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mordente A, et al. Anthracyclines and mitochondria. Adv Exp Med Biol. 2012;942:385–419. doi: 10.1007/978-94-007-2869-1_18. [DOI] [PubMed] [Google Scholar]
- 11.Andre F, Zielinski CC. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Ann Oncol. 2012;23(Suppl 6):vi46–51. doi: 10.1093/annonc/mds195. [DOI] [PubMed] [Google Scholar]
- 12.Abdallah HM, et al. P-glycoprotein inhibitors of natural origin as potential tumor chemosensitizers: A review. J Adv Res. 2015;6(1):45–62. doi: 10.1016/j.jare.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng J, et al. Elevated glutathione is insufficient to protect against doxorubicin-induced nuclear damage in heart in multidrug resistance associated protein 1 (Mrp1/Abcc1) null mice. J Pharmacol Exp Ther. 2015 doi: 10.1124/jpet.115.225490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Incalci M. DNA-topoisomerase inhibitors. Curr Opin Oncol. 1993;5(6):1023–8. [PubMed] [Google Scholar]
- 15.Xia L, et al. Identification of genes required for protection from doxorubicin by a genome-wide screen in Saccharomyces cerevisiae. Cancer Res. 2007;67(23):11411–8. doi: 10.1158/0008-5472.CAN-07-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol. 2011;76:91–9. doi: 10.1101/sqb.2012.76.010637. [DOI] [PubMed] [Google Scholar]
- 17.Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40(2):253–66. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11(8):579–92. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young JC. Mechanisms of the Hsp70 chaperone system. Biochem Cell Biol. 2010;88(2):291–300. doi: 10.1139/o09-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glover JR, Lum R. Remodeling of protein aggregates by Hsp104. Protein Pept Lett. 2009;16(6):587–97. doi: 10.2174/092986609788490087. [DOI] [PubMed] [Google Scholar]
- 21.Lum R, et al. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem. 2004;279(28):29139–46. doi: 10.1074/jbc.M403777200. [DOI] [PubMed] [Google Scholar]
- 22.Nagelkerke A, et al. The unfolded protein response as a target for cancer therapy. Biochim Biophys Acta. 2014;1846(2):277–84. doi: 10.1016/j.bbcan.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 23.Jego G, et al. Targeting heat shock proteins in cancer. Cancer Lett. 2013;332(2):275–85. doi: 10.1016/j.canlet.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 24.Mahalingam D, et al. Targeting HSP90 for cancer therapy. Br J Cancer. 2009;100(10):1523–9. doi: 10.1038/sj.bjc.6605066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoue Y, et al. Yeast prion protein New1 can break Sup35 amyloid fibrils into fragments in an ATP-dependent manner. Genes Cells. 2011;16(5):545–56. doi: 10.1111/j.1365-2443.2011.01510.x. [DOI] [PubMed] [Google Scholar]
- 26.Eisenman HC, Craig EA. Activation of pleiotropic drug resistance by the J-protein and Hsp70-related proteins, Zuo1 and Ssz1. Mol Microbiol. 2004;53(1):335–44. doi: 10.1111/j.1365-2958.2004.04134.x. [DOI] [PubMed] [Google Scholar]
- 27.Alani E, Reenan RA, Kolodner RD. Interaction between mismatch repair and genetic recombination in Saccharomyces cerevisiae. Genetics. 1994;137(1):19–39. doi: 10.1093/genetics/137.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tishkoff DX, et al. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell. 1997;88(2):253–63. doi: 10.1016/s0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- 29.Tkach JM, Glover JR. Nucleocytoplasmic trafficking of the molecular chaperone Hsp104 in unstressed and heat-shocked cells. Traffic. 2008;9(1):39–56. doi: 10.1111/j.1600-0854.2007.00666.x. [DOI] [PubMed] [Google Scholar]
- 30.Santoso A, et al. Molecular basis of a yeast prion species barrier. Cell. 2000;100(2):277–88. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 31.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272(33):20313–6. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 32.Szumiel I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol. 2015;91(1):1–12. doi: 10.3109/09553002.2014.934929. [DOI] [PubMed] [Google Scholar]
- 33.Havaki S, et al. The role of oxidative DNA damage in radiation induced bystander effect. Cancer Lett. 2015;356(1):43–51. doi: 10.1016/j.canlet.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 34.Monneret C. Recent developments in the field of antitumour anthracyclines. Eur J Med Chem. 2001;36(6):483–93. doi: 10.1016/s0223-5234(01)01244-2. [DOI] [PubMed] [Google Scholar]
- 35.Langer SW. Dexrazoxane for the treatment of chemotherapy-related side effects. Cancer Manag Res. 2014;6:357–63. doi: 10.2147/CMAR.S47238. [DOI] [PMC free article] [PubMed] [Google Scholar]