

SUMMARY
Organogenesis during embryonic development occurs through the differentiation of progenitor cells. This process is extraordinarily accurate, but the mechanisms ensuring high fidelity are poorly understood. Coronary vessels of the mouse heart derive from at least two progenitor pools, the sinus venosus and endocardium. We find that the ELABELA (ELA)-APJ signaling axis is only required for sinus venosus-derived progenitors. Because they do not depend on ELA-APJ, endocardial progenitors are able to expand and compensate for faulty sinus venosus development in Apj mutants, leading to normal adult heart function. An upregulation of endocardial SOX17 accompanied compensation in Apj mutants, which was also seen in Ccbe1 knockouts, indicating that the endocardium is activated in multiple cases where sinus venosus angiogenesis is stunted. Our data demonstrate that by diversifying their responsivity to growth cues, distinct coronary progenitor pools are able to compensate for each other during coronary development, thereby providing robustness to organ development.
In Brief
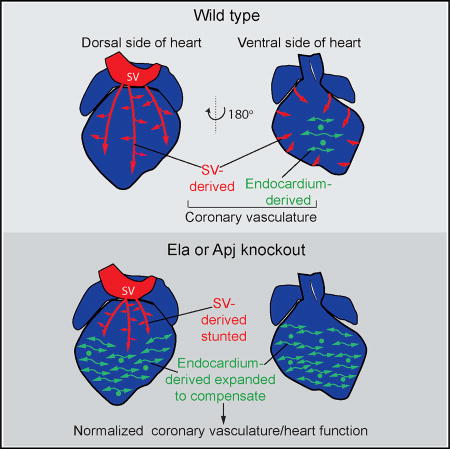
Coronary blood vessels of the heart are formed from two progenitor sources: the sinus venosus (SV) and the endocardium. Sharma et al. show that ELA-APJ is only required for growth from the SV and that endocardial-derived CVs can compensate for loss of SV-derived vessels to restore heart function, ensuring developmental robustness.
INTRODUCTION
Organogenesis is a complex process by which multiple cell types from different lineages come together to form functional tissues and organs. This process is highly invariable, only rarely giving rise to congenital malformations that compromise tissue function. How the embryo carries out organogenesis with such fidelity is not well understood, but knowledge on the underlying mechanisms could identify new avenues for reparative medicine.
One possible means by which the embryo could ensure successful development is to provide alternative pathways and/or progenitor cells that can be utilized when the original is compromised, but whether this occurs is not known. There is growing evidence that multiple organs (pancreas, liver, heart) possess more than one progenitor pool for the same cell type, and that these differentiate and contribute to mature tissues (Das and Red-Horse, 2017; Zaret, 2008). Our laboratory and others have identified dual progenitor populations for the endothelial layer of the coronary vasculature (Chen et al., 2014b; Red-Horse et al., 2010; Tian et al., 2014; Wu et al., 2012), which is the blood circulation that feeds the heart. One progenitor is the venous blood vessel located on the dorsal (or back) side of the developing heart called the sinus venosus (SV). The SV vascularizes the heart by sending out sprouts that colonize ventricular heart muscle from the outside in (Figure 1A). A second progenitor population is the endocardium, the endothelial cell layer that lines the lumen of the heart. This source contributes vessels that migrate into the heart muscle from the inside out (Figure 1A). The SV- and endocardial-derived coronary vessel populations invest complementary regions of the heart, meet in the middle, and together form a functional coronary circulation (Chen et al., 2014b; Tian et al., 2014). It is not known why the coronary vasculature and other organ systems are endowed with multiple progenitors, but studying the interactions between distinct cellular sources could lead to new strategies for organ repair and regeneration.
Figure 1. ELA-APJ Signaling Is Required for Coronary Vessel Development.

(A) Schematic of the heart showing migration paths of two different coronary vessel progenitors: (1) sinus venosus (SV) derived and (2) endocardium (endo) derived.
(B) Apj is specifically expressed in SV-derived coronary vessels as determined by ApjCreER labeling at E16.5 in the indicated regions of the heart.
(C–H) Whole-mount imaging and quantification of coronary coverage on the dorsal aspects of embryonic hearts immunostained to label endothelial cells (VE-cadherin) and cardiomyocytes (cTnT). Coronary growth is decreased in endothelial-specific Apj KOs (C and D), but increase in the absence of Apelin (E and F). Ela-deficient hearts phenocopy Apj mutants (G and H). E13.5 hearts are shown in (C), (E), and (G) where solid lines demarcate ventricle and dotted lines indicate the leading front of vascular growth. In (D), (F), and (H), dots are individual hearts and error bars are mean ± SD. ns, not significant.
(I) In situ hybridization localizing Ela transcripts show that it is expressed in the epicardium of E11.5 hearts. Adjacent sections were immunostained for the epicardial cell (epi) marker, integrin α4.
(J) Schematic showing the proposed interaction between ELA, secreted by epicardial cells, and its receptor APJ, expressed by SV-derived sprouts, during early coronary development.
cHET, conditional heterozygous; cKO, conditional knockout; liv, liver; WT, wild-type; ao, aorta; pt, pulmonary trunk.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; ns, not significant (p > 0.05). Scale bars, 200 µm (C, E, and G) and 100 µm (I). See also Figures S1–S3.
Although there is now clear evidence that the SV and endocardium give rise to the coronary vasculature (Chen et al., 2014b; Red-Horse et al., 2010; Tian et al., 2014, 2013; Wu et al., 2012; Zhang et al., 2016), there is still much to learn about the molecular signals that cause these progenitors to grow onto the heart and mature into arteries, capillaries, and veins. One emerging theme is that, despite the SV and endocardium producing similar cell types, they appear to do so through different mechanisms. SV-derived coronary vessels undergo sprouting angiogenesis (Red-Horse et al., 2010; Tian et al., 2013), whereas it has been proposed that the endocardium buds out and/or becomes trapped during compaction of the heart wall (Red-Horse et al., 2010; Tian et al., 2014). Furthermore, vascular endothelial growth factor C (VEGF-C) is required for SV sprouting but not for endocardial migration on the heart (Chen et al., 2014b), while the latter responds specifically to VEGF-A (Wu et al., 2012). There is also differential expression of angiogenic receptors between the two progenitors. The G-protein-coupled receptor, APJ, is highly expressed in the SV but not the endocardium, which has aided lineage-tracing experiments of the SV using the ApjCreER transgene (Chen et al., 2014b, 2014a). These data suggest that the distinct progenitor of the coronary vasculature could respond to vastly different developmental programs.
Recent evidence has expanded our understanding of the APJ signaling axis. It is now known to bind to two peptide hormone ligands, APELIN (APLN) and ELABELA (ELA) (Chng et al., 2013; Helker et al., 2015; Ho et al., 2015; Pauli et al., 2014). Apln is expressed by the endothelial cells themselves, and, in other organs, stimulates endothelial cell migration (del Toro et al., 2010; Papangeli et al., 2016). Ela is expressed non-cell autonomously in zebrafish to stimulate cell motility and nodal signaling during early embryogenesis (Chng et al., 2013; Deshwar et al., 2016; Pauli et al., 2014; Perez-Camps et al., 2016), and to activate angioblast migration toward the midline during vasculogenesis (Helker et al., 2015). Ela also has cardioprotective functions during heart failure (Sato et al., 2017) and is important for suppressing Pre-eclampsia during pregnancy (Ho et al., 2017). The role of ELA in mammalian development has not been reported. Given that agonists of APJ are being considered for therapeutic agents to regulate cardiovascular homeostasis in the adult (Yang et al., 2015), a full understanding of how Ela affects developing tissues and organs is an important goal.
Here, we show that the ELA-APJ signaling axis specifically regulates the SV-to-coronary migratory pathway in mice and that, in its absence, the endocardium will compensate for the lack of coronary vessels. In both Apj- and Ela-deficient mouse hearts, blood vessel migration onto regions of the heart populated by the SV is stunted, whereas regions derived from the endocardium are not. This results in region specific tissue hypoxia. Apj mutants exhibiting this phenotype survive to adulthood with normal heart function, a time when vascular density is normalized. Lineage tracing of endocardial cells in Apj mutants reveals that they dramatically expand to fill the regions affected by the decrease in SV sprouting. Finally, we present data suggesting the SV sprouts in response to developmentally timed signals while the endocardium is stimulated by the physiological cue of hypoxia. Thus, we propose that the acquisition of two developmental pathways, one genetically regulated and the other in response to microenvironmental cues, has equipped the mammalian heart with a backup mechanism ensuring a functional coronary vasculature. Given the identification of multiple progenitors in other tissues (Das and Red-Horse, 2017; Zaret, 2008), this may be a widespread mechanism providing robustness to organ development.
RESULTS
SV Sprouting Relies on ELA-APJ Signaling
To begin to understand how dynamics between the SV and endocardial progenitor cells faithfully construct the coronary vasculature, we sought to block one while keeping the other intact. Investigations into the molecular mediators of coronary development revealed that SV and endocardial sprouting onto the heart use distinct signaling pathways (Chen et al., 2014b). We found Apj (also known as apelin receptor [Aplnr]), a G-protein-coupled receptor that is involved in cardiovascular development and homeostasis (Charo et al., 2009; del Toro et al., 2010; Inui et al., 2006; Kang et al., 2013; Papangeli et al., 2016; Scott et al., 2007; Zeng et al., 2007), to be expressed specifically in the SV but not in the endocardium (Chen et al., 2014b). Remarkably, the differential expression of Apj was maintained throughout late gestation after the SV and endocardial progenitors had become coronary vessels, and after they had merged and become a single circulation consisting of arteries, capillaries, and veins. Specifically, labeling ApjCreER expressing cells at embryonic day 16.5 (E16.5) revealed its expression in vessels from SV-derived regions, but not endocardial-derived regions (Figures 1B and S1A). In contrast, coronary endothelial cells in adult hearts all expressed Apj, regardless of their origin, indicating that the vessels become phenotypically more similar as the animals age (Figure S1B). Thus, the expression pattern of Apj suggests that, during early heart development, it may play a functional role specifically in the SV-to-coronary conversion.
To determine the function of APJ in coronary development, we analyzed coronary vessels in embryonic hearts from Apj knockout (KO) animals. We performed whole-mount immunofluorescence using antibodies against endothelial cells (anti-VE-cadherin) and cardiomyocytes (anti-cardiac troponin T [cTnT]) to visualize the coronary vasculature and ventricular myocardium, respectively. Confocal images showed that the initiation of coronary sprouting from the SV at E11.5 was unaffected, but that coronary vessel growth at subsequent stages, when they progressively migrate down the dorsal side of the heart, was significantly stunted in Apj KOs (Figures S2A and S2B). Heterozygous animals were also significantly delayed indicating a haploinsufficiency for Apj (Figure S2B). Next, hearts containing an endothelial-specific deletion of Apj using a Tie2Cre-deletor line crossed to Apj floxed mice were similarly analyzed to assess whether the vessel defect was cell autonomous. Tie2Cre+;Apjfl/KO (Apjecko) hearts had significantly reduced coronary growth compared with Tie2Cre−;Apjfl/+ controls (Figures 1C, 1D, and S2C), while heterozygous animals had an intermediate phenotype (Figures 1D and S2C). In addition to delayed progression of vessel growth onto the heart, the developing coronary plexus in Apjecko animals had reduced branching in both the subepicardial (surface) and intramyocardial (deep) layers (Figures S2D and S2E), indicating defective angiogenesis. The negative effect on coronary expansion was not secondary to defective myocardial growth, as thickness of ventricular muscle was not dramatically affected in Apjecko hearts (Figure S2F). Similarly, all mutants formed coronary artery stems (orifices) by E15.5, although they were slightly delayed when compared with controls (Figure S2G). These data show that the APJ receptor is required in SV-derived endothelial cells as they migrate into the heart and form the coronary vasculature.
We next aimed to identify the possible endogenous stimuli responsible for APJ-mediated SV angiogenesis. APJ can be activated upon binding peptide ligands (del Toro et al., 2010; Inui et al., 2006; Kang et al., 2013; Papangeli et al., 2016; Scott et al., 2007; Zeng et al., 2007) or through a ligand-independent stretch activated pathway (Scimia et al., 2012). One of APJ’s ligands, APELIN (APLN), is expressed in sprouting coronary endothelial cells (Red-Horse et al., 2010; Tian et al., 2013), but Apelin KO animals did not phenocopy the coronary defect seen in Apj KOs (Figures 1E and 1F). In fact, Apelin-deficient hearts displayed a phenotype opposite to that in Apj mutants with an increase in coronary growth so that the heart was fully covered at developmental time points earlier than wild-type controls (Figures 1E and 1F). ELABELA (ELA, also known as APELA) is a recently identified early APJ endogenous ligand that promotes endoderm differentiation and stimulates migration of angioblasts during vasculogenesis (Chng et al., 2013; Helker et al., 2015; Ho et al., 2015; Pauli et al., 2014). Ela-deficient hearts displayed a phenotype identical to that of Apj mutants, indicating that this endogenous peptide is the bona fide ligand that stimulates coronary growth (Figures 1G and 1H). Sections through E18.5 hearts showed that Ela also had normal myocardial thickness at E18.5, although the heart apexes were rounded when compared with controls (Figure S2H). In situ hybridization showed that Ela was robustly expressed by the surface mesothelium (epicardium) of developing hearts, the location where SV sprouts initially grow (Figures 1I and S3). The opposing phenotypes in Apln and Ela KOs indicate that these two ligands may have opposing functions vis-a-vis APJ in the context of coronary vessel formation. ELA appears to be the endogenous agonist for APJ since they exhibit the same phenotype, suggesting that APLN may function to dampen and/or balance ELAAPJ signaling. Taken together, these data demonstrate that APJ signaling activated by ELA binding stimulates SV-derived coronary vessel development (Figure 1J).
Endocardial Sprouting Does Not Require ELA-APJ
The next question was to ascertain whether ELA-APJ signaling was required in both coronary progenitor populations or specifically in those from the SV. Previous lineage-tracing experiments from our laboratory and others have shown that SV- and endocardial-derived vessels normally colonize distinct regions of the heart (Chen et al., 2014b; Tian et al., 2014). This compartmentalization is readily visualized on the ventral (front) aspect of the developing heart where the vessels in the lateral regions derive largely from the SV and the central portions from the endocardium (Figures 1A and S4A). To assess an SV versus endocardial requirement, we analyzed coronary vessels in both of these areas in Apjecko hearts. As expected, the SV-derived region displayed absent or poorly developed coronary vessels in mutants compared with wild-type controls (Figure 2A). In contrast, coronary vessels in the endocardial-derived region were normal in Apjecko hearts (Figure 2A). Identical results were obtained in Ela KO hearts (Figure 2B). As a functional readout of vessel function, we assessed tissue hypoxia using Hypoxyprobe. Injection of Hypoxyprobe into Apj KO animals at E15.5 revealed robust labeling of SV-derived regions compared with littermate controls while endocardial-derived regions were less affected (Figures 2C and 2D). These data suggest that the ELA-APJ signaling axis functions solely in SV-derived coronary progenitors to stimulate their migration onto the heart so that they can fully oxygenate developing cardiac muscle.
Figure 2. ELA-APJ Signaling Is Specifically Required for SV-Derived Coronary Growth.

(A and B) Whole-mount immunostaining of the ventral aspect of E15.5 Apj- and Ela-deficient hearts immunolabeled for VE-cadherin (endothelial cells) and cTnT (cardiomyocytes) show that vessels are specifically affected in regions derived from the sinus venosus (SV-derived, box 1), but not the endocardium (Endo-derived, box 2). Boxed regions show high magnification views of the vasculature. Blood islands (arrowheads) replace the branched vessels normally found at Apj- and Ela-dependent SV-derived regions.
(C and D) Hypoxyprobe labeling (C) and quantification (D) in wild-type (WT) and Apj KO hearts show a functional deficiency of coronary vessels (tissue hypoxia) that is more pronounced in SV-derived regions. In (D), dots represent fields of view (FOV); error bars are mean ± SD.
(E) Depletion of Apj specifically in the SV using ApjCreER decreases vascular coverage on the dorsal side of the heart.
(F) Depletion of Apj specifically in the endocardium using Nfatc1Cre does not affect coronary vessel density where endocardial (EC)-derived vessels predominate. Dots are individual hearts.
(G) Ectopic blood islands found in SV-derived regions of Apj-deficient hearts (arrowheads in A) are lineage labeled by Nfatc1Cre; RosamTmG and localize near hypoxic cells. Dach1 is a nuclear marker for coronary endothelial cells.
**p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; ns, not significant (p > 0.05). Scale bars, 200 µm (A–C) and 20 µm (G). See also Figure S4.
To confirm specificity for SV-derived angiogenesis, we deleted Apj in either the SV or endocardial compartment using ApjCreER (Chen et al., 2014b) or Nfatc1Cre (Wu et al., 2012) deletor lines, respectively. Resulting hearts were then analyzed for coronary vascular growth in the relevant regions of the heart (Figures 1A and S4A). As expected, ApjCreER;Apjfl/ko mice displayed a significant delay in coronary growth on the dorsal aspect of the heart (Figure 2E). In addition, hearts from both total heterozygous and conditional heterozygous (one copy deleted only in Cre-expressing cells) displayed an intermediate phenotype (Figure 2E). Consistent with regional defects in Apjecko (using the Tie2Cre deletor), deletion of Apj from the endocardium using Nfatc1Cre did not compromise coronary growth onto the ventral aspect of the heart to where endocardial-derived vessels migrate (Figures 2F and S4B). Thus, ELA-APJ signaling functions specifically to drive coronary angiogenesis from the SV but not the endocardium.
Analysis of coronary vessels in the various Apj and Ela mutants revealed abnormal vascular structures in SV-derived regions that were indicative of endocardial angiogenesis. During normal coronary vascularization, a number of endocardial progenitors arrive on the heart wall through the process of budding, which initially forms round structures termed blood islands (Hutchins et al., 1988; Red-Horse et al., 2010; Yzaguirre et al., 2015). Blood islands are normally found on the central portion of the ventral heart where endocardial migration is most pronounced. However, these blood islands were abundant in the peripheral regions of the ventral aspect of Apjecko, Ela KO, and Apj KO hearts where they were deficient in SV-derived vessels (Figures 2A and 2B, arrowheads and data not shown). To test whether the blood island-like structures at the periphery in Apj-deficient mice were endocardially derived, we lineage traced the endocardium in Apj KO hearts by crossing this line with Nfatc1Cre and the RosamTmG reporter allele, which labels endocardial-derived vessels. Indeed, these blood island structures at the periphery were lineage labeled, indicating the ectopic migration of endocardial cells when SV-derived vessels are absent (Figure 2G). Furthermore, the ectopic blood islands were prominent in the most hypoxic regions (Figure 2G). This observation implied that endocardial-derived coronary vessels could expand into SV-derived regions in response to hypoxia to compensate for local delays in coronary growth.
Embryonic Defects Are Recovered in Adult Hearts
Given the specific effects on SV-derived coronary development, Apj mutants provided a model with which to test how the developing heart responds to defects in a single progenitor source. Analyzing the number of mutants recovered in Apjecko crosses revealed no or only very minimal lethality at various stages up to weaning age (Figure 3A), despite the severe functional defects during development (Figures 2A and 2C). In fact, analyzing vascular density in regions populated by the SV (outer ventricle wall) showed that the significant decrease in endothelial cell number seen in late gestation was corrected in adult stages (Figure 3B). Accordingly, we found measures of cardiac function to be normal in Apj KO animals (Figure 3C). Mean ejection fraction (Figure 3D), mean fractional shortening (Figure 3E), left ventricular internal diameter at diastole (LVIDd), (Figure 3F), and left ventricular internal diameter at systole (LVIDs) (Figure 3G) were not significantly different between wild-type and KO when measured at 6–10 weeks. These data suggest that the developmental coronary defect in Apj KO animals is normalized during postnatal growth.
Figure 3. Coronary Vessel Defects in Apj-Deficient Embryos Are Recovered in Adults.

(A) Absence of lethality with endothelial-specific deletion of Apj (Apjecko).
(B) Intramyocardial coronary vessel density in the outer wall of the left ventricle is decreased in Apjecko at E16.5, but not different in adult hearts, indicating recovery. Dots are individual fields of view (FOV) (n = 6 hearts/genotype), and error bars are mean ± SD.
(C–G) Echocardiography (C) reveal normal heart function in adult Apjecko animals. Measurements of mean ejection fraction (D), fractional shortening (E), left ventricular internal diameter at diastole (LVIDd) (F), and left ventricular internal diameter at systole (LVIDs) (G) are not significantly different among the genotypes.
ESD, end-systolic diameter; EDD, end-diastolic diameter; HET, heterozygous;WT, wild-type. Dots are individual animals. ***p≤0.001; ns, not significant (p > 0.05).
The Endocardium Compensates for Disrupted SV Angiogenesis
We next tested the hypothesis that endocardial progenitor cells can compensate for the loss of SV-derived coronary vessels in Apj mutant hearts. To this end, endocardial-derived coronary vessels were lineage traced in Apj KO hearts by crossing this line to the Nfatc1Cre and RosamTmG Cre reporter alleles. In wild-type animals, Nfatc1Cre lineage-labeled cells were most numerous in the central portion of the heart where the SV contributes to very few coronary vessels (Figure 4A). In contrast, Apj mutant hearts had a greatly expanded number of endocardial-derived coronary vessels on the ventral side of the heart (Figure 4A). Heterozygous animals displayed an intermediate level of expansion, further indicating the importance of gene dosage in APJ signaling (Figure 4A). In Apj mutant hearts, Nfatc1Cre lineage labeling extended into regions normally derived mostly from the SV in wild-type hearts (Figure 1A, boxed region). Measuring the total area occupied by Nfatc1Cre lineage-labeled cells on the ventral aspect showed an average value of 55% ± 9% in wild-type while heterozygous and KO values were 71% ± 9% and 83% ± 6%, respectively (Figure 4B), which represents a significant expansion. The percentage of Nfatc1Cre lineage-labeled endothelial cells in optical sections was also quantified for four different regions of the heart. The right lateral, dorsal, left lateral, and central portion of the ventral side contain differential amounts of SV-derived vessels, which are listed from highest to lowest levels of contribution (Figure 4C) (Chen et al., 2014b). Accordingly, the fold increase in percentage of Nfatc1-Cre lineage-traced vessels in KO hearts was largest in the areas of greater SV contribution. These were also the regions more affected by the absence of Apj. For example, endocardial-derived vessels increased by an average of 84% on the right lateral side, 52% on the dorsal side, and 35% on the left lateral side (Figure 4C). No increase was seen on the central portion of the ventral side where the number of SV-derived vessels is very low (Figure 4C) (Chen et al., 2014b). Therefore, endocardial compensation is progressively more prominent as the dependence on SV-derived vessels increases.
Figure 4. Endocardial Coronary Progenitor Cells Compensate for SV Angiogenesis Defects in Apj Mutant Hearts.

(A–C) Nfatc1Cre;RosamTmG lineage tracing in Apj KO hearts reveals a dose-dependent expansion of endocardial-derived vessels as levels of Apj decrease (WT > HET > KO). (A) Confocal images labeling all coronary vessels (Dach1+, red) and those lineage-labeled with Nfatc1Cre (green). (B) Quantification of vessel coverage on the ventral side of the heart by Nfatc1Cre;RosamTmG-labeled cells. Dots are individual hearts, and error bars are mean ± SD. (C) Quantifying the percentage of Nfatc1Cre lineage-labeled cells in different regions of the heart reveal that as dependence on the SV increases, the percent increase in endocardial-derived vessels from WT to KO is higher. Dots are individual fields of view (FOV) from n = 4 (WT), n = 14 (HET), and n = 5 (KO) animals. Error bars are mean ± SD.
(D) Native SV-derived (wild-type) and ectopic endocardial-derived (Apj KO) vessels in the dorsal region of the heart display different morphologies. SV, sinus venosus; endo, endocardium.
HET, heterozygous; WT, wild-type. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; ns, not significant (p > 0.05). Scale bars, 200 µm (A) and 10 µm (D). See also Figure S4.
It is clear from Apj expression that coronary vessels retain some memory of their progenitor source (Figure 1B), but whether there are additional differences is not known. We found that, in addition to Apj expression, SV- and endocardial-derived plexus vessels and capillaries display a dramatically different morphology. SV-derived vessels are more slender and elongated, whereas endocardial-derived vessels are more dilated and bulbous (Figures 4D and S4C). We found that compensating endocardial-derived vessels displayed the dilated and bulbous morphology even when invading regions normally populated mostly by the SV (Figure 4D). These differing morphologies and their localization in wild-type and KO hearts further support the expanded endocardial lineage tracing in Apj mutant animals.
SOX17 Is Upregulated in Endocardial Cells when SV Angiogenesis Is Stunted
We next aimed to discover a protein that would mark endocardial cells that were activated to undergo coronary angiogenesis, which could be used to investigate the dynamics of compensation, and whether it occurs in mutants other than Apj. One possibility was that angiogenic molecules that are expressed in coronary vessels, but not the endocardium, might be induced in a subset of endocardial cells as they transform into coronary vessels. Screening candidate molecules for which robust and specific antibodies existed identified SOX17 as an endocardial activation marker. DACH1 can be used to distinguish coronary endothelial cells from the endocardium (Chen et al., 2014b). This allowed us to ascertain that SOX17 was present in the majority of coronary endothelial cells, but was not expressed in the bulk of the endocardium (Figures 5A and S5). This selective expression was consistent with published RNA-sequencing data comparing the endocardium with coronary vessels (Zhang et al., 2016). We found that DACH1-negative endocardial cells directly adjacent to the compact myocardium at the apex of the heart expressed SOX17 (Figures 5A and S5). This is the region farthest away from the SV and the last to be invested with SV-derived coronary vessels. Furthermore, these SOX17-positive endocardial cells extended into the compact myocardium in a morphology that resembled sprouting vessels (Figures 5A and S5). Quantification of the number of SOX17-positive endocardial cells showed that it increased in areas where coronary vessel numbers were lower, i.e., at the apex (Figure 5B). These data support the notion that SOX17 is a coronary vessel marker expressed in the subset of endocardial cells activated to develop into coronary vessels.
Figure 5. SOX17 Is a Marker of Activated Endocardium that Is Expanded in Mutants with Deficient SV Sprouting.

(A) Immunostaining to label VE-cadherin, DACH1, and SOX17 in E13.5 hearts. DACH1 and SOX17 are widely expressed in coronary vessels (CV); SOX17 is also expressed in a subset of endocardial cells near the compact myocardium at the apex where they produce coronary vessels (activated endocardium).
(B) Quantification of the number of coronary (DACH1+, SOX17+) and activated endocardial (DACH1−, SOX17+) cells in the indicated regions of the heart (n = 6 hearts). Error bars are mean ± SD.
(C–F) SOX17 immunofluorescence in Apj (C and D) and Ccbe1 mutants (E and F) show that activated endocardium is expanded where SV-derived coronary vessels are absent. Arrowheads indicate the leading front of coronary migration. Quantifications shown in (D) and (F) where error bars represent SDs. Apj (n = 4 wild-type, n = 5 KO hearts); Ccbe1 (n = 3 wild-type, n = 4 KO hearts).
Endo, endocardium; Sep, septum; Act. Endo, activated endocardium; RV, right ventricle; CV, coronary vessels; FOV, field of view; WT, wild-type.
*p ≤ 0.05, **p ≤ 0.01. Scale bars, 200 µm. See also Figures S5 and S6.
We next used this marker to analyze the extent of endocardial activation in mutants with stunted SV-derived coronary sprouting. In Apj mutants, SOX17 was upregulated in regions of the heart wall that lacked SV-derived coronary vessels and where the endocardium compensates (Figures 5C and 5D). Previous data have established that VEGF-C, like ELA/APJ, is specifically required for SV sprouting (Chen et al., 2014b). We investigated SOX17 expression in mice lacking a VEGF-C pathway member, CCBE1, which is required for its function (Bui et al., 2016; Hogan et al., 2009). Ccbe1 KO mice displayed the same coronary phenotype as Vegfc mutants (Figure S6), and SOX17 was increased in coronary-deficient regions (Figures 5E and 5F). Notably, the upregulation was not activated throughout the entire endocardium, but instead was localized to areas that lacked SV sprouts (Figures 5C–5F). These data show that endocardial cell activation toward a coronary phenotype is increased in multiple mutants (Apj and Ccbe1 KOs) lacking proper SV-derived coronary sprouting.
Hypoxia Is Differentially Associated with SV and Endocardial Coronary Sprouts
Hypoxyprobe labeling experiments showed that compensating endocardial cells bud into highly hypoxic regions in Apj KO hearts, suggesting that this state might trigger the response (see Figure 2G). To understand whether the signals activating compensatory growth were similar or different from normal endocardial-to-coronary development, we analyzed Hypoxyprobe labeling and hypoxia-inducible factor 1α (HIF-1α) localization in wild-type hearts. The results showed that regions of initial SV sprouting onto the heart were not hypoxic (Figure 6A) and were not labeled with nuclear HIF-1α (Figures 6B and 6C). In contrast, areas where endocardial sprouts emerge (septum) were heavily hypoxic (Figure 6D) and positive for HIF-1α (Figures 6C and 6E). This is in agreement with hypoxia being associated with postnatal endocardial conversion to coronary vessels (Tian et al., 2014) and observations that the SV specifically required VEGF-C (Chen et al., 2014b) while the endocardium relies on VEGF-A (Wu et al., 2012; Zhang and Zhou, 2013). These data support a model whereby initial outgrowth of the SV onto the heart is a developmentally timed event, while tissue expansion stimulates endocardial angiogenesis through environmental signals (Figure 6F). Thus, removal of the developmentally timed program triggers a hypoxic environment that stimulates compensation by the physiologically responsive coronary source.
Figure 6. Hypoxia Is Specifically Associated with Endocardial Sprouting.

(A, B, D, and E) Immunofluorescent confocal images labeling coronary vessels and endocardium (Endo) with VE-cadherin and hypoxic regions with either Hypoxyprobe (A and D) or HIF-1α (B and E). Hearts are E12.5. Hypoxyprobe (A) and nuclear HIF-1α (B) staining is absent on the surface of the heart where SV-derived vessels migrate. Hypoxyprobe (D) and HIF-1α (E) heavily label the septum where endocardial sprouts emerge.
(C) Quantification of HIF-1α expression in different regions of the heart (n = 5 hearts). Error bars represent SDs.
(F) A schematic summary of distinct signaling cues that guide SV- versus endocardial-derived coronary vessel growth. SV vessels are dependent on developmentally timed cues (ELA/APJ, VEGF-C) whereas endocardial vessels rely upon physiological cues (hypoxia). In Ela/Apj KO (absence of timed cues), the endocardium can compensate for defective SV vessels, possibly in response to hypoxia.
ven, ventricle; FW, free wall; WT, wild-type. ***p ≤ 0.001, ****p ≤ 0.0001. Scale bars, 50 µm.
DISCUSSION
Here we show that, in the presence of genetic changes, compensation by alternative progenitor cells is a mechanism providing fidelity to coronary development. Both heterozygous and homozygous deletions of Apj and Ela cause significant defects in coronary vessel development due to deficiencies in the migration of Apj-expressing SV-derived endothelial cells beneath the Ela-expressing epicardium. Remarkably, these defects are compensated by the endocardial progenitor population, which does not depend on ELA-APJ. Consequently, endocardial-derived coronary vessels expand their territory in Apj mutants and heart function is normal in the adult. We also find evidence of endocardial activation in compensating regions of Apj mutant hearts in that they begin expressing SOX17. Upregulation of SOX17 is also seen in Ccbe1 mutant hearts, which, similar to Apj, have defects in SV angiogenesis. This suggests that compensation is not specific to the Apj phenotype. Instead it is a general feature of the heart, which can respond to a lack of SV-derived vessels. Existing data suggest that coronary compensation occurs in other species. When avian coronary development is inhibited by epicardial ablation, ectopic connections form between the endocardium and coronary arteries (Eralp et al., 2005). Thus, there exists a dynamic interplay between two coronary vessel sources.
Embryogenesis consistently produces organs and structures of the same size and shape with few mistakes, even in outbred animals and human populations with polymorphic DNA, which involves the mechanisms of regulative development. Through these mechanisms, embryos can sense and compensate for genetic disturbances in multiple ways. For example, opposing transcriptional networks in the Xenopus embryo allow cells to sense when bone morphogenetic proteins are lacking and restore normal development by inducing anti-dorsalizing morphogenetic protein transcription (Reversade and De Robertis, 2005). Removing a fraction of cells from a tissue during development can result in an increase in divisions that compensate to restore organ size (Bort et al., 2006; de Beco et al., 2012; Sturzu et al., 2015). In this case, cells may be triggered in part by mechanical cues, which when relayed through the HIPPO pathway regulate cell proliferation and organ size (Yu et al., 2015). During Drosophila and mammalian development, genetically manipulated cells communicate their fitness level to their neighbors through secreted factors resulting in removal of the less fit “loser” population via apoptosis. This can lead to healthier adult tissues and extended lifespan in the adult (Clavería et al., 2013; de Beco et al., 2012; Meyer et al., 2014). In our system, endocardial cells appeared to sense the absence of SV-derived coronary vessels and responded by upregulating SOX17. Future studies will further investigate the triggers and drivers of the endocardial compensatory response. In total, our findings add to the list of methods by which a tissue can compensate for genetic deficiencies: the existence of alternative progenitor pathways that respond to different signals so as to diversify developmental potential.
Our data and that of other groups support an intriguing model where SV outgrowth is developmentally timed while endocardial angiogenesis is physiologically triggered. The SV sprouts into a tissue that is not initially hypoxic and is dependent on VEGF-C and ELA-APJ, which are not known to be hypoxia inducible (Chen et al., 2014b, 2014a). In contrast, endocardial cells emerge from the midline of the heart near the highly hypoxic ventricular septum, and require the hypoxia-inducible gene VEGF-A (Wu et al., 2012; Zhang and Zhou, 2013). We have found that VEGF-A does not play a predominant role in SV sprouting (K.R.-H., unpublished data); however, intramyocardial VEGF-A might play a role in the full migration of SV-derived sprouts throughout the deep layers of the myocardium that occurs at later stages in development (van den Akker et al., 2008). It is interesting to note that, unlike mice, zebrafish do not develop coronary vessels on a specific day in development, but over a range of days during their juvenile phase that is directly correlated to animal size (Harrison et al., 2015). Furthermore, zebrafish coronary vessels have been proposed to arise primarily from the endocardium and in response to the hypoxia-inducible receptor ligand pair, CXCR4/CXCL12 (Harrison et al., 2015). These observations suggest that the endocardial contribution to the heart is primarily responsive to physiological stimuli such as tissue hypoxia. The CXCR4/CXCL12 axis is also required for mouse coronary development, but the role of this ligand-receptor pair is different. Mutants do not have defects in initial migration of the coronary vasculature onto the heart, but rather in their connection to aortic blood flow and subsequent maturation, further suggesting that SV-derived vessel establishment is not regulated by hypoxia-inducible molecules (Cavallero et al., 2015; Ivins et al., 2015). We propose that the addition of the SV to the mammalian heart could have added the extra feature of a more complex system that is regulated by both genetic timing and the environment, which gives the system more flexibility in response to deleterious mutations and better ensures the establishment of the thick compact heart wall required for mammalian physiology (Figure 6F). Further understanding of how the embryonic heart repairs itself in the face of genetic assaults will provide knowledge on how we can repair this critical organ during injury and disease, and suggest additional ways by which organogenesis is made so robust.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
- EXPERIMENTAL MODELS AND SUBJECT DETAILS
-
◦Mice
-
◦
- METHODS DETAILS
-
◦Immunostaining, Imaging, and Image Processing
-
◦Hypoxia Analysis
-
◦In Situ Hybridization
-
◦Echocardiography
-
◦
- QUANTIFICATION AND STATISTICAL ANALYSIS
-
◦Vascular Phenotypic Analysis
-
◦Hypoxia Quantification
-
◦Lineage Tracing
-
◦Endocardial Activation
-
◦Hif-1α Expression
-
◦Statistical Analysis
-
◦
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| VE-cadherin | BD Pharmingen | Cat# 550548; RRID: AB_2244723 |
| Cardiac troponin T (cTnT) | DSHB | Cat# CT3; RRID: AB_528495 |
| Dach1 | Proteintech | Cat# 10914-1-AP; RRID: AB_2230330 |
| Integrin α4 | BD Pharmingen | Cat# 553314; RRID: AB_39477 |
| SOX17 | R&D systems | Cat# AF1924; RRID: AB_355060 |
| HIF-1α | Novus | Cat# NB100-479SS; RRID: AB_790147 |
| Alexa Fluor Conjugated Secondary Antibodies (488,594,633,635,647) | Life Technologies (ThermoFisher Scientific) | N/A |
| Anti-digoxigenin antibody | Roche diagnostics | Product# 11093274910; RRID: AB_514497 |
| Anti-hypoxyprobe-1 Mouse Dylight 549 Mab | HPI | HP7-100 Kit |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PFA | Electron Microscopy Sciences | Cat# 15714 |
| NBT-BCIP | Roche | Product# 11681451001 |
| Proteinase K | New England Biology (NEB) | Cat# P8107S |
| Acetic anhydride | Sigma-Aldrich | Lot# SHBC1816V |
| Formamide | Sigma-Aldrich | Product# F 9037 |
| Saline Sodium Citrate (SSC, 20×) | Sigma-Aldrich | Product# 93017-10L-F |
| Danhardt’s solution | Sigma-Aldrich | Product# D 2532 |
| CHAPS | Sigma-Aldrich | Product# C5070 |
| Yeast RNA | Sigma-Aldrich | Product# R 6750 |
| Triethanolamine | Sigma-Aldrich | Lot# BCBK5944V |
| Critical Commercial Assays | ||
| Centrisep Spin Column | Princeton Separations | Cat# CS-900 |
| Qiaquick gel extraction kit | Qiagen | Cat# 28704 |
| Platinum PCR SuperMix High Fidelity | Invitrogen | Cat# 12532-016 |
| Dig Labeling In-Vitro Transcription Kit (SP6/T7) | Roche | Product# 11175025910 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6 | The Jackson Laboratory | Stock# 000664 |
| Mouse: CD1 | Charles River | Strain# 022 |
| Mouse: FVB | Charles River | Strain# 207 |
| Mouse: Tie2Cre | The Jackson Laboratory | Stock# 004128 |
| Mouse: RosamTmG Cre reporter | The Jackson Laboratory | Stock# 007676 |
| Mouse: ApjCreER | Dr. Kristy Red-Horse, Stanford University (Chen et al., 2014b) | N/A |
| Mouse: Nfatc1Cre | Dr. Bin Zhou, Albert Einstein College of Medicine (Wu et al., 2012) | N/A |
| Mouse: Apj KO, Apelin KO, Apj Flox | Dr. Thomas Quertermous, Stanford University (Charo et al., 2009; Papangeli et al., 2016) | N/A |
| Mouse: Ela KO | Dr. Bruno Reversade, A*STAR (Ho et al., 2017) | N/A |
| Mouse: CCBE1KO | Dr. Mark L. Kahn, University of Pennsylvania (Bos et al., 2011) | N/A |
| Oligonucleotides | ||
| ELA ISH Forward Primer | 5’-TCTGAGTTCTGGCCATAGGA-3’ | NCBI Accession: NM_001297554.1 |
| ELA ISH Reverse Primer | 5’ AATTAATACGACTCACTATAGGG-CATAGGACGTGATGTACTGGTATG-3’ | NCBI Accession: NM_001297554.1 |
| T7 Promoter Sequence | 5’-TAATACGACTCACTATAGGG-3’ | |
| Software and Algorithms | ||
| Image J | NIH (https://www.nih.gov/ij/) | N/A |
| GraphPad Prism 7 | GraphPad Software (https://www.graphpad.com/scientific-software/prism) | N/A |
| Zen Microscope and Imaging Software | Zeiss (https://www.zeiss.com/microscopy/us/downloads/zen) | N/A |
| Adobe Photoshop | Adobe (https://www.adobe.com/products/photoshop) | N/A |
| Vevo 770 Standard Measurement Package | Visualsonics (https://www.visualsonics.com/product/software) | N/A |
| Other | ||
| Vectashield for fluorescence | Vector Labs | H-1000 |
| Vectashield with DAPI | Vector Labs | H-1200 |
| Hypoxyprobe-1 | HPI | HP7-100 Kit |
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kristy Red-Horse (kredhors@stanford.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Use of all the mouse lines in this study followed Stanford IACUC guidelines. Purchased mouse strains used in this study were: CD1 (Charles River, strain 022), FVB (Charles River, strain 207), C57bl/6 (The Jackson Laboratory, stock 000664), Tie2Cre (The Jackson Laboratory, stock 004128), and RosamTmG Cre reporter (The Jackson Laboratory, stock 007676). The following lines were described previously: ApjCreER (Chen et al., 2014b), Nfatc1Cre (Wu et al., 2012), Apj KO (Charo et al., 2009), Apelin KO (Charo et al., 2009), Apj flox (Papangeli et al., 2016), Ela KO (Ho et al., 2017), and Ccbe1 (Bos et al., 2011). Apj KO, Apj flox, and Ela KO mice were maintained on pure C57bl/6 background throughout this study. All other mouse lines used in this study were maintained and analyzed in a mixed background.
METHODS DETAILS
Immunostaining, Imaging, and Image Processing
Embryos harvested from timed pregnancies (morning of plug designated E0.5) were fixed in 4% paraformaldehyde (PFA) for 1 hour rotating at 4°C. Hearts were subjected to either cryo-sectioning and immunofluorescence or whole-mount immunofluorescence staining. Immunostaining was performed in either 1.5 ml tubes with constant rotation (whole mount) or on microscope slides (tissue sections). Primary antibodies in blocking solution (5% goat or donkey serum, 0.5% Triton X-100 in PBS) were incubated overnight at 4°C followed by several PBT (PBS with 0.5% Triton X-100) washes for 6h. Secondary antibodies diluted in blocking solution were incubated overnight at 4°C and washed several times with PBT.
Samples were imaged in Vectashield (Vector Labs) using either a Zeiss LSM-700 or Axioimager A2 epifluorescence microscope. Processing software such as Zen (from Zeiss), ImageJ (NIH), and Photoshop (Adobe Systems) were utilized for image processing.
Antibodies used are: VE-cadherin (BD Pharmingen, 550548; 1:125); cTnT (DSHB, CT3; 1:500); Dach1 (Proteintech, 10914-1-AP; 1:1000); Integrin α4 (BD Pharmingen, 553314; 1:100); SOX17 (R&D systems, AF1924, 1:500); and HIF-1α (Novus, NB100-479SS, 1:100). Secondary antibodies were Alexa Fluor conjugates (488, 555, 594, 633, 635, 647, Life Technologies; 1:250).
Hypoxia Analysis
Pregnant females at e15.5 from timed pregnancies were injected intraperitoneally with 2.5 mg (200 ul from 12.5 mg/ml working solution) Hypoxyprobe-1 (HPI, HP7-100 Kit). After 3 hours, embryos were harvested and fixed. Hearts from fixed embryos were subjected to whole mount immunofluorescence using mouse Dylight 549 Mab (HPI, HP7-100 Kit, 1:200) antibody to detect hypoxyprobe-1 labeling and a VE-cadherin antibody (BD Pharmingen, 550548, 1:125) to visualize coronary vessels.
In Situ Hybridization
Dig-labelled antisense ELA probe was designed by in-vitro transcription of ELA cDNA fragment amplified by PCR using Forward: 5’-TCTGAGTTCTGGCCATAGGA-3’and Reverse: 5’-AATTAATACGACTCACTATAGGGCATAGGACGTGATGTACTGGTATG-3’ primers (NCBI accession: NM_001297554.1). In-vitro transcription was performed using Roche Dig-Labeling In-Vitro Transcription Kit (Roche, 11175025910). Probe was filtered using Centrisep Column Separation (Princeton Separations, Cat# CS-900) before use.
Embryos were fixed in 4% PFA (pH7.0) for 1–2 hours at 4°C, washed in PBS for 30 minutes (changing the wash every 10 minutes), and cryoprotected in 30% sucrose for 1 hour before snap freezing and sectioning. Sample processing and hybridizations steps were performed as previously described (Chen et al., 2014b). Briefly, samples were treated with proteinase K, acetylated, blocked with pre-hybridization buffer, and hybridized with denatured digoxigenin-labeled (Roche) antisense probes in hybridization solution and incubated overnight at 60°C in an airtight container. Next day, after series of washes, samples were blocked with 2% bovine serum albumin in MABT for 2–3 hours at room temperature and incubated overnight at 4°C with alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche). Signal was detected with NBT-BCIP (Roche).
Echocardiography
Left ventricular geometry and function were evaluated in 10–15 week-old mice (Apj WT, n=6; Apj KO, n=6) with a high-resolution (30 MHz) Vevo 2100 transthoracic echocardiography system (VisualSonics). Chest hair was removed with depilatory cream, and mice were sedated with 1–3% inhaled isoflurane titrated to a heart rate between 350–450/minute. Images were obtained through a parasternal short-axis view with M-mode ultrasound at the level of the papillary muscle and mid-way between the papillary muscle and apex. Ejection fraction, fractional shortening, and internal ventricular dimensions were computed with the Vevo 770 Standard Measurement Package. All analyses were performed by a single investigator blinded to the genotype.
QUANTIFICATION AND STATISTICAL ANALYSIS
Vascular Phenotypic Analysis
Vascular coverage was calculated as the total area of heart ventricles occupied by coronary vessels. To calculate this, ImageJ was used to circumscribe the whole heart and area containing coronary vessels. The region containing vessels was expressed as percentage.
Branch points were counted within a 500 µm squared box, defined as field of view (FOV), at the dorsal plexus on the surface (sub-epicardium) and intramyocardial layer of the left and right ventricles.
Myocardial thickness was measured in ImageJ from cryosections of the heart. Four measurements were taken from the lateral wall of the right ventricle and averaged. Results were reported using Prism 7 (GrpahPad).
Hypoxia Quantification
Tissue hypoxia was quantified by measuring mean fluorescence intensity of hypoxyprobe-1 labeling in 3 different FOVs per sample using Zen software (Zeiss Zen, 2011).
Lineage Tracing
Hearts from Apj WT, HET, and KO animals possessing the Nfatc1Cre and RosamTmG alleles were isolated at E16.5. Hearts were fixed and subjected to whole mount immunofluorescence. Hearts were immunostained with anti-Dach1 and anti-VE-cadherin; recombination was detected by direct fluorescence. The percentage of Dach1-positive lineage labeled cells was quantified from at least two FOV in each regions of the heart. Hearts from ApjCreER; RosamTmG mice were quantified in a similar manner. ImageJ and Zen (from Zeiss) softwares were used for quantifications.
Endocardial Activation
Quantification of endocardial activation was performed by counting Sox17+ ECs that were negative for Dach1 (coronary marker) in two different fields of view (FOV) per heart in the endocardium-myocardium border regions. Double positive endothelial cells were counted as coronary Ecs.
Hif-1α Expression
HIF-1α expression was quantified by measuring mean fluorescence intensity (mean grey value) of HIF-1α staining in 2 different fields of view (FOV) for each region per heart using Image J software.
Statistical Analysis
Unpaired t-tests were used to determine the two-tailed P-value for each comparison. Quantification graphs were reported using Prism 7 (GraphPad).
Supplementary Material
Highlights.
ELA-APJ is required for coronary vessel (CV) sprouting from the sinus venosus (SV)
Endocardial-derived CVs can expand to rescue defective SV sprouting
The existence of two progenitor sources provides robustness to coronary development
Endocardial cells activated to become CVs express Sox17
Acknowledgments
We would like to extend our sincere thank you to all the members of the Red-Horse laboratory for their constructive comments and feedback during the preparation of the manuscript. K.R.-H. is supported by the NIH (R01HL12850301) and New York Stem Cell Foundation (Robertson Investigator) and G.H.F. is supported by NIH (R25-GMO48972) and California Institute of Regenerative Medicine (CIRM) (EDUC2-08391).
Footnotes
Supplemental Information includes six figures and can be found with this article online at http://dx.doi.org/10.1016/j.devcel.2017.08.008.
AUTHOR CONTRIBUTIONS
B.S. and K.R.-H. conceptualized, designed, and analyzed the experiments and wrote the manuscript. B.S. performed the majority of the experiments. G.H.F. contributed in the characterization of Ccbe1 mutant phenotype. H.I.C. contributed in collecting in situ hybridization data. L.H. and B.R. provided Elabela KO mouse, provided invaluable discussions, and edited the manuscript. T.Q. provided Apj KO, Apelin KO, and Apj flox mouse lines. A.B.G. and Y.J.W. performed adult cardiac function analyses.
References
- Bort R, Signore M, Tremblay K, Martinez Barbera JP, Zaret KS. Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev. Biol. 2006;290:44–56. doi: 10.1016/j.ydbio.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Bos FL, Caunt M, Peterson-Maduro J, Planas-Paz L, Kowalski J, Karpanen T, van Impel A, Tong R, Ernst JA, Korving J, et al. CCBE1 is essential for mammalian lymphatic vascular development and enhances the lymphangiogenic effect of vascular endothelial growth factor-C in vivo. Circ. Res. 2011;109:486–491. doi: 10.1161/CIRCRESAHA.111.250738. [DOI] [PubMed] [Google Scholar]
- Bui HM, Enis D, Robciuc MR, Nurmi HJ, Cohen J, Chen M, Yang Y, Dhillon V, Johnson K, Zhang H, et al. Proteolytic activation defines distinct lymphangiogenic mechanisms for VEGFC and VEGFD. J. Clin. Invest. 2016;126:2167–2180. doi: 10.1172/JCI83967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallero S, Shen H, Yi C, Lien C-L, Kumar SR, Sucov HM. CXCL12 signaling is essential for maturation of the ventricular coronary endothelial plexus and establishment of functional coronary circulation. Dev. Cell. 2015;33:469–477. doi: 10.1016/j.devcel.2015.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo DN, Ho M, Fajardo G, Kawana M, Kundu RK, Sheikh AY, Finsterbach TP, Leeper NJ, Ernst KV, Chen MM, et al. Endogenous regulation of cardiovascular function by apelin-APJ. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1904–H1913. doi: 10.1152/ajpheart.00686.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HI, Poduri A, Numi H, Kivela R, Saharinen P, McKay AS, Raftrey B, Churko J, Tian X, Zhou B, et al. VEGF-C and aortic cardiomyocytes guide coronary artery stem development. J. Clin. Invest. 2014a;124:4899–4914. doi: 10.1172/JCI77483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HI, Sharma B, Akerberg BN, Numi HJ, Kivela R, Saharinen P, Aghajanian H, McKay AS, Bogard PE, Chang AH, et al. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development. 2014b;141:4500–4512. doi: 10.1242/dev.113639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev. Cell. 2013;27:672–680. doi: 10.1016/j.devcel.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Clavería C, Giovinazzo G, Sierra R, Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500:39–44. doi: 10.1038/nature12389. [DOI] [PubMed] [Google Scholar]
- Das S, Red-Horse K. Cellular plasticity in cardiovascular development and disease. Dev. Dyn. 2017;246:328–335. doi: 10.1002/dvdy.24486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beco S, Ziosi M, Johnston LA. New frontiers in cell competition. Dev. Dyn. 2012;241:831–841. doi: 10.1002/dvdy.23783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker JS, Larrivee B, Breant C, Duarte A, Takakura N, Fukamizu A, et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood. 2010;116:4025–4033. doi: 10.1182/blood-2010-02-270819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshwar AR, Chng SC, Ho L, Reversade B, Scott IC, Robertson E. The Apelin receptor enhances Nodal/TGFβ signaling to ensure proper cardiac development. Elife. 2016;5:e13758. doi: 10.7554/eLife.13758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eralp I, Lie-Venema H, DeRuiter MC, van den Akker NMS, Bogers AJJC, Mentink MMT, Poelmann RE, Gittenberger-de Groot AC. Coronary artery and orifice development is associated with proper timing of epicardial outgrowth and correlated Fas-ligand-associated apoptosis patterns. Circ. Res. 2005;96:526–534. doi: 10.1161/01.RES.0000158965.34647.4e. [DOI] [PubMed] [Google Scholar]
- Harrison MRM, Bussmann J, Huang Y, Zhao L, Osorio A, Burns CG, Burns CE, Sucov HM, Siekmann AF, Lien C-L. Chemokine-guided angiogenesis directs coronary vasculature formation in zebrafish. Dev. Cell. 2015;33:442–454. doi: 10.1016/j.devcel.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helker CSM, Schuermann A, Pollmann C, Chng SC, Kiefer F, Reversade B, Herzog W. The hormonal peptide Elabela guides angioblasts to the midline during vasculogenesis. Elife. 2015;4:2653. doi: 10.7554/eLife.06726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Tan SYX, Wee S, Wu Y, Tan SJC, Ramakrishna NB, Chng SC, Nama S, Sczerbinska I, Chan Y-S, et al. ELABELA is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell. 2015;17:435–447. doi: 10.1016/j.stem.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Ho L, Van Dijk M, Chye STJ, Messerschmidt DM, Chng SC, Ong S, Yi LK, Boussata S, Goh GH-Y, Afink GB, et al. ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science. 2017;8:eaam6607. doi: 10.1126/science.aam6607. [DOI] [PubMed] [Google Scholar]
- Hogan BM, Bos FL, Bussmann J, Witte M, Chi NC, Duckers HJ, Schulte-Merker S. Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat. Genet. 2009;41:396–398. doi: 10.1038/ng.321. [DOI] [PubMed] [Google Scholar]
- Hutchins GM, Kessler-Hanna A, Moore GW. Development of the coronary arteries in the embryonic human heart. Circulation. 1988;77:1250–1257. doi: 10.1161/01.cir.77.6.1250. [DOI] [PubMed] [Google Scholar]
- Inui M, Fukui A, Ito Y, Asashima M. Xapelin and Xmsr are required for cardiovascular development in Xenopus laevis. Dev. Biol. 2006;298:188–200. doi: 10.1016/j.ydbio.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Ivins S, Chappell J, Vernay B, Suntharalingham J, Martineau A, Mohun TJ, Scambler PJ. The CXCL12/CXCR4 axis plays a critical role in coronary artery development. Dev. Cell. 2015;33:455–468. doi: 10.1016/j.devcel.2015.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Kim J, Anderson JP, Wu J, Gleim SR, Kundu RK, McLean DL, Kim J-D, Park H, Jin S-W, et al. Apelin-APJ signaling is a critical regulator of endothelial MEF2 activation in cardiovascular development. Circ. Res. 2013;113:22–31. doi: 10.1161/CIRCRESAHA.113.301324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SN, Amoyel M, Bergantiños C, la Cova de C, Schertel C, Basler K, Johnston LA. An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science. 2014;346:1258236. doi: 10.1126/science.1258236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papangeli I, Kim J, Maier I, Park S, Lee A, Kang Y, Tanaka K, Khan OF, Ju H, Kojima Y, et al. MicroRNA 139-5p coordinates APLNR-CXCR4 crosstalk during vascular maturation. Nat. Commun. 2016;7:11268. doi: 10.1038/ncomms11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Norris ML, Valen E, Chew G-L, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343:1248636. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Camps M, Tian J, Chng SC, Sem KP, Sudhaharan T, Teh C, Wachsmuth M, Korzh V, Ahmed S, Reversade B. Quantitative imaging reveals real-time Pou5f3-Nanog complexes driving dorsoventral mesendoderm patterning in zebrafish. Elife. 2016;5:e11475. doi: 10.7554/eLife.11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464:549–553. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reversade B, De Robertis EM. Regulation of ADMP and BMP2/4/7 at opposite embryonic poles generates a self-regulating morphogenetic field. Cell. 2005;123:1147–1160. doi: 10.1016/j.cell.2005.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Sato C, Kadowaki A, Watanabe H, Ho L, Ishida J, Yamaguchi T, Kimura A, Fukamizu A, Penninger JM, et al. ELABELA-APJ axis protects from pressure overload heart failure and angiotensin II-induced cardiac damage. Cardiovasc. Res. 2017;113:760–769. doi: 10.1093/cvr/cvx061. [DOI] [PubMed] [Google Scholar]
- Scimia M-C, Hurtado C, Ray S, Metzler S, Wei K, Wang J, Woods CE, Purcell NH, Catalucci D, Akasaka T, et al. APJ acts as a dual receptor in cardiac hypertrophy. Nature. 2012;488:394–398. doi: 10.1038/nature11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott IC, Masri B, D’Amico LA, Jin S-W, Jungblut B, Wehman AM, Baier H, Audigier Y, Stainier DYR. The g protein-coupled receptor agtrl1b regulates early development of myocardial progenitors. Dev. Cell. 2007;12:403–413. doi: 10.1016/j.devcel.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Sturzu AC, Rajarajan K, Passer D, Plonowska K, Riley A, Tan TC, Sharma A, Xu AF, Engels MC, Feistritzer R, et al. Fetal mammalian heart generates a robust compensatory response to cell loss. Circulation. 2015;132:109–121. doi: 10.1161/CIRCULATIONAHA.114.011490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Hu T, Zhang H, He L, Huang X, Liu Q, Yu W, He L, Yang Z, Yan Y, et al. De novo formation of a distinct coronary vascular population in neonatal heart. Science. 2014;345:90–94. doi: 10.1126/science.1251487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Hu T, Zhang H, He L, Huang X, Liu Q, Yu W, He L, Yang Z, Zhang Z, et al. Subepicardial endothelial cells invade the embryonic ventricle wall to form coronary arteries. Cell Res. 2013;23:1075–1100. doi: 10.1038/cr.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Akker NMS, Caolo V, Wisse LJ, Peters PPWM, Poelmann RE, Carmeliet P, Molin DGM, Gittenberger-de Groot AC. Developmental coronary maturation is disturbed by aberrant cardiac vascular endothelial growth factor expression and Notch signalling. Cardiovasc. Res. 2008;78:366–375. doi: 10.1093/cvr/cvm108. [DOI] [PubMed] [Google Scholar]
- Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno-Rodriguez RA, Markwald RR, O’Rourke BP, Sharp DJ, et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell. 2012;151:1083–1096. doi: 10.1016/j.cell.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Maguire JJ, Davenport AP. Apelin, Elabela/Toddler, and biased agonists as novel therapeutic agents in the cardiovascular system. Trends Pharmacol. Sci. 2015;36:560–567. doi: 10.1016/j.tips.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F-X, Zhao B, Guan K-L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yzaguirre AD, Padmanabhan A, de Groh ED, Engleka KA, Li J, Speck NA, Epstein JA. Loss of neurofibromin Ras-GAP activity enhances the formation of cardiac blood islands in murine embryos. Elife. 2015;4:e07780. doi: 10.7554/eLife.07780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS. Genetic programming of liver and pancreas progenitors: lessons for stem-cell differentiation. Nat. Rev. Genet. 2008;9:329–340. doi: 10.1038/nrg2318. [DOI] [PubMed] [Google Scholar]
- Zeng X-XI, Wilm TP, Sepich DS, Solnica-Krezel L. Apelin and its receptor control heart field formation during zebrafish gastrulation. Dev. Cell. 2007;12:391–402. doi: 10.1016/j.devcel.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Zhang H, Pu W, Li G, Huang X, He L, Tian X, Liu Q, Zhang L, Wu SM, Sucov HM, Zhou B. Endocardium minimally contributes to coronary endothelium in the embryonic ventricular free walls. Circ. Res. 2016;118:1880–1893. doi: 10.1161/CIRCRESAHA.116.308749. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhou B. Accelerated coronary angiogenesis by vegfr1-knockout endocardial cells. PLoS One. 2013;8:e70570. doi: 10.1371/journal.pone.0070570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.