

Abstract
Hepatocyte apoptosis has been documented in both clinical and experimental alcoholic liver disease. This study was undertaken to examine the effect of dietary zinc supplementation on hepatic apoptosis in mice subjected to a long-term ethanol exposure. Male adult 129S6 mice fed an ethanol-containing liquid diet for 6 months developed hepatitis, as indicated by neutrophil infiltration and elevation of hepatic keratinocyte chemoattractant (KC) and monocyte chemoattractant protein-1 (MCP-1) levels. Apoptotic cell death was detected in ethanol-exposed mice by a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and was confirmed by the increased activities of caspase-3 and -8. Zinc supplementation attenuated alcoholic hepatitis and reduced the number of TUNEL-positive cells in association with inhibition of caspase activities. Ethanol exposure caused oxidative stress, as indicated by reactive oxygen species accumulation, mitochondrial glutathione depletion, and decreased metallothionein levels in the liver, which were suppressed by zinc supplementation. The mRNA levels of tumor necrosis factor (TNF)-α, TNF-R1, FasL, Fas, Fas-associated factor-1, and caspase-3 in the liver were upregulated by ethanol exposure, which were attenuated by zinc supplementation. Zinc supplementation also prevented ethanol-elevated serum and hepatic TNF-α levels and TNF-R1 and Fas proteins in the liver. In conclusion, zinc supplementation prevented hepatocyte apoptosis in mice subjected to long-term ethanol exposure, and the action of zinc is likely through suppression of oxidative stress and death receptor-mediated pathways.
Keywords: zinc, apoptosis, oxidative stress, death receptors, alcoholic liver disease
Introduction
Hepatocyte apoptosis has been well documented in alcoholic liver disease from both clinical (1–4) and experimental studies (5, 6). Mechanistic studies have demonstrated that oxidative stress and death receptors are the major mediators in ethanol-induced hepatocyte apoptosis (7). Ethanol exposure induces cytochrome P450 2E1 (CYP2E1) expression in the liver, through which ethanol is metabolized to generate reactive oxygen species (ROS) (8). ROS accumulation in the liver may cause oxidative damage, including mitochondrial dysfunction, leading to cytochrome c release, caspase activation, and ultimately hepatocyte apoptosis (7, 8). Over-expression of CYP2E1 in human hepatoma cell lines generated more ROS and cell death after ethanol exposure (9, 10). Up-regulation of death receptors and their ligands, such as TNF-α receptor 1 (TNF-R1)/TNF-α and Fas (APO1, CD95)/Fas ligand (FasL) was also associated with hepatocyte apoptosis in patients with alcoholic hepatitis (1, 3, 11, 12). Blocking death receptor/ligand interaction by knocking out the TNF-R1 or neutralizing by FasL antibody in mice attenuated hepatocyte apoptosis after ethanol exposure; this provides direct evidence to show the importance of death receptors in ethanol-induced apoptosis (9, 13). A recent study has shown that ethanol exposure enhances death receptor-induced apoptosis through both CYP2E1-dependent and -independent pathways (14).
Zinc deficiency has been documented in alcoholic liver disease in both patient and animal studies (15–17). Zinc is known to participate in multiple cellular functions, including metabolism, cell signaling, and gene regulation. Zinc deprivation in cell culture studies has been shown to induce apoptosis in diverse cell lines, including HepG2 cells (18, 19). Zinc deprivation also potentiates death receptor-mediated apoptosis (20). Our recent studies showed that zinc supplementation attenuates apoptotic cell death induced by D-galactosamine/TNF-α or acute ethanol intoxication (6, 21, 22). However, neither D-galactosamine/TNF-α nor acute ethanol intoxication induced hepatitis, which is characterized in alcoholic patients (1, 2). In the present study, long-term (6 months) dietary ethanol exposure to mice was conducted to mimic alcoholic hepatitis, and the effect of zinc supplementation on ethanol-induced hepatocyte apoptosis was determined.
Materials and Methods
Animals and Treatments
Male adult 129S6 mice (>25 g) were obtained from Taconic (Germantown, NY). All the mice were treated according to the experimental procedures approved by the Institutional Animal Care and Use Committee. Animals were pair-fed with the Lieber-DeCarli liquid diet (Bio-Serv, Frenchtown, NJ), containing either ethanol or isocaloric maltose dextrin. Animals were randomly assigned to 4 groups by using a 2 × 2 factorial design of ethanol × zinc. The content of ethanol in the diet (%, w/v) was gradually increased over a 6-month feeding period, starting at 3.1%, increasing 0.3% every month, and reaching 4.6% at the end. Zinc supplementation was conducted by adding zinc sulfate to the liquid diet at 75 mg elemental zinc/L. Food intake and body weight were recorded daily and weekly, respectively. At the end of the experiment, the mice were anesthetized with sodium pentobarbital (50 mg/kg), and plasma and liver tissue samples were harvested for assays.
Histopathology
The histopathological changes of the liver were observed by light microscopy. Liver tissues were fixed in 10% formalin, and sections at 5 μM were stained with hematoxylin and eosin staining.
Alanine Aminotransferase (ALT) Activities
The ALT activities in the serum were measured by following a colorimetric procedure (23).
Enzyme-Linked Immunosorbent Assay (ELI-SA)
The serum levels of TNF-α and hepatic levels of TNF-α, keratinocyte chemoattractant (KC), and monocyte chemoattractant protein-1 (MCP-1) were measured by using ELISA assay kits from R&D System (Minneapolis, MN) by following the manufacturer’s instructions.
Enzymatic Assay of Caspase-3 and Caspase-8
Fresh liver tissues were homogenized in the extraction buffer (25 mM HEPES buffer, pH 7.4, containing 5 mM EDTA, 2 mM dithiothreitol, 0.1% CHAPS, and 1% protease cocktail). The homogenate was centrifuged at 20,000 g for 30 min. The resulting supernatants were diluted with the assay buffer (50 mM HEPES, 10 mM dithiothreitol, 1.0 mM EDTA, 100 mM NaCl, 0.1% CHAPS, and 10% glycerol, pH 7.4) and incubated at 37°C with caspase-3 substrate (Ac-DEVD-pNA) or caspase-8 substrate (Ac-IETD-pNA); p-Nitroaniline was used as the standard. Cleavage of the substrate was monitored at 405 nm, and the specific activity was expressed in picomoles of the product, nitroaniline, per minute per milligram of protein.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
Apoptotic cell death in the liver was assessed by detection of DNA fragmentation using an ApopTag Peroxidase in situ Apoptosis Detection Kit (Chemicon, Temecula, CA). In brief, liver tissue slides were pretreated with proteinase K and H2O2 and were incubated with the reaction mixture containing terminal deoxynucleotidyl transferase (TdT) and digoxigenin-conjugated dUTP for 1 hr at 37°C. The labeled DNA was visualized with HRP-conjugated anti-digoxigenin antibody with DAB as the chromagen followed by counterstaining with methyl green. The TUNEL-positive cells were counted under ×40 objective, and the data were expressed as the number of TUNEL-positive cells per 100 hepatocytes.
Fluorescent Microscopy of ROS
Dihydroethidium was used for in situ detection of ROS in the liver. Nonfluorescent dihydroethidium is oxidized by ROS to yield red fluorescent product that binds to nucleic acids, staining the nucleus a bright fluorescent red. Cryostat sections of liver were cut in 5-μm portions and incubated with 5 μM dihydroethidium (Molecular Probes, Eugene, OR) at 37°C for 30 min. The red fluorescence from dihydroethidium was detected using a Nikon 2000S fluorescence microscope.
Measurement of Mitochondrial GSH
Liver tissues were homogenized in 10 mM HEPES buffer (pH 7.4) containing 200 mM mannitol, 50 mM sucrose, 10 mM KCl, and 1 mM EDTA by using a Dounce homogenizer. The crude homogenate was centrifuged at 700 g, and the supernatant was centrifuged further at 15,000 g. The mitochondrial pellet was washed twice and resuspended in the same buffer. Meta-phosphoric acid was used for deproteinization at a final concentration of 5%. After centrifuging at 15,000 g, the supernatants were used for GSH assay using a Bioxytech GSH-400 assay kit (OXIS, Portland, OR).
Measurement of Metallothionein (MT)
Hepatic MT concentrations in the liver were determined by a cadmium-hemoglobin affinity assay. Briefly, liver tissues were homogenized in 4 volumes of 10 mM Tris-HCl buffer (pH 7.4) at 4°C. After centrifugation of the homogenate at 10,000 g for 15 min, 200 μl of supernatant was transferred to microtubes for MT analysis, as described previously (24).
Immunohistochemistry
Liver tissues were fixed with 4% paraformaldehyde and sectioned in 5 μm portions. The sections were incubated with rabbit anti–TNF-R1 or rabbit anti-Fas (Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight followed by incubation with a DAKO EnVision+ HRP conjugated goat anti-rabbit IgG (DAKO, Carpinteria, CA) for 30 min. Diaminobenzidine was used as HRP substrate for visualization. The negative controls were conducted by omitting the primary antibody.
Real-Time RT-PCR
The mRNA levels of death receptor–related genes were assessed by real-time RT-PCR. In brief, the total RNA was isolated and reverse transcribed with the Moloney murine leukemia virus reverse transcriptase and oligo-dT primers. The forward and reverse primers were designed using Primer Express Software and listed in Table 1. The SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA) was used for real-time RT-PCR analysis. The relative differences of gene expression among groups were evaluated using cycle time values and expressed as relative changes, setting the value of the control mice as one.
Table 1.
Primer Sequences for Real-Time RT-PCRa
| Gene | GeneBank accession no. | Sequences (forward/reverse) |
|---|---|---|
| TNF-α | XM_110221 | GACCCTCACACTCAGATCATCTTCTCCTCCACTTGGTGGTTTGCT |
| TNF-R1 | M59378 | TGCACTAAACAGCAGAACCGAGTTGCTCAGCCTCATGCACTG |
| FasL | V06948 | GGCTGGGTGCCATGCAGGCACTGCTGTCTACCCAGAA |
| FAS | M83649 | CTGCACCCTGACCCAGAATACTGGCTCAAGGGTTCCATGTT |
| FAF-1 | U39643 | TGCTGAGCCTGTTAGCAAGCTAAACGCCGTTCCAGGAACT |
| Caspase-3 | U19522 | TCCTGGTCTTTGTACGCTACCACCTGATGTCGAAGTTGAGGTAGCT |
RT-PCR, reverse transcriptase-polymerase chain reaction; TNF, tumor necrosis factor; FAF, Fas-associated factor.
Statistics
All data are expressed as mean ± SD. The data were analyzed by analysis of variance (ANOVA) and by Newman-Keuls’ Multiple-Comparison Test. Differences between groups were considered significant at P < 0.05.
Results
Food and Ethanol Intake, Body Weight Gain, and Liver/Body Weight Ratio in Mice Exposed to Ethanol for 6 Months
Daily food intake was monitored in the ethanol-exposed mice, and pair-fed mice were adjusted for their food supply accordingly. As shown in Figure 1, the calculated daily ethanol intake (g/kg) was gradually increased during the 6-month ethanol exposure, although the food intake showed a slight decrease in association with an increase in dietary ethanol levels. The average daily food intake was 405 g/kg/d, and the calculated ethanol intake was 15 g/kg/d. The calculated zinc intake was 30.4 mg/kg/d. Ethanol-fed mice showed a lower weight gain compared with the pair-fed control mice at the end of the experiment. Zinc supplementation to the ethanol-fed mice did not improve the weight gain but significantly inhibited ethanol-increased liver weight; the ethanol-fed mice showed a significant increase in the liver/body weight ratio, which was significantly reduced by zinc supplementation (Table 2). At the late stage of the feeding experiment, the ethanol-fed mice developed seizure behavior, which was not significantly improved by zinc supplementation.
Figure 1.
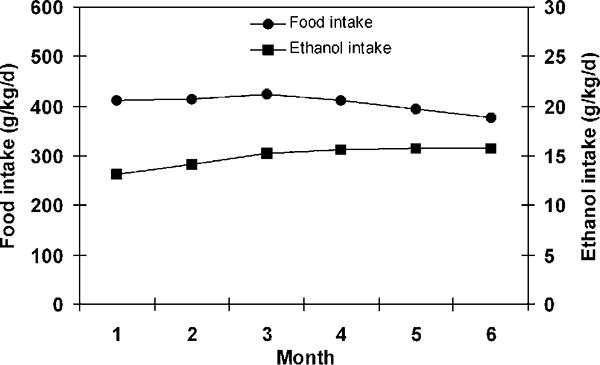
Food and ethanol intake in 129S6 mice that were chronically fed ethanol for 6 months. Food intake was recorded daily. Results are means ± SD (n = 4–6).
Table 2.
Body Weights and Liver Weights*
| Groups | Controls | EtOH | Zn | Zn/EtOH |
|---|---|---|---|---|
| Body weight | ||||
| Initial | 26.2 ± 0.4 | 26.4 ± 1.1 | 26.1 ± 0.7 | 26.5 ± 0.9 |
| End | 31.6 ± 0.6 | 27.1 ± 1.2 | 31.0 ± 1.0 | 27.7 ± 2.8 |
| Gain | 5.1 ± 0.5a | 0.6 ± 2.7b | 4.6 ± 1.6a | 0.9 ± 1.8b |
| Liver weight | 0.96 ± 0.10a | 1.25 ± 0.16b | 0.96 ± 0.04a | 1.05 ± 0.08a |
| Liver/body weight ratio | 3.11 ± 0.31a | 4.61 ± 0.38c | 3.11 ± 0.07a | 3.82 ± 0.31b |
EtOH, ethanol; Zn, zinc.
Significantly different (P < 0.05) among a, b, and c in each measurement.
Zinc Supplementation Attenuated Alcoholic Hepatitis
Light microscopy exam revealed that ethanol exposure caused hepatitis, as indicated by neutrophil infiltration in the liver (Fig. 2A). Ethanol exposure also elevated the hepatic levels of chemokines, including KC and MCP-1 (Fig. 2B). Zinc supplementation attenuated ethanol-elevated KC and MCP-1 levels, leading to a decrease in the number of neutrophils. Ethanol-fed mice showed a significant increase in serum ALT activities (74.1 ± 10.6 U/l) compared with control mice (33.6 ± 3.5 U/l) and zinc (28.1 ± 4.6 U/l), which was partially inhibited by zinc supplementation (47.6 ± 8.2 U/l).
Figure 2.
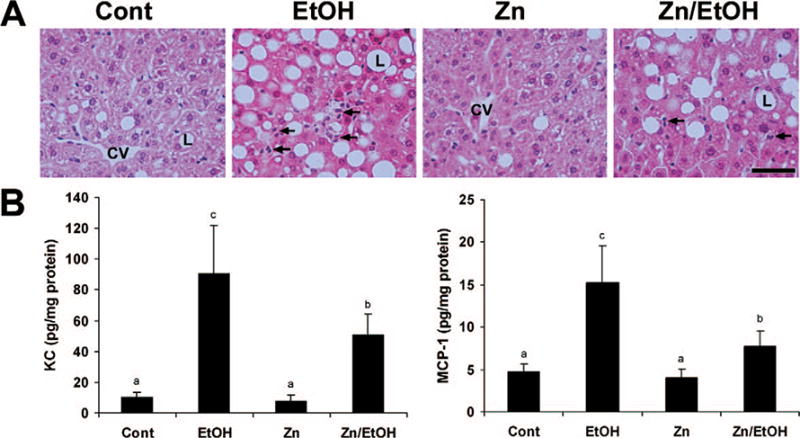
Effects of zinc supplementation on alcoholic hepatitis in 129S6 mice chronically fed ethanol for 6 months. A. Light microscopy with hematoxylin staining shows neutrophil infiltration (arrows) in the liver of ethanol-fed mice. The number of neutrophils (arrows) was decreased by zinc supplementation. CV, central vein; L, lipid droplet. Scale bar: 50 μM. B. Protein levels of KC and MCP-1 in the liver were measured by ELISA. Results are means ± SD (n = 4–6). Significantly different (P < 0.05) among a, b, and c. Cont, control; EtOH, ethanol. Color versions of the figure are available in the online version of the article.
Zinc Supplementation Inhibited Ethanol-Induced Apoptosis in the Liver
To determine whether or not zinc supplementation inhibits ethanol-induced apoptosis in the liver, the measurements of caspase activation and TUNEL assay were performed. As shown in Figure 3A, TUNEL assay showed increases in the positive labeling in the liver sections of all the ethanol-exposed mice. However, the number of apoptotic nuclei were reduced by zinc supplementation (Fig. 3B). The activities of caspase-3 and caspase-8 were increased by ethanol exposure but were normalized by zinc supplementation (Fig. 3C).
Figure 3.
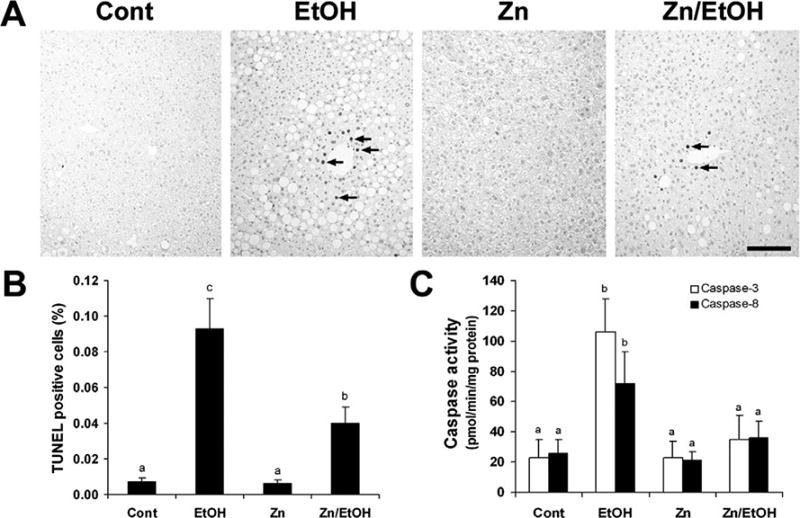
Effect of zinc supplementation on hepatocellular apoptosis in 129S6 mice chronically fed ethanol for 6 months. A. TUNEL assay of hepatocellular apoptosis was conducted using an ApoTag Apoptosis Detect kit. Scale bar: 100 μM. B. Quantitative measurements of TUNEL-positive cells in the liver. C. Activities of caspase-3 and -8 in the liver were colorimetrically measured by using Ac-DEVD-pNA as the caspase-3 substrate and Ac-IETD-pNA as the caspase-8 substrate. Results are means ± SD (n = 4–6). Significantly different (P < 0.05) among a, b, and c. Cont, control; EtOH, ethanol.
Zinc Supplementation Suppressed Ethanol-Induced Oxidative Stress in the Liver
Oxidative stress was assessed by measuring ROS and the antioxidants GSH and MT. Ethanol exposure caused ROS accumulation in the liver, as indicated by red fluorescence formation on the nuclei upon oxidation of dihydroethidium by ROS (Fig. 4A). Zinc supplementation remarkably suppressed ethanol-induced accumulations of ROS in the liver. Ethanol exposure decreased both the mitochondrial GSH levels and MT concentrations by about 50% (Fig. 4B). Zinc supplementation attenuated GSH depletion and normalized MT levels.
Figure 4.
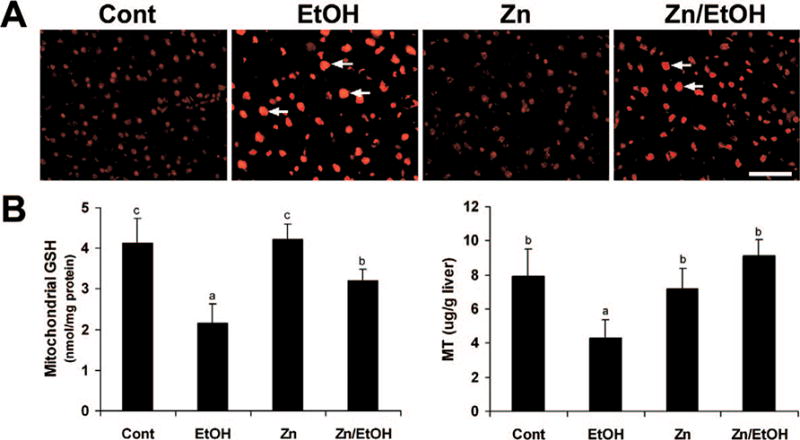
Effect of zinc supplementation on oxidative stress in the liver of 129S6 mice chronically fed ethanol for 6 months. A. accumulation in the liver was detected by dihydroethidium that yields red fluorescence (arrows) in the nuclei upon oxidation by ROS. Scale 50 μM. B. Quantitative measurements of mitochondrial GSH and MT in the liver. GSH was measured using a Bioxytech GSH-400 assay kit, and MT was determined by a cadmium-hemoglobin affinity assay. Results are means ± SD (n = 4–6). Significantly different (P < 0.05) among a, and c. Cont, control; EtOH, ethanol. A color version of the figure is available in the online version of the article.
Zinc Supplementation Modulated Hepatic Genes and Proteins Involved in Apoptotic Cell Death
To define the possible role of zinc supplementation in regulation of death receptor-mediated apoptosis, expression of death receptor-related genes was determined by a real time RT-PCR assay. As shown in Figure 5, the long-term ethanol exposure significantly elevated the mRNA levels of TNF-α, TNF-R1, FasL, Fas, FAF-1, and caspase-3. Zinc supplementation normalized the mRNA levels of TNF-α, TNF-R1, FasL, FAF-1, and caspase-3 and significantly decreased Fas mRNA levels. ELISA of protein levels demonstrated that ethanol exposure elevated TNF-α in both the serum and the liver, which was attenuated by zinc supplementation (Fig. 6A). Immunohistochemical staining of TNF-R1 and Fas protein in the liver showed that ethanol exposure increased the membrane labeling of both TNF-R1 and Fas (Fig. 6B). Zinc supplementation decreased the positive cell number and the staining intensity of both TNF-R1 and Fas in the liver.
Figure 5.
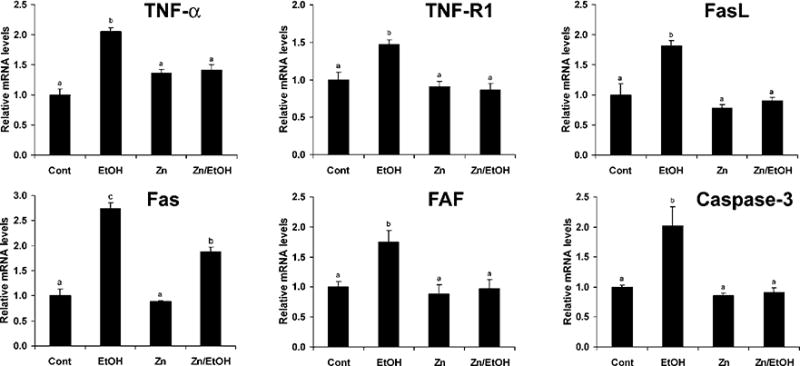
Effects of zinc supplementation on expression of death receptor-related genes in 129S6 mice chronically fed ethanol for 6 months. Gene expression was determined by real time RT-PCR assay. Total RNA was isolated, and real-time RT-PCR was performed as described in Materials and Methods. Results are means ± SD (n = 4–6). Significantly different (P < 0.05) among a, b, and c. Cont, control; EtOH, ethanol.
Figure 6.
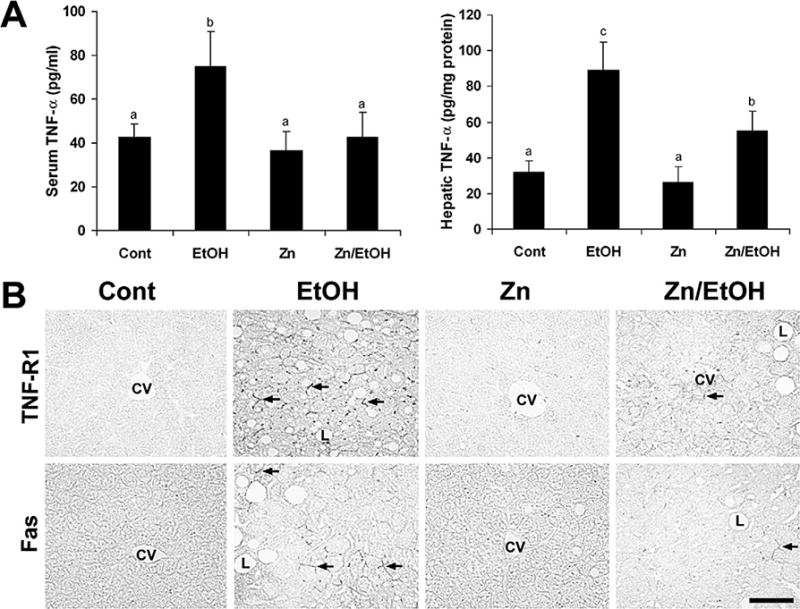
Effects of zinc supplementation on death receptor proteins in 129S6 mice chronically fed ethanol for 6 months. A. TNF-α protein levels in the serum and liver were measured by using ELISA kits. Results are means ± SD (n = 4–6). Significantly different (P < 0.05) among a, b, and c. B. TNF-R1 and Fas proteins (arrows) in the liver were examined by immunohistochemistry. Scale bar: 50 μM. Cont, control; EtOH, ethanol.
Discussion
The present study demonstrated that dietary zinc supplementation has an inhibitory effect on alcoholic hepatitis and hepatocyte apoptosis in mice subjected to long-term ethanol exposure. In addition to the antioxidant effect, which has been reported in previous studies (16, 17), another remarkable effect of zinc supplementation observed in the present study is the suppression of the elevated expression of death receptor–related genes and proteins. These results, thus, suggest that the anti-apoptotic effect of zinc supplementation is likely mediated through inhibition of ethanol exposure–induced oxidative stress and death receptor–related genes and proteins.
Hepatocyte apoptosis is a pathological feature of human alcoholic hepatitis (1–4) and of animal models of alcoholic liver disease (5, 6). One of the most well-recognized mechanisms underlying ethanol exposure-induced hepatocyte apoptosis is ethanol metabolism–associated ROS generation and oxidative stress, named the intrinsic pathway. Many studies have demonstrated that ethanol metabolism in the liver generates ROS through the CYP2E1 pathway (8). ROS accumulation in hepatocytes may cause mitochondrial dysfunction, leading to cytochrome c release and caspase activation (5, 7). Mitochondrial GSH plays a central role in the control of mitochondrial ROS generation, thereby modulating the sensitivity to cell death pathways (7). Ethanol exposure has been shown to decrease the mitochondrial GSH levels preferentially in pericentral hepatocytes, where most of the hepatocyte apoptosis occurs (8). Restoration of GSH has been shown to inhibit ethanol-induced liver injury (25, 26). MT is a highly conserved, low–molecular-weight, thiol-rich protein (27, 28). MT shares an important similarity with GSH, due to the fact that one third of their amino acids are cysteine. Importantly, the thiol groups in MT are preferential attacking targets for free radicals compared with the other sulfhydryl residues (29, 30). Our previous study demonstrated that over-expression of MT in the liver of MT-transgenic mice conferred resistance to acute ethanol-induced liver injury (31). The present study showed that ethanol-induced decreases in mitochondrial GSH and MT are prevented by zinc supplementation. Correspondingly, the hepatic levels of ROS were suppressed by zinc supplementation. These data suggest that the antioxidant action of zinc may contribute to the inhibitory effect of zinc on ethanol-induced apoptosis. MT is a well known protein inducible by metals, especially by zinc. Our previous study showed that 3 doses of zinc injection at 5 mg/kg increased the MT concentration in the liver by 10-fold (32). However, long-term zinc supplementation in the present study only maintained the normal MT level in the liver. These results suggest that maintaining a normal level of MT in the liver by long-term zinc supplementation may play a critical role in the prevention of chronic alcoholic liver disease.
Death receptor–induced apoptosis, namely the extrinsic pathway, is also involved in ethanol-induced hepatic cell death; in particular, the TNF-R1/TNF-α and Fas/FasL systems are highly involved in this process (6). Increased TNF-α production has been well documented in patients with alcoholic hepatitis (33, 34), and elevated expression of TNF-R1 was also found in alcoholic hepatitis and in ethanol-exposed hepatocytes (3, 35). A TNF-R1 knockout mice model further confirmed that abrogating TNF-α signaling leads to a reduction of apoptosis after 4 weeks of ethanol exposure (13). Although TNF-α has been shown to play an important role in ethanol-induced hepatocyte apoptosis, our studies and others’ reports showed that TNF-α killing requires a prior sensitization of the liver or hepatocytes by ethanol, D-galactosamine or S-adenosylhomocysteine (36–39). In contrast, the liver is very sensitive to the killing by FasL/Fas signaling, and administration of Jo2 (a FasL agonist) has been shown to cause massive apoptosis in the liver (40). In patients with alcoholic hepatitis, the Fas and FasL expression in the liver was significantly increased (1, 3). Our previous studies demonstrated that acute ethanol intoxication increased the protein level of FasL in a mouse model and that neutralizing FasL with antibody inhibited ethanol-induced caspase activation and apoptosis (5, 6). Although the mechanisms underlying ethanol-induced up-regulation of FasL/Fas signaling have not been fully understood, in vitro studies demonstrated that ethanol and its metabolite, acetaldehyde, increase the Fas protein level and the FasL mRNA level, respectively, in hepatoma cells and hepatocytes isolated from alcoholic rats (41, 42). A recent study demonstrated that hepatocyte apoptosis induced by FasL/Fas signaling could be independent of CYP2E1 activity (43). In the present study, oxidative stress and up-regulation of TNF-α, TNF-R1, FasL, and Fas simultaneously occur in the liver after long-term ethanol exposure. These results suggest that blocking a single death pathway may not be sufficient to abrogate the hepatocyte apoptosis following a chronic ethanol exposure.
Many investigations have been aimed at exploring therapeutic agents that could prevent ethanol-induced hepatocyte apoptosis. Some agents including polyenylphosphatidylcholine (PPC), lycopene, dilinoleoylphosphatidylcholine (DLPC), and betaine have been found to be effective in the prevention of hepatocyte apoptosis induced by ethanol exposure in both in vivo and in vitro studies (12, 44–49). The anti-apoptotic effect of PPC, lycopene, and DLPC was associated with antioxidant action via down-regulating ethanol-mediated CYP2E1 induction. Betaine is known to regulate methionine metabolism and maintain the S-adenosylhomocysteine/homocysteine balance (50). One possible mechanism underlying the anti-apoptotic action of betaine is the inhibition of ethanol-elevated S-adenosylhomocysteine concentrations in the liver, which could sensitize TNF-α killing (51). Our previous studies showed that zinc pretreatment prevented hepatocyte apoptosis induced by acute ethanol intoxication and that the action of zinc was associated with inhibition of endotoxemia, hepatic TNF-α, and FasL protein levels (6). The finding in the present study further demonstrated that dietary zinc supplementation had inhibitory effects on all of the major pathways involved in ethanol-induced apoptosis, including ROS generation and up-regulation of death receptor–related genes and proteins, in particular, TNF-α/TNF-R1 and FasL/Fas.
The body weight gains of mice after 6 months of ethanol exposure were significantly lower in the present study. We observed that 129S6 mice can only tolerate up to 4.6% dietary ethanol, even with a feeding protocol of gradually increasing ethanol content, which is apparently lower than the commonly used 5.4% ethanol diet for C57BL6 mice and rats. At the late period of feeding, the ethanol-fed 129S6 mice developed seizure behavior. Ethanol exposure has been found to cause hypoglycemia and glucose tolerance, suggesting a disturbance of insulin secretion and glucose metabolism (52). We postulate that the reduced weight gain and the seizure behavior in ethanol-fed mice could result from the ethanol-altered glucose metabolism. On the other hand, the mice after 6 months of ethanol exposure showed a higher liver weight, which was from fat accumulation. Zinc supplementation did not improve the weight gain but significantly inhibited the ethanol-increased liver weight. The results suggest that zinc may have significant effect on lipid metabolism in the liver but not on glucose metabolism.
In conclusion, the present study provides evidence that hepatocyte apoptosis occurred in the liver after long-term ethanol exposure and was attenuated by zinc supplementation. The anti-apoptotic effect of zinc likely is through inhibition of oxidative stress and death-receptor signaling. These results suggest that dietary zinc supplementation may have beneficial effects on alcoholic liver disease.
Acknowledgments
This research was supported in part by the National Institutes of Health Office of Dietary Supplements grants, by the Intramural Research Program of the National Institutes of Health, by the National Cancer Institute Center for Cancer Research, and by the Veterans Administration, Louisville, KY.
We thank Xinguo Sun for technical assistance. YJK and CJM are Distinguished University Scholars of the University of Louisville.
References
- 1.Natori S, Rust C, Stadheim LM, Srinivasan A, Burgart LJ, Gores GJ. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol. 2001;34:248–253. doi: 10.1016/s0168-8278(00)00089-1. [DOI] [PubMed] [Google Scholar]
- 2.Ziol M, Tepper M, Lohez M, Arcangeli G, Ganne N, Christidis C, Trinchet JC, Beaugrand M, Guillet JG, Guettier C. Clinical and biological relevance of hepatocyte apoptosis in alcoholic hepatitis. J Hepatol. 2001;34:254–260. doi: 10.1016/s0168-8278(00)00047-7. [DOI] [PubMed] [Google Scholar]
- 3.Ribeiro PS, Cortez-Pinto H, Solá S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME, Rodrigues CM. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 4.Ramalho RM, Cortez-Pinto H, Castro RE, Solá S, Costa A, Moura MC, Camilo ME, Rodrigues CM. Apoptosis and Bcl-2 expression in the livers of patients with steatohepatitis. Eur J Gastroenterol Hepatol. 2006;18:21–29. doi: 10.1097/00042737-200601000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Z, Sun X, Kang YJ. Ethanol-induced apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3 activation pathway. Am J Pathol. 2001;159:329–338. doi: 10.1016/S0002-9440(10)61699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambert JC, Zhou Z, Kang YJ. Suppression of Fas-mediated signaling pathway is involved in zinc inhibition of ethanol-induced liver apoptosis. Exp Biol Med. 2003;228:406–412. doi: 10.1177/153537020322800411. [DOI] [PubMed] [Google Scholar]
- 7.Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front Biosci. 2005;10:3093–3099. doi: 10.2741/1765. [DOI] [PubMed] [Google Scholar]
- 8.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Cederbaum AI. Cytotoxicity and apoptosis produced by cytochrome P450 2E1 in HepG2 cells. Mol Pharmacol. 1998;53:638–648. doi: 10.1124/mol.53.4.638. [DOI] [PubMed] [Google Scholar]
- 10.Wu D, Cederbaum AI. Ethanol-induced apoptosis to stable HepG2 cell lines expressing human cytochrome P-4502E1. Alcohol Clin Exp Res. 1999;23:67–76. [PubMed] [Google Scholar]
- 11.Taieb J, Mathurin P, Poynard T, Gougerot-Pocidalo MA, Chollet-Martin S. Raised plasma soluble Fas and Fas-ligand in alcoholic liver disease. Lancet. 1998;351:1930–1931. doi: 10.1016/S0140-6736(05)78614-1. [DOI] [PubMed] [Google Scholar]
- 12.Seth D, Gorrell MD, Cordoba S, McCaughan GW, Haber PS. Intrahepatic gene expression in human alcoholic hepatitis. J Hepatol. 2006;45:306–320. doi: 10.1016/j.jhep.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Cederbaum AI. Acute ethanol pretreatment increases FAS-mediated liver injury in mice: role of oxidative stress and CYP2E1-dependent and -independent pathways. Free Radic Biol Med. 2007;42:971–984. doi: 10.1016/j.freeradbiomed.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McClain CJ, Antonow DR, Cohen DA, Shedlofsky S. Zinc metabolism in alcoholic liver disease. Alcohol Clin Exp Res. 1986;10:582–589. doi: 10.1111/j.1530-0277.1986.tb05149.x. [DOI] [PubMed] [Google Scholar]
- 16.Kang YJ, Zhou Z. Zinc prevention and treatment of alcoholic liver disease. Mol Aspects Med. 2005;26:391–404. doi: 10.1016/j.mam.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z, Wang L, Song Z, Saari JT, McClain CJ, Kang YJ. Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am J Pathol. 2005;166:1681–1690. doi: 10.1016/S0002-9440(10)62478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakatani T, Tawaramoto M, Opare Kennedy D, Kojima A, Matsui-Yuasa I. Apoptosis induced by chelation of intracellular zinc is associated with depletion of cellular reduced glutathione level in rat hepatocytes. Chem Biol Interact. 2000;125:151–163. doi: 10.1016/s0009-2797(99)00166-0. [DOI] [PubMed] [Google Scholar]
- 19.Cao J, Bobo JA, Liuzzi JP, Cousins RJ. Effects of intracellular zinc depletion on metallothionein and ZIP2 transporter expression and apoptosis. J Leukoc Biol. 2001;70:559–566. [PubMed] [Google Scholar]
- 20.Bao S, Knoell DL. Zinc modulates airway epithelium susceptibility to death receptor-mediated apoptosis. Am J Physiol Lung Cell Mol Physiol. 2006;290:L433–L441. doi: 10.1152/ajplung.00341.2005. [DOI] [PubMed] [Google Scholar]
- 21.Meerarani P, Ramadass P, Toborek M, Bauer HC, Bauer H, Hennig B. Zinc protects against apoptosis of endothelial cells induced by linoleic acid and tumor necrosis factor alpha. Am J Clin Nutr. 2000;71:81–87. doi: 10.1093/ajcn/71.1.81. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Z, Kang X, Jiang Y, Song Z, Feng W, McClain CJ, Kang YJ. Preservation of hepatocyte nuclear factor-4alpha is associated with zinc protection against TNF-alpha hepatotoxicity in mice. Exp Biol Med. 2007;232:622–628. [PubMed] [Google Scholar]
- 23.Bergmeyer HU, Bernt E. Glutamate-oxaloacetate transaminase: Colorimetric assay of Reitman and Frankel. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. 2nd. Vol. 2. New York: Academic Press; 1974. pp. 760–764. [Google Scholar]
- 24.Eaton DL, Cherian MG. Determination of metallothionein in tissues by cadmium-hemoglobin affinity assay. Methods Enzymol. 1991;205:83–88. doi: 10.1016/0076-6879(91)05089-e. [DOI] [PubMed] [Google Scholar]
- 25.Fermandez-Checa JC, Morales A, Colell A, Bellesta A, Rodes J, Kaplowitz N. S-adenosyl-L-methionine prevents both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dysfunction in periportal and perivenous rat hepatocytes. Hepatology. 1995;21:207–214. doi: 10.1002/hep.1840210133. [DOI] [PubMed] [Google Scholar]
- 26.Iimuro Y, Bradford BU, Yamashita S, Rusyn I, Nakagami M, Enomoto N, Kono H, Frey W, Forman D, Brenner D, Thurman RG. The glutathione precursor L-2-oxothiazolidine-4-carboxylic acid protects against liver injury due to chronic enteral ethanol exposure in the rat. Hepatology. 2000;31:391–398. doi: 10.1002/hep.510310219. [DOI] [PubMed] [Google Scholar]
- 27.Sato M, Bremner I. Oxygen free radicals and metallothionein. Free Radical Biol Med. 1993;14:325–337. doi: 10.1016/0891-5849(93)90029-t. [DOI] [PubMed] [Google Scholar]
- 28.Kang YJ. The antioxidant function of metallothionein in the heart. Proc Soc Exp Biol Med. 1999;222:263–273. doi: 10.1046/j.1525-1373.1999.d01-143.x. [DOI] [PubMed] [Google Scholar]
- 29.Thornalley PJ, Vasak M. Possible role for metallothionein in protection against radiation-induced oxidative stress. Kinetics and mechanism of its reaction with superoxide and hydroxyl radicals. Biochim Biophys Acta. 1985;827:36–44. doi: 10.1016/0167-4838(85)90098-6. [DOI] [PubMed] [Google Scholar]
- 30.Abel J, de Ruiter N. Inhibition of hydroxyl-radical-generated DNA degradation by metallothionein. Toxicol Lett. 1989;47:191–196. doi: 10.1016/0378-4274(89)90075-1. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Z, Sun X, Kang YJ. Metallothionein protection against alcoholic liver injury through inhibition of oxidative stress. Exp Biol Med. 2002;227:214–222. doi: 10.1177/153537020222700310. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Z, Sun X, Lambert JC, Saari JT, Kang YJ. Metallothionein-independent zinc protection from alcoholic liver injury. Am J Pathol. 2002;160:2267–2274. doi: 10.1016/S0002-9440(10)61174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McClain C, Hill D, Schmidt J, Diehl AM. Cytokines and alcoholic liver disease. Semin Liver Dis. 1993;13:170–182. doi: 10.1055/s-2007-1007347. [DOI] [PubMed] [Google Scholar]
- 34.McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D. Cytokines in alcoholic liver disease. Semin Liver Dis. 1999;19:205–219. doi: 10.1055/s-2007-1007110. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez DA, Moncada C, Nunez MT, Lavandero S, Ponnappa BC, Israel Y. Ethanol increases tumor necrosis factor-alpha receptor-1 (TNF-R1) levels in hepatic, intestinal, and cardiac cells. Alcohol. 2004;33:9–15. doi: 10.1016/j.alcohol.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Osawa Y, Nagaki M, Banno Y, Yamada Y, Imose M, Nozawa Y, Moriwaki H, Nakashima S. Possible involvement of reactive oxygen species in D-galactosamine-induced sensitization against tumor necrosis factor-alpha-induced hepatocyte apoptosis. J Cell Physiol. 2001;187:374–385. doi: 10.1002/jcp.1088. [DOI] [PubMed] [Google Scholar]
- 37.Song Z, Zhou Z, Uriarte S, Wang L, Kang YJ, Chen T, Barve S, McClain CJ. S-adenosylhomocysteine sensitizes to TNF-alpha hepatotoxicity in mice and liver cells: a possible etiological factor in alcoholic liver disease. Hepatology. 2004;40:989–997. doi: 10.1002/hep.20412. [DOI] [PubMed] [Google Scholar]
- 38.Song Z, Zhou Z, Song M, Uriarte S, Chen T, Deaciuc I, McClain CJ. Alcohol-induced S-adenosylhomocysteine accumulation in the liver sensitizes to TNF hepatotoxicity: possible involvement of mitochondrial S-adenosylmethionine transport. Biochem Pharmacol. 2007;74:521–531. doi: 10.1016/j.bcp.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Kang X, Jiang Y, Song Z, Feng W, McClain CJ, Kang YJ. Preservation of hepatocyte nuclear factor-4alpha is associated with zinc protection against TNF-alpha hepatotoxicity in mice. Exp Biol Med. 2007;232:622–628. [PubMed] [Google Scholar]
- 40.Galle PR, Hofmann WJ, Walczak H, Schaller H, Otto G, Stremmel W, Krammer PH, Runkel L. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med. 1995;182:1223–1230. doi: 10.1084/jem.182.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minana JB, Gomez-Cambronero L, Lloret A, Pallardo FV, Del Olmo J, Escudero A, Rodrigo JM, Pelliin A, Vina JR, Vina J, Sastre J. Mitochondrial oxidative stress and CD95 ligand: a dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology. 2002;35:1205–1214. doi: 10.1053/jhep.2002.32969. [DOI] [PubMed] [Google Scholar]
- 42.McVicker BL, Tuma DJ, Kubik JL, Tuma PL, Casey CA. Ethanol-induced apoptosis in polarized hepatic cells possibly through regulation of the Fas pathway. Alcohol Clin Exp Res. 2006;30:1906–1915. doi: 10.1111/j.1530-0277.2006.00235.x. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Cederbaum AI. Acute ethanol pretreatment increases FAS-mediated liver injury in mice: role of oxidative stress and CYP2E1-dependent and -independent pathways. Free Radic Biol Med. 2007;42:971–984. doi: 10.1016/j.freeradbiomed.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mak KM, Wen K, Ren C, Lieber CS. Dilinoleoylphosphatidylcholine reproduces the antiapoptotic actions of polyenylphosphatidylcholine against ethanol-induced hepatocyte apoptosis. Alcohol Clin Exp Res. 2003;27:997–1005. doi: 10.1097/01.ALC.0000071901.62432.09. [DOI] [PubMed] [Google Scholar]
- 45.Katz GG, Shear NH, Malkiewicz IM, Valentino K, Neuman MG. Attenuation of alcohol-induced apoptosis of hepatocytes in rat livers by polyenylphosphatidylcholine (PPC) Alcohol Clin Exp Res. 2000;24:207–212. [PubMed] [Google Scholar]
- 46.Xu Y, Leo MA, Lieber CS. Lycopene attenuates alcoholic apoptosis in HepG2 cells expressing CYP2E1. Biochem Biophys Res Commun. 2003;308:614–618. doi: 10.1016/s0006-291x(03)01435-9. [DOI] [PubMed] [Google Scholar]
- 47.Xu Y, Leo MA, Lieber CS. DLPC attenuates alcohol-induced cytotoxicity in HepG2 cells expressing CYP2E1. Alcohol Alcohol. 2005;40:172–175. doi: 10.1093/alcalc/agh142. [DOI] [PubMed] [Google Scholar]
- 48.Cabrales-Romero Mdel P, Márquez-Rosado L, Fattel-Fazenda S, Trejo-Solís C, Arce-Popoca E, Alemán-Lazarini L, Villa-Treviño S. S-adenosyl-methionine decreases ethanol-induced apoptosis in primary hepatocyte cultures by a c-Jun N-terminal kinase activity-independent mechanism. World J Gastroenterol. 2006;12:1895–1904. doi: 10.3748/wjg.v12.i12.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barak AJ, Beckenhauer HC, Mailliard ME, Kharbanda KK, Tuma DJ. Betaine lowers elevated s-adenosylhomocysteine levels in hepatocytes from ethanol-fed rats. J Nutr. 2003;133:2845–2848. doi: 10.1093/jn/133.9.2845. [DOI] [PubMed] [Google Scholar]
- 50.Purohit V, Abdelmalek MF, Barve S, Benevenga NJ, Halsted CH, Kaplowitz N, Kharbanda KK, Liu QY, Lu SC, McClain CJ, Swanson C, Zakhari S. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: summary of a symposium. Am J Clin Nutr. 2007;86:14–24. doi: 10.1093/ajcn/86.1.14. [DOI] [PubMed] [Google Scholar]
- 51.Kharbanda KK, Rogers DD, 2nd, Mailliard ME, Siford GL, Barak AJ, Beckenhauer HC, Sorrell MF, Tuma DJ. Role of elevated S-adenosylhomocysteine in rat hepatocyte apoptosis: protection by betaine. Biochem Pharmacol. 2005;70:1883–1890. doi: 10.1016/j.bcp.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 52.Bunout D. Nutritional and metabolic effects of alcoholism: their relationship with alcoholic liver disease. Nutrition. 1999;15:583–589. doi: 10.1016/s0899-9007(99)00090-8. [DOI] [PubMed] [Google Scholar]