

Abstract
We are developing whole, heat-killed, recombinant Saccharomyces cerevisiae yeast, engineered to encode target proteins, which stimulate immune responses against malignant cells expressing those targets. This phase 1 trial, enrolling patients with advanced colorectal or pancreas cancer, was designed to evaluate safety, immunogenicity, response, and overall survival of ascending doses of the GI-4000 series of products, which express 3 different forms of mutated Ras proteins. The study enrolled 33 heavily pretreated subjects (14 with pancreas and 19 with colorectal cancer), whose tumors were genotyped before enrollment to identify the specific ras mutation and thereby to identify which GI-4000 product to administer. No dose limiting toxicities were observed and no subject discontinued treatment due to a GI-4000 related adverse event (AE). The majority of AEs and all fatal events were due to underlying disease progression and AE frequencies were not significantly different among dose groups. GI-4000 was immunogenic, as Ras mutation-specific immune responses were detected on treatment in ∼60% of subjects. No objective tumor responses were observed but based on imaging, clinical status and/or biochemical markers, stable disease was observed in 6 subjects (18%) on day 29, while 1 subject had stable disease at days 57 and 85 follow-up visits. The median overall survival was 3.3 months (95% confidence interval, 2.3–5.3 mo), and 5 subjects survived past the 48-week follow-up period. No significant dose-dependent trends for survival were observed. This first clinical trial in humans with GI-4000 demonstrated a favorable safety profile and immunogenicity in the majority of subjects.
Key Words: Saccharomyces cerevisiae, yeast, ras, pancreas cancer, colon cancer
Cancer immunotherapy and vaccines use a variety of strategies to generate immune responses against malignant cells1–3 and several of these approaches are being tested as promising therapeutic interventions4–9 either singly or in combinations.8,9 However, conventional vaccines and immunotherapies have generally been more effective at activating the humoral immune system, but less effective at generating cell-mediated responses, particularly CD8+ cytotoxic T lymphocytes, which are required to kill cancer cells. In addition, many earlier tumor vaccines and immunotherapy approaches have proven unsuccessful due to neutralization by antibodies, complexity of manufacturing, and the need for patient-specific products.
The ras oncogene, and its Ras protein gene product, contain the most common oncogene-related mutations in human cancer, with 30% of all human tumors harboring mutant Ras proteins.10–12 Ras mutations are very frequent in many solid tumors including colorectal, pancreas, ovarian, non–small cell lung cancer (NSCLC) and malignant melanoma. Ras oncoproteins are ideal candidates for cancer immunotherapy because aberrant signaling through Ras contributes to uncontrolled cell proliferation and tumorigenesis. Furthermore, mutations in the ras oncogene occur in conserved locations, specifically codons 12, 13, and 61,13 and the number of mutations that can occur at each codon is limited to a few predominant amino acid substitutions.
Tarmogen (targeted molecular immunogen) technology is an immunotherapy platform based on the use of whole, heat-killed, recombinant Saccharomyces cerevisiae yeast as vectors that are engineered to express one or more target protein antigens. These yeast-based products activate dendritic cells14–16 and generate T cell immune responses that kill target cells expressing a number of viral and cancer antigens in human and murine models14–22 and in an antigen-specific CD8+ T cell-mediated manner.14,18,19,22 Tarmogens are efficiently manufactured, are not neutralized by antibodies, and therefore can be used for both priming and boosting, and do not require a subject-specific approach.
The GI-4000 product series consists of 4 different yeast-based products. Three products were available for use in the study described herein: GI-4014, GI-4015, and GI-4016, which target the 3 most common mutations at codon 12 in the ras gene and the 2 most common mutations at codon 61.17 All of these mutations result in constitutive activation of Ras and therefore unregulated activation of those growth factor pathways in which Ras is a second messenger, such as the epidermal growth factor pathway.
Products from the GI-4000 series have previously been shown to elicit remission of tumors in animal models and to do so in a mutation-specific manner.17 Improved responses occurred with repeated dosing, confirming that the yeast vector was not neutralized by the host immune system. These findings support the use of the GI-4000 series for priming and boosting of the immune response, and show that the approach elicits specific and robust immune-mediated control of diseased cells in animal models of cancer.
Because of the central role for Ras activation in tumor proliferation, targeted destruction of cells harboring mutant Ras proteins with GI-4000 could result in therapeutic benefit in a broad range of human cancers. The current first-in-human study was therefore designed to evaluate the GI-4000 series with regard to safety, immunogenicity, disease progression, and overall survival of patients with late stage pancreas, colorectal or NSCLC. Before enrollment, patient tumors were first genotyped to establish the presence/identity of ras mutations and thereby identify which GI-4000 yeast product would activate T cell immunity against the matched mutant Ras protein (ie, GI-4014, GI-4015 or GI-4016). A dose-escalation scheme was also included in the trial to establish a maximum tolerated dose (MTD). The results of this first clinical trial in humans with GI-4000 demonstrated a favorable safety profile, with positive immunogenicity results.
MATERIALS AND METHODS
Study Design
This study was a first-in-human, phase I, open-label, nonrandomized, multicenter, dose-escalation trial in subjects with a diagnosis of an advanced stage solid tumor known to harbor frequent mutations in ras (pancreas, colorectal, and NSCLC). The trial was conducted at 4 investigational sites in the United States.
Following informed consent, subjects who met inclusion and exclusion criteria had tumor tissue from biopsy, fine-needle aspiration or surgical resection specimens subjected to genomic sequencing of the ras gene. Subjects with mutations at either the codon 12 or 61 positions, which were represented in one of the GI-4000 products (GI-4014: G12V, Q61L, and Q61R; GI-4015: G12C, Q61L, and Q61R; or GI-4016: G12D, Q61L, and Q61R), were eligible for study enrollment. Eligible subjects were also required to meet the following inclusion criteria: have progressive and radiographically evaluable disease with evidence of recovery from previous chemotherapy, Eastern Cooperative Oncology Group (ECOG) performance score (PS) of 0 or 1 and expected survival >6 months. A tumor was considered adequately evaluable if the Response Evaluation Criteria in Solid Tumors (RECIST 1.0)23 were met, or if the presence of a reliable biomarker was adequate to define response, stability or progression.
Additional eligibility criteria evaluated before enrollment included absolute neutrophil count of >1500 cells/mm3, platelet count of >100,000/mm3, hematocrit of >30%, and alanine aminotransferase/aspartate aminotransferase levels <3 times the upper limit of normal. In addition, a negative urine or serum pregnancy test was required for women of child-bearing potential. A full medical history and a physical examination were both conducted before day 1 and ECOG PS were recorded. Blood was drawn for laboratory tests, biochemical tumor markers and immunologic assays, and skin testing was performed for immediate hypersensitivity to S. cerevisiae and delayed-type hypersensitivity to recall antigens Candida albicans and Trichophyton.
Objectives
The primary objective of the study was to evaluate the safety of weekly administration of ascending doses of GI-4000 for 28 days in subjects with solid tumors expressing mutations in the ras oncoprotein. Secondary objectives were to evaluate the following: immunogenicity as measured by lymphocyte proliferation assay (LPA) and by other measures of immune function; effect on disease progression; and effect on overall survival.
Variables
Demographic and baseline characteristics included age, sex, ethnic origin, weight, height, vital signs, time since diagnosis, number of previous chemotherapy regimens, tumor type, stage and grade, tumor imaging result, tumor biomarker levels, and codon position 12 or 61 ras gene mutations. Efficacy was evaluated on days 29, 57, and 85 unless disease progression had occurred or subsequent anticancer therapy had started. The following efficacy variables were assessed: tumor biomarker levels in serum; tumor response as defined by RECIST; time to disease progression, defined as the time from study drug start to the first observation of disease progression; and overall survival, defined as the time from study drug start until death due to any cause. Safety was assessed before administration of each dose and on days 57 and 85 by evaluating adverse events (AEs), serious adverse events (SAEs), safety laboratory assessments, physical examinations and vital signs. Blood for immunological assays was obtained before administration of each dose and on days 57 and 85. For complement activation assays (Bb, C3a, and C5a assays) blood samples were obtained predose and 1 hour postdose on days 1 and 15.
Tumor Tissue Sequencing
Cellular genomic DNA was extracted from biopsy material and amplified by polymerase chain reaction (PCR) using DNA primers flanking exon 1 (containing codon 12 sequences) and exon 2 (codon 61 sequences) of K-ras, H-ras, and N-ras genes. Amplified PCR products were resolved on agarose gels, then extracted and subjected to double-stranded DNA sequence analysis to identify ras mutations.
Interventions
The study drug, GI-4000 (GlobeImmune Inc.), is an investigational product being developed under an IND application with the US FDA. GI-4000 consisted of 3 different yeast-based immunotherapy products targeting the 3 most common mutations at codon 12 in the ras gene and the 2 most common mutations at codon 61 (GI 4014: G12V, Q61L, and Q61R; GI-4015: G12C, Q61L, and Q61R; GI-4016: G12D, Q61L, and Q61R). The version of GI-4000 containing a specific ras mutation identified in a particular subject’s tumor was selected for use. The yeast strains were engineered to express the ras mutants as previously described,17 using consensus sequences lacking the variable carboxyl-terminal signaling domains that distinguish the different ras family members (K-ras, N-ras, and H-ras).
Subjects received 5 weekly doses of study drug on days 1, 8, 15, 22, and 29 and underwent in-clinic follow-up assessments on days 57 and 85. Thereafter, subjects were followed for a total of 48 weeks after enrollment with monthly telephone contacts to assess long-term tolerance to study drug via AEs and overall health. To avoid the immunosuppressive effects of simultaneous chemotherapy, subsequent chemotherapy was withheld until the end of study treatment (day 29).
At least 3 subjects were assigned to the following ascending dose levels of GI-4000 on a cohort basis: 0.1, 0.3, 1.0, 10, 20, and 40 YU (yeast units; 1 YU=1×107 yeast). Study drug was administered as a subcutaneous injection into the lateral upper arm alternated between left and right arms at sequential visit days. The 20 YU and 40 YU dose levels were split into multiple doses. In the 20 YU dose level, an injection of 10 YU was given into each upper arm for a total of 20 YU. In the 40 YU dose level, an injection of 10 YU was given into each upper arm and each upper thigh for a total of 40 YU. Escalation to the subsequent dose level was initiated after at least 3 subjects had received 5 weekly doses and were observed for at least 1 week posttreatment for dose limiting toxicity (DLT). Dose escalation was to be continued either until the maximum target dose of 40 YU was reached or until DLT was observed. DLT was defined as any grade 3 or 4 AE that was possibly, probably or definitely study drug related; or a grade 3 injection site reaction. All toxicity was graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 3.0.
Immunology
LPA
Peripheral blood was collected in the presence of heparin at baseline, immediately before each product administration and also on days 57 and 85. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque density gradient centrifugation and washed by centrifugation 3 times in Dulbecco’s phosphate buffered saline. To evaluate levels of T cell activation in subjects treated with GI-4000, freshly isolated PBMCs (2×105 per well) were cultured in Roswell Park Memorial Institute/5% heat-inactivated human AB+ serum in 96 U-well microtiter plates for 6 days in a humidified 5% CO2 incubator at 37°C in the presence of Ras peptide pools of 19 or 20 amino residues in length containing the relevant mutations at Ras position 12 or 61. Each pool contained 3–5 peptides, with a final concentration of each peptide in the assay of 2 μg/mL. Stimulation with the T cell mitogen, phytohemagglutinin, 5 μg/mL, and with C. albicans yeast extracts (Greer, Lenoir, NC), 20 μg/mL, were used as positive controls of proliferative activity and medium alone was used as a negative control. All conditions were set up in replicates of 3 or 4 as permitted based on the recovery of harvested PBMCs. Tritiated thymidine was added 24 hours before harvest and thymidine incorporation was measured by liquid scintillation.
A result was considered positive if any of the antigen-specific conditions had a stimulation index (SI) of at least 2-fold over the appropriate control condition. A subject was deemed a responder if an SI ≥2 was exhibited for at least 1 postbaseline timepoint. In addition, for all conditions where an SI ≥2 was obtained, a Student t test was applied to ascertain statistical significance of the response. Extreme outliers (identified by the extreme Studentized deviate method24) were removed from the analysis.
Exploratory Gene Expression Analysis
PBMCs from days 1 (pretreatment), 8, 15, 22, and 29 were collected as described above and cryopreserved in liquid nitrogen until the day of use. Thawed PBMCs were plated at 106 cells/well in 96-well plates in Roswell Park Memorial Institute/10% human AB serum and cells were incubated overnight before stimulation. Peptide pools were added at 2 μg/mL final concentration (V12 20-mer peptide pool) or a peptide pool consisting of CD8+ T cell epitopes from cytomegalovirus, Epstein-Barr virus and influenza virus (CEF).25 Staphylococcal enterotoxin B (Sigma-Aldrich) at 1 μg/mL was added as a positive control and medium alone was used as a negative (untreated) control. After 6 hours, the cells were harvested, RNA was extracted and then analyzed by real-time quantitative PCR analysis (performed by Source MDx, Boulder, CO) using primers specific for interferon (IFN)-γ, granulocyte-macrophage colony stimulating factor (GM-CSF), tumor necrosis factor (TNF)-α and GAPDH, which was used as an internal control.
The averages of the cycle threshold number for triplicate samples for IFN-γ, GM-CSF or TNF-α cDNA were reported as the relative fold increase in mRNA expression compared with the untreated control using the 2-∆∆ cycle threshold method.
Complement Activation Assays
Complement activation assays were performed at the Complement Analysis Testing Facility, National Jewish Health, Denver, CO. Plasma was collected predose and 1 hour postdose on days 1 and 15 and shipped on dry ice to the testing facility. Samples were stored at −70°C until assayed. The split products, factor Bb, C3a and C5a, were measured by enzyme-linked immunobsorbent assays that utilized monoclonal antibodies against the specific human split product (Quidel Corporation, San Diego, CA). Standards, controls and test specimens were tested in duplicate.
Sample Size
As this was a phase I study primarily designed to evaluate safety and immunogenicity, formal power calculations would not have been meaningful and, therefore, were not performed. The study was sized to allow for at least 3 subjects to be enrolled at each dose level before dose escalation, in order to give adequate experience at each dose level. The study targeted a maximum of 20–30 subjects in the event that the highest dose (40 YU) was reached.
Statistical Methods
Demographic and baseline characteristics were listed for all subjects and summarized by treatment. The baseline value of a variable was the last value recorded before the first dose of study drug.
All subjects who received at least 1 dose of GI-4000 were included in the tumor biomarker analyses. All subjects who had at least 1 measurable lesion at baseline and received at least 1 dose of GI-4000 were considered evaluable for response. Subjects had their response classified as complete response, partial response, stable disease (SD) or progressive disease according to the RECIST 1.0 definitions23 or to biomarker or clinical assessment. The response rate was estimated as the proportion of subjects evaluable for response who met the criteria for complete response or partial response.
All subjects who received at least 1 dose of GI-4000 were included in the time to disease progression and overall survival analyses. Time to disease progression was defined as the time from study drug start to the first observation of disease progression. Subjects whose disease had not progressed at the final analysis were censored at their last contact date.
Overall survival was defined as the length of time from study drug start until death due to any cause. Subjects who were alive at the final analysis were censored at their last contact date. Kaplan-Meier curves of time to disease progression and overall survival were generated, and 95% confidence intervals for the median times to disease progression and overall survival were computed using the method of Brookmeyer and Crowley,26 as implemented in PROC LIFETEST in SAS, Version 8.2.
All subjects who received at least 1 dose of GI-4000 were included in the safety analyses. Safety data, including AEs, SAEs, discontinuations due to AEs, laboratory findings, concomitant medications and other safety evaluations were listed and summarized descriptively. The MTD also was identified. The MTD was defined as the highest dose without DLT.
Each NCI CTCAE term (or, if no term were available, the Investigator verbatim) was coded using MedDRA, Version 8.0 preferred terms, and summarized by dosage of study drug and worst severity grade. Treatment-emergent AEs (referred to as AEs) were defined as any abnormal clinical observation that occurred on study drug, which was not present before exposure to study drug, or any clinical sign or symptom that worsened in either intensity or frequency after exposure to study drug. AEs occurring after administration of the first dose of study drug through study termination were summarized by body system, preferred term within each body system, dosage of study drug, and CTCAE severity, and relationship to study drug (possibly, probably or definitely related) according to the Investigator. The following data were also summarized: SAEs regardless of causality and SAEs considered related to study drug, deaths on study or in the follow-up period, and the reason for study discontinuation, including AEs resulting in study discontinuation.
Results of laboratory data were listed by subject and values outside the reference ranges were flagged. Descriptive summaries of selected laboratory test parameters were generated by dose level. Laboratory test abnormalities comparing baseline and worst NCI CTCAE grades on study were performed. Vital sign measurements at each timepoint were listed by subject. If applicable, vital signs were analyzed descriptively by dose level.
Interim Analysis
No formal interim analyses were planned and none was conducted. GlobeImmune’s Medical Monitor conducted reviews of safety and tolerability parameters on an ongoing basis during the study. Reviews of safety labs and evaluation of AEs for each subject were completed before dose escalation.
RESULTS
Participants
Study disposition is diagramed in Figure 1. The planned sample size for this study was 20–30 subjects, with a follow-up time of 48 weeks. From the 169 screened subjects, 33 subjects were enrolled from 4 US sites between August 2004 and March 2006. The last subject’s final visit occurred on June 12, 2006 and database lock occurred on February 10, 2009. More than 3 subjects were enrolled in most cohorts, as some subjects withdrew early from therapy and were replaced for accurate toxicity assessment of the dose level. In addition, because this was a multicenter study, some investigational sites enrolled subjects on the same date, thus increasing enrollment in some dose groups.
FIGURE 1.
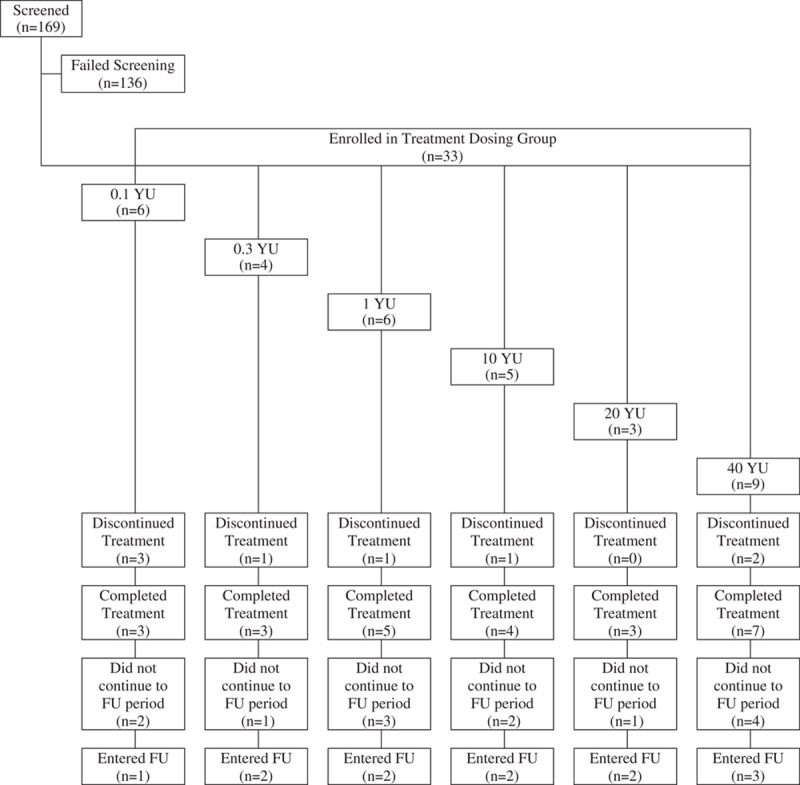
Subject disposition. FU indicates follow-up; YU, yeast units.
A total of 25 subjects (76%) completed all 5 subcutaneous injections of GI-4000. In each cohort, at least 3 subjects completed the protocol-specified treatment course of 5 weekly injections and 7 of 9 subjects at the highest dose (40 YU) received all 5 injections. The 20 and 40 YU dose groups were added by protocol amendment since subjects receiving the 1 YU dose showed no evidence of DLTs and, in addition, there was preliminary evidence of antigen-specific immune responses even at the lowest dose group (0.1 YU).
The majority of treatment discontinuations (6/8 subjects, 75%) were due to study withdrawal because subjects were thought to be unlikely to survive (including 1 subject for treatment failure/lack of efficacy). Other reasons were the AE of increased fatigue (1 subject) and death (1 subject), both of which were deemed unrelated to study treatment.
A total of 12 subjects (36%) participated in the follow-up phase of the trial. The main reason for not participating in follow-up was because subjects were thought to be unlikely to survive (13/21 subjects, 62%). Additional reasons were: consent withdrawal (2 subjects), lost to follow-up (2 subjects), death (1 subject), AE of increased fatigue (1 subject) and investigational site errors (2 subjects).
Baseline Characteristics
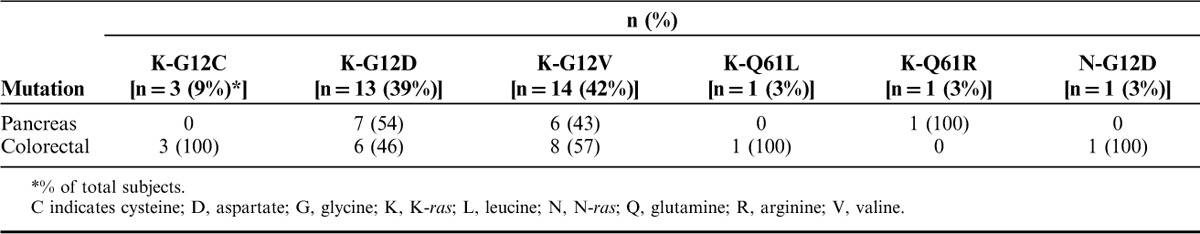
Table 1 summarizes the baseline demographic characteristics for all subjects by dose group. The mean age for subjects was 61 years, 61% were male and the majority were white (82%). In addition, baseline ECOG PS was 0–1 for all subjects except 1, and subjects had either colorectal cancer (58%) or pancreas cancer (42%); none of the subjects screened with NSCLC had the necessary ras mutations represented in one of the versions of GI-4000. All subjects had product-related K-ras mutations except for one subject with a G12D N-ras mutation. As shown in Table 2, the most frequently observed mutations were G12V (42%) and G12D (42%, including 1 subject with an N-ras mutation), with an incidence that was almost evenly distributed between subjects with pancreas and colorectal cancer. All subjects had advanced disease at study entry and had received at least one previous chemotherapy regimen. Subjects with colorectal cancer (n=19) received more prior therapies than subjects with pancreas cancer (n=14), as would be expected due to the poorer prognosis and shorter survival of subjects with pancreas cancer. Although the eligibility criteria allowed only 1–3 prior regimens, the median number of prior systemic regimens for subjects with colorectal cancer was 3 (range from 2 to 11) and the median number of prior systemic regimens for subjects with pancreas cancer was 2 (range from 1 to 4).
TABLE 1.
Summary of Baseline Demographic Characteristics for All Enrolled Subjects
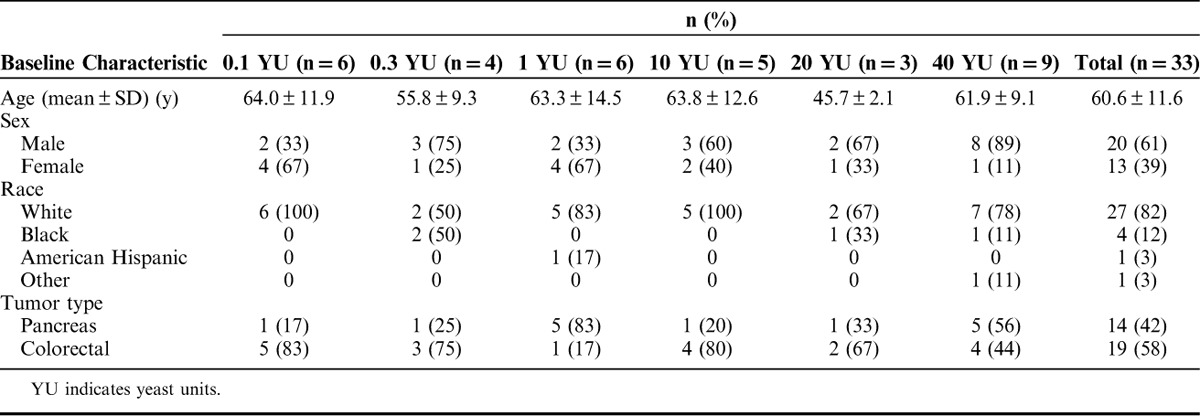
TABLE 2.
Baseline Summary of Product-related Tumor Ras Mutations for Enrolled Subjects by Cancer Type
Safety
Given that at least 3 subjects in each cohort received all 5 injections, the safety of GI-4000 could be fully assessed in this study. Table 3 summarizes AEs occurring in at least 10% of subjects. The dose escalation was successfully performed without the occurrence of any DLTs throughout the study, and none of the subjects discontinued treatment due to a GI-4000 related AE. In addition, none of the deaths within 30 days of the last dose was considered related to study drug, and none of the SAEs was considered related to GI-4000.
TABLE 3.
Adverse Events Occurring in At Least 10% of Total Number of Subjects Regardless of Causality
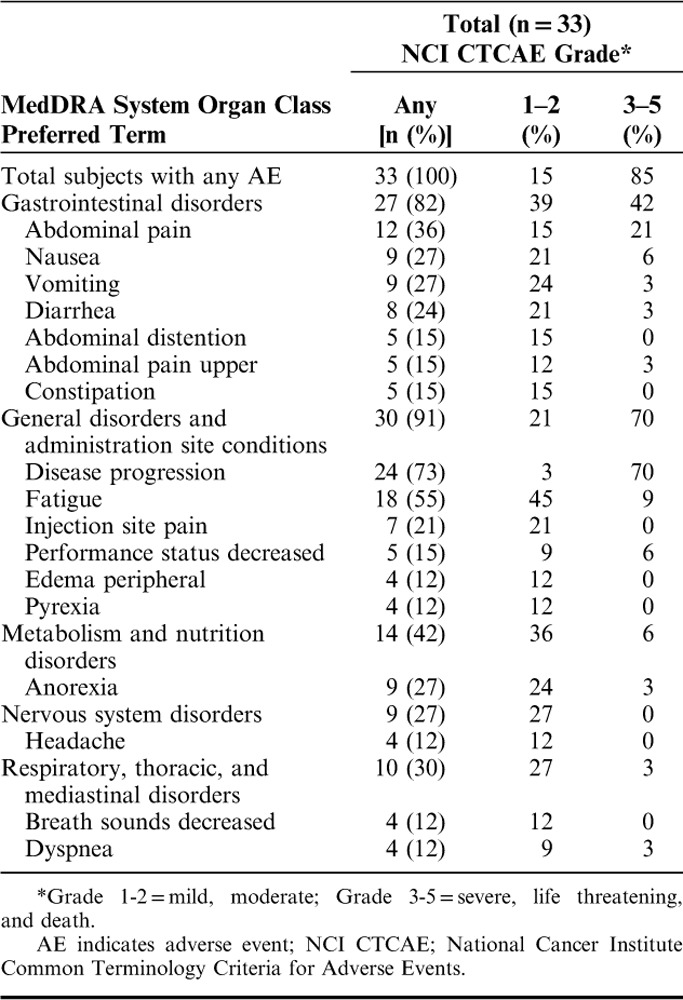
In each dose cohort, all subjects experienced one or more AEs regardless of causality. A total of 85% of subjects experienced grade 3–5 AEs, with 49% experiencing grade 3 events, 9% grade 4 events and 27% fatal events. Disease progression was, however, captured and reported as an AE and contributed to the high number of subjects with grade 3–5 AEs. As expected in this population of subjects with advanced cancer, the underlying disease was the main contributing factor for many grade 3–5 events, from fatigue to gastrointestinal symptoms, such as abdominal pain, nausea, vomiting, diarrhea, and constipation. The incidence of these AEs did not increase with increasing dose levels of GI-4000.
Injection site-related AEs were all mild in intensity, and the incidence was associated with receiving the highest local dose of 10 YU GI-4000 per injection site (10, 20, and 40 YU dose groups).
A total of 8 subjects (24%) experienced AEs during the poststudy period. None of the AEs in this reporting period was considered to be at least possibly related to GI-4000, but all were considered expected for subjects with advanced cancer.
SAEs were reported for 31 subjects (94%), with the worst severity being grade 3 in 67% and grade 5 in 27% of subjects. None of the SAEs was deemed to be related to study treatment. Most events were sporadic occurrences that were to be expected in a subject population with advanced and symptomatic pancreas and colon cancer (eg, gastrointestinal symptoms, thrombotic events). All 9 reported fatal events (27%) were due to disease progression. The incidence of SAEs regardless of causality was not significantly different for the respective dose groups.
The effect of GI-4000 on hematology, chemistry, urinalysis, physical examinations, and vital signs was not clinically significant.
Immunogenicity
A total of 31 subjects were evaluated by LPA, which is primarily a measure of antigen-specific CD4+ T cell activation. Nineteen of 31 subjects (61%) responded to Ras peptide stimulation in LPA either by maintenance or improvement in their baseline response or by a treatment-emergent response. A summary of the data is shown in Tables 4 and 5 and responses for 14 individual subjects are shown in Figure 2. Eight subjects (26%) exhibited detectable Ras peptide antigen-specific responses at baseline before treatment, and all but one of these subjects (in the 1 YU group) sustained or improved the response for at least 1 timepoint following GI-4000 administration (7/8, 87%). Of those who were negative at baseline (n=23), 12 subjects developed antigen-specific LPA responses following GI-4000 therapy (12/23, 52%). There were LPA responses across all dose groups with no obvious effect of dose on the response although the 40 YU group had the lowest frequency of responders (29%). However, approximately one third (13/40) of the PBMC samples tested from 40 YU group subjects did not proliferate in response to C. albicans indicating a diminished immunocompetence, and probably reflecting the overall health status of these subjects. In contrast, PBMC samples from subjects in all other groups had far fewer negative responses to C. albicans [14% (4/28), 20% (4/20), 12% (4/32), 18% (5/28) and 0% (0/18) for 0.1, 0.3, 1, 10 and 20 YU groups, respectively].
TABLE 4.
Summary of Immune Responders: Overall Immune Responders
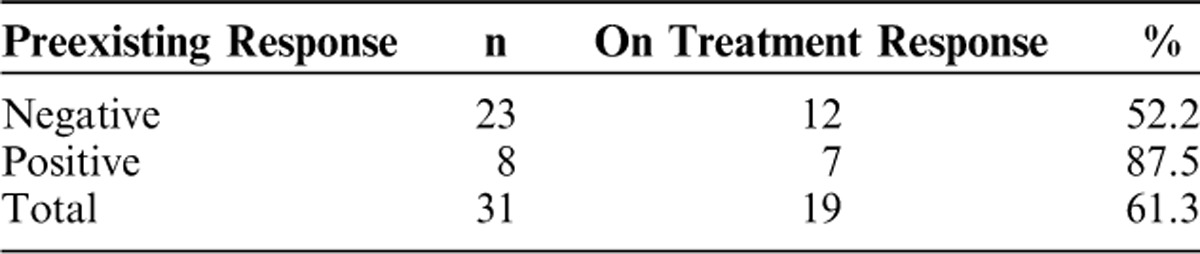
TABLE 5.
Summary of Immune Responders: Immune Responders by Dose Group

FIGURE 2.
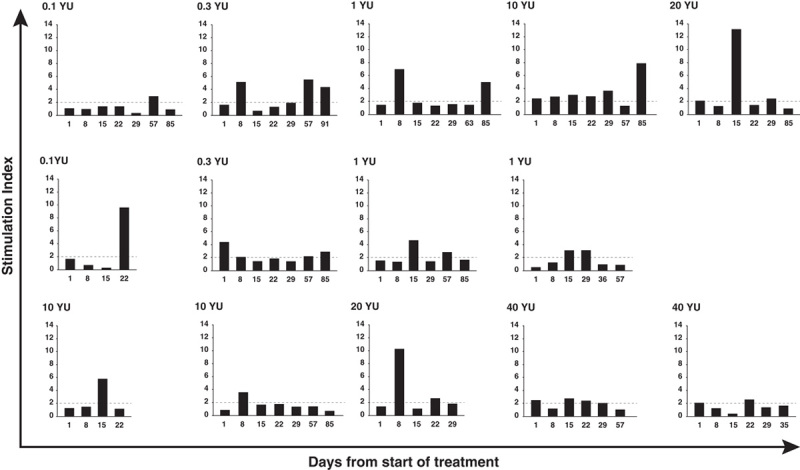
Representative lymphocyte proliferation assay (LPA) responses. LPA responses are shown as stimulation indices for 3H-thymidine uptake in peripheral blood mononuclear cells following stimulation with Ras peptide pools at baseline (day 1) and at up to 6 timepoints following initiation of treatment with GI-4000. Dotted line indicates a stimulation index of 2, the cut off for responsiveness. Fourteen of 19 subjects with proliferative responses ≥2 are shown. The additional 5 subjects deemed responders by LPA are not shown either because only 1 on treatment sample day was available (n=3) or because the baseline response was not exceeded by the on treatment responses (n=2). Top row: responses of 5 long-term survivors who survived beyond the 48 week follow-up period. YU indicates yeast unit.
One subject in the 20 YU group (with pancreas cancer), who survived past the 48-week follow-up period (as shown), was additionally evaluated by an exploratory cytokine gene expression assay in which PCR amplification was used after ex vivo stimulation of PBMCs with Ras G12V peptide pools. Treatment emergent increases in GM-CSF mRNA expression were observed (Fig. 3A) following initiation of treatment with GI-4000. In contrast, when PBMCs were stimulated with a CEF peptide pool, high GM-CSF mRNA expression was seen at baseline but expression did not increase following GI-4000 treatment (Fig. 3B). TNF-α and IFN-γ mRNA expression increased to a lesser extent on treatment in response to Ras peptide pool stimulation and IFN-γ mRNA expression was also increased at day 8 following stimulation with the CEF peptide pool but thereafter did not exceed baseline levels (Figs. 3A, B). This subject was also a responder by LPA (Fig. 2: top row, far right).
FIGURE 3.
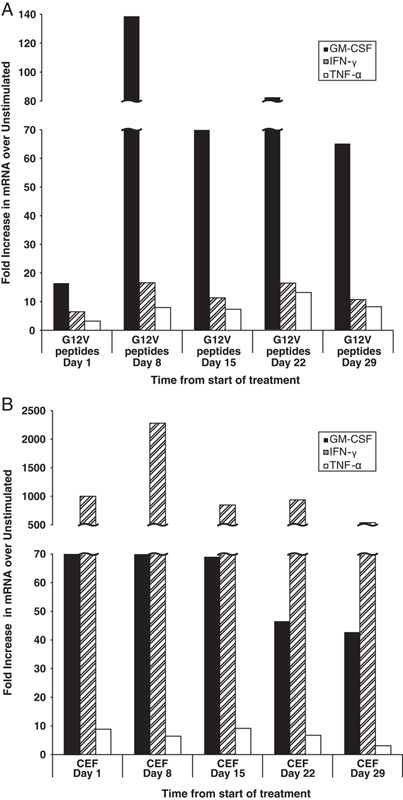
mRNA expression in peripheral blood mononuclear cells restimulated with Ras peptide pools. One subject in the 20 YU group (with G12V ras mutation) survived beyond the 48 week follow-up period. Bars show relative increases in expression of GM-CSF, IFN-γ, and TNF-α mRNA compared with unstimulated controls following stimulation of peripheral blood mononuclear cells with Ras G12V peptide pools (A) or CEF peptides (B) at 2 µg/mL final concentration. GM-CSF indicates granulocyte-macrophage colony stimulating factor.
Complement Activation
Complement activation was carried out as a safety assessment to evaluate whether GI-4000 activated the complement pathway. Although there were individual subjects who appeared to have dose-related changes in their complement split products, there was no consistent pattern that could be attributed either to activation or to decreased levels of complement fragments after treatment (data not shown). Of note, 1 subject (0.1 YU group) had greatly elevated C3a (13,777 ng/mL) and C5a (33.05 ng/mL) on day 15 before receiving study drug but these values dropped to 621 and 10.59 ng/mL, respectively, posttreatment.
Efficacy
Analyses of efficacy were performed using the intent-to-treat population, which included all 33 subjects enrolled.
Tumor response was assessed using RECIST, biomarkers or clinical evaluation by the investigator and an evaluation of response was performed for all subjects on days 29, 57, and 85, unless disease progression had occurred or subsequent anticancer therapy had started. No objective tumor responses were observed, but based on radiographic imaging, clinical status, and/or biochemical markers, SD was observed in 6 subjects (18%) on day 29, while 1 subject (3%) in the 20 YU dose group had SD at the day 57 and 85 visits.
No significant trends for the time to disease progression were observed based on the analyses per dose group.
Overall survival was defined as the length of time from the start of study drug until death due to any cause. Subjects alive at the end of study were censored in the survival analysis. Table 6 summarizes the subject deaths at database lock: 31 deaths (94%) had been confirmed in the overall population, while the status of 2 subjects (6%) could not be confirmed. After database lock, a search of the Social Security Death Registry confirmed date of death for both subjects. The median overall survival, estimated from the univariate Kaplan-Meier curves (Fig. 4), was 3.3 months (95% confidence interval, 2.3–5.3 mo). There was a negligible difference in median overall survival between subjects with colorectal and pancreas cancer (98 vs. 90 d, respectively) mainly due to 1 long-term survivor with pancreas cancer in the 20 YU group (as previously shown). Five subjects survived past the 48-week follow-up period, including 1 subject whose disposition at time of database lock could not be confirmed, and all 5 subjects exhibited antigen-specific LPA responses on treatment (see Fig. 2, top row). No significant dose-dependent trends for survival were observed.
TABLE 6.
Summary of Subject Deaths by Dose Group and Cancer Type

FIGURE 4.
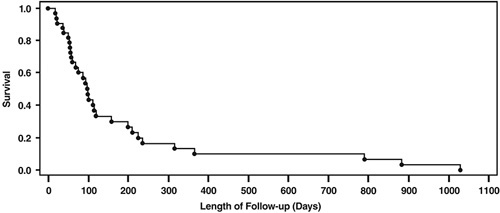
Overall survival based on Kaplan-Meier analyses of all enrolled subjects.
DISCUSSION
This study was conducted to characterize the safety profile, immunogenicity and effect on disease progression of ascending doses of GI-4000 in subjects with solid tumors expressing mutations in the Ras oncoprotein. Baseline characteristics were reflective of this population of subjects with advanced pancreas or colorectal cancer. The results demonstrated that GI-4000 was well-tolerated and immunogenic in this subject population. No DLTs, therapy-related SAEs or therapy-related clinically significant laboratory abnormalities were observed. Injection site reactions were only mild, even at the highest locally administered dose per injection of 10 YU, which was used for the 3 highest dose levels of 10, 20, and 40 YU (each dose of 20 YU was administered as 10 YU×2 injections, and each dose of 40 YU as 10 YU×4 injections). A treatment emergent immune signal was detected in over half of the subjects (defined by an SI ≥2 in the proliferation assay). One subject with stage IV pancreas cancer had prolonged SD and on-treatment Ras-specific immune responses. Five subjects survived past the formal 48-week follow-up of the study and all 5 exhibited Ras mutation-specific immune responses (measured by LPA) on treatment. As longitudinal analysis was not performed (ie, baseline samples were not assayed on the same day as on-treatment samples), apparent on-treatment increases in responses of baseline positive subjects cannot be definitively identified. For baseline negative subjects, the emergence of an immune response at later timepoints is more indicative of the ability of GI-4000 to elicit de novo immune responses. Although there were no obvious effects of dose on the immune response, the highest dose group (40 YU) had the lowest frequency of immune responders. This could be attributed to the poor quality of the samples, related presumably to health status of the subjects in this dose group. There were no survivors past the 48-week point in this group and approximately one third of samples tested from 40 YU group subjects did not proliferate in response to C. albicans by LPA (data not shown), indicating a diminished immunocompetence. In contrast, the majority of samples from all other groups responded to C. albicans in LPA.
In conclusion, this trial presented a favorable safety and immunologic profile of GI-4000 in a population of subjects with advanced pancreas and colorectal cancer. The highest dose tested (40 YU) in this trial was selected for use in subsequent phase 2 trials: GI-4000-02, which evaluated GI-4000 in subjects with newly diagnosed, resected pancreas cancer and GI-4000-03, a phase 2b study in NSCLC patients.27
CONFLICTS OF INTEREST/FINANCIAL DISCLOSURE
GlobeImmune Inc. sponsored and funded the study.
At the time of study conduct, C.C., J.F., and D.A. were employees of GlobeImmune Inc. and T.C.R was CEO of GlobeImmune Inc. D.B. is a founder of GlobeImmune Inc. All current and former GlobeImmune authors held or hold stock and/or stock options in the Company. All remaining authors have declared that there are no financial conflicts of interest with regard to this work.
Footnotes
Present address: Bert O’Neil, Indiana University Simon Cancer Center, Indianapolis, IN.
GlobeImmune personnel were involved in the design and conduct of the study and provided logistical support during the trial. Employees of the sponsor worked with the investigators and statistician to prepare the statistical analysis plan, but the analyses were performed by Lynn Shemanski, PhD, of C3 Research Associates, Seattle, WA. Statistical analyses of immune responses were performed by QST Consultations Ltd, Allendale, MI. The manuscript was prepared by GlobeImmune Inc. personnel and by Julie Gelderloos, PhD, of Gelderloos Medical Writing, LLC.
REFERENCES
- 1.Wood CG. Adjuvant approaches to renal cell carcinoma. Clin Adv Hematol Oncol. 2008;6:19–21. [PubMed] [Google Scholar]
- 2.Van Poppel H, Joniau S, Van Gool SW. Vaccine therapy in patients with renal cell carcinoma. Eur Urol. 2009;55:1333–1342. [DOI] [PubMed] [Google Scholar]
- 3.Cha E, Fong L. Therapeutic vaccines for prostate cancer. Curr Opin Mol Ther. 2010;12:77–85. [PubMed] [Google Scholar]
- 4.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. [DOI] [PubMed] [Google Scholar]
- 5.Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520–3526. [DOI] [PubMed] [Google Scholar]
- 6.Oudard S, Rixe O, Beuselinck B, et al. A phase II study of the cancer vaccine TG4010 alone and in combination with cytokines in patients with metastatic renal clear-cell carcinoma: clinical and immunological findings. Cancer Immunol Immunother. 2011;60:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quoix E, Ramlau R, Westeel V, et al. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. Lancet Oncol. 2011;12:1125–1133. [DOI] [PubMed] [Google Scholar]
- 8.Antonia SJ, Larkin J, Ascierto PA. Immuno-oncology combinations: a review of clinical experience and future prospects. Clin Cancer Res. 2014;20:6258–6268. [DOI] [PubMed] [Google Scholar]
- 9.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 11.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. [DOI] [PubMed] [Google Scholar]
- 12.Cox AD, Der CJ. Ras family signaling: therapeutic targeting. Cancer Biol Ther. 2002;1:599–606. [DOI] [PubMed] [Google Scholar]
- 13.Knauf JA, Ouyang B, Knudsen ES, et al. Oncogenic RAS induces accelerated transition through G2/M and promotes defects in the G2 DNA damage and mitotic spindle checkpoints. J Biol Chem. 2006;281:3800–3809. [DOI] [PubMed] [Google Scholar]
- 14.Stubbs AC, Martin KS, Coeshott C, et al. Whole recombinant yeast vaccine activates dendritic cells and elicits protective cell-mediated immunity. Nat Med. 2001;7:625–629. [DOI] [PubMed] [Google Scholar]
- 15.Bernstein MB, Chakraborty M, Wansley EK, et al. Recombinant Saccharomyces cerevisiae (yeast-CEA) as a potent activator of murine dendritic cells. Vaccine. 2008;26:509–521. [DOI] [PubMed] [Google Scholar]
- 16.Remondo C, Cereda V, Mostböck S, et al. Human dendritic cell maturation and activation by a heat-killed recombinant yeast (Saccharomyces cerevisiae) vector encoding carcinoembryonic antigen. Vaccine. 2009;27:987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu Y, Bellgrau D, Dwyer-Nield LD, et al. Mutation-selective tumor remission with Ras-targeted, whole yeast-based immunotherapy. Cancer Res. 2004;64:5084–5088. [DOI] [PubMed] [Google Scholar]
- 18.Haller AA, Lauer GM, King TH, et al. Whole recombinant yeast-based immunotherapy induces potent T cell responses targeting HCV NS3 and Core proteins. Vaccine. 2007;25:1452–1463. [DOI] [PubMed] [Google Scholar]
- 19.Wansley EK, Chakraborty M, Hance KW, et al. Vaccination with a recombinant Saccharomyces cerevisiae expressing a tumor antigen breaks immune tolerance and elicits therapeutic antitumor responses. Clin Cancer Res. 2008;14:4316–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bui MR, Hodson V, King T, et al. Mutation-specific control of BCR-ABL T315I positive leukemia with a recombinant yeast-based therapeutic vaccine in a murine model. Vaccine. 2010;28:6028–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanaka A, Jensen JD, Prado R, et al. Whole recombinant yeast vaccine induces antitumor immunity and improves survival in a genetically engineered mouse model of melanoma. Gene Ther. 2011;18:827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King TH, Kemmler CB, Guo Z, et al. A whole recombinant yeast-based therapeutic vaccine elicits HBV X, S and core specific T cells in mice and activates human T cells recognizing epitopes linked to viral clearance. PLoS One. 2014;9:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–216. [DOI] [PubMed] [Google Scholar]
- 24.Nair KR. The distribution of the extreme deviate from the sample mean and its studentized form. Biometrika. 1948;35:118–144. [PubMed] [Google Scholar]
- 25.Currier JR, Kuta EG, Turk E, et al. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J Immunol Methods. 2002;260:157–172. [DOI] [PubMed] [Google Scholar]
- 26.Brookmeyer R, Crowley J. A confidence interval for the median survival time. Biometrics. 1982;38:29–41. [Google Scholar]
- 27.Chaft JE, Litvak A, Arcila ME, et al. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin Lung Cancer. 2014;15:405–410. [DOI] [PubMed] [Google Scholar]